")
Back to Journals » Journal of Blood Medicine » Volume 10
Evaluating ibrutinib in the treatment of symptomatic Waldenstrom’s macroglobulinemia
Authors Papanota AM , Ntanasis-Stathopoulos I , Kastritis E , Dimopoulos MA, Gavriatopoulou M
Received 9 February 2019
Accepted for publication 6 August 2019
Published 27 August 2019 Volume 2019:10 Pages 291—300
DOI https://doi.org/10.2147/JBM.S183997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Aristea-Maria Papanota,* Ioannis Ntanasis-Stathopoulos,* Efstathios Kastritis, Meletios A Dimopoulos, Maria Gavriatopoulou
Department of Clinical Therapeutics, National and Kapodistrian University of Athens, School of Medicine, Alexandra General Hospital, Athens, Greece
Correspondence: Maria Gavriatopoulou
Department of Clinical Therapeutics, National and Kapodistrian University of Athens, School of Medicine, Alexandra General Hospital, 80 Vas. Sofias Avenue, Athens 11528, Greece
Tel +30 213 216 2547
Fax +30 213 216 2511
Email [email protected]
*These authors contributed equally to this work
Abstract: Waldenstrom’s macroglobulinemia (WM) is a rare lymphoplasmacytic lymphoma with indolent course and prolonged disease course. The first-in-class Bruton’s tyrosine kinase inhibitor, ibrutinib, has shown significant activity and a distinct adverse event profile among both newly diagnosed and relapsed/refractory WM patients. Interestingly, clinical responses to ibrutinib have been shown to be dependent on patients’ MYD88 and CXCR4 mutational status. The recent outcomes of the Phase III iNNOVATE trial showed that the combination of ibrutinib with rituximab resulted in a significantly prolonged progression-free survival compared with rituximab monotherapy, which provides a novel therapeutic option in the clinical practice especially for the rituximab-refractory WM patients. However, the need for continuous drug administration along with the unique toxicity manifestations may render the patient management challenging. Furthermore, our understanding of the underlying resistant mechanisms to ibrutinib is currently being evolved.
Keywords: Waldenstrom’s macroglobulinemia, IgM, ibrutinib, Bruton’s tyrosine kinase
Introduction
Waldenström’s macroglobulinemia (WM) constitutes a rare lymphoplasmacytic lymphoma, which is primarily characterized by bone marrow invasion by a population of monoclonal, small lymphocytes showing evidence of plasmacytoid differentiation along with the presence of IgM monoclonal gammopathy. The infiltration pattern is predominantly intertrabecular.1
WM diagnosis is not always indicative of the need for immediate therapy. Criteria for therapy initiation include the presence of symptoms related to bone marrow infiltration, such as anemia and constitutional symptoms, which are by far the most common reasons to initiate treatment. Furthermore, several patients present with symptoms related to monoclonal IgM, such as hyperviscosity and peripheral neuropathy. Cold agglutinin disease, cryoglobulinemia, bulky disease and organomegaly are less common indications for therapy initiation in WM.1,2
Currently, there is no standard of care for WM, but the anti-CD20 monoclonal antibody rituximab alone or in combination is commonly used both in the US and Europe.2,3 Rituximab monotherapy is mostly considered in frail patients or those with immunologic complications related to the monoclonal paraprotein, such as neuropathy. Monotherapy should be avoided in patients presenting with high baseline IgM levels, due to the possibility of symptomatic hyperviscosity following the rituximab-related IgM flare. Rituximab-based chemoimmunotherapy regimens including rituximab-cyclophosphamide-dexamethasone, rituximab-bendamustine or rituximab-bortezomib (a proteasome inhibitor)-dexamethasone have a tolerable toxicity profile and provide durable responses; thus, they constitute a good therapeutic option for most patients.2 However, during the course of the disease, the malignant cell population will inevitably become refractory to rituximab, leading to an imperative need for new therapeutic choices.4 Furthermore, the need for novel treatment approaches is indicated by the fact that a greater proportion of WM patients has been reported to present with rituximab intolerance compared with other B-cell malignancies.5 Ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor, is a novel therapeutic option approved for the treatment of WM by the Food and Drug Administration (FDA) on January 2015.6 The aim of this review is to provide an overview of the current role of ibrutinib in WM treatment algorithm and critically discuss challenging issues with profound implications in the clinical practice.
Ibrutinib mechanism of action and indications
Ibrutinib is a small molecule that acts through BTK inhibition. More specifically, the target of ibrutinib and its active metabolite PCI-45227 is a cysteine residue located on site 481 within the ATP binding domain of BTK. Both molecules bind covalently and irreversibly to this residue providing potent and sustained inhibition of the BTK enzymatic activity.7
B-cell receptor (BCR) signaling, antigen-dependent or independent, plays a crucial role in B-cell malignancies.8,9 BTK is a member of the non-receptor tyrosine kinases that are found early in the molecular cascade following BCR activation.10 The B-cell linker protein (BLNK) binds BTK and phospholipase C gamma 2 (PLCγ-2) resulting in hydrolysis of membrane PIP2 and production of IP3 leading to calcium release by the endoplasmic reticulum.11 The increased intracellular Ca2+ concentration leads to the activation of PKCβ which in turn activates the transcriptional factors NF-κΒ and NFAT that promote B-cell survival and differentiation.12 Ibrutinib blocks the aforementioned signaling pathways by inhibiting BTK.7
Ibrutinib is currently approved by the FDA for the treatment of mantle cell lymphoma (MCL) after at least one prior line of therapy, chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia with or without 17p deletion, marginal zone lymphomaafter at least one prior anti-CD20 therapy, chronic graft versus host disease after failure of one or more lines of systemic therapy and for symptomatic WM regardless of line of therapy.13
Molecular background and predictive factors in WM
In 2012, Treon et al performed whole genome sequencing of CD19+ bone marrow cells from WM patients in an attempt to clarify the mutational background of the disease. The most common somatic mutation identified in 91% of the patients with lymphoplasmacytic lymphoma was MYD88 L265P.14 MYD88 is part of the signaling cascade following toll-like receptor and interleukin-1 receptor activation.15 After stimulation of the abovementioned receptors, MYD88 binds as a homodimer to the activated receptor complex. The homodimer recruits interleukin-1 receptors-associated kinase (IRAK4) and triggers its autophosphorylation. Phosphorylated IRAK4 activates IRAK1 and IRAK2.16 IRAK1 activates tumor necrosis factor receptor-associated factor 6, which leads to IκBα and NF-κΒ activation. The importance of these findings lies to the fact that the growth and survival of the malignant clone in WM is strongly dependent on NF-κB signaling.17
In WM, BTK forms a complex with the mutated MYD88 and, thus, it is constantly activated.18 In contrast, this has not been observed in wild-type MYD88 WM cells.19 A recent study showed that the NF-κB pathway mutations mainly observed in MYD88 wild-type patients are downstream to BTK. This provides a possible explanation for the resistance of those patients to ibrutinib.20 All the above provide a strong rationale for BTK inhibition as therapeutic target in patients with MYD88 mutated WM.
Furthermore, an increased expression of hematopoietic cell kinase (HCK) has been identified in MYD88 L265P mutated WM cells. HCK is a member of the SRC family of protein tyrosine kinases and triggers pro-survival signaling including BTK. Interestingly, HCK has been also found to be a highly relevant target molecule of ibrutinib.21
The second most common class of somatic mutations in WM are those in the CXCR4 gene, which are observed in 30% of WM patients.22 These somatic mutations are identified in the C terminus of CXCR4 in WM cells and are similar to those found in the germline of patients with WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome, a congenital immunodeficiency disorder.23,24 Roccaro et al have shown that 28.2% of WM patients present with the C1013G/CXCR4 mutation, which acts as an activating mutation and an inducer of drug resistance to mTOR, PI3K, and BTK inhibitors. Interestingly, the use of an anti-CXCR4 monoclonal antibody led to tumor reduction independently of the mutational status.25 Furthermore, Xu et al developed highly sensitive allele-specific polymerase chain reaction assays in order to detect the most common CXCR4WHIM mutations in WM; their approach revealed more mutations compared with Sanger sequencing. They also demonstrated that CXCR4 mutations are more common in WM than previously thought. Interestingly, CXCR4 mutations are mainly subclonal following MYD88 mutation. Different CXCR4 mutations may be found in the same patient suggesting an underlying genomic instability.26 Two categories of CXCR4 mutations affecting the C terminus have been mainly described in WM patients; the nonsense CXCR4WHIM/NS mutations that shorten the distal region of CXCR4 by 15–20 amino acids and the frameshift mutations CXCR4WHIM/FS that include a region of approximately 40 amino-acids in the C-terminal site. Both mutations are encountered in approximately the same relative frequency among WM patients with somatic mutations of the CXCR4.27 The mutations in the C-terminal domain of the CXCR4 receptor lead to a permanently activated state by blocking the internalization of the receptor that normally occurs after SDF-1α stimulation.28 CXCR4 activation promotes AKT kinase and extracellular-regulated kinase (ERK) function, which may be associated with resistance to ibrutinib therapy.29,30
The molecular basis of acquired resistance to ibrutinib
Disease progression can occur while on active ibrutinib therapy; however, the underlying mechanisms are rather unclear. Ibrutinib resistance is associated with poor prognosis.31 Therefore, understanding the biological background and determining novel therapeutic molecules that may result in remission among ibrutinib resistant patients represent an active area of investigation.
Ibrutinib binds to the Cysteine 481 residue of BTK. BTKCys481 mutations were identified in MYD88 mutated WM cells derived from patients progressing while on ibrutinib therapy. It is interesting that these mutations are mainly subclonal and demonstrate a variable clonal distribution. When compared with baseline tumor cells from the same patients, those mutations were absent indicating that they evolved de novo during the course of ibrutinib therapy.32 Another study showed that ibrutinib resistance as a result of BTKCys481 mutation is mediated through sustained ERK1/2 activation. ERK1/2 activation may also provide protection from BTK inhibition through a paracrine mechanism to BTK wild-type WM cells.20 Those findings suggest ERK1/2 inhibitors as possible therapeutic molecules to overcome ibrutinib resistance.
The development of ibrutinib-resistant WM cell lines that lack BTKCys481 mutation or CXC4WHIM-like mutations has been also reported, which means that those patients exhibit BTK-signaling independent survival. AKT and Bcl-2 associated pathways may play a role in inducing survival of ibrutinib-resistant WM cells.33
Overview of clinical data
Ibrutinib was initially tested for its safety in a Phase I study including patients with relapsed or refractory B-cell malignancies. Four of 56 patients had been diagnosed with WM (Table 1). The enrolled patients received escalating doses of oral ibrutinib. The maximum tolerated dose was not reached and full occupancy of the BTK active site occurred at 2.5 mg/kg. Dose escalation continued until 3 dose levels above full BTK occupancy (12.5 mg/kg). The drug was well tolerated without dose-limiting events. The most common adverse eventswere grade 1 or 2. Objective response rate was 60%. Three of four WM patients responded to ibrutinib.34
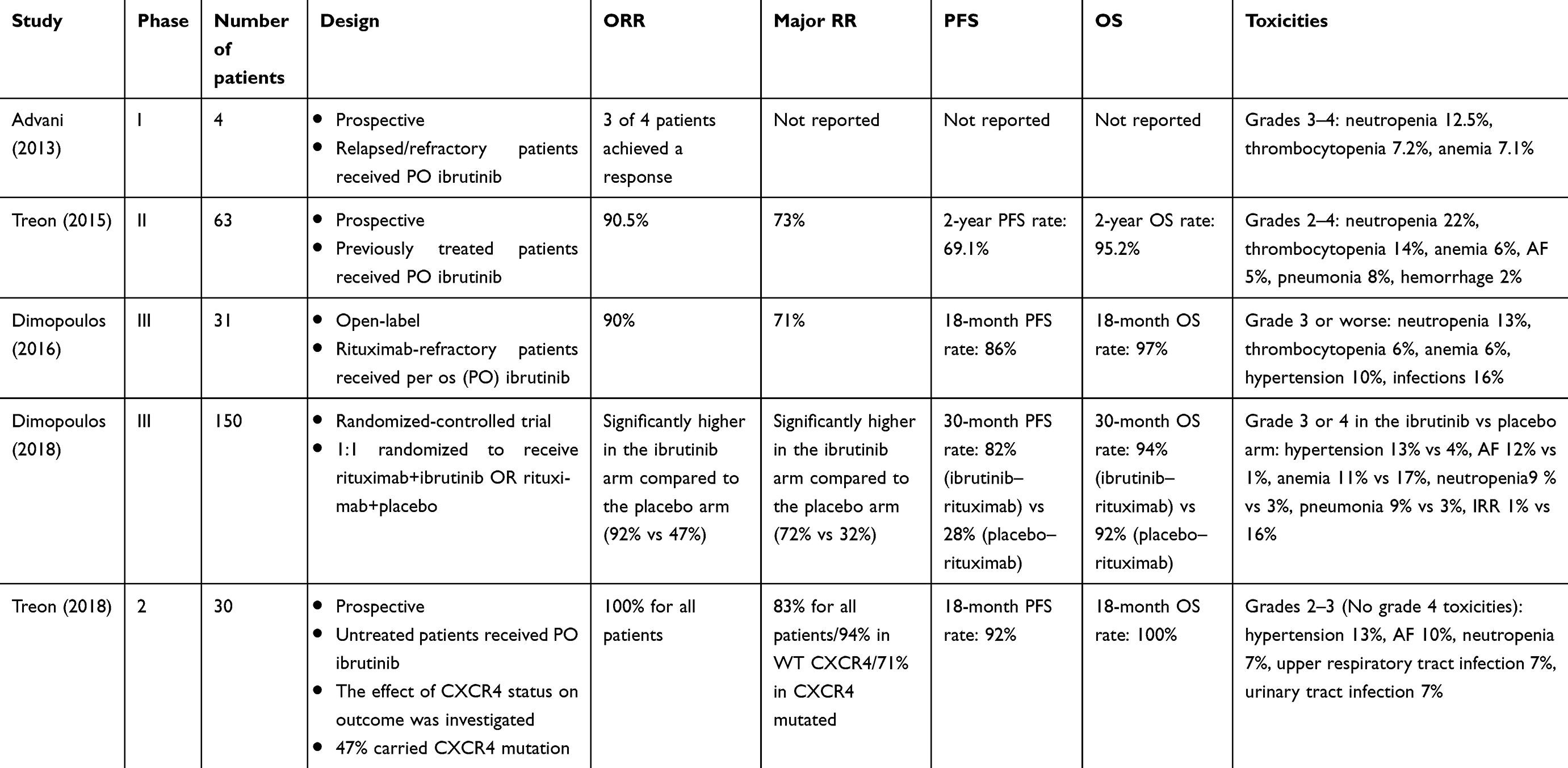 |
Table 1 Overview of clinical studies evaluating ibrutinib-based regimens among patients with WM |
Efficacy of ibrutinib in symptomatic WM patients that have received at least one prior therapy was tested in a landmark Phase II study conducted by Treon et al (Table 1).35 Sixty-three patients were enrolled in the study and received ibrutinib orally at a daily dose of 420 mg until disease progression or unacceptable toxicity. The primary objective of the study was to determine the overall response rate (ORR), which includes minor response, partial response, very good partial response and complete response and the rate of major response (partial response or greater). Progression-free survival (PFS) and safety were the secondary endpoints. Results showed an ORR of 90.5% (95% CI 80.4–96.4) and a major response rate of 70.3% (95% CI 60.3–83.4). The median time to at least minor response was 4 weeks. At the time of best response, the median serum IgM decreased from 3520to 880 mg/dL, the median hemoglobin increased from 10.5 to 13.8 g/dL, and the median bone marrow infiltration decreased from 60% to 25%. Response rates were estimated among the three genomic groups of WM: MYD88L265PCXCR4WT, MYD88L265PCXCR4WHIM, and MYD88WTCXCR4WT. Both overall and major response rates were highest in MYD88L265PCXCR4WT, 100% and 91.2%, respectively. The lowest response rates (71.4% and 28.6%) were observed among patients with the MYD88WTCXCR4WT genotype. MYD88L265PCXCR4WHIM patients achieved an ORR of 85.7% and a major response rate of 61.9%. The estimated 2-year overall survival rate was 95.2% and the estimated 2-year PFS rate was 69.1%. Toxic effects related to the investigational product were moderate. Those findings supported the approval of ibrutinib by the FDA and European Medicines Agency (EMA) as the first ever approved drug for WM.
An update of the abovementioned study was presented at the 59th American Society of Hematology annual meeting. According to this update, response rates improved with prolonged treatment. ORR was 90.4% and major response rate was 77.7%. At best response, the median IgM levels reduced to 821 mg/dL, the median bone marrow infiltration declined to 20% ,and the median hemoglobin was 14.2 g/dL. With a median follow-up of 47.1 months, median PFS for all patients was not reached. Median PFS has also not been reached for MYD88MUTCXCR4WT patients. For MYD88MUTCXCR4MUT patients, the median PFS was 45 and 21 months for MYD88WTCXCR4WT patients.36
Ibrutinib has also been tested among rituximab-refractory WM patients. Currently, the reported outcomes are based on a substudy of the randomized Phase III trial iNNOVATE (Table 1). The substudy was non-randomized and included a single group of rituximab-refractory patients that were treated with ibrutinib monotherapy (n=31).36 The study defined rituximab-refractory disease as the failure to achieve at least a minor response after the last rituximab-containing therapy or as disease progression <12 months after the last rituximab dose of the last rituximab-containing regimen. The primary and secondary objectives of the study were the ORR, PFS, overall survival, time to next therapy, hemoglobin level improvement, and patient recorded outcomes based on two validated questionnaires. Ninety percent of the patients achieved an overall response and 71% had a major response. It has to be highlighted that 61% of them were refractory or non-responsive to their last treatment. The median time to response was 1 month. At a median follow-up of 18.1 months, the 18-month PFS was 86% and overall survival 97%. Median hemoglobin levels rised to 12.7 g/dL at week 49 from 10.3 g/dL at baseline. MYD88L265PCXCR4WT patients had a similar response rate to MYD88L265PCXCR4WHIM patients; however, the small patient number, the missing data and the fact that the applied technique was not the standard one, should be considered as important limitations during the interpretation of these results.
The primary results of the placebo-controlled, double-blind randomized trial iNNOVATE were published recently.37 One hundred and fifty newly diagnosed and relapsed-refractory patients were randomized with a 1:1 ratio to receive rituxima–ibrutinib or rituximab–placebo. Forty-five percent of the patients were previously untreated. The primary endpoint was PFS, while secondary endpoints included overall survival, response rates, hematologic improvement, and safety. The 30-month PFS rate was 82% for the ibrutinib–rituximab arm and median PFS was not reached, while PFS rate for the rituximab–placebo arm was 28% and median PFS was 20.3 months. As far as 30-month overall survival rate is concerned, the value was 94% for the ibrutinib arm and 92% for the placebo arm. ORR was significantly higher for the rituximab–ibrutinib arm compared to rituximab–placebo arm (92% vs 47%) and the same applies for major response rate (72% vs 32%). It is also important that the very good partial response (VGPR) rate was lower in CXCR4 mutated patients. Median IgM levels decreased more rapidly in the rituximab–ibrutinib arm suggesting the addition of ibrutinib to rituximab as a way to prevent rituximab-related IgM flare. Regarding the safety profile of the combination, adverse events were predictable, according to the established toxicity profile of each drug separately. Interestingly, PFS rates among different genotypes were similar.
In the 60th annual meeting of the American Society of Hematology, an abstract comparing ibrutinib–rituximab with real-world treatments for WM was also presented. In the absence of randomized controlled trials comparing ibrutinib with other treatment regimens currently used for WM, an adjusted comparison was conducted between patient data from the iNNOVATE trial and real-world data from the Lyon-Sud database in France. Ibrutinib–rituximab significantly improved both progression-free and overall survival compared to real-world physician’s choice regimens both as upfront treatment or in the relapsed/refractory setting.38
Recently, results from a prospective study of ibrutinib monotherapy in treatment-naïve patients have been published.39 A total of 30 patients received ibrutinib until disease progression or unacceptable toxicity. All were MYD88 mutated and 47% were CXCR4 mutated. ORR for all patients was 100% and major response rate was 83%. CXCR4 wild-type patients demonstrated higher major response rate compared to CXCR4 mutated patients (94% vs 71%, respectively). The 18-month PFS rate was 92%. No grade 4 toxicities were described.
Updated follow-up data from the iNNOVATE study were presented in the 60th ASH annual meeting. Following a prolonged follow-up period of 36 months, the ORR (≥MR) were 95% in the rituximab–ibrutinib arm versus 48% in the rituximab–placebo arm, while the major response rates (≥PR) were 77% versus 33%, respectively. Major responses with rituximab–ibrutinib were shown regardless of the MYD88/CXCR4 genotype; however, the MYD88L265P/CXCR4WT patients had the shortest time to major response. Regarding survival data, the median PFS was not reached in the rituximab–ibrutinib arm versus 20.3 months in the rituximab arm, whereas the estimated 30-month PFS rates were 79% versus 41%, respectively. In the rituximab–ibrutinib group, no differences in PFS were observed among different genotypes. The 30-month OS estimate was 93% in the rituximab–ibrutinib arm vs 90% in the rituximab–placebo arm. Regarding the non-randomized arm C including rituximab-refractory patients that received ibrutinib monotherapy, the 36-month PFS and OS rates were 61% and 84%, respectively. ORR in this arm was 90% and the major response rate was 77%.40
Current role of ibrutinib in WM
Ibrutinib is approved by the EMA for the treatment of WM patients who are not candidates for chemoimmunotherapy and by the FDA for all adult patients with WM. The eighth International Workshop on WM panel suggested that MYD88 mutational status should be tested before initiation of treatment with ibrutinib. MYD88 and CXCR4 status impact should be further tested in clinical trials and currently is not universally applied for tailoring treatment options.2
Dosing
Ibrutinib is administered orally at a daily dose of 420 mg. Continuous daily dosing is indicated. The drug is administered until disease progression or unacceptable toxicity. Ibrutinib is metabolized in the liver by CYP3A. Dose modifications should be considered when ibrutinib is administered on patients with hepatic impairment or receiving concomitant drugs that act as strong CYP3A inhibitors or inducers.41
Adverse events
Ibrutinib is generally well tolerated. As a first-generation BTK inhibitor, it is non-selective and its off-target effects are responsible for most of the adverse events. The commonest side effects include gastrointestinal disorders, fatigue, rash, dyspnea, and upper respiratory tract infections.42
One of the most important side effects of ibrutinib is the increased risk of bleeding. According to a systematic review and pooled analysis of four randomized controlled trials, ibrutinib therapy was associated with a statistically significant higher incidence of all grade bleeding (RR: 2.93, 95% CI: 1.14–7.52). However, no statistically significant difference in the risk of major bleeding was observed.43 Ibrutinib is also associated with unstable thrombus formation in vitro. Ibrutinib does not ablate adhesion to collagen, but inhibits integrin αIIbβ3 inside-out signaling.44 According to the treatment guidelines from the Eighth International Workshop on WM, testing for von Willebrand activity should be considered in patients with a history of bleeding diathesis before initiation of ibrutinib therapy. Ibrutinib should be held for 3–7 days before and after surgery, depending on the type of the intervention.2
Atrial fibrillation (AF) emerged as a side effect of ibrutinib therapy in the clinical trials setting.45 A systematic review and pooled analysis of four randomised controlled trial (RCT)s in CLL demonstrated a significantly higher risk of AF among patients receiving ibrutinib.43 The underlying mechanism remains to be elucidated. The inhibition of PI3K-Akt cardioprotective pathway by ibrutinib is a possible hypothesis for the explanation of the underlying mechanism of AF in ibrutinib receiving patients.46 However, more data are needed in order to further elucidate the underlying pathogenesis. Thromboprophylaxis with novel anticoagulants is recommended.45 Discontinuation of ibrutinib due to AF is avoided with prompt cardiologic consultation and dose modifications in almost all patients.47
Dose modifications
Ibrutinib treatment should be interrupted in the case of grade 4 hematologic toxicity, grade 3 or greater febrile neutropenia and grade 3 or greater non-hematologic toxicities. Resolution of toxicity or improvement to at least grade 1 is needed in order to resume ibrutinib in the daily dose of 420 mg. In the case of toxicity recurrence, ibrutinib should be withheld and reinitiated at a reduced dose level. This applies for up to two recurrences. Further recurrences indicate the need for treatment discontinuation.13
Special considerations
Taking into consideration all the above, ibrutinib therapy may be interrupted to manage toxicities or to prevent bleeding preoperatively. Twenty percent of WM patients develop withdrawal symptoms upon treatment interruption. Symptoms usually arise within 2 days and immediately resolve after reinitiation of ibrutinib.48 The underlying mechanism is only partially understood. Ibrutinib induces suppression of inflammatory cytokines and exhibits a downregulatory effect on macrophages and T-cells.49–51 It is possible that re-activation of malignant cells and tumor microenvironment following ibrutinib hold promotes a state similar to cytokine release syndrome that could be responsible for the withdrawal symptoms observed.48
Furthermore, an IgM rebound is observed in most patients after ibrutinib discontinuation. In a study by Gustine et al, 73% of the patients discontinuing ibrutinib had an IgM rebound.52 The incidence of IgM rebound was 48% at 4 weeks and 79% at 8 weeks. Sixteen percent of those patients developed symptomatic hyperviscosity requiring plasmapheresis. These findings indicate that close monitoring of IgM levels should follow ibrutinib discontinuation, whereas the discrimination between IgM flare and disease progression may be challenging.
The relationship between ibrutinib dose intensity (DI) and patient outcomes was examined recently in a retrospective study.53 Overall DI is the proportion of administered versus planned ibrutinib doses from the beginning of ibrutinib therapy until the last dose administered. Mean overall DI lower than 97% was related to shorter PFS and threefold higher risk of progression compared with mean overall DI >97%. Moreover, a 4 times higher risk of progression was associated with interruption of ibrutinib for more than 7 days at any time during the treatment course. Transient increases in serum IgM and even disease progression can be observed during temporary holds of ibrutinib. However, response is usually restored when ibrutinib therapy is reinitiated.
As it has been aforementioned, treatment with ibrutinib continues until disease progression. Furthermore, the high cost of ibrutinib brings into the foreground the issue of cost-effectiveness that should be taken into consideration by the public health policies.
Furthermore, there are still unanswered questions regarding the role of ibrutinib in the therapeutic algorithm of WM. Although the iNNOVATE trial showed the superiority of ibrutinib–rituximab compared with rituximab, there are no data available regarding the comparison of ibrutinib–rituximab with ibrutinib. Further studies evaluating the efficacy of ibrutinib–rituximab compared to other established regimens for WM both in the upfront and in the relapsed/refractory setting should be also conducted. Last but not least, the potential strategy of interrupting ibrutinib administration and assuring persistence of disease response has to be determined in future studies.
Ongoing clinical trials
Novel combinations of ibrutinib with rituximab and bendamustine (NCT01479842), pevonedistat (NCT03479268), lenalidomide (NCT01955499), pembrolizumab (NCT02332980) (NCT02950220), daratumumab (NCT03679624), bortezomib and rituximab (NCT03620903), and ulocuplumab (NCT03225716) are under clinical development. Ongoing clinical trials of ibrutinib in WM are presented in Table 2 (clinicaltrials.gov).
 |
Table 2 Major ongoing trials of ibrutinib-based regimens in WM |
Novel BTK inhibitors
Acquired resistance to ibrutinib and adverse events related to ibrutinib led to the development of more specific novel BTK inhibitors. Zanubrutinib (BGB-3111) is an oral next-generation BTK inhibitor with greater selectivity for BTK than ibrutinib. Zanubrutinib is a highly potent BTK inhibitor that binds to Cys 481 residue of BTK like ibrutinib. Zanubrutinib exhibits minimal inhibition of TEC and EGFR family off-target kinases, which is related to lower toxicity related to ibrutinib.54 A Phase I trial of zanubrutinib in WM included 42 patients; 9 of them were previously untreated and 33 had relapsed or refractory disease. Zanubrutinib exhibited satisfactory tolerability and activity in WM population. The best objective response rate for the whole trial population was 90% and the major response rate was 76%. Forty-three percent of the patients achieved a VGPR, which is higher than the reported VGPR rate of ibrutinib (27%). The 1-year PFS rate was 91.7%.55 A Phase III trial of zanubrutinib versus ibrutinib is currently ongoing in order to assess the safety and efficacy of zanubrutnib compared to ibrutinib (NCT03053440).54
Acalabrutinib (ACP-196) is another novel BTK inhibitor. Acalabrutinib binds irreversibly to BTK and shows no activity against interleukin-2-inducible kinase (ITK) and EGFR.56 Acalabrutinib is in an advanced stage of clinical development for CLL and MCL, whereas its safety and efficacy in WM is currently under investigation in a Phase II trial with promising primary results including an ORR rate of 93% (NCT02180724).57–59
Vecabrutinib (SNS-062) is a noncovalent second-generation BTK inhibitor. The special characteristic of vecabrutinib lies in its binding ability to both to wild type and Cys481 mutated BTK.60 Vecabrutinib is currently being tested in patients with B-cell malignancies including both wild type and mutated BTK in a Phase Ib study (NCT03037645).
Conclusion
Given the pivotal role of BTK activation in B-cell malignancies, ibrutinib is being established in the treatment of WM. Importantly, it is currently the only drug approved by FDA and EMA for the treatment of symptomatic WM, which is considered an orphan disease. The importance of ibrutinib in WM is highlighted by the fact that regulatory authorities provided approval for its use in the frontline setting based on the results from a clinical trial that had included only relapsed/refractory patients. Clinical data support its efficacy either as monotherapy or in combination with rituximab. Ibrutinib is a safe and efficacious alternative for patients who are not candidates for chemoimmunotherapy, due to frailty, comorbidities, unacceptable, toxicity or intolerance. Importantly, an individualized approach should be followed when choosing ibrutinib as a therapeutic option, given its distinct adverse event profile. MYD88 and CXCR4 mutations may have a predictive role for the response to ibrutinib. Further clinical investigation in the field is needed in order to determine the additive value of adding rituximab to ibrutinib compared with ibrutinib monotherapy. Additionally, there is a need for optimizing the therapeutic combinations in order to provide deep (high CR rates) and durable responses. Then, we could formulate a strategy for interrupting ibrutinib and reduce treatment costs without compromising disease response. The development of new molecules that may overcome resistance mechanisms to ibrutinib remains a challenge, as well. Currently, novel BTK inhibitors are under clinical development in an attempt to overcome resistance to ibrutinib and to achieve less off-target inhibition and an even more favorable toxicity profile.
Disclosure
Efstathios Kastritis reports honoraria from Genesis Pharma, Takeda, Janssen, Amgen. Meletios Athanasios Dimopoulos reports consultancy and honoraria from Novartis, Janssen, Celgene, Takeda, Amgen, BMS. Maria Gavriatopoulou reports honoraria from Amgen, Janssen, Celgene, and Takeda. The authors report no other conflicts of interest in this work.
References
1. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):110–115. doi:10.1053/sonc.2003.50082
2. Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenstrom’s Macroglobulinemia. Blood. 2016;128(10):1321–1328. doi:10.1182/blood-2016-04-711234
3. Buske C, Seymour JF. Immunochemotherapy in Waldenstrom macroglobulinemia - still the backbone of treatment. Leuk Lymphoma. 2015;56(9):2489–2490. doi:10.3109/10428194.2015.1058938
4. Gavriatopoulou M, Ntanasis-Stathopoulos I, Kastritis E, Dimopoulos MA. How I treat rituximab refractory patients with WM. Oncotarget. 2018;9(96):36824–36825. doi:10.18632/oncotarget.26411
5. Castillo JJ, Kanan S, Meid K, Manning R, Hunter ZR, Treon SP. Rituximab intolerance in patients with Waldenstrom macroglobulinaemia. Br J Haematol. 2016;174(4):645–648. doi:10.1111/bjh.13794
6. FDA expands approved use of Imbruvica for rare form of non-Hodgkin lymphoma: first drug approved to treat Waldenström’s macroglobulinemia. [press release]. 2015.
7. Charalambous A, Schwarzbich MA, Witzens-Harig M. Ibrutinib. Recent results in cancer research. 2018;212:133–168. doi:10.1007/978-3-319-91439-8_7
8. Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi:10.1038/nature08638
9. Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–4320. doi:10.1182/blood-2011-06-338855
10. Sideras P, Muller S, Shiels H, et al. Genomic organization of mouse and human Bruton’s agammaglobulinemia tyrosine kinase (Btk) loci. J Immunol. 1994;153(12):5607–5617.
11. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193–205. doi:10.1111/ejh.12427
12. Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–127.
13. Imbruvica (ibrutinib) Capsules [prescribing Information]. Sunnyvale, CA: Pharmacyclics, Inc; March 2016.
14. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi:10.1056/NEJMoa1200710
15. Watters TM, Kenny EF, O’Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol. 2007;85(6):411–419. doi:10.1038/sj.icb.7100095
16. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–890. doi:10.1038/nature09121
17. Leleu X, Eeckhoute J, Jia X, et al. Targeting NF-kappaB in Waldenstrom macroglobulinemia. Blood. 2008;111(10):5068–5077. doi:10.1182/blood-2007-09-115170
18. Shinners NP, Carlesso G, Castro I, et al. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179(6):3872–3880. doi:10.4049/jimmunol.179.6.3872
19. Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122(7):1222–1232. doi:10.1182/blood-2012-12-475111
20. Chen JG, Liu X, Munshi M, et al. BTK(Cys481Ser) drives ibrutinib resistance via ERK1/2 and protects BTK(wild-type) MYD88-mutated cells by a paracrine mechanism. Blood. 2018;131(18):2047–2059. doi:10.1182/blood-2017-10-811752
21. Yang G, Buhrlage SJ, Tan L, et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood. 2016;127(25):3237–3252. doi:10.1182/blood-2016-01-695098
22. Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi:10.1182/blood-2013-09-525808
23. Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem. 2010;285(10):7805–7817. doi:10.1074/jbc.M109.091173
24. Dotta L, Tassone L, Badolato R. Clinical and genetic features of Warts, Hypogammaglobulinemia, Infections and Myelokathexis (WHIM) syndrome. Curr Mol Med. 2011;11(4):317–325.
25. Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120–4131. doi:10.1182/blood-2014-03-564583
26. Xu L, Hunter ZR, Tsakmaklis N, et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenstrom Macroglobulinaemia. Br J Haematol. 2016;172(5):735–744. doi:10.1111/bjh.13897
27. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791–2796. doi:10.1182/blood-2014-01-550905
28. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768(4):952–963. doi:10.1016/j.bbamem.2006.11.002
29. Cao Y, Hunter ZR, Liu X, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P) -directed survival signalling in Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;168(5):701–707. doi:10.1111/bjh.13200
30. Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia. 2015;29(1):169–176. doi:10.1038/leu.2014.187
31. Martin P, Maddocks K, Leonard JP, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood. 2016;127(12):1559–1563. doi:10.1182/blood-2015-10-673145
32. Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood. 2017;129(18):2519–2525. doi:10.1182/blood-2017-01-761726
33. Paulus A, Akhtar S, Yousaf H, et al. Waldenstrom macroglobulinemia cells devoid of BTK(C481S) or CXCR4(WHIM-like) mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer J. 2017;7(5):e565. doi:10.1038/bcj.2017.40
34. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. doi:10.1200/JCO.2012.42.7906
35. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. doi:10.1056/NEJMoa1501548
36. Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab-refractory Waldenstrom’s macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18(2):241–250. doi:10.1016/S1470-2045(16)30632-5
37. Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenstrom’s macroglobulinemia. N Engl J Med. 2018;378(25):2399–2410. doi:10.1056/NEJMoa1802917
38. Karlin L, Besson H, Tapprich C, Garside J, Salles G. Efficacy of Ibrutinib-Rituximab versus Real-World (RW) treatments for patients with Waldenström’s Macroglobulinemia (WM): adjusted comparison of iNNOVATE and the Lyon-Sud RW database. Blood. 2018;132(Suppl 1):1604. doi:10.1182/blood-2018-04-848028
39. Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naive patients with Waldenstrom macroglobulinemia. J Clin Oncol. 2018;36(27):2755–2761. doi:10.1200/JCO.2018.78.6426
40. Buske C, Tedeschi A, Trotman J, et al. Ibrutinib treatment in Waldenström’s Macroglobulinemia: follow-up efficacy and safety from the iNNOVATETM study. Blood. 2018;132(Suppl 1):149.
41. de Zwart L, Snoeys J, De Jong J, Sukbuntherng J, Mannaert E, Monshouwer M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Ther. 2016;100(5):548–557. doi:10.1002/cpt.419
42. de Weerdt I, Koopmans SM, Kater AP, van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica. 2017;102(10):1629–1639. doi:10.3324/haematol.2017.164103
43. Yun S, Vincelette ND, Acharya U, Abraham I. Risk of atrial fibrillation and bleeding diathesis associated with ibrutinib treatment: a systematic review and pooled analysis of four randomized controlled trials. Clin Lymphoma Myeloma Leuk. 2017;17(1):31–37 e13. doi:10.1016/j.clml.2016.09.010
44. Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib inhibits platelet integrin alphaiibbeta3 outside-in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326–2335. doi:10.1161/ATVBAHA.115.306130
45. Thorp BC, Badoux X. Atrial fibrillation as a complication of ibrutinib therapy: clinical features and challenges of management. Leuk Lymphoma. 2018;59(2):311–320. doi:10.1080/10428194.2017.1339874
46. McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829–3830. doi:10.1182/blood-2014-10-604272
47. Gustine JN, Meid K, Dubeau TE, Treon SP, Castillo JJ. Atrial fibrillation associated with ibrutinib in Waldenstrom macroglobulinemia. Am J Hematol. 2016;91(6):E312–E313. doi:10.1002/ajh.24366
48. Castillo JJ, Gustine JN, Meid K, Dubeau T, Severns P, Treon SP. Ibrutinib withdrawal symptoms in patients with Waldenstrom macroglobulinemia. Haematologica. 2018;103(7):e307–e310. doi:10.3324/haematol.2017.186908
49. Niemann CU, Herman SE, Maric I, et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib–findings from an investigator-initiated Phase II study. Clin Cancer Res. 2016;22(7):1572–1582. doi:10.1158/1078-0432.CCR-15-1965
50. Ping L, Ding N, Shi Y, et al. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget. 2017;8(24):39218–39229. doi:10.18632/oncotarget.16836
51. Ren L, Campbell A, Fang H, et al. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcgamma Receptor (FcgammaR) function. J Biol Chem. 2016;291(6):3043–3052. doi:10.1074/jbc.M115.687251
52. Gustine JN, Meid K, Dubeau T, et al. Ibrutinib discontinuation in Waldenstrom macroglobulinemia: etiologies, outcomes, and IgM rebound. Am J Hematol. 2018;93(4):511–517. doi:10.1002/ajh.25023
53. Castillo JJ, Gustine JN, Meid K, et al. Impact of ibrutinib dose intensity on patient outcomes in previously treated Waldenstrom macroglobulinemia. Haematologica. 2018;103(10):e466–e468. doi:10.3324/haematol.2018.191999
54. Tam CS, LeBlond V, Novotny W, et al. A head-to-head Phase III study comparing zanubrutinib versus ibrutinib in patients with Waldenstrom macroglobulinemia. Future Oncol. 2018;14(22):2229–2237. doi:10.2217/fon-2018-0163
55. Trotman J, Opat S, Marlton P, et al. Bruton’s Tyrosine Kinase (BTK) inhibitor BGB-3111 demonstrates high very good partial response (VGPR) rate in patients with Waldenström macroglobulinemia (WM). Hematol Oncol. 2017;35(S2):70–71. doi:10.1002/hon.2404
56. Harrington BK, Gulrajani M, Covey T, et al. ACP-196 is a second generation inhibitor of bruton tyrosine Kinase (BTK) with enhanced target specificity. Blood. 2015;126(23):2908.
57. Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–332. doi:10.1056/NEJMoa1509981
58. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–667. doi:10.1016/S0140-6736(17)33108-2
59. Owen R, McCarthy H, Rule S, et al. Acalabrutinib in Patients (pts) with Waldenström Macroglobulinemia (WM) ASCO 2018 Abstract 7501.
60. Neuman LL, Ward R, Arnold D, et al. First-in-human Phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of the noncovalent Bruton Tyrosine Kinase (BTK) inhibitor SNS-062 in healthy subjects. Blood. 2016;128(22):2032.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.