")
Back to Journals » OncoTargets and Therapy » Volume 12
Evaluating Acalabrutinib In The Treatment Of Mantle Cell Lymphoma: Design, Development, And Place In Therapy
Authors Girard J , Reneau J, Devata S, Wilcox RA, Kaminski MS, Mercer J, Carty S, Phillips TJ
Received 3 July 2019
Accepted for publication 16 September 2019
Published 30 September 2019 Volume 2019:12 Pages 8003—8014
DOI https://doi.org/10.2147/OTT.S155778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Jennifer Girard, John Reneau, Sumana Devata, Ryan A Wilcox, Mark S Kaminski, Jessica Mercer, Shannon Carty, Tycel J Phillips
Department of Internal Medicine, Division of Hematology-Oncology, Rogel Cancer Center University of Michigan, Ann Arbor, MI, USA
Correspondence: Tycel J Phillips
Department of Internal Medicine, Division of Hematology-Oncology, Rogel Cancer Center University of Michigan, 1500 East Medical Center Drive, Ann Arbor, MI 48109, USA
Tel +1 734 232 2883
Email [email protected]
Abstract: Mantle cell lymphoma (MCL) is an incurable intermediate-grade lymphoma representing 5–6% of non-Hodgkin’s lymphomas diagnosed in the United States. The introduction of inhibitors of Bruton’s tyrosine kinase (BTK) into targeted therapy for MCL has significantly improved outcomes in patients with relapsed/refractory (R/R) disease. Since the initial approval of the first-generation inhibitor, ibrutinib, several second-generation inhibitors have been explored. Acalabrutinib, a second-generation BTK inhibitor, has demonstrated impressive efficacy in clinical trials along with a safety profile that thus far appears improved compared to ibrutinib. The results of a Phase II trial in patients with R/R MCL led to the approval of acalabrutinib in this patient population while fueling further exploration of acalabrutinib in several ongoing clinical trials.
Keywords: mantle cell lymphoma, Bruton’s tyrosine kinase, long-term safety, combinations
Introduction
Mantle cell lymphoma (MCL) is an incurable intermediate-grade lymphoma representing 5–6% of non-Hodgkin’s lymphomas (NHLs) diagnosed in the United States. MCL has a variable presentation and clinical course, with a median overall survival (OS) of approximately 5 years and a median age at diagnosis of 65.1,2 The disease is characterized by lymphocytes that are immunophenotypically similar to the lymphocytes in the mantle cells of normal germinal follicles: sIg M+, sIgD+ with CD5+ CD20+ CD10± and CD43+. A hallmark of MCL is the presence of t(11;14), a reciprocal chromosomal translocation t(11;14)(q13;32) that involves CCND1, PRAD1, and bcl1, resulting in overexpression of CCND1 which encodes cyclin D1.3,4 The detection of this translocation or overexpression of the cyclin D1 protein is specific to MCL compared with other B cell lymphomas and is detectable in approximately 90% of all diagnosed cases. Other genetic abnormalities occur in MCL with lower frequency; alternations in p53 are more common in the pleomorphic and blastoid variants.5–8
Treatment for this B cell lymphoma has evolved over the last several decades with treatment of newly diagnosed patients dichotomized based on age and/or performance status (PS).9–12
For most patients under 65 and those who are considered fit, meeting criteria for treatment, the following approach is frequently used: chemotherapy with rituximab plus a cytarabine-based regimen followed by consolidation with autologous stem cell transplant (ASCT) followed by maintenance rituximab. One of the more commonly utilized approaches based on the Phase II MCL-2 trial which evaluated induction chemotherapy with the rituximab, cyclophosphamide, vincristine, doxorubicin, prednisone alternating with rituximab and high-dose cytarabine (The Nordic Regimen) and reported 6-year progression-free survival (PFS) was 66% and 6-year overall survival (OS) was 70% with a median PFS of 8.5 years and OS of 12.7 years.13–24 Additional Phase II studies utilizing rituximab, bendamustine, and cytarabine followed by ASCT at Dana-Farber Cancer Institute reported a 96% complete response (CR) and 96% PFS at a median follow-up of 13 months.25 A Phase III trial evaluated rituximab maintenance therapy vs observation after ASCT. All patients received R-DHAP induction (rituximab, dexamethasone, cytarabine, cisplatin) followed by ASCT; those randomized to rituximab maintenance every 8 weeks for 3 years with median follow-up of 50.2 months demonstrated a 4-year event-free survival (EFS) of 79% vs 62%, p=0.001, with PFS of 83% vs 64%, p<0.001, and OS of 89% vs 80%, p=0.04.26 Overall, the utilization of rituximab as maintenance therapy has demonstrated a survival benefit and should be utilized after most induction regimens.
Many patients diagnosed with MCL are not candidates for transplant due to advanced age and comorbidities in this age group. Despite response rates of 50–70% with chemoimmunotherapeutic regimens, the median time to treatment failure in this patient group is 18 months with a median survival time of approximately 3 years. In older patients and those not fit enough for transplant, chemoimmunotherapy utilizing bendamustine and rituximab should be considered the standard approach; this is based on two pivotal trials that demonstrated the superiority of BR over R-CHOP in this patient population. The role of maintenance rituximab after BR is controversial as the only trial to evaluate this modality did not demonstrate any benefit, but this result has been criticized based on the trial’s statistical design.27 The LYM-3002 Phase III trial evaluated whether replacing vincristine with the proteasome inhibitor bortezomib (VR-CAP) in newly diagnosed patients could improve outcomes. This randomized trial demonstrated that after a median follow-up 40 months VR-CAP increased PFS to 24.7 months vs 14.4 months (p<0.001) with OS at 4 years at 64% vs 54%. Attempts to limit toxicity by utilizing noncytotoxic approaches include a multicenter Phase II trial evaluating lenalidomide + rituximab induction followed by RL maintenance. The 2-year PFS was 85% and OS was 97%. Responses were durable at 4 years with a PFS of 70% and OS of 83%, and 36% patients of were in remission beyond 5 years.9–12,28 ECOG E1411 is on ongoing Phase II trial investigating four first-line approaches to treatment in patients with MCL. Patients are randomized to BR ± bortezomib. A second randomization after induction evaluates the benefit of lenalidomide to rituximab maintenance. In spite of the abundant clinical trial data and introduction of multiple new agents into clinical practice, OS, while improved compared to published data from the last several decades, remains poor. MCL remains an incurable disease, and all patients with MCL will relapse and require salvage therapy with poor response to therapy. Prior to the introduction of Bruton’s tyrosine kinase inhibitors (BTKi), responses to salvage therapy were limited.
Development Of Bruton’s Tyrosine Kinase Inhibitors
BTK is a kinase exclusively expressed in B cells and myeloid cells. BTK is of the TEC family of nonreceptor cytoplasmic tyrosine kinases that include the tyrosine kinase expressed in hepatocellular carcinoma (TEC), interleukin-2-inducible T cell kinase (ITK), resting lymphocyte kinase (RLK) and bone marrow expressed kinase (BMX). BTK contains five protein domains: pleckstrin (PH), TEC, SRC homologies as well as the kinase domain. BTK is transiently recruited from the cytoplasm to the cell membrane through interaction with PIP3 and the pleckstrin domain. BTK activation results from phosphorylation of Y551 in the kinase domain by SYK or SRC family kinases and results in autophosphorylation of Y223 in the SRC domain. In B cells, the initial event in BTK signaling is binding of IgM BCR to antigen and subsequent phosphorylation of Igα and Igβ via LYN/SRC. This ultimately results in activation of 1) mitogen-activated protein kinase (MAPK) 2) nuclear factor kappa light chain enhancer (NFκB), and 3) the serine/threonine kinase AKT, resulting in cellular proliferation, differentiation, survival, and inhibition of pro-apoptotic FOXO proteins.29–35
BTK is vital for B cells. Mutations in BTK can result in incomplete B cell differentiation with almost no mature B cells in the peripheral blood. BTK-deficient B cells cannot produce immunoglobulins resulting in the condition of X-linked agammaglobulinemia.36–42 In MCL, BTK is overexpressed and phosphorylated at Y223. BTK is essential for retention of MCL cells in lymphoid tissues, since BTK inhibition induces an egress of malignant cells into the peripheral blood.43
The critical role of BTK prompted its investigation as a therapeutic target in mature B cell malignancies in the relapsed and refractory setting. Initial drug development efforts led to the development of first-generation BTKi that bind covalently to the C481 residue of the ATP-binding pocket within the kinase domain. This binding prevents BTK from phosphorylating its substrate PLCγ, thereby halting its ability to affect downstream activation of MAPK, NFκB, and AKT pathways.
Spebrutinib (Celgene) was the first BTKi in clinical trials. It is a potent but not fully specific BTKi that covalently bonds to C481 with IC50 2 nM.44 A Phase Ib clinical trial evaluated spebrutinib in patients with (R/R) B-cell malignancies and demonstrated overall response rate (ORRs) of 31% at 750 mg once daily, 50% at 1000 mg once daily, and 66.7% at 375 mg twice daily with no patient achieving a CR. In a preclinical MCL model, the agent was additionally demonstrated to have efficacy and potential for synergy with various agents.45
Ibrutinib (J&J AbbVie) was the first BTKi approved for the treatment of B cell malignancies; it was approved as second-line therapy for R/R MCL in 2013.
Ibrutinib contains an electrophilic acrylamide moiety that covalently binds Cys481 causing inhibition of BTK. Although effective at inhibiting BTK due to its interaction with other kinases that possess an analogous binding site including TEC, EGFR, etc., ibrutinib has several off-target effects and also interacts with other kinases such as TEC, EGFR, etc. that include rash, diarrhea from inhibition of EGFR as well as atrial fibrillation, bleeding, and infection.46 Preclinical data suggest that ibrutinib can impact immune function through off-target interactions. For example, ibrutinib can alter T cell function through inhibition of ITK and TEC/TXK46 and has been noted to antagonize rituximab-induced antigen-dependent cell-mediated cytotoxicity (ADCC) through the inhibition of FcR-stimulated NK cell function.47
A pivotal Phase II clinical trial treated 115 patients with single-agent ibrutinib at 560 mg daily until disease progression or intolerable AEs. The median age was 68 years and median prior therapy was three lines. 46 (41%) patients remained on therapy for 15.3 months; 50 patients discontinued due to progression, and 8 discontinued due to AEs (diarrhea, fatigue, and nausea). Mild-to-moderate neutropenia and thrombocytopenia occurred in 18% of patients, and grade 3 bleeding events requiring transfusion occurred in 4.5% of patients. 16 patients died due to progression and four due to AEs. ORR was 68% (47% partial response and, 21% CR) with a median response duration of 17.5 months (95% CI 15.8 not reached). Median OS was not reached, and OS was 58% at 18 months. These data were the basis for accelerated approval of ibrutinib in November 2013.48,49 Updated data reported at a median follow-up of 26.7 months with a median treatment duration of 8.3 months, whereby 46% were treated for >12 months and 22% for >2 years. The median duration of response was 17.5 months with PFS and OS rates of 31% and 47%, respectively, at 2 years and are the highest reported for a single agent in R/R MCL.50 Long-term pooled analysis of clinical trials utilizing ibrutinib in MCL has demonstrated continued efficacy as well as improved outcomes in patients with fewer prior therapies.51
Primary or acquired resistance to ibrutinib has been noted in MCL, although the etiology of either mechanism is not currently known unlike CLL were the primary cause of drug resistance to BTK has mostly been related to mutations at the covalent binding site on BTK. Another known acquired mechanism of resistance to ibrutinib is a gain of function mutation at R665W and L845F of the BTK substrate, PLCγ2 resulting in autonomous downstream signal activation through the MAPK, NFκB and AKT pathways.52–54
Ibrutinib is currently in clinical trial as frontline induction therapy for young fit patients with MCL. The triangle Phase III study EudraCT 2014-001363-12 by the European MCL consortium is randomizing patients to three arms: 6 cycles R-CHOP/R-DHAP followed by ASCT vs 6 cycles RCHOP +ibrutinib/RDHAP followed by ASCT and 2 years ibrutinib (MI) vs 6 cycles RCHOP plus ibrutinib/RHAP and 2 years MI NCT02858258. The SHINE trial is evaluating the combination of ibrutinib with BR vs BR alone in patients 65 and older (NCT01776840).
Development Of Acalabrutinib
Acalabrutinib (ACP-196) is a second-generation BTK inhibitor that forms an irreversible covalent bond with BTK at (Cys481 in the kinase) domain. It has a higher selectivity for BTK as compared with other kinases, including MEC, PDGFR, EGFR, ITK, and others (Figure 1). Pharmacodynamic (PD) modeling in vivo using mice treated with 0.1 to 30 mg/kg acalabrutinib vs ibrutinib demonstrated a half-maximal effective concentration (ED50) of 1.3 mg/kg for acalabrutinib vs 2.9 mg/kg for ibrutinib with prolonged PD effect with acalabrutinib compared to ibrutinib. Acalabrutinib being a more selective BTKi is believed to have less off-target inhibition and side effects compared to ibrutinib. In vitro studies using a competitive binding assay (DiscoverX) on wild-type and mutant kinases showed that acalabruitinib at 1 µM inhibited fewer off-target kinases compared to ibrutinib, with only 1.5% of the nonmutant protein kinases being inhibited to a level of ≥65% compared to 9% for ibrutinib. Additionally, acalabrutinib did not demonstrate any significant inhibitory effects on EGFR, the TEC kinase, or ITK signaling.55–57 While ibrutinib was reported to antagonize rituximab-induced ADCC,46 this issue was not noted with acalabrutinib, suggesting acalabrutinib has less impact on T cells or cellular mediated immune defense compared to ibrutinib.
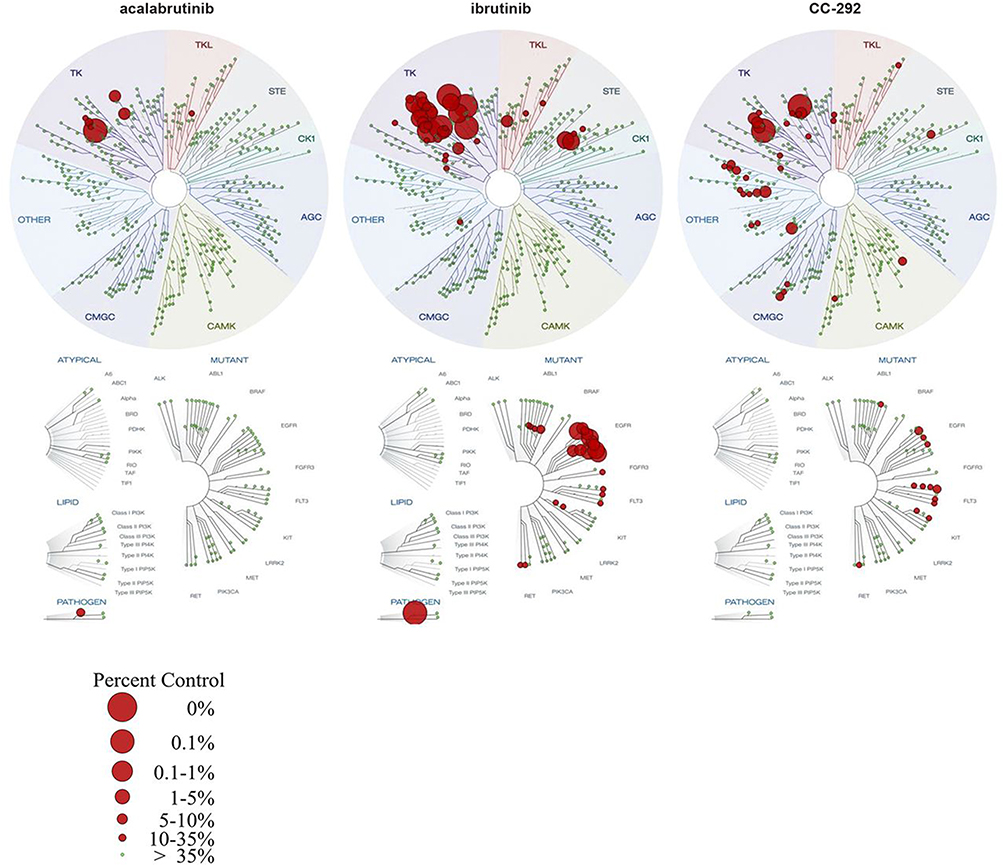 |
Figure 1 Comparison of acalabrutinib, ibrutinib, and spebrutinib in competitive binding assays on wild-type and mutant kinases (DiscoverX).56 |
The improved selectivity of acalabrutinib is thought to be due to its propiolamide side group versus ibrutinib’s acrylamide side group. The propiolamide side group may allow fewer off-target interactions with other kinases such as ITK, EGFR, ERBB2/4, JAK3, LYN, and SRK. While acalabrutinib’s half-maximal inhibitory concentration (IC50) for BTK is roughly 3 times that of ibrutinib, the IC50 against other kinases is much higher.58
In an early phase study evaluating different dosing frequencies of acalabrutinib, patients treated with 100 mg twice daily demonstrated median BTK occupancy of 99% at 4-hr post-dose and 97% at trough pre-dose/12-hr post-dose. In comparison, patients receiving acalabrutinib 200 mg once daily demonstrated BTK occupancy of 92% (p<0.01). Thus, twice-daily dosing was chosen as it resulted in lower interpatient variability.56,57
As of today, acalabrutinib is the only second-generation BTK inhibitor approved by the FDA for use in R/R MCL.
Approximately 40 clinical trials are evaluating the use of acalabrutinib; the majority are open for use in CLL (14), while others are investigating its use in nonhematologic malignancies. The remaining clinical trials are for NHL either as a single agent or in combination with for first-line or refractory indications (see Table 1).
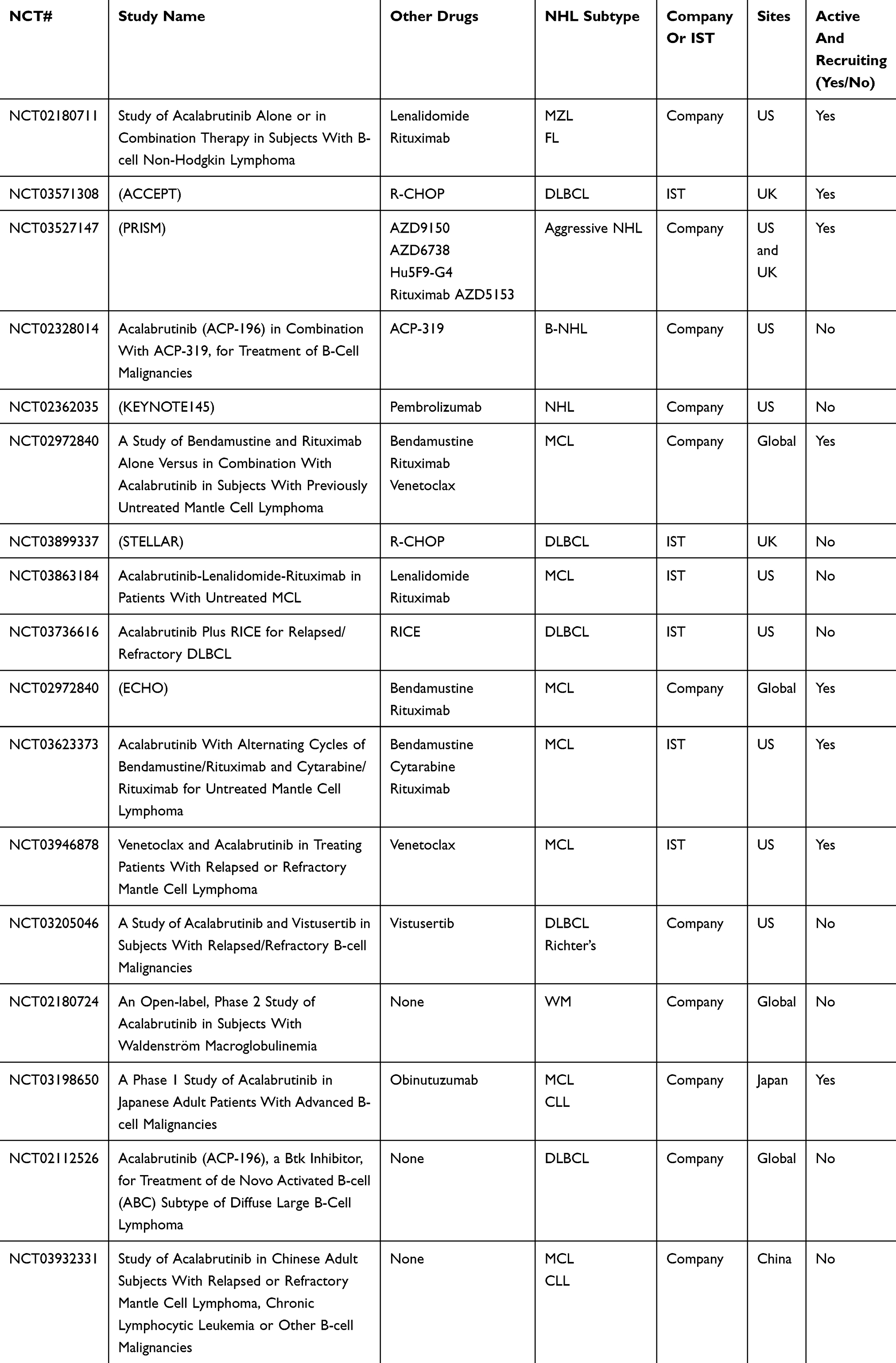 |
Table 1 Ongoing Clinical Trial With Acalabrutinib In Patients With NHL |
Acalabrutinib’s Place In The Treatment Of MCL
Acalabrutinib was first studied in R/R MCL in a Phase 2, multicenter international open-label study (ACE-LY-004). The study took place from March 2015 until January 2016 at 40 sites across 10 countries with enrollment of 124 patients with R/R MCL who had received up to 5 prior lines of therapy. Patients received acalabrutinib 100 mg BID PO in 28-day cycles. The primary study endpoint was ORR as assessed by the investigators using the Lugano classification. The secondary endpoint was ORR as assessed by an independent review committee (IRC), in addition to the duration of response (DOR), progression-free survival (PFS), overall survival (OS), safety, pharmacokinetics, and pharmacodynamics. Previous treatment with BTKi was not allowed. The median age of enrollment was 68% and 80% was male. Most had advanced-stage disease, ECOG ≤ 1(93%), low- or intermediate-risk MCL international prognostic index (MIPI) scores (83%), and a median of 2 prior lines of therapy. Most patients (95%) had previously received rituximab as a single agent or part of a combination regimen. In addition to rituximab, some patients had received CHOP-based regimen (52%), bendamustine (22%), hyperCVAD (21%), bortezomib/carfilzomib (18%), lenalidomide (7%) and ASCT (18%). The mean plasma concentration of acalabrutinib was 1000 ng/mL 1-hr post-dose with a BTK occupancy of 99%. BTK occupancy was noted to be 95–97% prior to the next dose. Primary endpoint ORR (CR+PR) was 81%, with 40% CR vs 80% and 40% CR as assessed by the IRC. The median time to response was 1.9 months, the median DOR was not reached with a reported 12-month DOR rate of 72% (CI 95%: 62–80%). At a median follow-up at 15.2 months, 54 (44%) patients had discontinued therapy, mostly due to progression of the disease (39 patients). The other most common reason for discontinuation was AEs in 7 patients. The most observed AEs were grade 1 or 2. The most common AEs of any grade were headache (38%), diarrhea (31%), fatigue (27%), and myalgia (21%). Although common, headache events were mostly grade 1 (64% of 47 patients) with a median time to onset of approximately 5 days. The median duration of headache events was reported to be 11 days, and most were self-resolving with only two patients having to discontinue therapy due to this event. With regard to diarrhea, the median time to onset of was 50 days with a median time to resolution of 7 days. There were few grade 3 or higher AEs which included neutropenia (10%), anemia (9%), and pneumonia (5%). SAEs were considered treatment related in 10% of patients; none were reported in more than one patient. At time of initial publication, no cases of atrial fibrillation were reported; bleeding events, the most frequent of which were contusion and petechiae, occurred in 31% of patients and were all grade 1 or 2 except for one grade 3 gastrointestinal hemorrhage in a patient with previous history of gastric ulcer.59 Outcomes from this study led to the accelerated approval of acalabrutinib (Calquence, AstraZeneca Pharmaceuticals Inc. under license of Acerta Pharma BV) for use in R/R MCL.
An update to the study was presented at the American Society of Hematology Annual Meeting in December 2018. The investigators reported that as of February 2018 the median time on study was 26.3 months (range, 0.3–35.1 months), and 40% of patients remain on treatment. Median relative dose intensity (ratio of actual to planned cumulative dose during drug exposure period) was 99% (range, 27–100%). Investigator-assessed ORR was not significantly different from the original publication. Median DOR was updated and noted to be 25.7 months (95% CI: 17.5 months, not reached). Median PFS was 19.5 months (95% CI: 16.5 months, 27.7). Median OS had not yet been reached with an estimated 24-month OS rate of 72% (95% CI: 64%, 80%). The safety profile was updated to report 13 patients (10%) with 16 cardiac events, including four grade 3/4 events (3%) in one patient each (acute coronary syndrome, acute myocardial infarction, cardiorespiratory arrest, and coronary artery disease). Four patients had hypertension events (3%); one event was grade 3. Three grade 3 bleeding events (gastrointestinal hemorrhage, hematuria, and hematoma) were reported. Grade 3/4 infections occurred in 15% of patients and none were grade 5; there was one case of cytomegalovirus viremia (CMV) and one case of pneumocystis jiroveci pneumonia (PCP; both grade 2). Again, progressive disease was the most likely reason for treatment discontinuation with an additional 15 patients coming off treatment since the Lancet article. Three additional patients came off trial due to AEs, bringing the total number of discontinuations from AEs to 10 (8%). Forty-three deaths (35%) were reported, most commonly from progressive disease (n=29; 23%) or AEs (n=6; 5%). Deaths due to AEs included bilateral pulmonary embolism, critical aortic stenosis, myelodysplastic syndrome, pneumonia, suicide, and non-small cell lung cancer; none were considered to be related to acalabrutinib.60
Currently, BTKi are the treatment of choice for most patients with MCL in first relapse based on recently reported data.51 The choice of BTKi is less clear based on available data. Currently, acalabrutinib has a safety profile that thus far appears improved compared to ibrutinib. It does require twice-daily dosing which can impact compliance and possibly outcome, while ibrutinib is dosed once daily. Moving forward patient comorbidities, compliance and physician preference will likely dictate which agent is utilized most in the second-line setting.
Clinical Trials Supporting The Use Of Acalabrutinib In Treatment Of MCL
There are several additional studies that have been conducted in MCL with acalabrutinib. Most of the ongoing trials have recapitulated studies that have already been conducted with ibrutinib.
LY-106 is a Phase I clinical trial that was designed to evaluate the combination of acalabrutinib with bendamustine and rituximab (BR), in patients with MCL. This multicenter open-label study enrolled patients from May 2016 through March 2017 at 15 sites across 3 countries. Study design included two cohorts: treatment naïve (TN) and R/R patients. Cohort 1 enrolled 18 TN, while cohort 2 enrolled 18 R/R MCL patients who had a median of 2 lines of therapy. All patients received acalabrutinib 100 mg PO BID plus bendamustine 90 mg/m2 on days 1, 2 and rituximab 375 mg/m2 for six 28-day cycles. Treatment-naïve (TN) patients then received acalabrutinib 100 mg PO BID daily plus rituximab 375 mg/m2 IV every other month as maintenance therapy for cycles 7–30, and then daily acalabrutinib 100 mg PO BID until disease progression or end of treatment. R/R patients received acalabrutinib 100 mg PO BID until disease progression after induction. The primary endpoint was safety of acalabrutinib in combination with bendamustine and rituximab with AEs graded using NCI CTACE v4.03. Secondary endpoints were investigator-assessed efficacy of acalabrutinib in combination with BR using ORR, DOR, and PFS assessed using Lugano criteria. Median time on study for the TN cohort was 20.6 months (range 0.6 −2.61) vs 16.9 months (range 1.2–26.6) for the RR patients. 72% of the TN patients completed 6 cycles of therapy vs 50% of the R/R cohort. No TN patients experienced progression of disease, while two R/R patients experienced disease progression. All three study drugs were discontinued in 44% of patients in cohort 1 vs 55% in cohort 2. Death occurred in 3 (17%) enrolled into the TN cohort vs 4 (20%) patients in the R/R cohort. Two patients (1%) withdrew from study in the TN cohort due to AEs vs 1 patient (5%) in the R/R cohort. AEs occurred in ≥20% of all patients enrolled; 58% experienced grade 1 or 2 nausea as the most frequent side effect, with more TN patients 14 (78%) reporting this event than in the R/R cohort (8, 40%). Grade 1 or 2 fatigue occurred in 53% of patients overall, with one R/R patient experiencing grade 3 fatigue. Grade 3/4 neutropenia was observed in 45% of patients overall; 22% experienced grade 3 and 17% experienced grade 4 in the TN cohort vs 25% grade 3 and 25% grade 4 neutropenia in the R/R cohorts. One TN patient experienced major hemorrhage, reported as pulmonary alveolar hemorrhage, leading to discontinuation of study treatment. Grade 3 or 4 AEs occurring in ≥ two patients included neutropenia, pneumonia, thrombocytopenia, abdominal pain, acute kidney injury, anemia, diarrhea, hypertension, hyperuricemia, and hypotension. Of note, no patients experienced atrial fibrillation, CMV, PCP, or tumor lysis syndrome during this study. In terms of secondary outcomes, ORR (CR + PR) was 94% (CI 73–100%) in the TN cohort vs 85% (CI 62–97%) in the R/R cohort. Thirteen (72% of evaluable) patients achieved a CR in the TN cohort vs 13 (65% of evaluable patients) in the R/R cohort. Four patients in both cohorts achieved a partial response (22%) in the TN cohort vs (20%) in the R/R cohort. The median time to initial response was 1.9 months (range, 1.6–2.8) in the TN cohort vs 1.8 months (range, 1.6–2.3) in the R/R cohort. No patients had progression of disease while on study. The median PFS was not reached in either cohort. In terms of exploratory outcomes, the steady-state concentration of acalabrutinib did not differ with or without BR.61 This Phase I study demonstrated that acalabrutinib in combination with BR has an acceptable safety profile with high response rates, which leads to the ongoing Phase III randomized double-blind placebo-controlled trial of acalabrutinib plus BR vs BR in patients 65 and older who are diagnosed with TN MCL (NCT02972840). The primary endpoint is PFS per the (Lugano classification for NHL) over 48 months and crossover to single-agent acalabrutinib is allowed for those who progress on the placebo-controlled arm. Additionally, study LY-106 continues to enroll TN patients in an additional cohort that is evaluating the safety and efficacy of acalabrutinib in combination with venetoclax and rituximab (AVR). The results of this cohort are pending.
Washington University in collaboration with Acerta Pharma is sponsoring a pilot study of acalabrutinib with alternating cycles of bendamustine/rituximab and cytarabine/rituximab for untreated MCL NCT03623373 which opened August 9, 2018, with an estimated enrollment of 15 patients. Patients are scheduled to receive six 28-day cycles of bendamustine on days 1 and 2, rituximab on day 1, acalabrutinib PO BID days 1–28 on odd cycles and cytarabine every 12 hrs. on days 1 and 2, acalabrutinib PO BID on days 1 through 7 and days 22 through 28 on even cycles with granulocyte-colony stimulating factor support and then undergo leukapheresis after cycle 6 for collection of >2 x 106 CD34+ stem cells/kg. The primary endpoint is stem cell mobilization success rate in subjects with MCL treated with this regimen. The secondary outcome measures include safety and tolerability of the regimen measured by treatment-related nonhematologic toxicity grade 3 or higher; ORR (CR+PR) over 6 months per Lugano classification; pre-transplant CR rate of over 6 months; and PFS of subjects and OS.
Several other trials evaluating acalabrutinib in additional ethnic populations or in combination with various agents are currently planned as listed in Table 1. These include 1) NCT03863184, a study of acalabrutinib, lenalidomide, and rituximab in patients with untreated MCL; 2) NCT03932331, a study of acalabrutinib in Chinese adult subjects with R/R MCL or other B-cell malignancies; and 3). NCT03946878, a study of venetoclax and acalabrutinib in patients with R/R MCL.
NCT02362035 and NCT02328014 are additional combination trials evaluating acalabrutinib in combination with pembrolizumab and ACP-319, respectively. Neither trial is specific for MCL, but both allow for enrollment of patients with R/R NHL.
Long-Term Safety
While the updated safety and efficacy data of acalabrutinib in MCL presented at ASH 2018 did not demonstrate any new safety information, there have been additional reports of bleeding and reports of atrial fibrillation in other NHL subtypes.62,63 With respect to the risk of bleeding, platelet aggregation studies have suggested that there is some clotting dysfunction due to actions of acalabrutinib. Dysfunctional aggregation responses to collagen and collagen-related peptide suggest that acalabrutinib inhibits platelet Btk and Tec at physiological concentrations. Additionally, newer PK data suggest that acalabrutinib impacts TEC to a similar degree as ibrutinib.64,65 However, unlike ibrutinib, acalabrutinib does not lead to dysfunctional thrombus formation nor does it inhibit Src family kinases, which are critical to platelet adhesion to collagen.64 Thus, acalabrutinib does not induce major platelet dysfunction as has been associated with ibrutinib. It is unclear how the maturing safety information will impact the utilization of acalabrutinib. Given that the cost and efficacy of acalabrutinib and ibrutinib are similar, acalabrutinib’s safety profile may be the deciding factor.
Economic Implications
While the advent of novel therapies such as acalabrutinib has led to improvement in patient outcomes, the financial consequences for patients can be enormous. As currently approved, all BTKi in the R/R setting are given until progression of disease or intolerance. As a result, patients can remain on treatment for several years. Mato et al (2019) published a review regarding AEs, resource use and economic burden associated with the treatment of MCL with cost data reported in 2016 dollars. Mean monthly (SD) all-cause costs during treatments with first-line therapy using BR and RCHOP were $12,958 ($12,687) and $24,719 ($44,996), respectively. The monthly cost of ibrutinib was estimated to be $21,690 ($24,773). Part of the cost estimate relates to AEs associated with treatment, which would explain the higher cost associated with R-CHOP as this is commonly utilized as part of intensive chemotherapy regimens. These regimens generally have more toxicity, leading to more frequent hospitalizations for AE management.66,67 While firm data on the comparative cost of acalabrutinib in MCL is not known at this time, a cost analysis of the two drugs published in 2018 reported an annual cost ibrutinib in the United States of approximately $146,000 a year with an estimated monthly cost of $12,180. While the wholesale cost for acalabrutinib to date is not known, the estimated cost is $14,064 per month resulting in a yearly cost of close to $169,000.68 With the slow but inevitable introduction of acalabrutinib and ibrutinib into the frontline setting, we will need to be cognizant of balancing benefits with expected costs especially when given in combination with rituximab or chemotherapy as costs will be expected to almost double the values reported by Mato et al. Ways to mitigate the cost of the drugs either through free drug programs, manufacturer-approved price reductions, or implementation of drug holidays will need to be explored.
Future Role Of Acalabrutinib In Treatment Of MCL
Given its current approval in R/R MCL, future studies utilizing acalabrutinib will undoubtedly focus on improving depth and duration of response in R/R through novel combinations. In the frontline setting, a large Phase III trial is evaluating regimens combining acalabrutinib with BR with the goal of improving outcomes in transplant ineligible patients. With the current trend to limit exposure to cytotoxic agents, we will likely see acalabrutinib combined with more targeted therapies such as the new arm of LY-106 (venetoclax + rituximab) and the upcoming study NCT03863184 (lenalidomide + rituximab). How these frontline studies will impact treatment will depend on response (radiographic and molecular), treatment duration (continuous vs fixed), and cost. Acalabrutinib should also be explored in the maintenance setting. Currently, in MCL, only rituximab has demonstrated a significant benefit in the maintenance setting, but even this can vary depending on the induction regimen.26,27 How acalabrutinib or other BTKi can impact this area of treatment is unknown. With respect to the R/R setting, while the current DOR of acalabrutinib is still maturing, the final median DOR is unlikely to differ significantly from what is currently reported with the use of ibrutinib. Finding ways to improve the DOR of BTKI remains paramount given the poor outcomes reported in patients who fail these agents.69,70 This is likely to be accomplished through combination studies utilizing other small molecules such as PI3K inhibitors, BH3 mimetics, etc. Additionally, given that a subset of MCL patients will present with tumors that harbor primary resistance to BTKi, further elucidation of resistance mechanisms to BTK inhibition will be important for the future development of acalabrutinib and other agents in this class.
Conclusions
Over the last several years, we have seen an explosion of new treatments for patients with MCL, but no other treatments to date have impacted the care of patients with MCL to the degree of BTKi. Since its approval in 2017 acalabrutinib has become a major asset in the treatment of patients with MCL. Initial studies with acalabrutinib demonstrated excellent response rates and suggested an improved safety profile as compared to the only other currently FDA-approved BTKi. The safety of the drug has stayed consistent in combination with chemotherapy and other targeted agents, allowing it to become a fixture in this patient population. In the future, we will look for verification of its long-term safety profile as current studies mature, future trials open, and more data are obtained from patients treated outside of clinical trials. With no reported differences in efficacy and no expectation that cost will differ among this drug class, we will depend on the safety and long-term tolerability for selecting among the various covalent BTKi agents moving forward.
Disclosure
TJP has received research support and participated on an advisory board for Pharmacyclics. The authors report no other conflicts of interest in this work.
References
1. Swerdlow SH WHO classification of tumours of haematopoietic and lymphoid tissues. World Health Organization Classification of Tumours. Lyon, France: IARC;
2. Banks PM, Chan J, Cleary ML, et al. Mantle cell lymphoma. A proposal for unification of morphologic, immunologic, and molecular data. Am J Surg Pathol. 1992;16(7):637–640. doi:10.1097/00000478-199207000-00001
3. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750–762. doi:10.1038/nrc2230
4. Welzel N, Le T, Marculescu R, et al. Templated nucleotide addition and immunoglobulin JH-gene utilization in t(11;14) junctions: implications for the mechanism of translocation and the origin of mantle cell lymphoma. Cancer Res. 2001;61(4):1629–1636.
5. Shiloh Y. ATM: ready, set, go. Cell Cycle. 2003;2(2):116–117. doi:10.4161/cc.2.2.342
6. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3(3):155–168. doi:10.1038/nrc1011
7. Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1(3):179–186. doi:10.1038/35043058
8. Kastan MB, Lim DS, Kim ST, Xu B, Canman C. Multiple signaling pathways involving ATM. Cold Spring Harb Symp Quant Biol. 2000;65:521–526. doi:10.1101/sqb.2000.65.521
9. Meusers P, Engelhard M, Bartels H, et al. Multicentre randomized therapeutic trial for advanced centrocytic lymphoma: anthracycline does not improve the prognosis. Hematol Oncol. 1989;7(5):365–380.
10. Rummel MJ, Niederle N, Maschmeyer G, et al. Study group Indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. doi:10.1016/S0140-6736(12)61763-2
11. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123(19):2944–2952. doi:10.1182/blood-2013-11-531327
12. Ruan J, Martin P, Christos P, et al. Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood. 2018;132(19):2016–2025. doi:10.1182/blood-2018-07-859769
13. Herrmann A, Hoster E, Zwingers T, et al. Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol. 2009;27(4):511–518. doi:10.1200/JCO.2008.16.8435
14. de Guibert S, Jaccard A, Bernard M, Turlure P, Bordessoule D, Lamy T. Rituximab and DHAP followed by intensive therapy with autologous stem-cell transplantation as first-line therapy for mantle cell lymphoma. Haematologica. 2006;91(3):425–426.
15. Khouri IF, Romaguera J, Kantarjian H, et al. Hyper-CVAD and high-dose methotrexate/cytarabine followed by stem-cell transplantation: an active regimen for aggressive mantle-cell lymphoma. J Clin Oncol. 1998;16(12):3803–3809. doi:10.1200/JCO.1998.16.12.3803
16. Lefrere F, Delmer A, Levy V, Delarue R, Varet B, Hermine O. Sequential chemotherapy regimens followed by high-dose therapy with stem cell transplantation in mantle cell lymphoma: an update of a prospective study. Haematologica. 2004;89(10):1275–1276.
17. Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005;23(28):7013–7023. doi:10.1200/JCO.2005.01.1825
18. Romaguera JE, Khouri IF, Kantarjian HM, et al. Untreated aggressive mantle cell lymphoma: results with intensive chemotherapy without stem cell transplant in elderly patients. Leuk Lymphoma. 2000;39(1–2):77–85. doi:10.3109/10428190009053541
19. Ganti AK, Bierman PJ, Lynch JC, Bociek RG, Vose JM, Armitage JO. Hematopoietic stem cell transplantation in mantle cell lymphoma. Ann Oncol. 2005;16(4):618–624. doi:10.1093/annonc/mdi107
20. Geisler CH, Kolstad A, Laurell A, et al.; for the Nordic Lymphoma Group. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood. 2008;112(7):2687–2693. doi:10.1182/blood-2008-03-140830
21. Le Gouill S, Callanan M, Macintyre E, et al. Clinical, metabolic and molecular responses after 4 courses of R-DHAP and after autologous stem cell transplantation for untreated mantle cell lymphoma patients included in the LyMa trial, a Lysa study. Blood. 2012;120:152.
22. Hermine O, Hoster E, Walewski. J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) is superior to 6 courses CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: results of the MCL younger trial of the European mantle cell lymphoma network (MCL net). Blood. 2010;116:110.
23. LaCasce A, Vandergrift JL, Rodriguez MA, et al. R-CHOP, followed by High Dose Therapy and Autologous Stem Cell Rescue (HDT/ASCR), and R-Hypercvad have equivalent progression-free survival and are superior to R-CHOP alone in younger patients with mantle cell lymphoma: a comparative effectiveness analysis from the National Comprehensive Cancer Network (NCCN) Non-Hodgkin’s lymphoma outcomes database project. Blood. 2009;114:403. doi:10.1182/blood-2009-04-215525
24. Pott C, Hoster E, Beldjord K, et al. R-CHOP/R-DHAP compared to R-CHOP induction followed by high dose therapy with autologous stem cell transplantation induces higher rates of molecular remission in MCL: results of the MCL Younger Intergroup Trial of the European MCL Network. Blood. 2010;116(21):965.
25. Armand P, Redd R, Bsat J, et al. A phase 2 study of Rituximab-Bendamustine and Rituximab-Cytarabine for transplant-eligible patients with mantle cell lymphoma. Br J Haematol. 2016;173(1):89–95. doi:10.1111/bjh.13929
26. Le Gouil S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017;377:1250–1260. doi:10.1056/NEJMoa1701769
27. Rummel MJ, Knauf W, Goerner M, et al. Two years rituximab maintenance vs. observation after first-line treatment with bendamustine plus rituximab (B-R) in patients with mantle cell lymphoma: first results of a prospective, randomized, multicenter phase II study (a subgroup study of the StiL NHL7-2008 MAINTAIN trial). JCO. 2016;34(15)_suppl,):7503.
28. Robak T, Jin J, Pylypenko H, et al. Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol. 2018;19(11):1449–1458. doi:10.1016/S1470-2045(18)30685-5
29. Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH. Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med. 2000;191(10):1735–1744. doi:10.1084/jem.191.10.1735
30. Fluckiger AC, Li Z, Kato RM, et al. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. Embo J. 1998;17(7):1973–1985. doi:10.1093/emboj/17.7.1973
31. Jiang A, Craxton A, Kurosaki T, Clark EA. Different protein tyrosine kinases are required for B cell antigen receptor-mediated activation of extracellular signal-regulated kinase, c-Jun NH2-terminal kinase 1, and p38 mitogen-activated protein kinase. J Exp Med. 1998;188(7):1297–1306. doi:10.1084/jem.188.7.1297
32. Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med. 2000;191(10):1745–1754. doi:10.1084/jem.191.10.1745
33. Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184(1):31–40. doi:10.1084/jem.184.1.31
34. Tomlinson MG, Woods DB, McMahon M, et al. A conditional form of Bruton’s tyrosine kinase is sufficient to activate multiple downstream signaling pathways via PLC Gamma 2 in B cells. BMC Immunol. 2001;2:4. doi:10.1186/1471-2172-2-4
35. Uckun FM, Waddick KG, Mahajan S, et al. BTK as a mediator of radiation-induced apoptosis in DT-40 lymphoma B cells. Science. 1996;273(5278):1096–1100. doi:10.1126/science.273.5278.1096
36. Tao L, Boyd M, Gonye G, Malone B, Schwaber J. BTK mutations in patients with X-linked agammaglobulinemia: lack of correlation between presence of peripheral B lymphocytes and specific mutations. Hum Mutat. 2000;16(6):528–529. doi:10.1002/1098-1004(200012)16:6<528::AID-HUMU12>3.0.CO;2-T
37. Conley ME, Mathias D, Treadaway J, Minegishi Y, Rohrer J. Mutations in btk in patients with presumed X-linked agammaglobulinemia. Am J Hum Genet. 1998;62(5):1034–1043. doi:10.1086/301828
38. Parslow TG, Stites DP, Terr AI, Imboden JB. Medical Immunology.
39. Virella G. Medical Immunology.
40. Paul WE. Fundamental Immunology.
41. Rich R. Clinical Immunology Principles and Practices.
42. Hyvönen M, Saraste M. Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. Embo J. 1997;16(12):3396–3404. doi:10.1093/emboj/16.12.3396
43. Pal SS, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57. doi:10.1186/s12943-018-0779-z
44. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. doi:10.1124/jpet.113.203489
45. Vidal-Crespo A, Rodriguez V, Matas-Cespedes A, et al. The Bruton tyrosine kinase inhibitor CC-292 shows activity in mantle cell lymphoma and synergizes with lenalidomide and NIK inhibitors depending on nuclear factor-κB mutational status. Haematologica. 2017;102(11):e447–e451. doi:10.3324/haematol.2017.168930
46. Pan Z, Scheerens H, Li SJ, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1):58–61. doi:10.1002/cmdc.200600221
47. Kohrt HE, Sagiv-Barfi I, Rafiq S, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–1960. doi:10.1182/blood-2014-01-547869
48. Salles GA, Gopal AK, Martin P, et al. An open-label phase II study of ibrutinib in patients with refractory follicular lymphoma. JCO. 2013;31(15_suppl):
49. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. doi:10.1056/NEJMoa1306220
50. Wang ML, Blum KA, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. Blood. 2015;126(6):739–745. doi:10.1182/blood-2015-03-635326
51. Rule S, Dreyling M, Goy A, et al. Outcomes in 370 patients with mantle cell lymphoma treated with ibrutinib: a pooled analysis from three open-label studies. Br J Haematol. 2017;179(3):430–438. doi:10.1111/bjh.14870
52. Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi:10.1056/NEJMoa1400029
53. Woyach JA, Johnson AJ. Targeted therapies in CLL: mechanisms of resistance and strategies for management. Blood. 2015;126(4):471–477. doi:10.1182/blood-2015-03-585075
54. Chiron D, Di Liberto M, Martin P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. doi:10.1158/2159-8290.CD-14-0098
55. Maly J, Blachly JS. Chronic lymphocytic leukemia: exploiting vulnerabilities with targeted agents. Curr Hematol Malig Rep. 2016;11(1):52–60. doi:10.1007/s11899-016-0299-0
56. Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240–252. doi:10.1124/jpet.117.242909
57. Byrd JC, Wierda W, Jones J, et al. The Bruton tyrosine kinase (Btk) inhibitor ACP-196: marked activity in relapsed/refractory CLL with a favorable safety profile. Blood. 2015;126:831.
58. Herman SEM, Motraveta A, Niemann CU, et al. The Bruton tyrosine kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. 2017;23(11):2831–2841. doi:10.1158/1078-0432.CCR-16-0463
59. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2018;391(10121):659–667. doi:10.1016/S0140-6736(17)33108-2
60. Wang M, Rule S, Zinzani PL, et al. Long-term follow-up of acalabrutinib monotherapy in patients with relapsed/refractory mantle cell lymphoma. Blood. 2018;132:2876.
61. Phillips TJ, Smith SD, Jurczak W, et al. Safety and efficacy of acalabrutinib plus Bendamustine and Rituximab (BR) in patients with Treatment-Naive (TN) or Relapsed/Refractory (R/R) Mantle Cell Lymphoma (MCL). Blood. 2018;132:4144.
62. Awan FT, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3:1553–1562. doi:10.1182/bloodadvances.2018030007
63. Jennifer Ann Woyach JA, Kerry Anne Rogers KA, Seema Ali Bhat SA, et al. Acalabrutinib with obinutuzumab (Ob) in treatment-naive (TN) and relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL): three-year follow-up. JCO. 2019;37(suppl; abstr 7500).
64. Bye AP, Unsworth AJ, Desborough MJ, et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1:2610–2623. doi:10.1182/bloodadvances.2017011999
65. Hopper M, Gururaja T, Kinoshita T, Dean JP, Hill R, Mongan A. Kinetic catalytic inhibition and cell-based analysis, not just target binding, are required to assess kinase selectivity of covalent inhibitors: comparable BTK vs TEC selectivity profile for ibrutinib and acalabrutinib. Blood. 2018;13:3498.
66. Karve S, Nagar S, Goyal R, Kaye JA, Mato AR. Adverse events, resource use, and economic burden in patients with mantle cell lymphoma in the United States. Blood. 2017;130:3442.
67. Goyal R, Nagar SP, Kabadi SM, Kaye JA, Seal B, Mato AR. Adverse events, resource use, and economic burden associated with mantle cell lymphoma: a real-world assessment of privately insured patients in the United States. Leukemia Lymphoma. 2019;60(4):955–963. doi:10.1080/10428194.2018.1509320
68. Alrawashdh N, Alkhatib N, McBride A, Persky D, Abraham I. Economic evaluation for the US of ibrutinib versus acalabrutinib for patients with relapsed or refractory mantle cell lymphoma. Blood. 2018;132:4829.
69. Martin P, Maddocks K, Leonard JP, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood. 2016;127:1559–1563. doi:10.1182/blood-2015-10-673145
70. Cheah CY, Chihara D, Romaguera JE, et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol. 2015;26(6):1175–1179. doi:10.1093/annonc/mdv111
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.