")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
European perspective on the management of rheumatoid arthritis: clinical utility of tofacitinib
Authors Kawalec P, Śladowska K , Malinowska-Lipień I , Brzostek T , Kózka M
Received 30 October 2017
Accepted for publication 24 November 2017
Published 21 December 2017 Volume 2018:14 Pages 15—29
DOI https://doi.org/10.2147/TCRM.S138677
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Paweł Kawalec,1 Katarzyna Śladowska,2 Iwona Malinowska-Lipień,3 Tomasz Brzostek,3 Maria Kózka4
1Drug Management Department, Institute of Public Health, Faculty of Health Sciences, Jagiellonian University Medical College, 2Department of Experimental Hematology, Institute of Zoology and Biomedical Research, Faculty of Biology and Earth Sciences, Jagiellonian University, Krakow, Poland; 3Department of Internal and Community Nursing, Institute of Nursing and Midwifery, Faculty of Health Sciences, Jagiellonian University Medical College, Krakow, Poland; 4Department of Clinical Nursing, Institute of Nursing and Midwifery, Faculty of Health Sciences, Jagiellonian University Medical College, Krakow, Poland
Abstract: Xeljanz® (tofacitinib) is an oral small-molecule inhibitor that reversibly inhibits Janus-activated kinase (JAK)-dependent cytokine signaling, thus reducing inflammation. As a result of these mechanisms, effects on the immune system such as a moderate decrease in the total lymphocyte count, a dose-dependent decrease in natural killer (NK) cell count, and an increase in B-cell count have been observed. Therefore, tofacitinib provides an innovative approach to modulating the immune and inflammatory responses in patients with rheumatoid arthritis (RA), which is especially important in individuals who do not respond to tumor necrosis factor inhibitors or show a loss of response over time. The aim of this article was to review studies on the pharmacology, mode of action, pharmacokinetics, efficacy, and safety of tofacitinib in patients with RA. Tofacitinib has been shown to reduce symptoms of RA and improve the quality of life in the analyzed groups of patients. Moreover, it showed high efficacy and an acceptable safety profile in Phase III randomized clinical trials on RA and was the first JAK inhibitor approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) in the RA therapy, thus providing a useful alternative treatment strategy. Randomized controlled studies revealed a significant benefit over placebo in efficacy outcomes (American College of Rheumatology [ACR] 20 and ACR50 response rates); accordingly, clinically meaningful improvements in patient-related outcomes compared with placebo have been reported. The safety profile seems acceptable, although some severe adverse effects have been observed, including serious infections, opportunistic infections (including tuberculosis and herpes zoster), malignancies, and cardiovascular events, which require strict monitoring irrespective of the duration of tofacitinib administration. As an oral drug, tofacitinib offers an alternative to subcutaneous or intravenous biologic drugs and should be recognized as a more convenient way of drug administration.
Keywords: JAK inhibitor, tofacitinib, effectiveness, rheumatoid arthritis, treatment
Introduction
RA is a chronic autoimmune inflammation of joints resulting in the severe destruction of cartilage and bones; the disease leads to severe disability, decreased quality of life, and reduced life expectancy;1–3 it occurs in ~1% of adults all over the world.1 Because RA is a chronic disease with potentially severe symptoms, therapy generates high health care utilization (direct costs) as well as productivity loss (indirect costs) over the lifetime of the disease as the onset of the condition is very often in young adults.2,4
The goal of treatment is to achieve remission or to slow down disease progression if remission is not possible; low disease activity is accepted as an important therapeutic goal.3,4
Response to RA therapy is often measured as American College of Rheumatology (ACR) scores (eg, ACR20 indicates that symptoms have improved 20% from baseline and ACR50 means that they have improved 50%) or as improvement from the baseline European League Against Rheumatism (EULAR response) scale scores.3,5
The current treatment of RA includes nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, as well as disease-modifying antirheumatic drugs (DMARDs), including conventional DMARDs (methotrexate [MTX], leflunomide, and sulfasalazine); the primary therapy in most patients is MTX, while individuals with poor response to MTX are often treated with subsequent drugs: biologic DMARDs such as tumor necrosis factor (TNF) inhibitors (infliximab, adalimumab, certolizumab pegol, etanercept, and golimumab), non-TNF biologics (abatacept, rituximab, and tocilizumab), and targeted synthetic DMARDs (such as Janus-activated kinase [JAK] inhibitors including tofacitinib, baricitinib);6,7 alternative treatment option is a combination of TNF inhibitors with MTX; in a number of RA patients TNF inhibitors are combined with MTX.3 Biologic drugs essentially changed the current paradigm of therapy providing new treatment options; they reduce the symptoms and activity of the diseases and improve the quality of life in the case of patients not responsive to conventional DMARDs therapy.
Remission is achieved with DMARDs in 30%–40% of cases only, and 30% of the patients showed either insufficient response or lack of response to treatment with TNF inhibitors.6 The lack of efficacy (primary nonresponse) or the loss of efficacy over time (secondary nonresponse) and the safety profile issues make it difficult to find a drug that would offer long-lasting efficacy in a significant group of the RA patients.
According to the current research, 41%–58% of patients receiving TNF inhibitors do not achieve the ACR20 response, while other patients lose their clinical response or suffer from adverse events (AEs) during the therapy.8–10 Patients with an inadequate response or intolerance to conventional DMARDs are generally prescribed biologic DMARDs, usually in combination with MTX.11 Patients whose disease activity remains moderate or high with DMARD monotherapy are recommended to use a combination of DMARDs or to add a TNF inhibitor, a non-TNF biologic, or tofacitinib (all with or without MTX). For patients whose disease activity remains moderate or high despite the use of a single TNF inhibitor, the ACR conditionally recommends using a non-TNF biologic over another TNF inhibitor (with or without MTX).3 According to the EULAR guidelines on the use of biologics in RA, in all treatments, biologics should be combined preferentially with MTX or with other immunosuppressive agents; moreover, if the therapy targets have not been achieved with the first conventional DMARD strategy, it is necessary to add a biologic DMARD or a small synthetic molecule such as tofacitinib or baricitinib.7 Patients who do not respond adequately to treatment with TNF inhibitors are generally prescribed another drug of the same class or biologic DMARD with an alternative mechanism of action such as tocilizumab (interleukin [IL]-6 receptor antagonist), rituximab (monoclonal antibody against B cells), and abatacept (blocker of T-cell activation).7,12
In this review, we search PubMed database until August 2017 to identify eligible English language trials evaluating the efficacy and safety of tofacitinib in RA patients. We analyzed randomized clinical trials, their extensions, and follow-up studies. For a more comprehensive assessment of the drug, an in-depth analysis of the safety profile was performed based on long-term extension (LTE) studies. The results of network meta-analyses (NMA) were also used to present the relative efficacy and safety of tofacitinib compared with active reference treatments. Efficacy was assessed on the basis of standard endpoints used in RA (ACR20 and ACR50), while safety outcomes included the adverse effects reported with tofacitinib treatment. A difference with P<0.05 was considered as statistically significant.
Emerging treatment options
Despite a wide spectrum of current pharmacotherapies, new drugs such as secukinumab, ixekizumab, baricitinib, filgotinib, peficitinib, and ocrelizumab have emerged, providing novel, promising treatment options.13,14
Secukinumab and ixekizumab are monoclonal antibodies that selectively bind to and neutralize interleukin-17A (IL-17A), in which overexpression leads to inflammation, joint destruction, and bone erosion in RA.15,16 Secukinumab has been recently approved by the US Food and Drug Administration (FDA) for moderate-to-severe plaque psoriasis, psoriatic arthritis (PsA), and ankylosing spondylitis (AS). A Phase II trial demonstrated that secukinumab reduced the signs, symptoms, and activity of disease during a 24-week follow-up in patients with RA with poor response to TNF inhibitors.15 Another Phase II trial showed that ixekizumab significantly improved clinical signs and symptoms in patients with RA who were naive to biologic treatment or had shown inadequate response to TNF inhibitors during a 12-week follow-up.16 Both secukinumab and ixekizumab had an acceptable safety profile.15,16
Other agents with promising results in Phase III or IIb studies include JAK inhibitors, such as baricitinib, filgotinib, and peficitinib. Recent clinical trials have shown baricitinib to be a safe and effective drug in the RA patients who fail to respond to existing background therapy (including MTX) during 52 weeks of treatment.17 Satisfactory efficacy and safety results were also reported for peficitinib and filgotinib monotherapies. The reduction in RA symptoms has been observed after a short-term treatment with peficitinib (12 weeks)18 and filgotinib (24 weeks).19
In patients with no significant response to MTX or TNF inhibitors, the inclusion of ocrelizumab, the monoclonal antibody against CD20 protein, as an add-on treatment was beneficial as it significantly reduced symptoms of RA. Moreover, it proved superior to placebo in all clinical and radiographic improvement scales, while the safety profile (including serious AEs [SAEs]) was similar between the compared groups.13,20
Review of pharmacology, mode of action, and pharmacokinetics of tofacitinib
Xeljanz® (tafacitinib) (CP-690550) is a JAK-targeted synthetic small-molecule inhibitor modulating cytokines critical to the progression of immune and inflammatory responses. It preferentially inhibits signaling by receptors associated with JAK3 as the main target, with JAK1 and JAK2 also being affected at higher concentrations due to a debilitating effect on the signaling pathways with nanomolar potency, resulting in the inhibition of transmigration of STAT molecules to the nucleus.21,22 The JAK family, consisting of JAK-1, JAK-2, and JAK-3, and tyrosine kinase 2 is essential for the signaling pathways of various cytokines and growth factors that have been implicated in the pathogenesis of RA, which are integral to lymphocyte function and inhibition of their signaling and thus modulate the multiple aspects of the immune response (eg, IL-2, IL-6, IL-7, IL-12, IL-15, IL-17, IL-23, granulocyte-macrophage colony-stimulating factor, and interferon [IFN]).23–25
Tofacitinib is a reversible, competitive inhibitor that binds to the adenosine triphosphate (ATP)-binding site in the catalytic cleft of the kinase domain of JAK and thus inhibits the phosphorylation and activation of JAK, thereby preventing the activation of STATs and the launching of gene transcription. This leads to a decreased cytokine production and modulation of the immune response. Tofacitinib has a short pharmacokinetic half-life; pharmacokinetic profile of tofacitinib in humans is characterized by rapid absorption and elimination, with a time to peak concentration (Tmax) of ~1 hour and a half-life (t1/2) of ~3 hours.25
The clearance mechanisms for tofacitinib in humans appear to be both nonrenal (hepatic metabolism, 70%) and renal (30%) excretions of the parent drug. The metabolism of tofacitinib is primarily mediated by cytochrome P450 3A4 (CYP3A4; 53%) with a minor contribution from CYP2C19 (17%).25
FDA approved Xeljanz® (tofacitinib) (Pfizer, Inc., New York, NY, USA) for the therapy of adult patients with moderate-to-severe RA in 2012; it was also authorized by the European Medicines Agency (EMA) in combination with MTX for the treatment of moderate-to-severe active RA in adult patients who have responded inadequately to or who are not tolerant to one or more DMARDs; in case of intolerance or contraindications to MTX, it can be given as monotherapy.26
Efficacy studies on tofacitinib in RA, including any comparative studies
The efficacy of tofacitinib 5 mg and 10 mg twice a day (BID) administered as monotherapy or in combination with conventional synthetic DMARDs (mainly MTX) in patients with active RA has been shown in Phase III studies24,27–36 in a follow-up of up to 24 months and in LTE trials with up to 105 months.37,38
Tofacitinib was assessed as monotherapy31,33,35 or in combination with conventional synthetic DMARDs24,27,29,30,32,34,36 in patients with RA who responded poorly to other biologic DMARDs28,29,33 and in MTX-naive patients with active RA.35
Tofacitinib demonstrated greater efficacy compared with placebo across clinical, functional, and structural measures of disease severity24,27–30,32–34,36 as presented in Table 1.
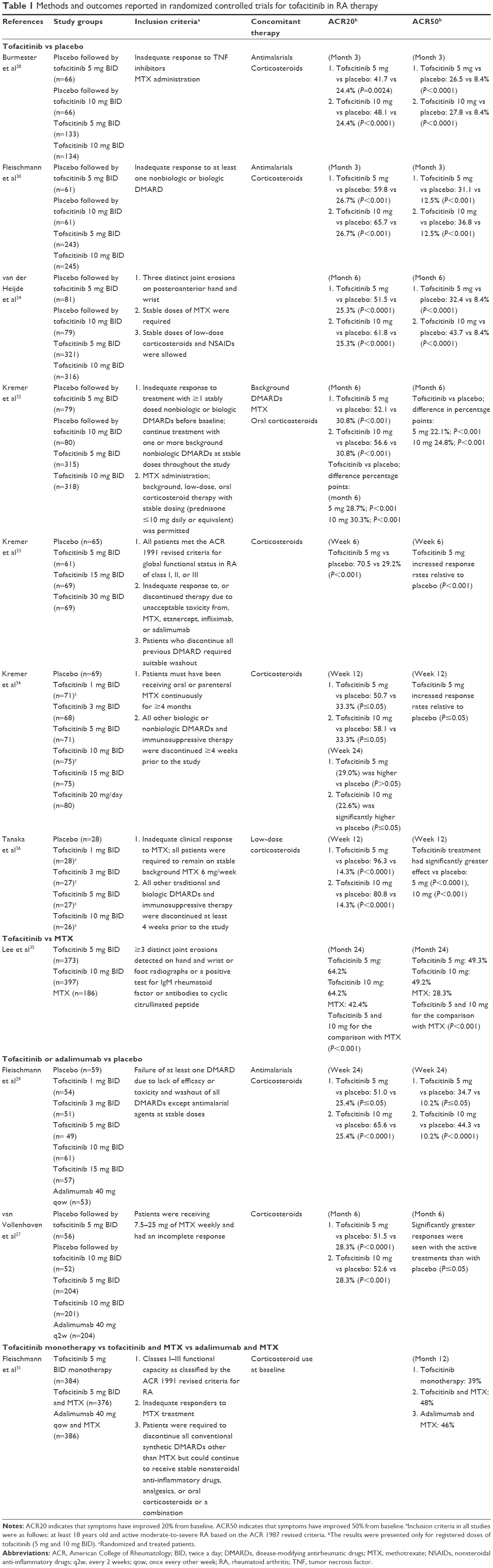 | Table 1 Methods and outcomes reported in randomized controlled trials for tofacitinib in RA therapy |
As presented in Table 1, the improvement in ACR20 and ACR50 responses was observed shortly (6 weeks) after starting tofacitinib treatment33 and the response levels remained significantly higher compared with placebo for up to 6 months.24,32,34 The highest ACR20 response (96.3%) was found at 12 weeks after the use of tofacitinib at a dose of 5 mg BID in a study by Tanaka et al.36 In most studies, the efficacy of tofacitinib was dose dependent, with a numerically higher response rate observed after a dose of 10 mg BID compared with a dose of 5 mg BID.24,27–30,32,34
In patients who had not previously received MTX over a 2-year period of treatment, tofacitinib monotherapy at a dose of 5 or 10 mg BID was correlated with a significant reduction in symptoms of RA, improvement in physical functioning, and delay of structural joint damage, as compared with MTX.35 Another study reported high efficacy of tofacitinib in patients who had shown an inadequate response to DMARDs.30
In treatment-refractory patients, of whom one-third had previously been treated with two or more TNF inhibitors, tofacitinib at doses of 5 and 10 mg BID showed rapid, significant, and clinically meaningful improvements compared with placebo. All primary and secondary outcomes improved with both doses vs placebo at 3 months.28
With regard to secondary endpoints related to disease activity, clinically meaningful and statistically significant reductions from baseline in disease activity score (DAS28)erythrocyte sedimentation rate (ESR) scores (indicating decreased disease activity) were observed in the tofacitinib group at 3 months, as compared with placebo.30 Patients receiving tofacitinib also demonstrated clinically meaningful improvements in the levels of fatigue and pain.24,30,32 Significant differences between the tofacitinib and placebo groups in both ACR and HAQ-DI responses were reported at 2 weeks.30 Tofacitinib in combination with nonbiologic DMARDs, primarily MTX, was superior to placebo in terms of the rates of ACR20 response and DAS28 (ESR)-defined remission and in improving HAQ-DI scores in patients with active RA.24,28,32,36
The results of a 6-week Phase IIa trial showed that tofacitinib administered at a dose of 5 mg BID improved the ACR20, ACR50, and ACR70 response rates in the treatment of active RA. In many patients, these improvements were already seen at 1 week.33
In a Phase IIb study, tofacitinib was investigated for the treatment of patients with active RA who had shown inadequate response to MTX monotherapy. Over 24 weeks, tofacitinib at doses >3 mg BID was effective in terms of improving the ACR response rates. The efficacy was already established at 2 weeks and was still observed at 24 weeks.34 Tofacitinib at doses of 5 and 10 mg BID showed rapid action, and a clinically significant change in ACR20 and ACR50 response rates from baseline was reported at 1 week, followed by an improvement in the quality of life scores.36
The efficacy of the 5 or 10 mg tofacitinib dose over the 12-month study period was significantly superior to placebo with respect to all clinical outcomes. The results were similar to those of adalimumab at a dose of 40 mg every 2 weeks.27,29
The ACR20 response rate at 12 weeks reached 88.6% and was maintained throughout the LTE study (up to 264 weeks). An increase in the ACR20 response rate was notable in patients who did not show adequate response to tofacitinib at a dose of 5 mg BID when the dose increased to 10 mg BID. The ACR50 and ACR70 response rates were 65.5 and 42.5%, respectively, at 12 weeks, and these results were also maintained throughout the study.38 These findings were confirmed by another LTE study, in which the ACR20, ACR50, and ACR70 response rates were maintained over the 48-month period.37
The Oral Rheumatoid Arthritis Trial (ORAL) Strategy aimed to assess the comparative efficacy of tofacitinib monotherapy, tofacitinib with MTX, and adalimumab with MTX for the treatment of RA in patients with a previous inadequate response to MTX.31 A combination therapy of tofacitinib and MTX was noninferior to a combination of adalimumab and MTX, while tofacitinib monotherapy was not shown to be noninferior to either combination.31
While direct comparisons for biologics in RA are quite rarely an option for clinical trials and no robust data are available, indirect comparisons (NMAs) could provide the most relevant and comprehensive data for the relative efficacy assessment. An NMA by Vieira et al12 revealed that tofacitinib at a dose of 5 mg BID combined with MTX was similar in terms of the relative risk (RR) of ACR20, ACR50, and ACR70 responses to abatacept, golimumab, rituximab, and tocilizumab combined with conventional DMARDs during the follow-up period of 12 and 24 weeks. The results of the NMA suggest that tofacitinib at a dose of 5 mg BID was more effective than placebo and comparable with the biologics in terms of efficacy, as measured by HAQ-DI improvement at 12 weeks.12
The above findings were confirmed by another NMA, which aimed to assess the efficacy of tofacitinib at a dose of 5 and 10 mg used as either monotherapy or combined with MTX or DMARDs, in comparison with biologic therapies at 24 weeks. The biologics included abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, infliximab, tocilizumab, and baricitinib.39
Tofacitinib 5 mg was comparable to the other monotherapies at 24 weeks in terms of ACR20 and ACR70 response rates, while tofacitinib 10 mg had more effective ACR20, ACR50, and ACR70 response rates compared to placebo and was comparable to other monotherapies. Tofacitinib 5 mg demonstrated both greater efficacy than placebo and comparability to other monotherapies for ACR50 responses. For all ACR20, ACR50, and ACR70 responses at 24 weeks, tofacitinib 5 or 10 mg combined with DMARDs was more effective than placebo accompanied by DMARDs or MTX and showed comparable responses to other combination therapies.39
In addition, for the ACR70 responses, tofacitinib 5 and 10 mg were more effective than certolizumab 400 mg, all combined with DMARDs or MTX. Tofacitinib 10 mg revealed much higher efficacy in ACR20 response than etanercept 50 mg, abatacept 10 mg/kg, and infliximab 3 mg/kg; all combined with DMARDs. The ACR50 response at 24 weeks indicated significantly higher efficacy rates of tofacitinib 5 and 10 mg compared to baricitinib (all administered with concomitant therapies), while tofacitinib 10 mg provided significantly higher ACR50 response compared to etanercept 50 mg, abatacept 125 mg, and infliximab 3 mg/kg; all administered with MTX or DMARDs. For the ACR70 response at 24 weeks, preponderance of tofacitinib over adalimumab 40 mg and abatacept 10 mg/kg combination treatments was revealed. In addition, tofacitinib 10 mg was more efficacious than etanercept 25 mg, etanercept 50 mg, infliximab 3 mg/kg, and baricitinib 2 mg; all administered with DMARDs.39
Comparative safety and tolerability of tofacitinib
Tofacitinib efficacy in RA is related to its strong immunosuppressive action reflected by inhibition of JAK-STAT activation, decrease in cytokine production, and modulation of lymphocyte proliferation;12 thus, a majority of AEs are related to reduced immunity status. The most common reported AEs in clinical trials by system organ class included: nasopharyn-gitis,28,29,31,32,34,36,37 upper respiratory tract infections,29–32,34,37 urinary tract infections,28,29,31,33,34 and bronchitis.28,29
Other most frequently revealed AEs were as follows: gastrointestinal disorders24,33–35 such as diarrhea28–30 and nausea,29,31,33,34 nervous system disorders33 including headache28–30,33,36 and dizziness,29 and abnormalities in laboratory parameters.24,30,31,33,34,36 Although the majority of AEs across all dosages of tofacitinib were mild to moderate in severity,29,31,34,36 some of them were serious, such as opportunistic infections (herpes zoster, tuberculosis, and cytomegalovirus),24,30,32,35 malignancies,31,33,35 cardiovascular (CV) events,29,30 abnormalities in clinical laboratory parameters,29,30 and gastrointestinal perforations.29
In the short-term observation period (6 weeks), the incidence of any AEs was 59% at both tofacitinib 5 mg BID and in the placebo groups, with the infection rate of 24.6% in tofacitinib 5 mg BID vs 26.2% in the placebo group.33
Between 0 and 3 months’ follow-up, treatment-emergent AEs (TEAEs) were reported with similar frequency in patients treated with tofacitinib at 5 mg BID (48.9%), tofacitinib at 10 mg BID (54.1%), and placebo (45.6%).24 In another study,36 five cases of SAEs were reported across all tofacitinib treated groups. SAEs occurred in numerically lower incidence in the tofacitinib 5 and 10 mg BID group (1.5%) than in the placebo group (4.5%), and no serious infection events were reported. Discontinuations rate due to TEAEs was similar in the tofacitinib group (5.2%) to the placebo group (5.3%).28
Between 3 and 6 months, 41.9% of the patients across all tofacitinib groups experienced TEAEs and 4% of the patients suffered from SAEs (serious infections).28 Similar results were reported at 6 months in a follow-up study by Fleischmann et al.29 Across all tofacitinib-treatment arms, 53.7% of the patients experienced TEAEs, while SAEs occurred in 2.9% of the patients treated with tofacitinib compared with 5.9% of the patients receiving placebo. The rate of infections with tofacitinib at a dose of 5 and 10 mg BID was 34.7 and 34.4%, respectively, compared with 17.6% of the patients in the placebo group. The most common significant infections in the tofacitinib groups were urinary tract infections (3.7%), pneumonia (1.5%), and sinusitis (0.7%).29 During the same follow-up period in a study by Kremer et al,34 21 patients (4.1%) experienced SAEs, including five cases of serious infections. An increased rate of serious infections with tofacitinib as compared with placebo was reported in a study by Fleischmann et al.30 Serious infections were reported in six patients treated with tofacitinib and included cellulitis, liver abscess, bronchitis, tuberculous pleural effusion, and pyelonephritis. In the placebo group, there were two incidences of cellulitis in one patient.30
During the 12-month follow-up, the incidence rates (IRs) of AEs calculated per 100 patient-years (py) of exposure were numerically lower in patients receiving tofacitinib 5 and 10 mg BID (IR =6.9 and 7.3, respectively) as compared with placebo (IR =10.9), with a similar tendency for the IR of SAEs (IR =171.9, 175.7, and 342.3 for patients receiving tofacitinib 5 and 10 mg and placebo, respectively).32 The IRs of serious infections per 100 py at 12 months in a study by van der Heijde et al24 were 4.17, 2.32, and 3.68 for the 5 and 10 mg doses and placebo, respectively. In another study, the IR of serious infections during a 12-month follow-up was numerically higher in patients receiving the 5 mg dose (3.4%) and the 10 mg dose (4%) than in the adalimumab group (1.5%).27 Finally, in a study by Fleischmann et al,31 combined MTX and tofacitinib monotherapy increased the incidence of AEs and TEAEs from 59% to 61% and from 26% to 30%, respectively, over 12 months.
A 24-month follow-up in a study by Lee et al35 revealed numerically similar data for the safety of tofacitinib and MTX treatment. The incidence of AEs was 79.6, 84.1, and 79.0%; the incidence rate of SAEs – 10.7, 10.8, and 11.8%, and of serious infections – was 3.0, 2.0, and 2.7% for tofacitinib 5 and 10 mg BID and MTX, respectively.35
An integrated analysis of Phase I–III clinical trials and LTE studies with tofacitinib in patients with RA showed that the IR of AEs, SAEs, and withdrawals due to AEs was numerically similar for both registered doses of tofacitinib (5 and 10 mg BID), regardless of whether the dose was average or constant. Infections, CV events, and malignancies were the most common causes of death.40
Pooled data from five Phase III studies suggested that younger patients (<65 years) had a lower risk of SEAs, serious infections, and withdrawal due to AEs than older patients (≥65 years). However, this could be related to comorbidities, which occur more often in older patients, and the results should therefore be interpreted with caution.41
Safety of tofacitinib relative to other biologic therapies
Due to the absence of direct comparisons of tofacitinib with biologic treatments in patients with RA, the only available safety results were based on NMA; findings indicated a comparable safety profile of tofacitinib and other biologic DMARDs used in RA.12,39 In patients treated with tofacitinib 5 mg BID, who had an inadequate response to TNF inhibitors, the risk of AEs, SAEs, and any discontinuations due to AEs was similar to other biologic DMARDs including abatacept, golimumab, tocilizumab, and rituximab; moreover, no significant differences were revealed compared to placebo. The rate of withdrawals due to limited efficacy was significantly reduced during tofacitinib treatment compared to abatacept, golimumab, tocilizumab, and placebo.12
The results of another NMA showed that tofacitinib monotherapy (5 or 10 mg BID) was associated with significantly lower risk of discontinuation due to AEs in patients with moderate or severe RA in relation to adalimumab 40 mg, every 2 weeks, and similar to other therapies (tocilizumab, adalimumab 40 mg once a week, etanercept, and certolizumab) and placebo. The rate of withdrawals due to AEs was higher during combination therapy of tofacitinib (5 or 10 mg BID) and DMARDs in comparison to placebo or abatacept added to DMARDs; for the remaining combination therapies, the rate was similar. Tofacitinib added to MTX did not have a significantly higher rate of discontinuation due to AEs compared to other biologic treatments or placebo, both in combination with MTX.39
Opportunistic infections
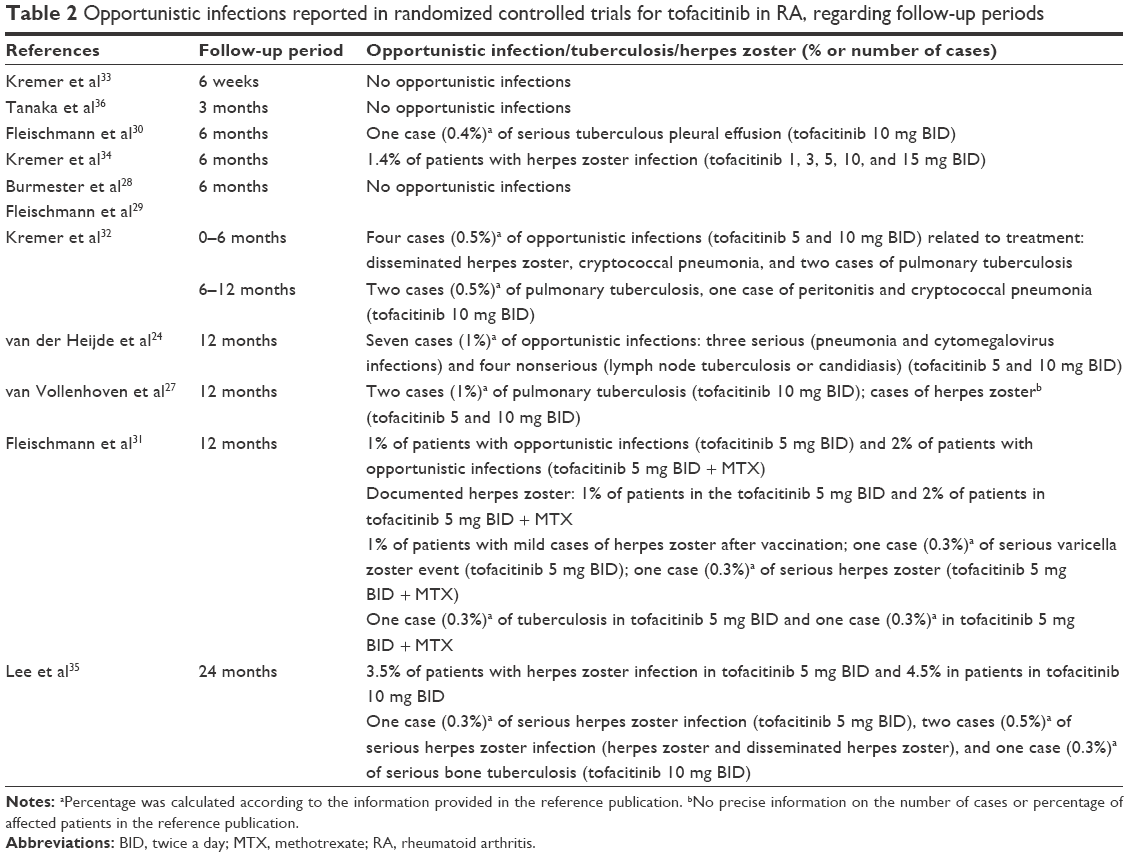
Clinical evidence suggests that tofacitinib treatment is associated with an increased risk of infections. Considering its mechanism of action affecting the immune system and the increased baseline risk of herpes zoster among patients with RA, an increased risk of opportunistic infections is quite an important issue.40,42,43 As expected, the cases of opportunistic infections were reported in main randomized controlled trials (RCT); the summary of these events is presented in Table 2.
 | Table 2 Opportunistic infections reported in randomized controlled trials for tofacitinib in RA, regarding follow-up periods |
In short-term therapy, no opportunistic infections were reported, but between 6 and 12 months, in tofacitinib 5 mg and 10 mg therapies cases appeared of opportunistic infections such as herpes zoster,24,31,32,34 tuberculosis,24,27,30–32 cryptococcal pneumonia,32 candidiasis,24 cytomegelovirus,24 and varicella zoster.31 At 24 months’ observation, the rate of herpes zoster infection was 4.5%.35
A review of Phase II, Phase III, and LTE studies from a tofacitinib program in RA showed an increase in the risk of opportunistic infections, which was numerically higher in patients receiving a dose of 10 mg BID than that of 5 mg BID.44 Another review of RCT and LTE studies revealed increased IRs of herpes zoster (IR =4.4/100 py) in patients treated with tofacitinib, particularly among Asian patients.45 The authors reported that complicated cases of herpes zoster among tofacitinib-treated patients were rare45 and visceral disease was not reported. The rates of herpes zoster were similar between the low- and high-dosage tofacitinib groups; although during the first few months after initiating the therapy, a higher risk was observed in the group receiving the 10 mg dose. Data suggest that preventive strategies for herpes zoster should be developed for patients with RA, given the high rate of morbidity observed regardless of RA therapy.45 The results from a meta-analysis of tofacitinib trials in RA also confirmed an increased risk of herpes virus infection (IR =3.87).46 As compared with other biologic DMARDs, the absolute rate differences for herpes zoster during tofacitinib treatment were approximately doubled per 100 py.46
The safety analysis of exposure to tofacitinib among 6,194 adult patients, with RA up to 8.5 years, from two Phase I, nine Phase II, six Phase III, and two LTE studies revealed that the rate of AEs was generally stable over time. The IR of serious infections was 2.7/100 py, and the IR of herpes zoster was 3.9/100 py. Moreover, the analysis by 6-month intervals did not reveal any notable increase in the risk of serious infections and herpes zoster with longer exposure to tofacitinib. The rates of complicated herpes zoster or serious herpes zoster infections were low and comparable with those of multidermatomal and ophthalmic herpes zoster reported with other DMARDs.40
A common opportunistic infection revealed in RCT trials (Table 2) was tuberculosis, although it was quite rare in the regions of low prevalence. This suggests that latent infections are incited by tofacitinib treatment.40,44 Patients with RA show a generally higher incidence of tuberculosis than the general population or patients receiving TNF inhibitors. However, a higher risk of tuberculosis infection after treatment with tofacitinib was shown to be within the range for biologic DMARDs.40
CV AEs
According to clinical studies, patients with RA have an increased risk of CV disease and CV-related death compared with the general population.47,48
One death attributed to a cerebrovascular accident was reported in a patient receiving tofacitinib at a dose of 15 mg BID, but no CV events were reported for tofacitinib at the doses of 5 mg and 10 mg during a 6-month follow-up. In the same study, hypertension developed in 2% of the patients receiving tofacitinib at a dose of 5 mg BID and 4.9% of those receiving with tofacitinib at a dose of 10 mg BID compared with 2.9% of the patients receiving placebo. However, none of these cases were classified as severe.29 In a study by Fleischmann et al,30 two patients in the 10 mg group developed congestive heart failure within 6 months of therapy.
During a 12-month follow-up, nonfatal major adverse events were not revealed in the groups receiving tofacitinib or a combination of tofacitinib and MTX groups, while there were two events (1%) in the group receiving adalimumab and MTX.31 During the same period in a study by van der Heijde et al,24 no patient had congestive heart failure; however, six patients treated with tofacitinib experienced six non-fatal CV events: three of them were classified as CV events (carotid artery stenosis, angina pectoris, and coronary artery disease) and the others as cerebrovascular events (lacunar infarction and cerebral infarction).
In another study, the following three CV events occurred during 12 months of tofacitinib therapy: transient ischemic attack, cerebrovascular accident (both at the 5 mg dose), and congestive heart failure (10 mg).32
CV safety findings in patients with RA were assessed in a pooled analysis, including 3,942 py of follow-up in Phase III studies and 8,699 py of follow-up in LTE studies on tofacitinib. Treatment with tofacitinib (5 or 10 mg BID) was associated with a low rate of CV events, despite short- and long-term increases in traditional levels. The rates of CV events with tofacitinib were generally similar to those observed for placebo, adalimumab, and MTX. Furthermore, the rates of CV events did not increase with longer treatment duration. Blood pressure was generally stable over time, and in Phase III studies, the number of hypertension-related AEs was low during tofacitinib treatment. These data were consistent with previously reported rates of CV events among patients with RA, including those treated with TNF inhibitors; however, analyses with longer follow-up are needed to confirm these findings.49
Malignancies
Chronic inflammation and autoimmune diseases such as RA, as well as clinical use of immunosuppressive drugs, are potentially correlated with an increased risk of malignancy.30,50
Malignancies were reported in some RCTs, and the following six cases of cancer in a 6-month follow-up were revealed: non-Hodgkin’s lymphoma, chronic lymphocytic leukemia (tofacitinib 5 mg), prostate cancer, Burkitt’s B-cell lymphoma, colon cancer (tofacitinib 10 mg), and only gastric cancer in the MTX group.35 A 12-month follow-up revealed cases of salivary gland neoplasm, benign hair follicle tumor, metastatic renal-cell carcinoma, and non-small-cell lung cancer diagnosed in the group receiving the 5 mg dose of tofacitinib and cholesteatoma, cervix carcinoma, and benign ovarian germ-cell teratoma in the group receiving the 10 mg dose of tofacitinib.27 However, in another study of 12 months’ duration, the number of malignancies was lower: one case (<1%) was reported in the tofacitinib monotherapy group, while two other patients (1%) developed nonmelanoma skin cancer (NMSC).31 In another study, nine patients were diagnosed with carcinomas: basal cell carcinoma (four cases), stomach adenocarcinoma (two cases), bone squamous cell carcinoma (one case), non-Hodgkin’s lymphoma (one case), and breast mucinous adenocarcinoma (one case) during 12 months of the 24 months’ follow-up. In addition, one patient in the tofacitinib-treated group (10 mg) had unconfirmed diagnosis of cervical squamous cell carcinoma.24
No cases of malignancies were revealed in other RCT studies;28,29,32–34,36 however, RCTs are inadequate to detect and evaluate rare events, such as malignancies, in comparison with LTE studies, which offer better chances to evaluate rare events.51
A meta-analysis and NMA of long-term RCTs (≥12 months) showed that the risk of all malignancies, solid malignancies, and hematological malignancies associated with the use of biologic DMARDs and tofacitinib was not increased compared with placebo or conventional synthetic DMARDs.51 A meta-analysis of RCT trials revealed a Peto odds ratio (OR) of overall malignancies not significantly higher compared with placebo (Peto OR =2.08; 95% CI: 0.50, 8.59), while the analysis of LTE studies demonstrated an IR of 1.78/100 py (95% CI: 1.50, 2.07) for tofacitinib treatment.51
Aggregated data from six Phase II, six Phase III, and two LTE studies involving tofacitinib (5 or 10 mg BID) revealed 107 cases of malignancy among 5,671 patients treated with tofacitinib (with the exception of NMSC). The most common malignancies were lung cancer (24 cases), breast cancer (19 cases), lymphoma (10 cases), and gastric cancer (six cases). The overall rates and types of malignancies remained stable over time irrespective of increasing the exposure to tofacitinib. Standardized IR for all malignancies (except NMSC) as well as lung cancer, breast cancer, lymphoma, and NMSC were within the expected range for patients with moderate or severe RA.50
Clinical trials and LTE studies involving tofacitinib-treated patients with RA reported a risk of NMSC of 1.16%, with the overall IR of 0.53/100 py (95% CI: 0.41, 0.67). The rates of NMSC in RCT studies were similar between patients receiving tofacitinib at a dose of 5 and 10 mg BID, while LTE studies suggested the dose-dependent risk of cancer.50 These findings were confirmed by a meta-analysis of 18 studies (two Phase I, eight Phase II, six Phase III, and two LTE studies), which revealed 83 (1.36%) cases of NMSC in a group of 6,092 tofacitinib-treated patients with RA. The risk of NMSC was generally small and stable (IR =0.55/100 py) during the 84-month follow-up.52 A numerically higher IR of NMSC in LTE studies was observed during therapy with the 10 mg dose in comparison with the 5 mg dose. However, further LTE studies are necessary to confirm these data.52
White blood cell parameters
Most clinical trials revealed a general tendency for a reduction in neutrophil29,34,35 and lymphocyte counts,35 but the disturbances usually stabilized during therapy.53 The mechanism by which tofacitinib decreases the circulating neutrophil count may be associated with the inhibition of JAK-dependent IL-6 signaling, which is involved in neutrophil activation, or with more general anti-inflammatory effects.53 In Phase II studies of tofacitinib in RA, the reduced neutrophil count was accompanied by a decrease in hemoglobin levels, which in some cases was clinically significant (>2 g/dL).54
A significant decrease in neutrophil and lymphocyte counts was observed in a group receiving tofacitinib at a dose of 10 mg compared to the MTX group during 24 months’ follow-up.35 The incidence of confirmed mild neutropenia was 9.1% with tofacitinib at a dose of 5 mg BID and 6.5% with tofacitinib at a dose of 10 mg BID, but neither was classified as severe or life threatening.29 Lower neutrophil count was observed with tofacitinib treatment in a dose-dependent manner in comparison with placebo at 12 months.34 In a number of studies, a decrease in neutrophil count was usually slight and none of the patients showed a potentially life-threatening reduction in absolute neutrophil count (febrile neutropenia).24,28,30,32,34
LTE studies reported a decreased mean neutrophil count during tofacitinib treatment (Table 3). The mean lymphocyte count increased slightly during the first 3 months of treatment with either dose of tofacitinib, and after that time, progressively decreased in Phase III studies. A further slow decrease was revealed up to month 48 in LTE studies with both doses of tofacitinib and then it stabilized.53
 | Table 3 Clinical laboratory results during tofacitinib treatment in RA |
Cholesterol levels
During Phase II studies, an increase in low-density lipoprotein (LDL) cholesterol and high-density lipoprotein (HDL) cholesterol levels was observed in patients with active RA receiving treatment with tofacitinib.29,32,34,36,55 In several studies, a dose-dependent elevation of LDL,30,32,33,35,36 HDL,32,33,35,36 and total cholesterol29,32,33,36 was observed. Generally, an increase in lipid levels occurred shortly after starting tofacitinib treatment (week 2), reached the highest value at 6–8 weeks, and then appeared to plateau, in a study by Kremer et al,34 while plateau was observed between 4 and 12 weeks in a study by Tanaka et al,36 and between 3 and 12 months in a study by Kremer et al32 (Table 3).
A study by Fleischmann et al29 reported increased levels of total, HDL, and LDL cholesterol in all tofacitinib-treated groups (41.2% [5 mg dose] and 56.4% [10 mg dose]) than in the placebo group (24.0%). During a 3-month follow-up in a study by Fleischmann et al,30 the mean LDL cholesterol level increased by 3.5% in the placebo group compared with 13.6% in the group receiving the 5 mg dose and 19.1% in the group receiving the 10 mg dose. In another study, an increase in HDL and LDL cholesterol levels was 10% in the tofacitinib groups and 7% in the placebo group.28 During the 24-month follow-up, the mean LDL cholesterol levels increased by 18.6, 21.6, and 3.9% in the tofacitinib 5 and 10 mg and MTX groups, respectively, while HDL cholesterol levels increased by 16.8, 17.4, and 7.0%, respectively.35 A meta-analysis of clinical trials on tofacitinib revealed the RR of hypercholesterolemia to be 1.70 (95% CI: 1.10, 2.63) and to be dose dependent.56
Hemoglobin
Some decrease in hemoglobin level during tofacitinib treatment was revealed in reference clinical trials.27,28,34 Mean changes from baseline in the hemoglobin level were low across all tofacitinib treatment groups at the 6 months’ period.29 In the follow-up period of 3 months, the rate of patients with decreased hemoglobin was no higher in the tofacitinib treatment groups (7.8% for 5 mg BID and 12.9% for 10 mg BID) than in the placebo group (10.2%) and most cases of decreased hemoglobin were mild to moderate in severity28 and did not impose treatment discontinuations29,34 or cause anemia34 except for in one study reporting 4.2% of cases of anemia in the tofacitinib 10 mg BID group.29 Changes were seen during months 0–3 of active treatment and stabilized in most cases in 6 months.28 Only one patient in the tofacitinib 10 mg group experienced potentially life-threatening hemoglobin decrease in the follow-up period of 12 months.27
Results of six RCT studies of 6–24 months duration, collected in a pooled analysis, revealed low incidence of clinically meaningful reductions in hemoglobin and incidence of anemia for tofacitinib treatment, similar to placebo or MTX. Reductions in fatigue and improvements in vitality demonstrated during tofacitinib treatment were largely independent of hemoglobin changes. Generally a numerically higher hemoglobin level was reported in tofacitinib 5 mg (0.47 g/dL) and 10 mg BID (0.28 g/dL) up to 24 months compared to placebo group (−0.01 g/dL) in 6 months follow-up. . Results of LTE studies showed that changes in hemoglobin were generally stable up to 66 months (Table 3), with similar percentages of patients with decreased level in both tofacitinib 5 and 10 mg BID groups.53
Creatine kinase
Recent evidence showed an increase in serum creatine level after initiation of tofacitinib treatment.24,29,31–36 Significant but not clinically meaningful increases in creatine level in tofacitinib groups were reported in a study,36 while elevation of creatine level was a cause of treatment discontinuation in four patients during 12 months of tofacitinib therapy.32 Persistent increase in serum creatine level was reported in six patients.29 A total of 51 patients had an elevation of serum creatine not fully resolved by the end of the study,33 but in most cases, the level returned to within 10% of their baseline. The elevation of serum creatine >50% from baseline occurred during a 12 months’ period in <1% of patients with RA treated with tofacitinib (5 and 10 mg); however, none of them experienced renal failure.24 In case of three patients (two in the tofacitinib 5 mg and one in the 10 mg group) an increase of at least 50% in creatine level was reported were reported during a 6 months’ period in a study,30 but it should be noted that the highest creatine level was within the normal range. Similar findings were found in Fleischmann et al29 and Kremer et al,34 in which four and five patients, respectively, experienced >50% increase in creatine level in the tofacitinib groups. At week 24, elevation of serum creatine >30% or 0.2 mg/dL was noted in 20.5 and 21.6% of patients treated with tofacitinib 5 and 10 mg, respectively.34 In a long-term observation period of 24 months, >33% elevation of creatine level was observed in 9.5 and 9.6% of patients treated with tofacitinib 5 and 10 mg, respectively, while >50% elevation of creatine level was observed in 1.6 and 2.8%.35 Continuing tofacitinib treatment in LTE study results in the stabilization of serum creatine level up to 288 weeks (Table 3).38
Other AEs of special interest
Although special attention was given to gastrointestinal perforations as AEs during DMARD treatment,40 only one case of temporary discontinuation of treatment due to gastrointestinal bleeding in a patient receiving tofacitinib at a dose of 3 mg BID was described.29 Data from LTE studies lasting up to 8.5 years also revealed a low risk of gastrointestinal perforations associated with tofacitinib treatment.40 In several studies, an increase in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels was reported among the RA24,27,34,35 patients treated with tofacitinib. In most RCT trials, an increase in AST or ALT levels three times above the normal range was rare,24,27,30,35 and occurred in single cases with a spontaneous return to normal levels, without association with study duration.24 A similar risk for increased aminotransferase levels was reported for patients treated with tofacitinib at a dose of 5 and 10 mg BID and MTX during long-term follow-up of 24 months.35 In contrast, elevated ALT and AST levels were the most common AEs reported up to 12 weeks (>5% in all treatment groups) in studies by Kremer et al34 and Tanaka et al,36 where the incidence of elevated ALT was up to 22.2% and the incidence of elevated AST was up to 14.8% in the group receiving tofacitinib at a dose of 5 mg BID. Discontinuation of treatment due to increased AST or ALT levels was rare and reported only in three cases.32
Patient-focused perspectives such as quality of life and patient’s satisfaction, acceptability, and adherence
Patients with active RA treated with tofacitinib combined with conventional DMARD therapy reported significant, sustained, and clinically meaningful improvements in patient-related outcomes (PROs) compared to placebo. Statistically significant improvements from baseline vs placebo were recorded at month 3 and sustained to 12 months after initiation of the therapy, in a number of domains relating to all eight SF-36 domains with tofacitinib 10 mg BID and seven SF-36 domains with tofacitinib 5 mg BID. Significantly, more tofacitinib-treated patients reported improvements greater than or equal to the minimum clinically important differences at month 3 vs placebo in all PROs, except the SF-36 role-emotional domain (results were significant for tofacitinib 10 mg BID).57
Two double blind, placebo-controlled studies58 evaluated patients with inadequate response to MTX who received tofacitinib 1–15 mg BID or 20 mg once daily or placebo, in a number of life quality domains; significant improvements vs placebo were observed at week 12. In the monotherapy study (n=384), significant improvements in the quality of life were reported at week 12 for tofacitinib 5 and 10 mg BID. In both studies, improvements in several domains of the SF-36 in the tofacitinib groups were seen at weeks 12 and 24. Tofacitinib, either in combination with MTX or as monotherapy, demonstrated rapid and sustained improvement in health-related quality of life.58
At month 3, tofacitinib 10 mg BID treatment resulted in significant changes from baseline vs placebo across all PROs, sustained to month 12, with the highest number of patients reporting improvements, at least of minimum clinically important differences, vs placebo. Changes from baseline at month 3 with tofacitinib 5 mg BID and adalimumab were similar and statistically significant compared to placebo across most patient-related outcomes; in the SF-36 Mental Component Score and Social Functioning, Role Emotional, and Mental Health domains, significantly more patients reported relevant improvements compared to placebo. Patients with moderate-to-severe RA and inadequate responses to MTX reported significant improvements across a broad range of PROs with tofacitinib 5 and 10 mg BID and adalimumab, that were significantly superior to placebo.59
In another study, it was revealed that tofacitinib was more commonly used as monotherapy and yielded at least comparable persistence and adherence as to adalimumab, etanercept, and abatacept.60
Tofacitinib, an oral drug, provides patients an alternative to the subcutaneously or intravenously administered biologic DMARDs route; it should be preferred as a more convenient and easier way of drug administration. It was confirmed that, in a survey completed by 380 patients with RA, the majority (56.4%) of patients prefer oral administration of drugs rather than parenteral and suggest that it could be a more attractive and accepted way of treatment among RA patients.61
Conclusions, place in therapy
Tofacitinib is a drug with a different mechanism of action than other products approved for the treatment of RA and may be considered as a therapeutic option in some patients. Regardless of its mechanism of action, the oral administration route might be preferred by some patients. Randomized controlled studies revealed a significant benefit over placebo in terms of the efficacy outcomes (ACR20 and ACR50 response rates), and clinically significant improvements in PROs were reported compared with placebo. Clinical trials on tofacitinib reported an increased risk of infections, including serious infections; therefore, strict and rigorous monitoring is required especially in patients older than 65 years. Further studies, especially in a clinical practice setting, are necessary to better understand the efficacy and safety profile of tofacitinib.
Disclosure
The authors report no conflicts of interest in this work.
References
Tobon GJ, Youinou P, Saraux A. The environment, geoepidemiology, and autoimmune disease: rheumatoid arthritis. J Autoimmun. 2010;35:10–14. | ||
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. | ||
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26. | ||
National Institute for Health and Clinical Excellence (NICE); National Collaborating Centre for Chronic Conditions. Rheumatoid Arthritis: The Management of Rheumatoid Arthritis in Adults. London (UK): Royal College of Physicians; 2009:79. | ||
Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38:727–735. | ||
Storage SS, Agrawal H, Furst DE. Description of the efficacy and safety of three new biologics in the treatment of rheumatoid arthritis. Korean J Intern Med. 2010;25:1–17. | ||
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. | ||
Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363(9410):675–681. | ||
Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50:1400–1411. | ||
Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343(22):1594–1602. | ||
Kiely PD, Deighton C, Dixey J, Ostor AJ. Biologic agents for rheumatoid arthritis–negotiating the NICE technology appraisals. Rheumatology (Oxford). 2012;51(1):24–31. | ||
Vieira MC, Zwillich S, Jansen JP, Smiechowski B, Spurden D, Wallenstein GV. Tofacitinib versus biologic treatments in patients with active rheumatoid arthritis who have had an inadequate response to tumor necrosis factor inhibitors: results from a network meta-analysis. Clin Ther. 2016;38(12):2628–2641. | ||
Kalden JR. Emerging therapies for rheumatoid arthritis. Reumathol Ther. 2016;3(1):31–42. | ||
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–243. | ||
Blanco FJ, Möricke R, Dokoupilova E, et al. Secukinumab in active rheumatoid arthritis: a phase III randomized, double-blind, active comparator- and placebo-controlled study. Arthritis Rheumatol. 2017;69(6):1144–1153. | ||
Genovese MC, Greenwald M, Cho CS, et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014;66(7):1693–1704. | ||
Taylor PC, Keystone EC, van der Heijde D, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:652–662. | ||
Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. 2016;75(6):1057–1064. | ||
Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. 2017;76(6):1009–1019. | ||
Abushouk AI, Ahmed H, Ismail A, et al. Safety and efficacy of ocrelizumab in rheumatoid arthritis patients with an inadequate response to MTX or tumor necrosis factor inhibitors: a systematic review and meta-analysis. Rheumatol Int. 2017;37(7):1053–1064. | ||
Kontzias A, Laurence A, Gadina M, O’Shea JJ. Kinase inhibitors in the treatment of immune-mediated disease. F1000 Med Rep. 2012;4:5. | ||
Maeshima K, Yamaoka K, Kubo S, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012;64(6):1790–1798. | ||
Meyer DM, Jesson MI, Li X, et al. Antiinflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm (Lond). 2010;7:41. | ||
van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving MTX twelve-month data from a twenty-four–month phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570. | ||
Hodge JA, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(2):318–328. | ||
EMA. Summary of Product Characteristic. 2017. Avaliable from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004214/WC500224911.pdf. Accessed September 28, 2017. | ||
van Vollenhoven R, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–519. | ||
Burmester GR, Blanco R, Charles-Schoeman CH, Wollenhaupt J. Tofacitinib (CP-690,550) in combination with MTX in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase III trial. Lancet. 2013;381:451–460. | ||
Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64(3):617–629. | ||
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367(6):495–507. | ||
Fleischmann R, Mysler E, Hall S, et al; ORAL Strategy Investigators. Efficacy and safety of tofacitinib monotherapy, tofacitinib with MTX, and adalimumab with MTX in patients with rheumatoid arthritis (ORAL Strategy): a phase IIIb/IV, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390(10093):457–468. | ||
Kremer J, Li ZG, Hall S, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159(4):253–261. | ||
Kremer JM, Bloom BJ, Breedveld FC, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60(7):1895–1905. | ||
Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background MTX in patients with active rheumatoid arthritis and an inadequate response to MTX alone. Arthritis Rheum. 2012;64(4):970–981. | ||
Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus MTX in rheumatoid arthritis. N Engl J Med. 2014;370:2377–2386. | ||
Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH; Tofacitinib Study Investigators. Phase II study of tofacitinib (CP-690,550) combined with MTX in patients with rheumatoid arthritis and an inadequate response to MTX. Arthritis Care Res (Hoboken). 2011;63:1150–1158. | ||
Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open label, long term extension studies. J Rheumatol. 2014;41:837–852. | ||
Yamanaka H, Tanaka Y, Takeuchi T, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background MTX, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. 2016;18:34. | ||
Bergrath E, Gerber R, Gruben D, Tatjana L, Makin C, Wallenstein G. Tofacitinib versus biologic treatments in moderate-to-severe rheumatoid arthritis patients who had an inadequate response to nonbiologic DMARDs: systematic literature review and network meta-analysis. Int J Rheumatol. 2017;2017:8417249. | ||
Cohen SB, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis. 2017;76:1253–1262. | ||
Curtis JR, Schulze-Koops H, Takiya L, et al. Efficacy and safety of tofacitinib in older and younger patients with rheumatoid arthritis. Clin Exp Rheumatol. 2017;35(3):390–400. | ||
Cohen S, Radominski SC, Gomez-Reino JJ, et al. Analysis of infections and all-cause mortality in phase II, phase III, and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66:2924–2937. | ||
Souto A, Maneiro JR, Salgado E, Carmona L, Gomez-Reino JJ. Risk of tuberculosis in patients with chronic immunemediated infammatory diseases treated with biologics and tofacitinib: a systematic review and meta-analysis of randomized controlled trials and long-term extension studies. Rheumatology (Oxford). 2014;53(10):1872–1885. | ||
Winthrop KL, Park SH, Gul A, et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75(6):1133–1138. | ||
Winthrop KL, Yamanaka H, Valdez H, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(10):2675–2684. | ||
Curtis JR, Xie F, Yun H, Bernatsky S, Winthrop KL. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75(10):1843–1847. | ||
Solomon DH, Goodson NJ, Katz JN, et al. Patterns of cardiovascular risk in rheumatoid arthritis. Ann Rheum Dis. 2006;65(12):1608–1612. | ||
Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a metaanalysis of observational studies. Ann Rheum Dis. 2012;71(9):1524–1529. | ||
Charles-Schoeman CH, Wicker P, Gonzalez-Gay MA, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum. 2016;46(3):261–271. | ||
Curtis JR, Lee EB, Kaplan IV, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. 2016;75(5):831–841. | ||
Maneiro JR, Soutoa A, Gomez-Reinoa JJ. Risks of malignancies related to tofacitinib and biological drugs in rheumatoid arthritis: systematic review, meta-analysis, and network meta-analysis. Semin Arthritis Rheum. 2017;47(2):149–156. | ||
Curtis JR, Lee EB, Martin G, et al. Analysis of non-melanoma skin cancer across the tofacitinib rheumatoid arthritis clinical programme. Clin Exp Rheumatol. 2017;35(4):614–622. | ||
Schulze-Koops H, Strand V, Nduaka CH. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase III and long-term extension studies. Rheumatology. 2017;56(1):46–57. | ||
Riese RJ, Krishnaswami S, Kremer J. Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes. Best Pract Res Clin Rheumatol. 2010;24(4):513–526. | ||
Souto A, Salgado E, Maneiro JR, Mera A, Carmona L, Gomez-Reino JJ. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta-analysis. Arthritis Rheumatol. 2015;67(1):117–127. | ||
Salgado E, Maneiro JR, Carmona L, Gomez-Reino JJ. Safety profile of protein kinase inhibitors in rheumatoid arthritis: systematic review and metaanalysis. Ann Rheum Dis. 2014;73:871–882. | ||
Strand V, Kremer JM, Gruben D, Krishnaswami S, Zwillich SH, Wallenstein GV. Tofacitinib in combination with conventional disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: patient-reported outcomes from a phase III randomized controlled trial. Arthritis Care Res (Hoboken). 2017;69(4):592–598. | ||
Wallenstein GV, Kanik KS, Wilkinson B, et al. Effects of the oral Janus kinase inhibitor tofacitinib on patient-reported outcomes in patients with active rheumatoid arthritis: results of two phase II randomised controlled trials. Clin Exp Rheumatol. 2016;34(3):430–442. | ||
Strand V, van Vollenhoven RF, Lee EB, et al. Tofacitinib or adalimumab versus placebo: patient-reported outcomes from a phase III study of active rheumatoid arthritis. Rheumatology (Oxford). 2016;55(6):1031–1041. | ||
Harnett J, Gerber R, Gruben D, Koenig AS, Chen C. Evaluation of real-world experience with tofacitinib compared with adalimumab, etanercept, and abatacept in RA patients with 1 previous biologic DMARD: data from a U.S. Administrative Claims Database. J Manag Care Spec Pharm. 2016;22(12):1457–1471. | ||
Louder AM, Singh A, Saverno K, et al. Patient preferences regarding rheumatoid arthritis therapies: a conjoint analysis. Am Health Drug Benefits. 2016;9(2):84–93. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.