")
Back to Journals » International Journal of Nanomedicine » Volume 15
Eudragit®-S100 Coated PLGA Nanoparticles for Colon Targeting of Etoricoxib: Optimization and Pharmacokinetic Assessments in Healthy Human Volunteers
Authors El-Maghawry E , Tadros MI , Elkheshen SA, Abd-Elbary A
Received 30 December 2019
Accepted for publication 11 May 2020
Published 8 June 2020 Volume 2020:15 Pages 3965—3980
DOI https://doi.org/10.2147/IJN.S244124
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Enas El-Maghawry,1 Mina I Tadros,2 Seham A Elkheshen,2 Ahmed Abd-Elbary2,†
1Department of Pharmaceutics & Pharmaceutical Technology, Faculty of Pharmaceutical Sciences and Pharmaceutical Industries, Future University in Egypt, Cairo, Egypt; 2Department of Pharmaceutics & Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt
†Ahmed Abd-Elbary passed away on August 18, 2019
Correspondence: Mina I Tadros
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt
Tel +2 1223620458
Fax +2 223628246
Email [email protected]
Aim: Etoricoxib is a selective inhibitor of COX-2 enzyme. It is proposed as a potent anti-inflammatory drug intended for the control of irritable bowel syndrome. The current work aimed at developing etoricoxib-loaded nanoparticles for colon- targeting.
Materials and Methods: PLGA nanoparticles were developed via nano-spray drying technique. The D-optimal design was adopted for the investigation of the influence of i) DL-lactide-coglycolide (PLGA) concentration, ii) polyvinylpyrrolidone K30 (PVP K30) concentration and iii) lactide:glycolide ratio in the copolymer chain on the yield%, the encapsulation efficiency (EE%), particle size (PS) and percentage of drug release after 2h (P2h), 4h (P4h) and 12h (P12h). To promote colon targeting of the systems, the best achieved system (M14) was either directly coated with poly(methacrylic acid-co-methyl methacrylate) [Eudragit®-S100] or loaded into hard gelatin capsules and the capsules were coated with poly(methacrylic acid-co-methyl methacrylate) (E-M14C). The pharmacokinetic parameters of etoricoxib following oral administration of E-M14C in healthy volunteers were assessed relative to commercial etoricoxib tablets.
Results: M14 system was prepared using PLGA (0.5% w/v) at a lactide:glycolide ratio of 100:0, in the presence of PVP K30 (2% w/v). M14 system was nano-spherical particles of 488 nm size possessing promising yield% (63.5%) and EE% (91.2%). The percentage drug released after 2, 4 and 12 hours were 43.41%, 47.34 and 64.96%, respectively. Following M14-loading into hard gelatin capsules and coating with poly(methacrylic acid-co-methyl methacrylate) [Eudragit-S100], the respective P2h, P4h and P12h were 10.1%, 28.60% and 65.45%. Significant (p < 0.05) differences between the pharmacokinetic parameter of E-M14C in comparison with the commercial product were revealed with a delay in Tmax (from 2.5h to 6h), a prolongation in MRT0-∞ (from 24.4h to 34.7h) and an increase in the relative oral bioavailability (4.23 folds).
Conclusion: E-M14C is a potential system for possible colon targeting of etoricoxib.
Keywords: colon targeting, etoricoxib, nano spray drying, PLGA, Eudragit-S100
Introduction
The colon is an organ that is vulnerable to major inflammatory bowel diseases (IBD). Two of these diseases are ulcerative colitis and Crohn’s disease, which are relapsing chronic inflammatory disorders of the intestinal mucosa.1 Ulcerative colitis affects the lining of the large intestine (colon) and rectum. Repeated inflammatory swelling leads to thickening of the intestinal wall and formation of scar tissues in the rectum. Death of colon tissue or severe infection with sepsis formation may occur in severe cases.2
Colon-specific drug delivery systems have gained increasing attention for the treatment of such diseases. A targeted colonic carrier of drugs provides local delivery of the drug to the colon.3 Some of the advantages of targeting a drug to the colon include lowering incidences of adverse side effects, delivering the drug to the target site and lowering the conventional dose.4 Many non-steroidal anti-inflammatory drugs for the treatment of colon diseases have been formulated into colon-targeted delivery systems as diclofenac,5 piroxicam,6 ibuprofen,7 ketoprofen,8,9 flurbiprofen,10 lornoxicam,11 celecoxib12 and etoricoxib.13
Compared to other NSAIDs, etoricoxib (5-chloro-6-methyl-3-[4-methylsulfonyl) phenyl]-2,3-bipyridine) is considered as an effective drug for the treatment of IBD.14,15 It is characterized by being a selective inhibitor of cyclooxygenase-2 (COX-2) enzyme leading to the production of fewer prostaglandins; hence, pain and inflammation are lessened and the side effects produced by blocking the COX-1 enzyme are reduced.16 Few attempts investigated the design of colon-targeting systems for etoricoxib including the preparation of tablets coated with three layers of Eudragit® E100, HPMC and Eudragit® L100 as enteric coat13 and capsules coated with a three-layered film consisting of an acid-soluble polymer, a water-soluble polymer, and cellulose acetate phthalate as enteric polymer.17
The dimensions of the delivery system greatly influence the targeting of the inflamed colon where nanosized particles are more readily taken up by macrophages or dendritic cells at the site of active inflammation.18 Furthermore, they have a better ability to adhere to the mucus layers, thus delivering higher amounts of the entrapped drugs to the inflamed areas, which result in better therapeutic efficacy and reduced systemic adverse effects.3,19 The spray drying technique is one of the advanced technologies to develop nanoparticles possessing narrow particle size distributions and relatively high yields and drug entrapment efficiencies.
PLGA is a highly biocompatible, biodegradable,20 mechanically processable polymer that degrades yielding water-soluble and non-toxic products of normal metabolism.21–24 Degradation of PLGA occurs through hydrolysis of its ester linkages in the presence of water.25 Through this hydrolytic attack, random chain scission occurs, causing its degradation into lactic and glycolic acids.26 PLGA monomers; lactic acid and glycolic acid are non-toxic and can be removed from the body by normal metabolic pathways.27 It has been shown that the time required for the degradation of PLGA is related to the ratio of monomers used in its production, ie, controlling the degradation rate can be achieved by adjusting the ratio of DL-lactide to glycolide in the copolymer.28 The degradation rate of PLGA decreases with increasing either the lactide or the glycolide content of the copolymer.29 Copolymers of PLGA were approved by the FDA for numerous applications including extended-release pharmaceuticals.30,31 Practically, it is possible to tune the overall physical properties of the drug-polymer matrix by controlling certain parameters such as molecular weight of the polymer, lactide to glycolide ratio as well as the nature and concentration of the drug.32–34
The current work aimed at developing and optimizing a promising etoricoxib-loaded nanosystem for colon targeting applying the D-optimal design via nano-spray drying technique using poly (lactide-co-glycolide) (PLGA) copolymers. To promote the colon targeting potential of PLGA nanoparticles, the best achieved nanosystem was optimized via either coating the nanoparticles with poly (methacrylic acid-co-methyl methacrylate) [Eudragit-S100] during the nanospray drying process or through loading the nanoparticles into hard gelatin capsules and coating the capsules with poly (methacrylic acid-co-methyl methacrylate). The pharmacokinetic parameters of etoricoxib following oral administration of the most optimized system were assessed in healthy volunteers relative to the market product. To the best of our knowledge, the colon targeting potential of etoricoxib was never explored using our optimized nanospray-dried system nor the colon targeting of the drug was pharmacokinetically validated in healthy volunteers.
Materials and Methods
Materials
Etoricoxib was purchased from Provizer Pharma (Gujarat, India). PURASORB PDLG 5002 (DL-lactide/Glycolide 50:50 copolymer; with an inherent viscosity midpoint of 0.20 dL/g), PURASORB PDLG 7502 (DL-lactide/Glycolide 75:25 copolymer; with an inherent viscosity midpoint of 0.22 dL/g) and PURASORB PDL 02 (DL-lactide/Glycolide 100:0 copolymer; with an inherent viscosity midpoint of 0.20 dL/g) were kindly supplied by PURAC Biomaterials (Gorinchem, Netherlands). Dialysis tubing cellulose membrane (Molecular weight cutoff 12,000–14,000 Da), acetonitrile (HPLC grade), ammonium acetate and tertiary-butyl methyl ether were purchased from Sigma Aldrich (St. Louis, Mo, USA). Eudragit®-S100; poly(methacrylic acid-co-methyl methacrylate) was a gift sample from Evonik Laboratory (Mumbai, India). Loratadine (Internal standard) was provided by UK Vet Chem (Mumbai, India). Polyvinylpyrrolidone K30 (PVP K30) was supplied by Morgan Chemical Industries (Cairo, Egypt). Concentrated hydrochloric acid and dibasic sodium phosphate were procured from El-Nasr Pharmaceutical Chemicals (Cairo, Egypt). The market product of Etoricoxib (Arcoxia®) is a product of Merck Sharp & Dohme Limited (Hertfordshire, UK).
Methods
Development of Etoricoxib-Loaded PLGA Nanoparticles
Etoricoxib-loaded PLGA nanoparticles were prepared via the spray drying technique. Accurate amounts of etoricoxib (2% w/v), PLGA (copolymer) and PVP K30 (carrier) were dissolved in acetonitrile. The solution was spray dried using B-90 Nano Spray Dryer (Büchi Labortechnik, Flawil, Switzerland). A mesh of 7 μm was used along with 60 Hz ultrasonic actuator frequency. The Nano Spray Dryer was operated in a closed-mode configuration. The instrument was connected to a cooling unit (the Inert Loop B-295) in order to offer the safe operation of solvents in a closed-mode configuration. An inert gas (Nitrogen) was used at 1.5 bars to prevent the explosion of gas mixture. An electric field was generated using CO2 gas at 1.5 bars for separation of the particles. The O2 concentration in the closed loop was kept below 4% v/v.35 The drying gas flow rate was 110 L/min, the inlet temperature was 85°C, the outlet temperature was 45°C, the pressure was 39 mbar and the spraying rate was 100%.36 The dried powders were collected from the particle collecting chamber using a scraper and were stored in a desiccator for further characterization.
Experimental Design
Design Expert software Ver. 7 (Stat-Ease Inc., Minneapolis, MN) was used for the design of experiments and assessment of results using the D-optimal design (DOD). The design maximizes the determinants obtained from the information matrix generated from all the possible combinations of the involved factors.37 It was involved in optimizing many nano-drug delivery systems38–41 and was proved to be better than the one-factor-at-a-time (OFAT) experiments.42,43
Twenty-two systems of etoricoxib-loaded PLGA nanoparticles were prepared, in triplicates (Table 1). Three independent (two quantitative and one qualitative) variables were investigated each at three levels. The quantitative (numerical) variables were: (i) the PLGA copolymer concentration (0.5%, 1.25%, 2%) in system solution, and (ii) the carrier concentration (PVP K30; 2%, 3.5–5%) in system solution, while the qualitative (categorical) variable was the lactide:glycolide ratio in the copolymer chain (50:50, 75:25, 100:0).
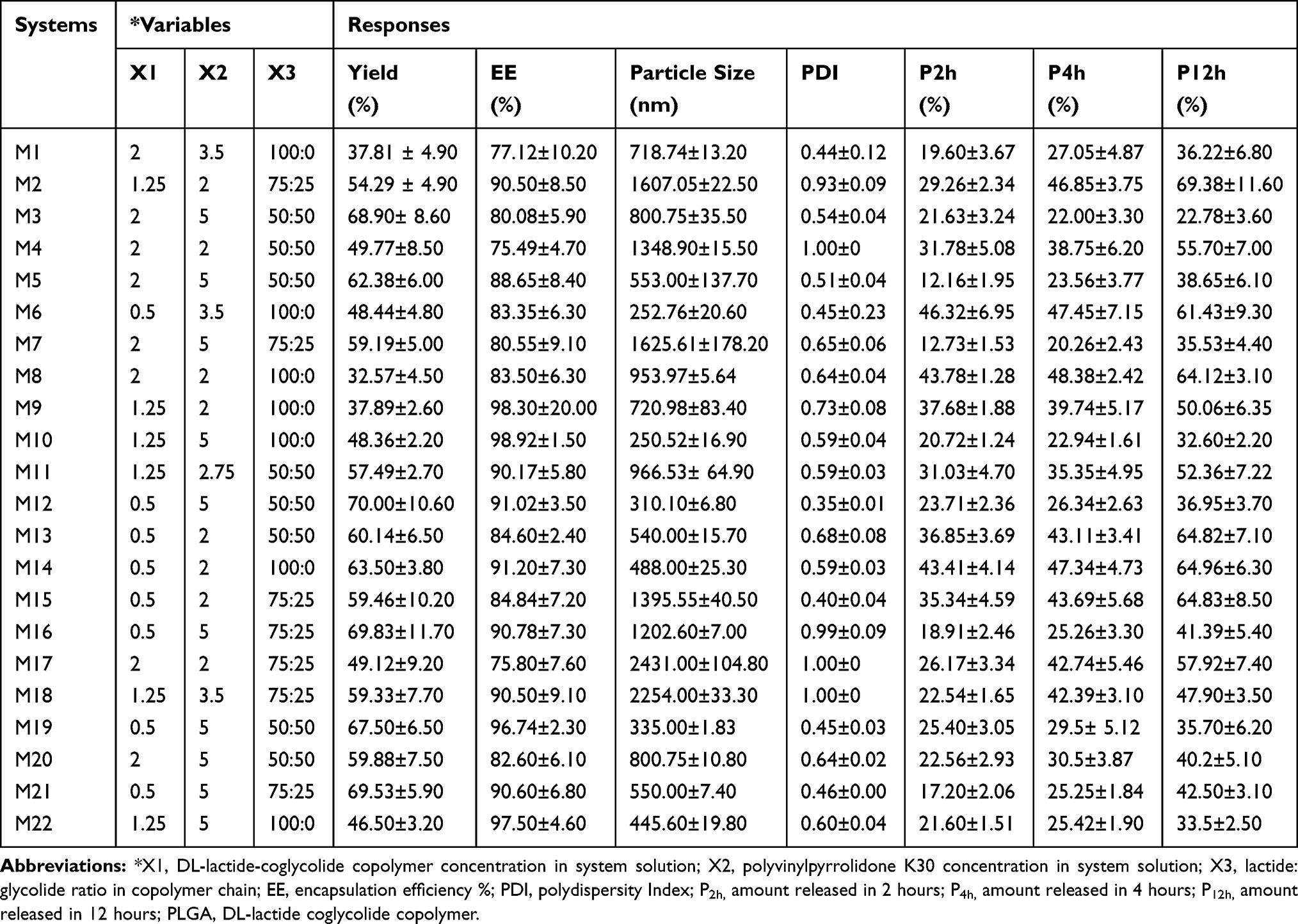 |
Table 1 The Variables and the Responses (Mean ± SD; n = 3) of the Investigated Etoricoxib-Loaded PLGA Systems; According to the D-Optimal Design |
Confirmation of the validity of the experimental design was checked by plotting a standard error of the design graph where the probability value (α) for determination of statistical significance was set at 0.05. Selection of models was based on sequential comparison and lack of fit test. Response surface 3D and contour 2D plots were constructed for the responses. The coefficients of equations were calculated using coded values. Hence, the various terms were compared directly; regardless of the magnitude. Values are given as mean ±SD. Statistical significance of the results was determined using one-way analysis of variance (ANOVA), employing a confidence interval of 95%. The numerical output of ANOVA includes the F-value, stating the magnitude of the impact of each factor and P-value as representative of the statistical significance.
Evaluation of the Developed Etoricoxib-Loaded PLGA Nanoparticles
Determination of the Percentage Yield
The percentage yield of each system was calculated by dividing the weight of the obtained nanoparticles by the total initial weights of drug, polymer and carrier.44
Determination of Drug Content and Encapsulation Efficiency %
A definite weight of the nanoparticles (which theoretically contains the equivalent of 10 mg of drug) was dissolved in 100 mL solution of 0.1 N HCl. After continuous shaking for 10 minutes, the solution was filtered through a 0.2-µm membrane filter. The solution was appropriately diluted and the drug concentration was measured spectrophotometrically (Shimadzu UV-1800 double beam spectrophotometer, Kyoto, Japan) at a predetermined λmax of 234 nm.45–47 The actual drug content was determined from which the drug encapsulation efficiency % (EE%) was calculated using the following equation:41,44,48
(1)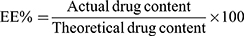
Determination of Particle Size and Polydispersity Index
The particle size and the polydispersity index (PDI) of the appropriately diluted water dispersions were measured using a Zetasizer (Nanoseries Instrument, Malvern-ZEN® 3600, Worcestershire, UK) via photon correlation spectroscopy. Measurements were conducted, in triplicates, at 25°C using an electric field strength of 23.2 V/cm.
In-vitro Drug Release Studies
The in-vitro drug release studies from the developed systems were conducted, in triplicates, in a USP dissolution tester apparatus (Vision G2 Elite 8TM, Hanson, CA) at 37 ± 0.5°C. The paddles were adjusted to rotate at a speed of 100 rpm. A definite weight of the nanoparticles (containing the equivalent of 60 mg of drug) was dispersed in 0.1N HCl (1 mL, pH 1.2) and loaded into the dialysis bag. The latter was secured by two clamps at each end. The dialysis bag was suspended in a dissolution medium of 0.1N HCl (600 mL, pH 1.2) for the first 2 hours. At the end of the second hour, the pH was raised to 6.8. Furthermore, it was raised to 7.4 at the end of the fourth hour and was maintained until the end of the study. The adjustments of the pH (pH meter; Griffin and George Ltd., London, England) were conducted using 0.235 M dibasic sodium phosphate solution. Perfect sink conditions prevailed during the studies. Five mL samples of the dissolution medium were withdrawn at predetermined time intervals, and immediately replaced with an equivalent volume of fresh medium to keep the volume constant. The drug concentration in the withdrawn samples was analysed spectrophotometrically at λmax of 234 nm. The cumulative percent of drug released (mean ± S.D.) after 2 h (P2h), 4 h (P4h) and 12 h (P12h) were determined.
Selection of the Best Achieved System
After confirming the polynomial equations relating the responses and the independent variables using the Design Expert software, the desirability function was activated to select the best achieved system with respect to the following constraints: maximizing the yield%, the EE% and P12h while minimizing the particle size.
Morphologic Examination
The topographic examination of the best achieved system (M14) was conducted using a scanning electron microscope fitted with an image analysis system (Quanta FEG250, Hillsboro, OR). The systems were fixed to brass grids and were sputter-coated (Edwards S-150A, Crawley, England) with a layer of gold (150 Å, 2 min) in a vacuum of argon gas (3 × 10−1 atm).46 Finally, samples were examined at 20 kV.
Differential Scanning Calorimetry (DSC)
The thermotropic properties of etoricoxib, PURASORB PDL 02, PVP K30, their physical mixture and M14 were explored to assess the degree of drug crystallinity in the nanoparticle. Three to four milligrams of each sample were encapsulated in an aluminium pan, heated up to a temperature of 300°C with 10 °C/min rate. Dry nitrogen was used as a carrier gas with a flow rate of 25 mL/min. The DSC thermogram was plotted using differential scanning calorimeter (DSC-60, Shimadzu, Kyoto, Japan).
Optimization of the Best Achieved System
To promote the colon targeting potential of M14 system, two approaches were used for coating the selected system with the pH-dependent coating polymer; poly (methacrylic acid-co-methyl methacrylate) [Eudragit-S100]. The first one involved coating the nanoparticles (M14) with poly (methacrylic acid-co-methyl methacrylate) solution (2%, w/v in a 1:1 solvent mixture of acetone: isopropyl alcohol) using spray drying technique. Two poly (methacrylic acid-co-methyl methacrylate): copolymer ratios were investigated; 1:1 (E-M14A system) and 2:1 (E-M14B system). The second approach involved loading the nanoparticles (M14 system) into hard gelatin capsules followed by coating the capsules with the same Eudragit solution applying the dip-coating method (E-M14C system).49,50 Briefly, each capsule was dipped for 90 seconds in a beaker containing 20 mL of the previously mentioned coating solution. The process was repeated fifteen times and the coat was dried with a stream of air after each dip.51
The particle size of E-M14A and E-M14B was compared to M14 while the weight gain % of E-M14C was evaluated relative to M14-loaded uncoated hard gelatin capsules.50,52 Each capsule was weighed before and after coating and the weight gain % was determined according to the following equation:
(2)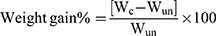
where Wc is the weight of the coated capsule and Wun is the weight of the uncoated capsule.
Furthermore, the in-vitro drug release studies of market product of etoricoxib, E-M14A, E-M14B and E-M14C were similarly conducted as previously noted. The percentages of drug released after 2 h (P2h), 4 h (P4h) and 12 h (P12h) were determined from the dissolution profiles and compared to the corresponding values for formula M14.
Pharmacokinetic Assessments of the Optimized System in Healthy Volunteers
Study Design
The studies were carried out to compare the pharmacokinetics of etoricoxib from E-M14C (test treatment) and market tablets (reference treatment) following oral administration of single 60 mg doses in healthy volunteers; under fasting conditions. This study was conducted according to a two-treatment, two-period, randomized, crossover design with a two-week washout period between the two phases. The protocol of the studies was reviewed and approved by the Research Ethics Committee of Faculty of Pharmacy, Cairo University, Cairo, Egypt [Serial no. PI 1902] and the Research Ethics Committee of Faculty of Pharmaceutical Sciences and Pharmaceutical Industries, Future University in Egypt, Cairo, Egypt [Serial no. REC-FPSPI-12/83]. The studies were carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
Administration of the Treatments
Six healthy male volunteers between 24 and 43 years old and weighing from 60 to 84 kg were invited to participate in the study after giving informed written consent. All subjects were judged to be healthy after conducting a general physical examination, reviewing their medical history and conducting the necessary biochemical and haematological laboratory assessments. The volunteers were instructed to avoid receiving any medication for one-week prior to- and during- the study period.
The volunteers were randomly divided into two equal groups. In Phase I, the first group of volunteers received the test treatment. The other group received the reference treatment. Following the washout period, the reverse of randomization took place in Phase II. Both products were administered with 150 mL of water in the morning after a 12-h overnight fasting. Food and drinks were withheld for at least 2 h after dosing.
Blood Sample Collection and Preparation
Blood samples (4 mL) were collected into heparinized tubes for the measurement of etoricoxib at zero time and at 30 min., 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 12, 24, 48 and 72 h post-dosing. The blood samples were centrifuged at 3000 rpm for 10 min, and the derived plasma samples were transferred to separate glass tubes and kept frozen at −80°C until analyses.
For the preparation of the plasma samples, each thawed sample (0.5 mL) was spiked with 100 µL of the internal standard solution (Loratadine; 200 ng/mL) and mixed using a vortex. For extraction of the drug, tertiary butyl methyl ether (4 mL) was added and mixed using a vortex. The organic layer was evaporated in a vacuum concentrator and finally, reconstituted in the mobile phase (0.5 mL).
Estimation of Etoricoxib Concentration in Plasma via LC-MS/MS
The determination of etoricoxib in the prepared plasma samples was conducted using a Shimadzu prominence HPLC system (Riverwood Drive, Columbia) consisted of HPLC pump (LC-20AD), a degasser (DGU-14A), an autosampler (SIL-20AC). The system was connected to a Triple Quad 4500 mass spectrometer (AB SCIEX instruments, Darmstadt, Germany) equipped with a turbo ion spray [pneumatically assisted electron spray ionization (ESI)] source]. The ESI interface was used in positive mode and the turbo ion spray source was heated to 399°C.
Chromatographic separation of extracted plasma samples was performed in the isocratic mode using an Agilent zorbax C18 column (50 mm × 5um diameter). The mobile phase consisted of a mixture of acetonitrile and 0.01% ammonium acetate (80:20). The system operated at a flow rate of 1 mL/min and the volume of injection was 10 μL.
The detector was operated in the multiple reaction monitoring mode with the precursor-to-product ion transitions were 358.96/279.90 Da for etoricoxib and 383.02/336.90 Da for Loratadine. A calibration curve was portrayed by plotting the peak area ratio of drug/IS against drug concentration in plasma over the concentration range of 10–5000 ng/mL. The calibration curve was found to be linear (y = 0.0017x + 0.0013). The estimated R2 value was found to be 0.995.
The analyst software Ver.1.6.3 (Applied Biosystems; Waltham, Massachusetts) was used to acquire the output signals from the detector, to integrate the peak areas, to calculate the drug/internal standard peak area ratios and to estimate the drug concentration.
Pharmacokinetic and Statistical Analyses
The pharmacokinetic parameters; Cmax (ng/mL), Tmax (h), MRT(0– ∞) (h), AUMC0- 72 (ng.h.2mL−1), AUMC0- ∞ (ng.h.2mL−1), AUC(0–72h) (ng.h/mL), AUC(0– ∞) (ng.h/mL) of the test and the reference treatments were estimated for each volunteer. The data were analysed via non-compartmental analysis using WinNonlin software Ver. 4.1 (Scientific consulting Inc., Cary, NC). The relative oral bioavailability of etoricoxib was estimated from the AUC(0–∞) values of the test and the reference treatments.
Statistical analysis of the parametric data was performed, using SPSS software Ver. 17 (SPSS Inc., Chicago, IL), applying two-way analysis of variance (ANOVA) at P values of <0.05. The data were expressed as mean values ± SD. Statistical analysis of Tmax values was performed using Wilcoxon signed-rank test and the data were expressed as median (range).
Results and Discussion
Development of Etoricoxib-Loaded Nanoparticles
Etoricoxib-loaded nanoparticles were successfully developed via the nano-spray drying technique. Three concentrations of PLGA were explored (0.5%, 1.25%, 2%), along with three ratios of lactide:glycolide in the copolymer chain (50:50, 75:25, 100:0). Preliminary studies revealed partial loss of the spray-dried powders; possibly due to stickiness on the outlet drying chamber walls. To minimize these problems, PVP K 30 was incorporated as a carrier at three concentrations (2%, 3.5, 5%).
Experimental Design
Twenty-two systems were suggested by the Design Expert Software (Ver. 7) according to the D-optimal design. The normal plot of residuals and the graphs of residual vs predicted and predicted vs actual of the investigated responses confirmed that there was no need for data transformation and/or further analysis. The generated model equations were statistically significant and were best fit for the prediction of the desired response outcomes. The goodness of fit of the models was checked via the determination of regression coefficient (r2) and adjusted coefficients (adj. r2). Relatively high values (>0.7) could indicate a high significance of fit of the model.53,54
Determination of Yield%
Nanoparticles were weighed after the spray drying process. The yield percentages of the developed systems ranged from 32.57% (M8) to 70% (M12) (Table 1). The statistical analysis of data (ANOVA) confirmed that the linear model is valid with a significant p-value and insignificant lack of fit value; F value = 20.33 (p< 0.0001) (Table 2). The most effective parameter on the yield (%) of nanoparticles was the Lactide:glycolide ratio in the copolymer chain with F-value of 19.87 (p< 0.0001). This was followed by the influence of the copolymer concentration in the system solution with F-value of 19.65 (p< 0.0004) and finally the contribution of PVP k30 concentration in the system solution with F-value of 12.51 (p< 0.002). The equation showing the relation between the independent variables and yield (%) of nanoparticles is presented in Table 2 (r2 = 0.8271). The predicted r2 (0.702) was in reasonable agreement with the adjusted r2 (0.7864). The adequate precision was 14.756; indicating an adequate signal. Thus, this linear model can be used to navigate the design space.
The response surface and the contour plots showing the effect of various factors on the yield (%) are shown in Figure 1A (A) and 1B (A), respectively. By observing the effect of one-factor plot on the percentage yield, and analysing the equation that describes the relation between the yield and parameters (Table 2), it was clear that the negative sign of the coefficient of the copolymer concentration in the system solution could indicate that the increase in the copolymer concentration would adversely affect the percentage yield. This could be ascribed to the increase in the viscosity of the solution and the resulting difficulties in spraying. This result agrees with Murakami et al,55 who used the modified spontaneous emulsification solvent diffusion method and found that the increase in PLGA copolymer concentration had a negative influence on the yield of the nanoparticles. On the other hand, the positive sign of the coefficient of the carrier concentration in the system solution could point out the positive impact of the carrier concentration on the yield (%) of the nanoparticles. This could justify the ability of PVP to prevent the aggregation of the nanoparticles and/or minimize their adherence to the wall of the drying chamber, and hence, facilitating their removal and collection. These findings were in line with those reported by Shakeri et al56 for the development of protein-loaded PLGA-PVP blend nanoparticles by the nanoprecipitation method. In a parallel line, the positive significant impact of lactide:glycolide ratio in the copolymer chain on the percentage yield was similarly revealed. The highest percentage yield was revealed with those systems prepared at a lactide:glycolide ratio of 100:0. Actually, the increase in the lactide:glycolide ratio in the copolymer chain is accompanied by an increase in the glass transition temperature (Tg) and in the yield percentage as well.57–59
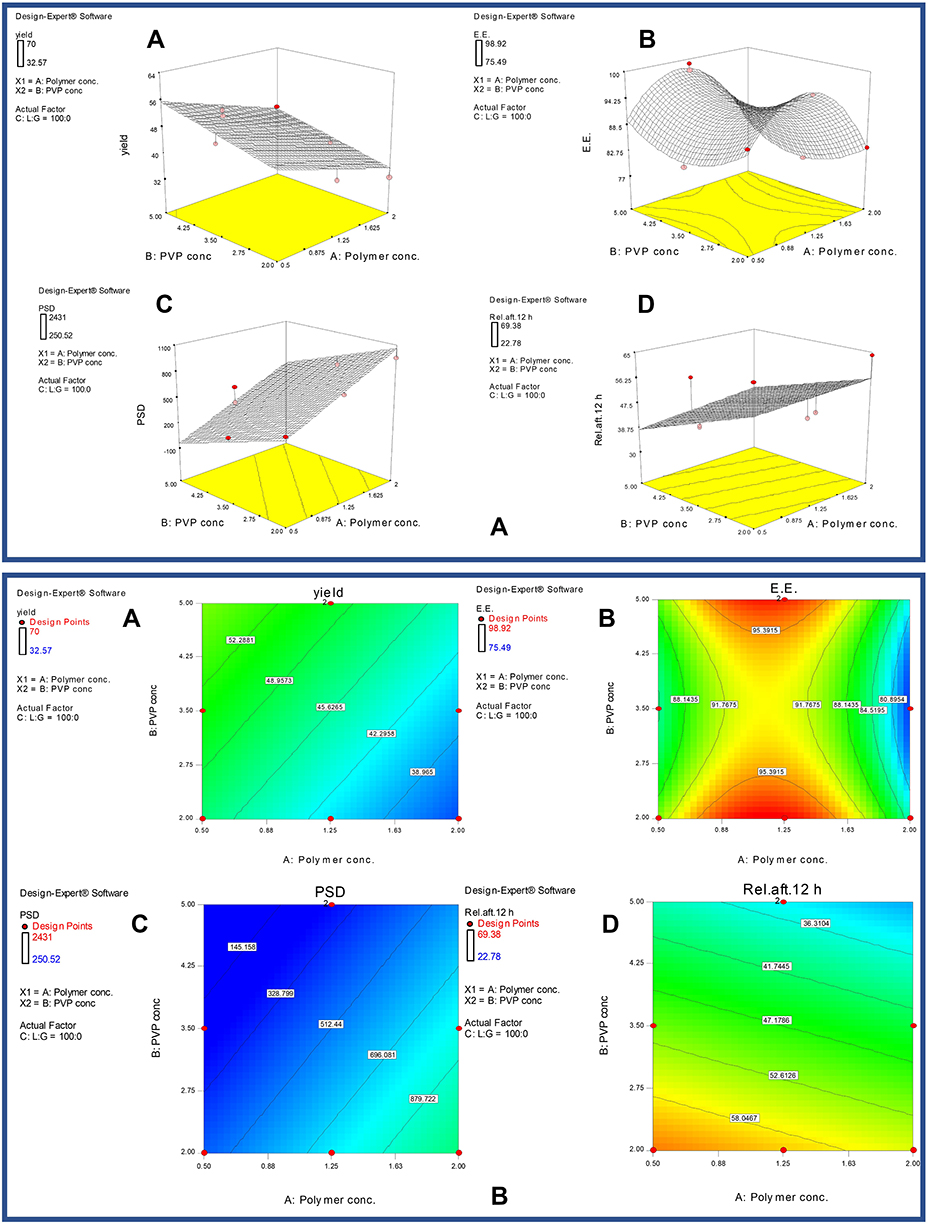 |
Figure 1 (A) Response surface plots showing the effects of copolymer concentration and Polyvinylpyrrolidone K30 concentration, at a lactide:glycolide ratio in the copolymer chain of 100:0, on (A) yield %, (B) encapsulation efficiency %, (C) particle size and (D) cumulative % of drug released after 12 h. (B) Contour plots showing the effects of copolymer concentration and Polyvinylpyrrolidone K30 concentration, at a lactide:glycolide ratio in the copolymer chain of 100:0, on: (A) yield %, (B) encapsulation efficiency %, (C) particle size and (D) cumulative % of drug released after 12 h. Abbreviations: PVP K30, polyvinylpyrrolidone K30; C:L:G, coded factor of lactide to glycolide ratio in copolymer chain; E.E., encapsulation efficiency %; PSD, particle size diameter; Rel. aft., release after; Conc., concentration. |
Estimation of Drug Encapsulation Efficiency (EE%)
The drug encapsulation efficiency (%) of the nanoparticles varied from 75.49% (M4) to 98.92% (M10) (Table 1). The quadratic equation was the best fitting model for EE% data with F value of 14.64 (p< 0.0001). The most effective parameter on the EE% of nanoparticle was the copolymer concentration; on squaring (A2) had an F-value of 75.08 (p<0.0001). The equation (r2 = 0.9415) that describes the relation between the independent variables and EE% of nanoparticles is presented in (Table 2). The predicted r2 (0.8772) was in reasonable agreement with the adjusted r2 (0.8301). The adequate precision was 12.703; indicating an adequate noise. It could be concluded that the quadratic equation could be used to navigate the design space.
Figure 1A(B) and 1B(B) represent the effect of the independent variables on the EE%; as represented by a quadratic model. It was clear that there is a direct correlation between the copolymer concentration and the drug EE%. This finding was previously demonstrated for rivastigmine-loaded PLGA nanoparticles, where the copolymer concentration was the major factor affecting the drug EE%.53 It could be inferred that higher amounts of the copolymer are available to entrap the loaded drug. Similar findings were reported by Bhambere et al60 for paclitaxel-loaded PLGA nanoparticles, by Kizilbey61 for rutin-loaded PLGA nanoparticles, by Nahata et al41 for aripiprazole-loaded PLGA microspheres and by Wagh et al62 for cyclosporine A-loaded PLGA nanoparticles.
Parallel to the effect of the copolymer concentration, the PVP k30 concentration showed a significant positive impact on EE%. PVP has proven history for improving the solubility of poorly soluble drugs and preventing their crystallisation.63 These results were in agreement with those reported by Shakeri et al56 and Coombes et al.63 Unlike the positive contributions of the aforementioned variables on drug EE%, the lactide:glycolide ratio in the copolymer chain showed a significant negative influence on the drug EE%. This finding does not match what was reported by Chung et al64 who revealed a positive contribution of the lactide:glycolide ratio in the copolymer chain on the EE% of albumin-loaded microspheres. At lower lactide:glycolide ratios, PLGA copolymers possessing low glass transition temperatures could be achieved; allowing for fast shell formation, effective encapsulation and better protection for the loaded drug.65 On the other hand, at higher lactide:glycolide ratios, more hydrophobic PLGA copolymers would be revealed; allowing for the development of small particles with more drug opportunities to escape from nanoparticles.
Measurements of Particle Size (PS)
The particle size of the developed nanoparticles varied significantly from 250.52 nm (M10) to 2431.00 nm (M17), while the PDI values of the systems ranged from 0.350 to 1 (Table 1).
The linear model was statistically proved to be the best fitting model for particle size data with F value of 42.75 (p< 0.0001). The most effective parameter on the size of the nanoparticle was the lactide:glycolide ratio in the copolymer chain with F-value of 54.27 (p<0.0001). The equation (r2 = 0.9415) that described the relation between the independent variables and the particle size is presented in (Table 2). The predicted r2 (0.888) was in reasonable agreement with the adjusted r2 (0.841). The precision was 20.972 indicating an adequate noise; suggesting that the linear model could be used to navigate the design space.
Figure 1A (C) and 1B(C) represent the effect of the independent variables on the particle size, as presented by a linear model. The polymer concentration had a significant positive effect on the particle size (Table 2). The increase in the particle size might be attributed to the increase in the viscosity of the sprayed solution which could retard the droplets subdividing. Similar findings were reported by Wagh et al62 for cyclosporine A-loaded PLGA nanoparticles and Joshi et al53 for rivastigmine-loaded PLGA nanoparticles. The subsequent slower nucleation rates would favour the generation of larger particles. On the other hand, the negative sign in the linear equation indicates that increasing the concentration of PVP K30 would lead to a decrease in the particle size. This might be attributed to the higher hydrophilicity associated with the use of higher amounts of this polymer. These findings are correlated to those of Quintanar-Guerrero et al66 who reported the production of smaller polymeric particles upon increasing PVP concentration. Furthermore, Zweers et al28 and Scholes et al67 reported that increasing the concentration of the carrier would decrease the size of the nanoparticles till an optimum carrier concentration. Beyond this level, it would positively influence the particle size.
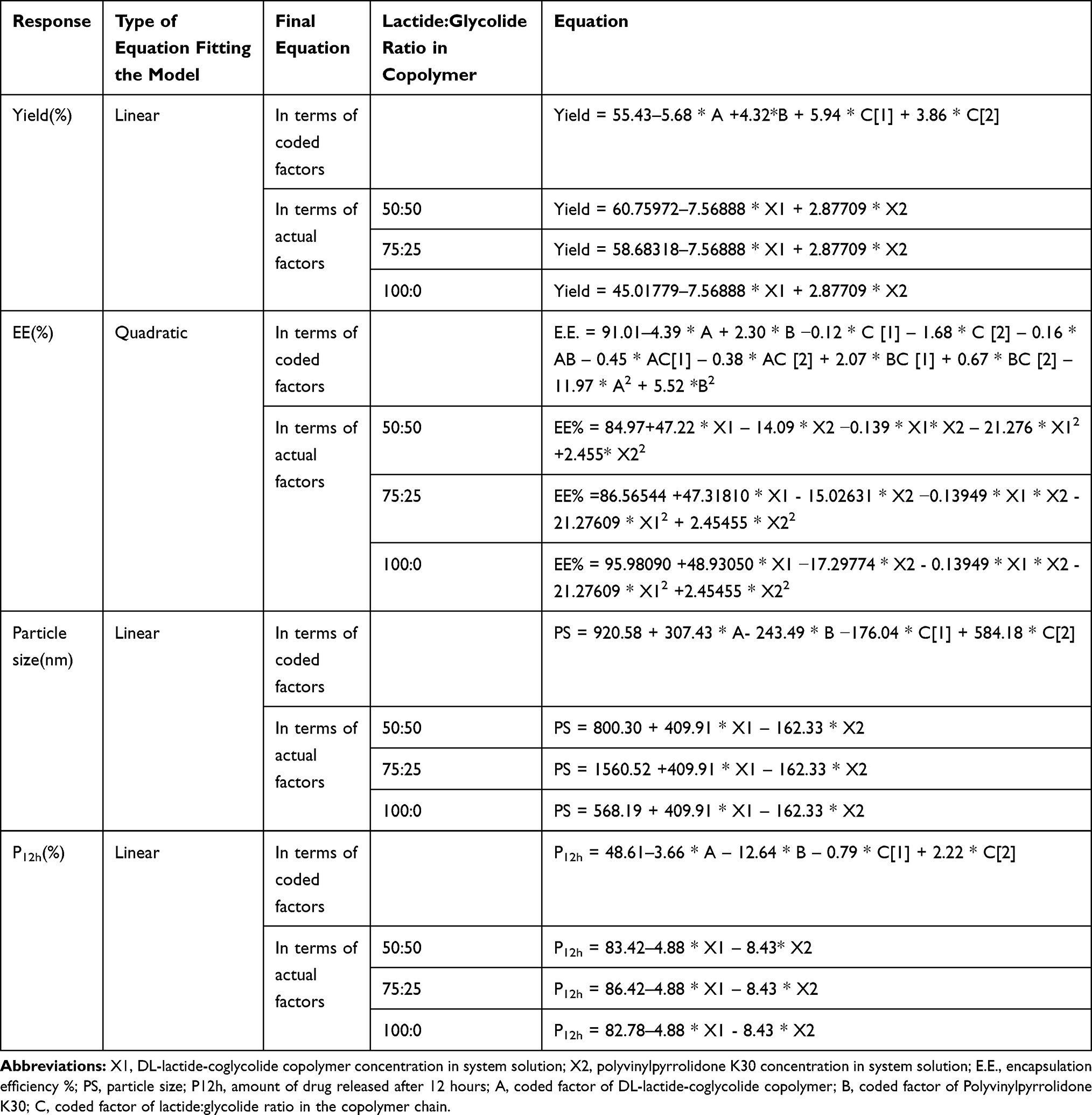 |
Table 2 Equations of Models that Best Fit the Investigated Responses |
The lactide:glycolide ratio had a significant effect on the particle size as indicated by its F-value of 54.27 (P< 0.0001). The positive and negative signs in the linear equation indicate that it can affect the particle size positively or negatively depending on the investigated ratio. Observing the one-way plot curve, it can be seen that increasing the ratio from 50:50 to 75:25 would increase the particle size. Interestingly, the particle size decreased upon raising the ratio to 100:0. Contrary to the current finding, Mehrotra et al68 reported that lactide:glycolide ratio had a non-significant influence on the particle size during developing lomustine-loaded PLGA nanoparticles via the interfacial deposition method.
The Cumulative Drug Released (%) After 12 h (P12h)
The cumulative drug released percentages from the developed nanoparticles after 12 h (P12h) ranged from 22.78% (M3) to 69.38% (M2). The linear model was the best fitting model for P12h data (p < 0.0001). The most effective parameter on the cumulative % release of the drug after 12 hours was the PVP K30 concentration with F-value of 77.60 (p <0.0001). The relation between independent variables and P12h was presented in the equation (r2 = 0.841) (Table 2). The predicted r2 (0.746) was in reasonable agreement with the adjusted r2 (0.803). The adequate precision was 12.509 indicating an adequate noise. It could be assumed that this linear model could be used to navigate the design space.
The drug release profiles (not shown) of the developed etoricoxib-loaded nanoparticles showed a biphasic pattern (Table 1). The initial fast release rate might be related to the presence of small particles, along with larger ones. The former would have smaller diffusion paths, so that the drug could rapidly access the solid/dissolution medium interface.25 The release rate in the second phase was assumed to be controlled by the drug diffusion rate across the polymer matrix.69 The negative sign indicates the inverse relationship between the copolymer concentration and P12h. This could be attributed to the formation of thicker coats of the copolymer around the drug at higher concentrations of the polymer thus, hindering the drug release percentages.
Figure 1A(D) and 1B(D) represent the effect of the independent variables on P12h; as represented by a linear model. As shown in the equation (Table 2), increasing the concentration of PVP K30 had a negative influence on P12h. These findings relatively match with the finding of Coombes et al63 who used PVP as a stabilizer for the development of ovalbumin-loaded microparticles and proved that its presence contributed to a reduction in the burst effect of the drug release as well as a prolongation of the drug release pattern.
An inverse correlation could be established between the lactide:coglycolide ratio and P12h. PLGA copolymers possessing a lactide:glycolide of 50:50 usually degrade more rapidly than PLA alone.70 PLGA copolymers possessing a lactide:glycolide ratio of 50:50 are more hydrophilic than those possessing a ratio of 75:25.71 The latter copolymers possess a greater number of the more hydrophobic lactic acid monomers which would degrade in a longer period of time. In other words, the absolute value of the degradation rate would be positively correlated to the lactide:glycolide ratio.72
Selection of the Best Achieved System
The desirability function was explored to allow proper selection of the best achieved system which possesses a compromise between the targeted constraints; maximizing the yield%, the EE% and P12h while minimizing the PS. The best achieved system, as suggested by the designed expert software, was M14. It had a promising yield (%) of 63.5%, EE% of 91.2%, P12h of 64.96% and PS of 488 nm.
Morphologic Characteristics
Figure 2 illustrates the scanning electron micrographs of the drug crystals (A) and the best achieved system; M14 (B). Spherical particles with smooth surfaces were revealed for the latter. The lack of the surface-adsorbed drug crystals could indicate the efficient entrapment of etoricoxib within the matrix of the nanoparticles.
 |
Figure 2 Scanning electron micrographs of: (A) drug crystals and (B) M14 system [nanoparticles prepared using poly-DL-lactide coglycolide coplymer (0.5%, w/v) at a lactide:glycolide ratio of 100:0, in presence of Polyvinylpyrrolidone K30 (2%, w/v)]. Abbreviations: HV, is the accelerating voltage for the electrons; Mag, is the magnification; WD, is the working distance between the sample surface and the lower end of the pole piece where the electrons are coming from; det, is the detector; BSED, Backscattered electron detector; HPW, high-performance waveform; PM, particulate matter; PLGA, DL-lactide coglycolide copolymer; PVP K30, polyvinylpyrrolidone K30. |
Differential Scanning Calorimetry (DSC)
Figure 3 shows the DSC thermograms of pure drug, PURASORB PDL 02 copolymer, PVP K30, their physical mixture and M14 system. Etoricoxib had a sharp endothermic peak at 139°C; corresponding to its melting point.73 The DSC thermograms of the copolymer (PURASORB PDL 02) and the carrier (PVP K30) showed endothermic peaks at 125°C.9,74 The permanence of the characteristic endothermic peak of the drug in the physical mixture and its disappearance in M14 system could indicate that the drug has changed from the crystalline to the more soluble amorphous state in the latter.
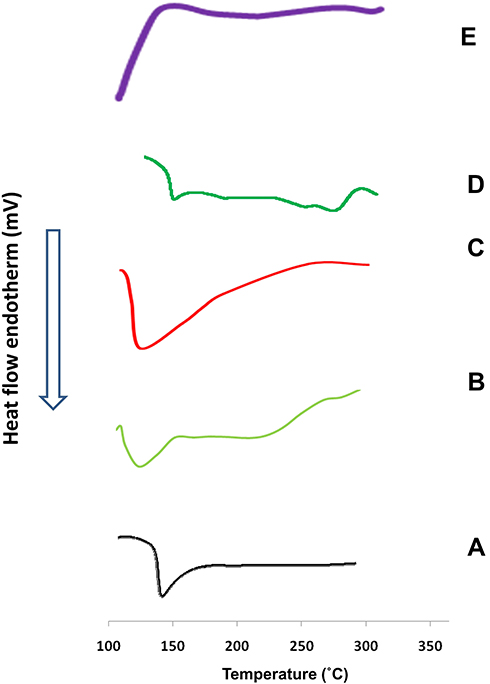 |
Figure 3 Differential scanning calorimetry thermograms of: (A) drug powder, (B) PURASORB PDL02 copolymer, (C) PVP K30, (D) their physical mixture and (E) M14 system [nanoparticles prepared using PLGA (0.5% w/v) at a lactide:glycolide ratio of 100:0, in presence of PVP K30 (2% w/v)]. Abbreviations: PLGA, DL DL-lactide coglycolide copolymer; PVP K30, polyvinylpyrrolidone K30; PDL02, DL-lactide coglycolide copolymer of lactide:glycolide ratio of 100:0 and inherent viscosity 0.2 dL/g. |
Optimization of the Best Achieved System
A successful colon targeted drug delivery should allow the release of minimum drug percentages during its transit in the stomach and in the upper intestine to ensure maximum drug delivery in the colon75. The respective P2h and P4h of M14 were relatively high; 43.41–47.34% (Table 1). To minimize the drug released percentages at the stomach (P2h) and the small intestine (P4h) and to promote the colon targeting potential of M14, two approaches were adopted for coating the system with poly (methacrylic acid-co-methyl methacrylate) [Eudragit-S100]. The first involved the incorporation of poly (methacrylic acid-co-methyl methacrylate) in a coating solution, at two poly (methacrylic acid-co-methyl methacrylate): copolymer ratios of 1:1 (E-M14A) and 2:1 (E-M14B) followed by coating of nanoparticles with spray drying. The second approach explored the dip coating of M14-loaded hard gelatin capsules with poly (methacrylic acid-co-methyl methacrylate) solution (2%, w/v) (E-M14C). The first approach led to an increase in the mean PS from 488 nm (M14) to 1250 nm (E-M14A) and 2699 nm (E-M14B). The larger PS of the E-M14B system could be related to the use of a higher poly (methacrylic acid-co-methyl methacrylate) concentration in the sprayed coating solution. On the other hand, the weight gain % of E-M14C system, relative to M14-loaded uncoated hard gelatin capsules, was ≈ 25%.
The in-vitro drug release profiles of market tablets of etoricoxib, M14, E-M14A, E-M14B and E-M14C systems are depicted in Figure 4. It was clear that the drug showed almost 100% release within 4 h from the commercial tablets. On the other hand, the investigated systems showed more retarded drug release profiles. The P2h of M14, E-M14A, E-M14B and E-M14C were 43.41%, 33.67%, 24.3% and 9.85%, respectively. According to the pharmacopeial specifications for enteric-coated systems, the P2h should not exceed 10%. It was clear that the implementation of the first approach (E-M14A, E-M14B) did not allow the development of coherent coating barriers which were unable to retard the rate of drug release into the acidic milieu; even with the higher poly (methacrylic acid-co-methyl methacrylate) concentration (E-M14B). On the other hand, a significantly lowered P2h was achieved following the adoption of the dip coating approach on the surface of the hard gelatin capsules filled with the nanoparticles (E-M14C).
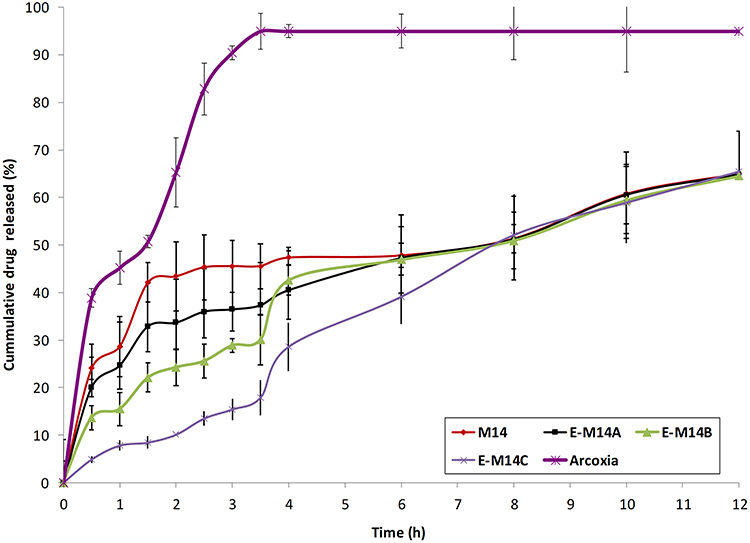 |
Figure 4 The in-vitro drug release profiles of Arcoxia® tablets, M14 (uncoated nanoparticles), E-M14A (nanoparticles with Eudragit®-S100 coat: PURASORB PDL02 ratio of 1:1), E-M14B (nanoparticles with Eudragit®-S100 coat: PURABORB PDL02 ratio of 2:1), E-M14C systems (M14 nanoparticles filled in Eudragit®-S100-coated hard gelatin capsules) (mean ± S.D., n = 3). Abbreviation: PDL02, DL-lactide coglycolide copolymer of Lactide:glycolide ratio of 100:0 and inherent viscosity 0.2 dL/g. |
The P4h of M14, E-M14A, E-M14B and E-M14C were 47.34%, 40.5%, 32.6 and 28.6%, respectively. The superiority of the dip coating approach to minimize the drug released percentages in the simulating small intestine medium could be inferred. Interestingly, no significant differences (p > 0.05) were observed between the P12h values for M14 (64.96%), E-M14A (64.82%), E-M14B (64.56%) and E-M14C systems (65.45%). It is worth to note that the respective cumulative percentages of drug released after 24 h were 81.8 %, 81.2%, 80.52% and 81.5%. The maximization of the percentages of drug release in the colon could be expected with E-M14C system. Furthermore, this latter system will preserve the nano-size of the PLGA nanoparticles which will allow the uptake by macrophages or dendritic cells at the site of active inflammation in the colon after the dissolution of the enteric-cote shield so it was selected for the In Vivo assessment.
Pharmacokinetic Assessments of the Optimized System in Healthy Volunteers
The mean (± S.D.) etoricoxib plasma concentration – time curves following oral administration of single 60 mg oral doses of market tablets of etoricoxib and E-M14C system in healthy volunteers, under fasting conditions, are portrayed in Figure 5.
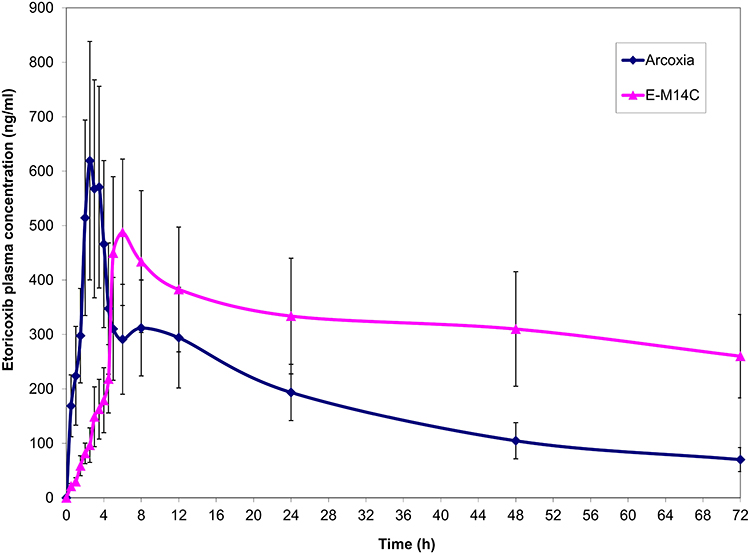 |
Figure 5 Plasma concentration–time curves of etoricoxib following oral administration of single oral doses (60 mg) of Arcoxia® tablets and E-M14C system (M14 nanoparticles filled in Eudragit®-S100-coated hard gelatin capsules) in healthy volunteers, under fasting conditions (mean ± S.D., n = 6). |
The Cmax values of the reference and the test treatments were 619.20 ± 212.46 ng/mL and 487.68 ± 134.63 ng/mL, respectively. The statistical analysis of data revealed the lack of significant differences (p = 0.205), Table 3.
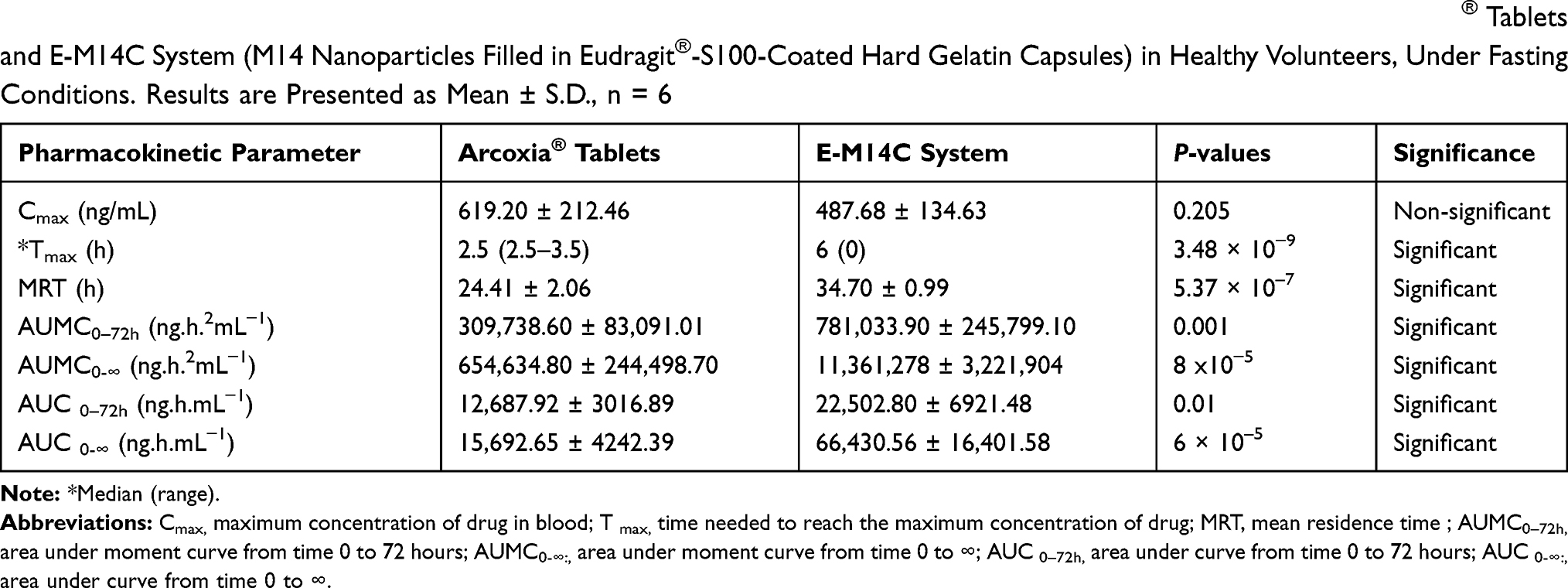 |
Table 3 Pharmacokinetic Parameters for Etoricoxib Following Oral Administration of Single Oral Doses (60 mg) of Arcoxia® Tablets and E-M14C System (M14 Nanoparticles Filled in Eudragit®-S100-Coated Hard Gelatin Capsules) in Healthy Volunteers, Under Fasting Conditions. Results are Presented as Mean ± S.D., n = 6 |
On contrary, the statistical analysis of the non-parametric data (Tmax) via Wilcoxon’s signed-rank test as well as the parametric data (MRT(0-∞), AUMC0–72h (ng.h.2mL−1), AUMC0-∞ (ng.h.2mL−1), AUC(0–72h) and AUC(0-∞)) via two-way ANOVA revealed the superiority of E-M14C system (Table 3). The respective median Tmax values for the market tablets of etoricoxib and E-M14C system were 2.5 and 6 h. These results are in correlation with the dissolution pattern of the drug from the two products in gradient pH medium simulating the GIT fluid. Both results confirm the ability of the latter system to minimize the percentages of drug released in the stomach and the small intestine, allowing for the delivery of most of its payload at the colon.
In a parallel line, the prolongation in MRT(0-∞) from 24.41 h to 34.70 h could point out the sustained drug release capabilities of E-M14C system. With respect to AUC(0–72h) values of both treatments, the relative oral bioavailability would be 177.35%. Taking into consideration AUC(0-∞) values of both treatments, the relative oral bioavailability would be raised to 423.32%.
Conclusions
Etoricoxib-loaded nanoparticles were successfully developed, according to the D-optimal design, via the nanospray drying technique. M14 system was prepared using PLGA (0.5% w/v) at a lactide:glycolide ratio of 100:0, in presence of PVP K30 (2% w/v). Further coating of M14-loaded hard gelatin capsules with poly (methacrylic acid-co-methyl methacrylate) (E-M14C) was necessary to minimize the percentages of drug released in the stomach and small intestine. The pharmacokinetic assessments of E-M14C in healthy human volunteers revealed its colon-targeting potential and proved its ability to increase the oral bioavailability of etoricoxib, relative to the marketed tablets. Further clinical studies are required to confirm the efficacy of this system in the management of irritable bowel syndrome.
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53(suppl 5):v1–v16. doi:10.1136/gut.2004.043372
2. Badhana S, Garud N, Garud A. Colon specific drug delivery of mesalamine using Eudragit S100-coated chitosan microspheres for the treatment of ulcerative colitis. Int Current Pharm J. 2013;2(3):42–48. doi:10.3329/icpj.v2i3.13577
3. Coco R, Plapied L, Pourcelle V, et al. Drug delivery to inflamed colon by nanoparticles: comparison of different strategies. Int J Pharm. 2013;440(1):3–12. doi:10.1016/j.ijpharm.2012.07.017
4. Rubinstein A. Colonic drug delivery. Drug Discov Today Technol. 2005;2(1):33–37. doi:10.1016/j.ddtec.2005.05.021
5. Orienti I, Cerchiara T, Luppi B, Bigucci F, Zuccari G, Zecchi V. Influence of different chitosan salts on the release of sodium diclofenac in colon-specific delivery. Int J Pharm. 2002;238(1–2):51–59. doi:10.1016/S0378-5173(02)00060-1
6. Lamoudi L, Chaumeil JC, Daoud K. PLGA nanoparticles loaded with the non-steroid anti-inflammatory: factor influence study and optimization using factorial design. Int J Chem Eng Appl. 2013;4(6):369. doi:10.7763/IJCEA.2013.V4.327
7. Salunkhe KS, Kulkarni MV. Formulation and in-vitro evaluation of dextrin matrix tablet of ibuprofen for colon specific drug delivery. Pak J Pharm Sci. 2008;21(1):17–20.
8. de Alencar RG, de Oliveira AC, Lima EM, da Cunha-filho MSS, Taveira SF, Marreto RN. Compacted multiparticulate systems for colon-specific delivery of ketoprofen. AAPS Pharm Sci Tech. 2017;18(6):2260–2268. doi:10.1208/s12249-016-0700-2
9. Abbas HK, Wais FMH, Abood AN. Preparation and evaluation of ketoprofen nanosuspension using solvent evaporation technique. Iraqi J Pharm Sci. 2017;41–55.
10. Orlu M, Cevher E, Araman A. Design and evaluation of colon specific drug delivery system containing flurbiprofen microsponges. Int J Pharm. 2006;318(1–2):103–117. doi:10.1016/j.ijpharm.2006.03.025
11. Hadi MA, Rao NR, Rao AS, Mahtab T, Tabassum S. A review on various formulation methods in preparing colon targeted minitablets for chronotherapy. J Applied Pharm Sci. 2018;8(03):158–164.
12. Krishnaiah Y, Satyanarayana V, Dinesh Kumar B, Karthikeyan R. Studies on the development of colon-targeted delivery systems for celecoxib in the prevention of colorectal cancer. J Drug Target. 2002;10(3):247–254. doi:10.1080/10611860290022697
13. Trivedi HD, Puranik PK. Formulation and development of colon specific etoricoxib CODESTM tablet: statistical optimization and in vivo roentgenography. Int J Pharm Pharm Res. 2016;7(1):185–215.
14. El Miedany Y, Youssef S, Ahmed I, El Gaafary M. The gastrointestinal safety and effect on disease activity of etoricoxib, a selective cox-2 inhibitor in inflammatory bowel diseases. Am J Gastroenterol. 2006;101(2):311. doi:10.1111/j.1572-0241.2006.00384.x
15. Miao XP, Li JS, Ouyang Q, Hu RW, Zhang Y, Li HY. Tolerability of selective cyclooxygenase 2 inhibitors used for the treatment of rheumatological manifestations of inflammatory bowel disease. Cochrane Database Systematic Rev. 2014;10.
16. Rodrigues AD, Halpin RA, Geer LA, et al. Absorption, metabolism, and excretion of etoricoxib, a potent and selective cyclooxygenase-2 inhibitor, in healthy male volunteers. Drug Metab Disposition. 2003;31(2):224–232. doi:10.1124/dmd.31.2.224
17. Kiran KGB, Naresh YS, Mohammed GA, Abdul Nasir K. Design and evaluation of a new capsule-type dosage form for colon-targeted delivery of etoricoxib. Int J Pharm Sci. 2013;3(1):147–151.
18. Lamprecht A, Schäfer U, Lehr C-M. Size-dependent bioadhesion of micro-and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–793. doi:10.1023/A:1011032328064
19. Chaudhury A, Das S. Recent advancement of chitosan-based nanoparticles for oral controlled delivery of insulin and other therapeutic agents. AAPS Pharm Sci Tech. 2011;12(1):10–20. doi:10.1208/s12249-010-9561-2
20. Sharma S, Parmar A, Kori S, Sandhir R. PLGA-based nanoparticles: a new paradigm in biomedical applications. TrAC Trends Anal Chem. 2016;80:30–40. doi:10.1016/j.trac.2015.06.014
21. Stevanović M, Savić J, Jordović B, Uskoković D. Fabrication, in vitro degradation and the release behaviours of poly (DL-lactide-co-glycolide) nanospheres containing ascorbic acid. Colloids Surf B Biointerfaces. 2007;59(2):215–223. doi:10.1016/j.colsurfb.2007.05.011
22. Taylor D, Thomas R, Penfold J. Polymer/surfactant interactions at the air/water interface. Adv Colloid Interface Sci. 2007;132(2):69–110. doi:10.1016/j.cis.2007.01.002
23. Budhian A, Siegel SJ, Winey KI. Controlling the in vitro release profiles for a system of haloperidol-loaded PLGA nanoparticles. Int J Pharm. 2008;346(1):151–159. doi:10.1016/j.ijpharm.2007.06.011
24. Fambri L, Migliaresi C, Kesenci K, Piskin E. Biodegradable polymers. Editor; Ronaldo Barbucci: Integrated Biomaterials Sci. Springer; 2002:119–187.
25. Dunne M, Corrigan O, Ramtoola Z. Influence of particle size and dissolution conditions on the degradation properties of polylactide-co-glycolide particles. Biomaterials. 2000;21(16):1659–1668. doi:10.1016/S0142-9612(00)00040-5
26. Raghuvanshi RS, Singh M, Talwar G. Biodegradable delivery system for single step immunization with tetanus toxoid. Int J Pharm. 1993;93(1–3):R1–R5. doi:10.1016/0378-5173(93)90188-L
27. Stevanović M, Jordović B, Uskoković D. Stereological analysis of DLPLG nanoparticles containing ascorbic acid during in vitro degradation process. 2007.
28. Zweers ML, Grijpma DW, Engbers GH, Feijen J. The preparation of monodisperse biodegradable polyester nanoparticles with a controlled size. J Biomed Mater Res B Appl Biomater. 2003;66(2):559–566. doi:10.1002/jbm.b.10046
29. Kaihara S, Matsumura S, Mikos AG, Fisher JP. Synthesis of poly (L-lactide) and polyglycolide by ring-opening polymerization. Nat Protoc. 2007;2(11):2767–2771. doi:10.1038/nprot.2007.391
30. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377
31. Jovanović I, Stevanović M, Nedeljković B, Ignjatović N The effect of processing parameters on characteristics of poly-l-lactide microspheres. Paper presented at: Materials science forum.2007
32. Allison SD. Effect of structural relaxation on the preparation and drug release behavior of poly (lactic‐co‐glycolic) acid microparticle drug delivery systems. J Pharm Sci. 2008;97(6):2022–2035. doi:10.1002/jps.21124
33. Mohamed F, van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci. 2008;97(1):71–87. doi:10.1002/jps.21082
34. Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly (D, L-lactide-co-glycolide) and its derivatives. J Controlled Release. 2008;125(3):193–209. doi:10.1016/j.jconrel.2007.09.013
35. Li X, Anton N, Arpagaus C, Belleteix F, Vandamme TF. Nanoparticles by spray drying using innovative new technology: the büchi nano spray dryer B-90. J Controlled Release. 2010;147(2):304–310. doi:10.1016/j.jconrel.2010.07.113
36. Gautier S, Arpagaus C, Schafroth N, Meuri M, Deschamps A, Maquet V. Very fine chitosan microparticles with narrow & controlled size distribution using spray-drying technologies. Drug Delivery Technol. 2010;10(8):30–37.
37. Triefenbach F Design of experiments: the D-optimal approach and its implementation as a computer algorithm. Bachelor’s Thesis in Information and Communication Technology. 2008.
38. Arai H, Suzuki T, Kaseda C, Takayama K. Effect of an experimental design for evaluating the nonlinear optimal formulation of theophylline tablets using a bootstrap resampling technique. Chem Pharm Bull. 2009;57(6):572–579. doi:10.1248/cpb.57.572
39. Dixit R, Nagarsenker M. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur J Pharm Sci. 2008;35(3):183–192. doi:10.1016/j.ejps.2008.06.013
40. Elnaggar YS, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1–2):133–141. doi:10.1016/j.ijpharm.2009.07.015
41. Nahata T, Saini TR. D-optimal designing and optimization of long acting microsphere-based injectable formulation of aripiprazole. Drug Dev Ind Pharm. 2008;34(7):668–675. doi:10.1080/03639040701836545
42. Czitrom V. One-factor-at-a-time versus designed experiments. Am Stat. 1999;53(2):126–131.
43. Abdel-Hafez SM, Hathout RM, Sammour OA. Towards better modeling of chitosan nanoparticles production: screening different factors and comparing two experimental designs. Int J Biol Macromol. 2014;64:334–340. doi:10.1016/j.ijbiomac.2013.11.041
44. Garud N, Garud A. Preparation and in-vitro evaluation of metformin microspheres using non-aqueous solvent evaporation technique. Trop J Pharm Res. 2012;11(4):577–583. doi:10.4314/tjpr.v11i4.8
45. Singh S, Mishra A, Verma A, Ghosh AK, Mishra AK. A simple Ultraviolet spectrophotometric method for the determination of etoricoxib in dosage formulations. J Adv Pharm Technol Res. 2012;3(4):237. doi:10.4103/2231-4040.104715
46. Elbary AA, Tadros MI, Alaa-Eldin AA. Sucrose stearate-enriched lipid matrix tablets of etodolac: modulation of drug release, diffusional modeling and structure elucidation studies. AAPS Pharm Sci Tech. 2013;14(2):656–668. doi:10.1208/s12249-013-9951-3
47. Yogesh P, Madhuri M, Vaishnavi M, Atul C, Pankaj G. Preparation, characterization, and evaluation of anti-inflammatory activity of etoricoxib loaded soluplusâ® nanocomposites. Int J Applied Pharm. 2018;10:6.
48. El-Bary AA, Aboelwafa AA, Al Sharabi IM. Influence of some formulation variables on the optimization of pH-dependent, colon-targeted, sustained-release mesalamine microspheres. AAPS Pharm Sci Tech. 2012;13(1):75–84. doi:10.1208/s12249-011-9721-z
49. Mehta R, Chawla A, Sharma P, Pawar P. Formulation and in vitro evaluation of Eudragit S-100 coated naproxen matrix tablets for colon-targeted drug delivery system. J Adv Pharm Technol Res. 2013;4(1):31. doi:10.4103/2231-4040.107498
50. Qureshi J, Ali J, Baboota S, Ahuja A, Mallikarjun C. Development and evaluation of chronotherapeutic drug delivery system for the management of nocturnal asthma. Trop J Pharm Res. 2012;11(5):703–712.
51. Alsharkas L, Kumar PV, Wei YS. Preparation and in vitro evaluation of enteric coated oral vancomycin hydrochloride sustained release formulation with mucoadhesive properties. Am J Adv Drug Delivery. 2018;06. doi:10.21767/2321-547X.1000029
52. Elbary AA, Tadros MI, Alaa-Eldin AA. Development and in vitro/in vivo evaluation of etodolac controlled porosity osmotic pump tablets. AAPS Pharm Sci Tech. 2011;12(2):485–495. doi:10.1208/s12249-011-9608-z
53. Joshi SA, Chavhan SS, Sawant KK. Rivastigmine-loaded PLGA and PBCA nanoparticles: preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur J Pharm Biopharm. 2010;76(2):189–199. doi:10.1016/j.ejpb.2010.07.007
54. Hinkelmann K, Kempthorne O. Design and Analysis of Experiments. Vol. 1. Wiley Online Library; 1994.
55. Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Preparation of poly (DL-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int J Pharm. 1999;187(2):143–152. doi:10.1016/S0378-5173(99)00187-8
56. Shakeri S, Roghanian R, Emtiazi G, Errico C, Chiellini E, Chiellini F. Preparation of protein-loaded PLGA-PVP blend nanoparticles by nanoprecipitation method: entrapment, initial burst and drug release kinetic studies. Nanomed J. 2015;2(3):175–186.
57. Muzaffar K, Wani SA, Dinkarrao BV, Kumar P. Determination of production efficiency, color, glass transition, and sticky point temperature of spray-dried pomegranate juice powder. Cogent Food Agriculture. 2016;2(1):1144444. doi:10.1080/23311932.2016.1144444
58. Fang Z, Bhandari B. Comparing the efficiency of protein and maltodextrin on spray drying of bayberry juice. Food Res Int. 2012;48(2):478–483. doi:10.1016/j.foodres.2012.05.025
59. Lee LY, Wang CH, Smith KA. Micro-porous paclitaxel-loaded PLGA foams–a new implant material for controlled release of chemotherapeutic agents. 2007.
60. Bhambere D, Shirivastava B, Sharma P, Gide P. Effect of polymer and formulation variables on properties of self-assembled polymeric micellar nanoparticles. J Nanomed Biotherapeutic Discov. 2014;4(129):2. doi:10.4172/2155-983X.1000129
61. Kızılbey K. Optimization of rutin-loaded PLGA nanoparticles synthesized by single-emulsion solvent evaporation method. ACS Omega. 2019;4(1):555–562. doi:10.1021/acsomega.8b02767
62. Wagh VD, Apar DU. Cyclosporine a loaded PLGA nanoparticles for dry eye disease: in vitro characterization studies. J Nanotechnol. 2014;2014.
63. Coombes A, Yeh M-K, Lavelle E, Davis S. The control of protein release from poly (DL-lactide co-glycolide) microparticles by variation of the external aqueous phase surfactant in the water-in oil-in water method. J Controlled Release. 1998;52(3):311–320. doi:10.1016/S0168-3659(98)00006-6
64. Chung TW, Tsai YL, Hsieh JH, Tsai WJ. Different ratios of lactide and glycolide in PLGA affect the surface property and protein delivery characteristics of the PLGA microspheres with hydrophobic additives. J. Microencapsulation. 2006;23(1):15–27.
65. Baysan U, Elmas F, Koç M. The effect of spray drying conditions on physicochemical properties of encapsulated propolis powder. J Food Process Eng. e13024.
66. Quintanar-Guerrero D, Fessi H, Allémann E, Doelker E. Influence of stabilizing agents and preparative variables on the formation of poly (D, L-lactic acid) nanoparticles by an emulsification-diffusion technique. Int J Pharm. 1996;143(2):133–141. doi:10.1016/S0378-5173(96)04697-2
67. Scholes P, Coombes A, Illum L, Daviz S, Vert M, Davies M. The preparation of sub-200 nm poly (lactide-co-glycolide) microspheres for site-specific drug delivery. J Controlled Release. 1993;25(1–2):145–153. doi:10.1016/0168-3659(93)90103-C
68. Mehrotra A, Pandit JK. Preparation and characterization and biodistribution studies of lomustine loaded PLGA nanoparticles by interfacial deposition method. J Nanomed Biotherapeutic Discov. 2015;5(138):2. doi:10.4172/2155-983X.1000138
69. Corrigan OI, Li X. Quantifying drug release from PLGA nanoparticulates. Eur J Pharm Sci. 2009;37(3–4):477–485. doi:10.1016/j.ejps.2009.04.004
70. Ratner BD, Hoffman AS, Schoen FJ, Lemons JE. Biomaterials Science: An Introduction to Materials in Medicine. Elsevier; 2004.
71. Rajapaksa TE, Lo DD. Microencapsulation of vaccine antigens and adjuvants for mucosal targeting. Curr Immunol Rev. 2010;6(1):29–37. doi:10.2174/157339510790231798
72. Park TG. Degradation of poly (lactic-co-glycolic acid) microspheres: effect of copolymer composition. Biomaterials. 1995;16(15):1123–1130. doi:10.1016/0142-9612(95)93575-X
73. Chauhan B, Shimpi S, Paradkar A. Preparation and characterization of etoricoxib solid dispersions using lipid carriers by spray drying technique. AAPS Pharm Sci Tech. 2005;6(3):E405–E409. doi:10.1208/pt060350
74. Erbetta CDAC, Alves RJ, Resende JM, de Souza Freitas RF, de Sousa RG. Synthesis and characterization of poly (D, L-lactide-co-glycolide) copolymer. J Biomater Nanobiotechnol. 2012;3(02):208. doi:10.4236/jbnb.2012.32027
75. Chandran S, Sanjay KS, Ali Asghar LF. Microspheres with pH modulated release: design and characterization of formulation variables for colonic delivery. J Microencapsul. 2009;26(5):420–431. doi:10.1080/02652040802424021
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.