")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Ethnic Diversity of DPD Activity and the DPYD Gene: Review of the Literature
Authors White C , Scott RJ, Paul C, Ziolkowski A, Mossman D, Ackland S
Received 1 September 2021
Accepted for publication 10 November 2021
Published 9 December 2021 Volume 2021:14 Pages 1603—1617
DOI https://doi.org/10.2147/PGPM.S337147
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Cassandra White,1,2 Rodney J Scott,1– 3 Christine Paul,1,2 Andrew Ziolkowski,3 David Mossman,3 Stephen Ackland1,2,4
1University of Newcastle, Newcastle, NSW, Australia; 2Hunter Cancer Research Alliance, Hunter Medical Research Institute, Newcastle, NSW, Australia; 3Division of Molecular Medicine, Pathology North John Hunter Hospital, Newcastle, NSW, Australia; 4Hunter Cancer Centre, Lake Macquarie Private Hospital, Gateshead, NSW, Australia
Correspondence: Cassandra White
Hunter Medical Research Institute, Lot 1, Kookaburra Circuit, New Lambton Heights, NSW 2305, Australia
Tel +61 2 4042 0000
Email [email protected]
Abstract: Pharmacogenomic screening can identify patients with gene variants that predispose them to the development of severe toxicity from fluoropyrimidine (FP) chemotherapy. Deficiency of the critical metabolic enzyme dihydropyrimidine dehydrogenase (DPD) leads to excessive toxicity on exposure to fluoropyrimidine chemotherapy. This can result in hospitalisation, intensive care admissions and even death. Upfront screening of the gene that encodes for DPD (DPYD) has recently been implemented in regions throughout Europe and the United Kingdom. Current screening evaluates DPYD variants that are well described within Caucasian patient populations and provides genotyped-guided dose adjustment recommendations based upon the presence of these variants. This article reviews the differences in DPYD gene variants within non-Caucasian populations compared to Caucasian populations, with regard to the implications for clinical tolerance of fluoropyrimidine chemotherapies and genotype guided dose adjustment guidelines.
Keywords: pharmacogenomics, non-Caucasian, DPYD gene, dihydropyrimidine dehydrogenase
Plain Language Summary
Fluoropyrimidine (FP; 5-FU, capecitabine) is a common chemotherapy used to treat many different cancers, including cancer of the colon and rectum, upper digestive tract, breast and head and neck. Cancer patients who receive FP chemotherapy are at risk of developing severe side effects from their treatment. A significant proportion of patients are deficient in the crucial enzyme (dihydropyrimidine, dehydrogenase DPD) that metabolises FP chemotherapies, and this can be assessed through testing the DPYD gene, the gene responsible for making DPD. Patients who are deficient in the DPD enzyme can have their chemotherapy dose adjusted in order to reduce side effects. This article explores the differences in DPYD gene variants between ethnic populations and the effect this has on DPD enzyme production and FP side effects.
Introduction
Fluoropyrimidine (FP) chemotherapy is frequently prescribed for treatment of a variety of cancers, including colorectal, upper gastrointestinal, breast and head and neck cancers. We estimate that these drugs are used in approximately 2 million people globally. Unfortunately, FPs have a narrow therapeutic window, meaning that patients frequently develop toxicity even at standard dosing. Severe toxicity can lead to hospitalisation, intensive care unit (ICU) admission and even death.1 Development of toxicity is in part determined by dihydropyrimidine dehydrogenase (DPD) the critical enzyme responsible for catabolism of FP chemotherapy. Deficiency in DPD is a risk factor for the development of severe toxicity. Genotyping of DPYD (which encodes DPD) can identify patients with DPYD variants who are DPD deficient and allow for prophylactic FP dose adjustments with initial treatment, thereby reducing the risks of FP therapy without compromising anticancer effect.2 While the variants that contribute to DPD deficiency are well described in Caucasian populations, there are less data about DPYD genetic variants within non-Caucasian populations. This article reviews studies that describe differences in DPD phenotyping and DPYD expression within non-Caucasian populations.
Fluoropyrimidine Metabolism and the Role of DPD
Fluoropyrimidine (FP) chemotherapies are synthetic antimetabolite agents that are prescribed to millions of patients globally each year. FP is the therapeutic backbone of colorectal cancer treatment and is also frequently a major component of therapies for curative, adjuvant and palliative management in upper gastrointestinal, head and neck and breast cancers. FP is delivered either as intravenous 5-fluorouracil (5-FU), or oral prodrugs, including capecitabine and tegafur. It is prescribed as both monotherapy and in combination with other chemotherapies, immunotherapies and biological agents for use in curative and palliative regimens, as well as chemoradiation protocols.
Dihydropyrimidine dehydrogenase (DPD) catalyzes the critical and rate limiting step in FP metabolism (Figure 1). Only 1–5% of 5-FU is converted to cytotoxic metabolites fluorodeoxyuridine (FUDR) and fluorouridine monophosphate (FUMP). These metabolites are further metabolized to fluorodeoxyuridine diphosphate (FdUDP), which damages DNA and RNA via incorporation of active metabolites fluorodeoxyuridine triphosphate (FdUTP) and flurouridine triphosphate (FUTP), respectively. FdUDP is also metabolised to fluorodeoxyuridine monophosphate (FdUMP), which inhibits DNA synthesis and repair via inhibition of thymidine synthase (TS).3,4 Approximately 80% of 5-FU is catabolised by DPD to dihydrofluorouracil (DHFU or FUH2) which in turn is converted to fluoro-beta-ureidopropionate (FUPA) and fluoro-beta-alanine (FBAL). These metabolites are excreted in the urine, along with the remaining 5–20% of unchanged 5-FU.5 Changes in DPD activity therefore have significant effect on the metabolic fate of available 5-FU (Figure 2). Profound inherited DPD deficiency can manifest as microcephaly, mental retardation and epilepsy.6
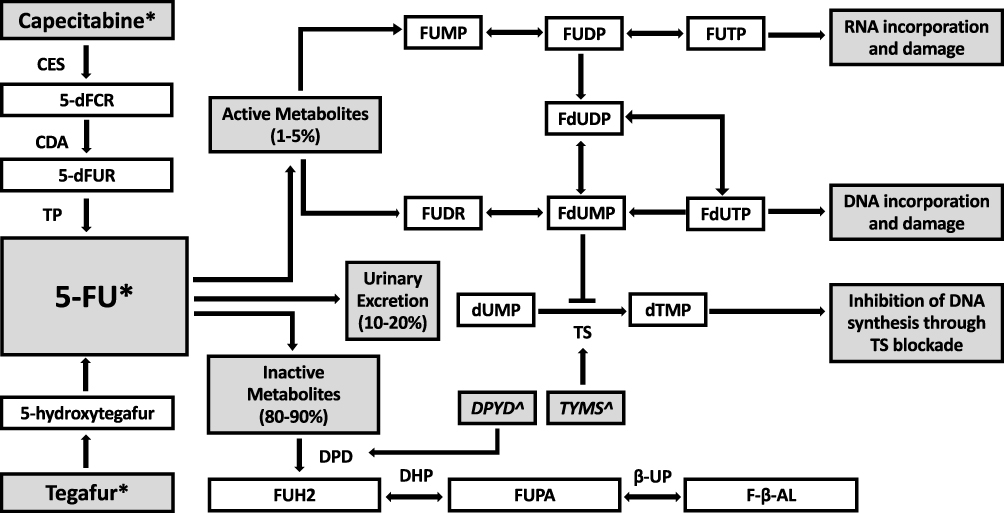 |
Figure 1 Metabolic pathway of fluoropyrimidines. Abbreviations: CES, carboxyl esterase; CDA, cytidine deaminase; TP, thymidine phosphorylase; TS, thymidylate synthase; DPD, dihydropyrimidine dehydrogenase; DHP, dihydropyrimidinase; B-UP, beta-ureidopropionase; 5ʹdFCR, 5ʹ-deoxyfluorocytidine riboside; 5ʹdFUR, 5ʹ-deoxyfluorouridine; FUH2, dihydrofluorouracil; FUPA, fluoro-beta-ureidopropionate; FBAL, fluoro-beta-alanine; FUMP, fluorouridine monophosphate; FUDP, fluorouridine diphosphate; FUTP, fluorouridine triphosphate; FUDR, fluorodeoxyuridine; FdUMP, fluorodeoxyuridine monophosphate; FdUDP, fluorodeoxyuridine diphosphate; FdUTP, fluorodeoxyuridine triphosphate; dUMP, deoxyuridine monophosphate; dTMP, deoxythymidine monophosphate. Notes: *Fluoropyrimidine chemotherapeutic agents, ^Genes. Genes: DPYD encodes DPD; TYMS encodes TS. |
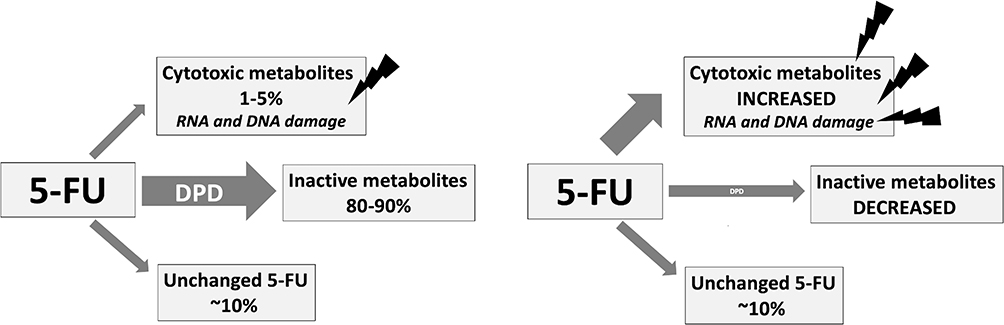 |
Figure 2 Effect of DPD deficiency on 5-FU metabolites. Notes: (left) 5-FU metabolism with normal DPD; (right) 5-FU metabolism with DPD deficiency. |
FP has a narrow therapeutic index, with a risk that a substantial proportion of patients may experience significant toxicity (Figure 2). Even at standard doses, approximately 30% of the patients develop grade 3 or higher toxicities, including myelosuppression, gastrointestinal effects (predominantly oral mucositis and diarrhoea), and palmar-plantar erythrodysesthesia (hand-foot syndrome, HFS).7–9 Up to 1% of the patients die from FP-related toxicity.4 It is estimated that DPD enzyme deficiency accounts for 39–61% of severe FP toxicity, which typically develops within the first 1–2 cycles of treatment.1,9 Between 3% and 5% of the people are partially DPD deficient within the Caucasian populations, and 0.02% are totally deficient.10,11
The DPD enzyme is encoded by DPYD, comprising 23 exons found on chromosome 1. While there are hundreds of variants that have been identified, a small group of well-described DPYD variants result in pathogenic DPD deficiency and severe toxicity on exposure to FP chemotherapy. Commonly described pathological DPYD variants in Caucasian populations include c.1905+1G>A (DPYD *2A, IVS14+1G>A, rs3918290), c.2846A>T (p.D949V, rs67376798), c.1236G>A/HapB3, (rs56038477/ rs75017182), and c.1679T>G (DPYD*13, p.I560S, rs55886062) existing either in trans or in cis configuration (Table 1). These variants are well described in various studies, mainly derived from patients of Caucasian ancestry.12–14 The combined incidence of these variants in Caucasian populations is as high as 12%.15–17
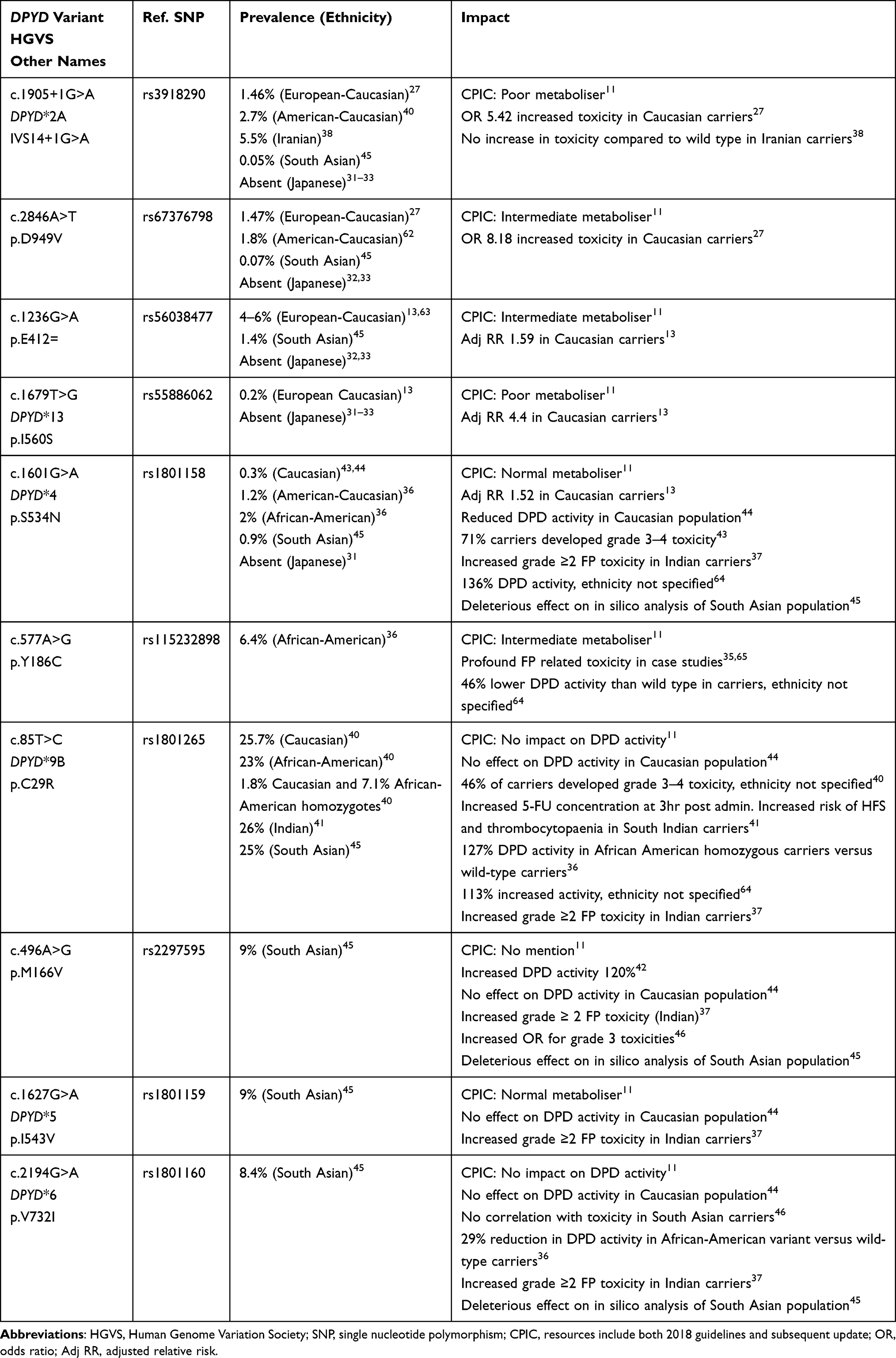 |
Table 1 Functional DPYD Variants in Caucasian and Non-Caucasian Populations |
Clinical Implications of DPYD Variants
The clinical utility of identifying patients with pathogenic DPYD variants is based upon dose adjustment of FP chemotherapy agents according to DPYD variant status to minimize the risk of toxicity. Prospective dose adjustment improves tolerance and safety of FP prescribing in patients who are expected to develop severe toxicity in the context of DPYD variant carrier status.12,15 Recommendations for genotype-guided dose adjustment are provided by two expert groups in pharmacogenetics; the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG). Both consortia recommend stratification of variant carriers into their metaboliser capability categories (poor metaboliser, intermediate/partial metaboliser and normal metaboliser) and agree that dose reductions of 25–50% should be initiated for patients who carry single variants of the above four predominant Caucasian variants (specific recommendations depend on both the individual variant and the gene activity score). Extreme caution (dose reductions 50–75% or more) and even FP omission are advised for compound heterozygotes or homozygotes.4,11 Clinical tolerance of dose-adjusted FP therapy should be closely evaluated and further adjustments made according to tolerability, including dose escalation for those who tolerate treatment well. Importantly, there are various examples of “normal metaboliser” variants as assigned by these guidelines that illustrate decreased DPD function and/or clinical toxicity in non-Caucasian groups. This review highlights some areas of published discrepancy between DPD activity, DPYD variant frequency and function.
DPYD Variants in Non-Caucasian Populations
Recently, several studies have explored the diversity of DPYD genotype variants outside of predominantly Caucasian populations, with attention focussed DPYD variants in African and Asian ancestries (including Eastern and Western Asian regions). Studies included in this review were those found by a literature search using PubMed and extensive reading around DPYD and DPD activity testing. Pivotal papers were examined in detail, and references were investigated for further articles pertaining to ethnic variability of DPYD and DPD investigations. The studies included in this review are not exhaustive; rather they offer an overview of novel DPYD variants within different ethnic groups, the phenotypic implications on both DPD activity and surrogate marker testing, and the impact of pre-treatment genotyping and phenotyping on clinical tolerance and toxicity in relation to FP exposure.
DPD Activity in Non-Caucasian Populations
Many studies over several decades have investigated DPD activity as an indicator of 5-FU toxicity.18–20 The preferred methodology employed across various studies for measuring phenotypic activity is high performance liquid chromatography (HPLC) on peripheral blood mononuclear cells (PBMC) cytosol in vitro using 14C-5-FU as substrate to ascertain the range and median activity in nmol/min/mg. From this, reduced activity can be categorised as “partial” and “profound” deficiency. “Profound” deficiency in this context does not refer to the neurological disability described by Braakehekke et al, but rather the capacity for 5-FU metabolism following FP administration.6 An alternate technique evaluates the ratio of the natural substrate uracil to the product of enzyme activity, dihydrouracil, in plasma.21
The concept of measuring DPD as a means of predicting FP toxicity was described by Lu et al in 1993.18 They evaluated 124 volunteers and 25 selected cancer patients who had experienced moderate-to-severe FP toxicities (62% Caucasian and 38% non-Caucasian), measuring DPD activity by HPLC on PBMCs. DPD activity was categorised as normal, partial or profound deficiency. Mean DPD activity across all patients was 0.425 ± 0.124 (SD) nmol/min/mg. Six out of 25 cancer patients who had developed grade 2–3 toxicity were found to be partially deficient, with a DPD level ≤30% of that in healthy controls. An additional 3 patients, who died from FP-related toxicity, were found to have profound DPD deficiency, <10% of normal. No significant difference in mean DPD activity was identified between Caucasian and non-Caucasian participants.
In contrast, a larger study of Caucasian versus non-Caucasian DPD activity found ethnic variation. Morsman et al explored in vitro DPD activity in a population of 296 volunteers from a variety of ethnic ancestries; South-West Asian (including Sri Lankan, Pakistani and Indian), Kenyan, Ghanaian and British-Caucasian.22 PBMC samples were analysed by HPLC and mean DPD activities were calculated and compared between subgroups, and against reference cohorts of French-Caucasian and Korean populations.23,24 The median DPD activity was similar in South-West Asian, Kenyan and British Caucasian populations (0.192, 0.194 and 0.215 nmol/min/mg, respectively). By comparison, Ghanaian participants had a significantly lower mean activity (0.119 nmol/min/mg). With regard to the reference cohorts, the French-Caucasian population illustrated a mean of 0.227 nmol/min/mg, and a significantly higher mean was found within the Korean population.23 Sohn et al, the original investigators of this Korean population found a mean DPD activity of 0.280 nmol/min/mg within their 114 healthy patient cohort.24 Unlike Lu, Morsman and Sohn studied healthy volunteers, and so could not correlate DPD activity with FP toxicity as these individuals were not exposed to FP chemotherapy agents.
Further DPD activity variation was described in people with African-American ancestry. Mattison et al evaluated DPD activity in PBMC samples of 258 healthy individuals of both African-American (n=149) and Caucasian (n=109) ancestry showing a DPD deficiency in 5.8% (15/258) across the entire cohort.25 Overall, 8% of African-American patients were deficient, compared to 2.8% of Caucasian individuals (P=0.07). All four individuals who were found to be profoundly deficient were African-American. Median DPD activity level of African-American individuals was lower than that of Caucasian individuals (0.26 ± 0.006 and 0.29 ± 0.007 nmol/min/mg, respectively; P=0.002). Although statistically significant, it is unclear if a difference of 0.03 mmol/min/mg in median activity confers clinical difference. Interestingly, these median levels are higher than those reported by Morsman et al for both Caucasian and African ancestry (Kenyan and Ghanaian) groups, 0.215, 0.194 and 0.119 nmol/min/mg, respectively.22 This may be a reflection of the variability of DPD activity measurement across centres or could reflect further variability within ethnic subgroups of similar ancestry.
Frequency of Typical and Novel DPYD Variants in Non-Caucasian Populations
The c.1905+1G>A variant was the first functionally significant DPYD variant to be described by Meinsma et al in a Dutch family with an inherited inborn error of pyrimidine metabolism.26 Several studies ensued, mainly retrospective, providing data to determine the significance of the other functional DPYD variants predominant in Caucasians, c.2846A>T, c.1236G>A and c.1679T>G.13,27 While these variants can predict the development of FP toxicity in heterozygous or homozygous Caucasian carriers, there is significant variability in the expected effects of these variants in non-Caucasian carriers.
The four prominent variants are not specifically limited to Caucasian populations and show variability in population and allelic frequency across ethnic groups (Tables 1 and 2). Uzunkoy et al evaluated the frequency of c.1905+1G>A using Restriction Fragment Length Polymorphism (RFLP) PCR in 218 Turkish participants, including 56 colorectal cancer patients undergoing FP chemotherapy.28 They found two heterozygote carriers in healthy participants and calculated a Minor Allele Frequency (MAF) of 0.6%, compared to 0.9% in Caucasian data. Uzunkoy et al then reviewed the literature for MAF in other ethnic populations, identifying a similar MAF in a Portuguese population, and an absolute absence of detection of the c.1905+1G>A variant in various studies within Asian and African populations. Unfortunately, there was no synchronous DPD activity evaluation to correlate the c.1905+1G>A genotype with DPD phenotype to explore the functional effects of ethnic diversity, nor was there an opportunity to correlate the variant with clinical toxicity, since the identified carriers were healthy participants. Uzunkoy suggested that the frequency alone supported inclusion of c.1905+1G>A in screening of Turkish cancer patients; however, there is certainly scope to investigate this further and correlate both DPD activity and clinical phenotypes in order to confirm that current dose adjustment guidelines are applicable to Turkish carriers. There is no data to further delineate the racial ancestry of this Turkish population.
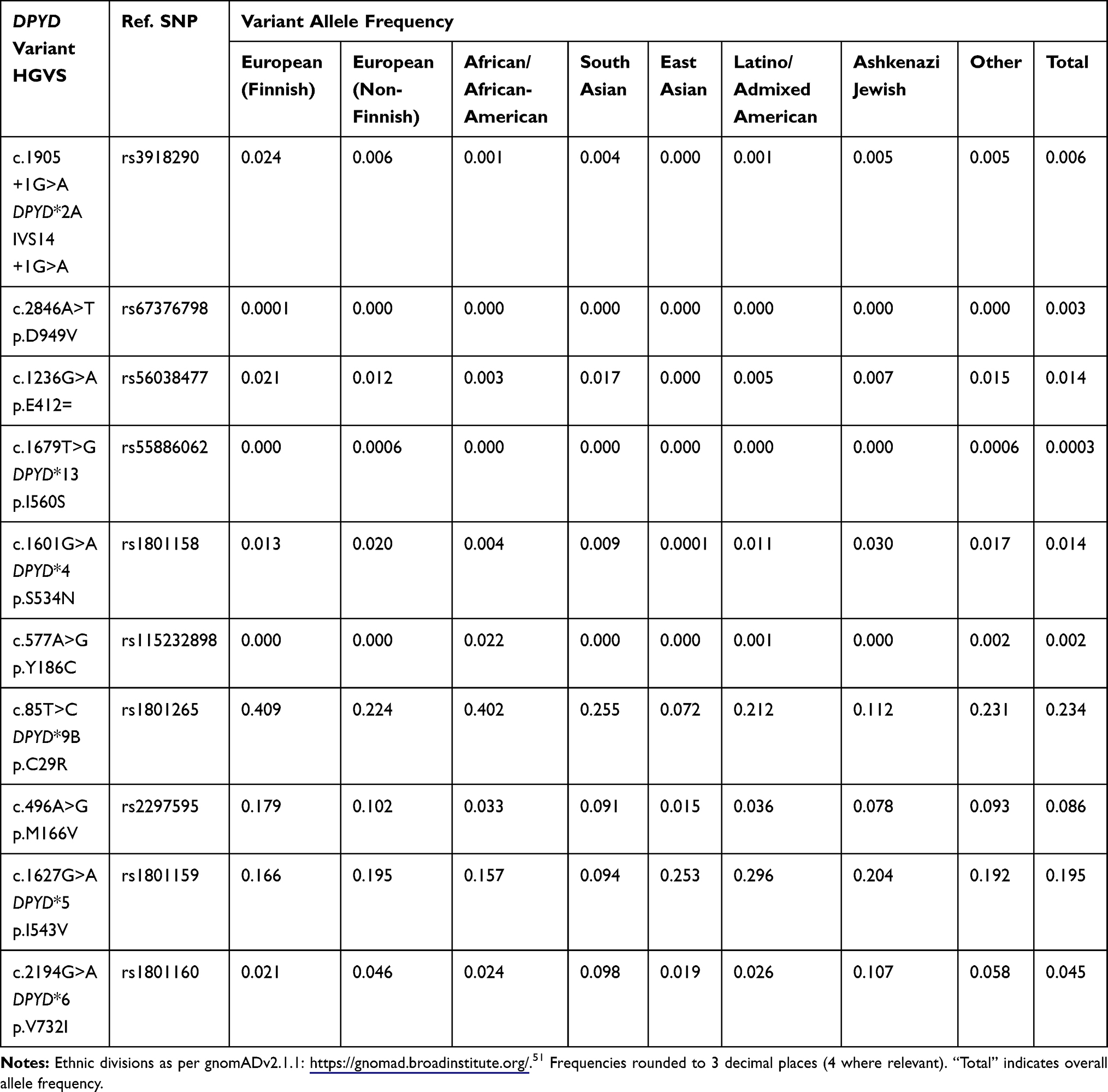 |
Table 2 Allele Frequency of DPYD Variants in Ethnic Populations |
Several studies of other non-Caucasian populations indicate the absence of the Caucasian DPYD variants. Studies of patients from East Africa (Elraiyah, 2017), China (He, 2008) and Japan (Maekawa, 2007) all indicate an absence of the c.1905+1G>A, c.1679T>G, and c.1601G>A variants.29–31 Variant c.1236G>A was also absent in the East African study of 588 individuals by Elraiyah et al, predominantly comprised of Somali and Kenyan subjects. Given the low frequency of these variants even within Caucasian populations, this could be explained by low prevalence coupled with small sample populations. Hishinuma et al further confirmed the differences in DPYD gene variants within the Japanese population, conducting a Sanger sequence analysis of 1070 healthy Japanese individuals, previously subjected to whole-genome sequencing (WGS).32 Twenty-one novel variants were identified and 12 of these exhibited decreased DPD enzymatic activity in vitro compared to wild-type controls. They confirmed the absence of the four common Caucasian DPYD variants. While this analysis shows a unique array of DPYD variants within the Japanese population that deliver reduced DPD expression in vitro, a clinical correlation in Japanese patients undertaking FP chemotherapies is needed to confirm significance.
Further example of ethnic heterogeneity within the Japanese population has been demonstrated by Yokoi et al, who investigated DPYD variants in 301 patients undergoing FP-based chemotherapy (among other exploratory gene variants).33 They found 15 DPYD variants, 7 of which were further evaluated in silico and were found to have probable pathogenic effects. Clinically, heterozygous carriers of each of these 7 variants developed grade 3 toxicities, including neutropaenia and/or diarrhoea and vomiting. Grade 3 toxicity incidence in the remaining wild-type cohort was 17%, 50/294.33 Yokoi identified that each pathogenic variant carrier was different and that this would pose an issue for pre-treatment screening panels in Japanese populations. Caucasian variants and others described within this review were also not detected in this Japanese cohort.
Linkage disequilibrium (LD) is the inheritance of alleles at closely situated loci occurring at a frequency that is higher than random chance, suggesting a non-random association.34 LD can be used to assist with gene mapping of ethnic groups. Maekawa et al investigated the linkage disequilibrium of DPYD variants and found strong LD between several novel variants (c.85T>C, c.496A>G and c.1627G>A) paired with lesser-known variants.31 While the clinical implications of these pairs are unclear, Maekawa illustrates that LD is present and possibly common in larger genes such as DPYD, further complicating the expected phenotypic effects of individual variants.
Clinical Impact of Novel Variants in Non-Caucasian Populations
Variants that are either not present or not clinically significant in Caucasian cohorts can have a profound impact in non-Caucasian populations. Saif et al first described the c.577A>G (aka. p.Y186C, rs115232898) variant in an African-American patient who developed profound toxicity on exposure to FP chemotherapy.17 Due to significant mononuclear cell depletion in the context of severe myelosuppression, DPD activity assayed by HPLC of PBMCs was not obtained. Similarly, Zaanan et al also described the development of severe toxicity following FP exposure in an African-Caribbean patient who was also identified as a heterozygote carrier of c.577A>G (in addition to c.85T>C and c.1627A>G).35 Offer et al studied DPD activity in PBMCs of 94 African-American and 81 European-American healthy participants to measure the correlation of c.577A>G carriers with DPD activity.36 Targeted DPYD exome sequencing identified 30 variants, 6 of which were found only in African-American participants, and 3 found only in European-Americans. The DPYD variant c.1905+1G>A was not identified in any participants. DPYD variant c.577A>G was identified in 6.4% of African-American participants and no European-American participants. The mean DPD activity for c.577A>G carriers was 46% lower than for wild-type carriers (0.279 versus 0.514 nmol 5-FU/min/mg, p=0.00029). Reduced DPD activity of 29% was also found among 6 African-American carriers of c.2194G>A (p.V732I), compared to wild-type carriers (0.361 versus 0.508 nmol/min/mg respectively, p=0.049). There was no associated reduction in DPD activity in European-American participants carrying the same variant. Carriers of c.85T>C who were not carriers of c.577A>G were evaluated for DPD activity, which revealed that homozygous c.85T>C carriers had 27% more DPD activity than wild-type participants (0.609 versus 0.480 nmol/min/mg, respectively p=0.013). It is worth clarifying that Offer et al uses a modified calculation technique that demonstrates the conversion of 5-FU per mg of protein, rather than FUH2 production per mg of protein as used by Morsman et al and Mattison et al.22,25,36 Offer et al explored the effect of linkage disequilibrium (LD) on DPD activity given that many participants with low DPD activity were carriers of more than one non-synonymous variant. They found weak LD between c.577A>G and c.2194G>A. Despite the stratified assessment of c.85T>C in carriers who were wild type for c.577A>G, no LD correlation was found. There is phenotypic discordance between the measured DPD activity reduction of 46% as measured by Offer et al and the assignment of “intermediate metaboliser” by the CPIC addendum (2020, https://cpicpgx.org/guidelines/guideline-for-fluoropyrimidines-and-dpyd/). A reduction of almost 50% in DPD function suggests that a variant allele that is essentially non-functional may require aggressive reduction in FP chemotherapy dosing in African-American patient carriers. This data is limited by small cohort size and warrants evaluation of variant status, DPD activity and 5-FU toxicity together in larger populations to gain a clearer understanding of the true clinical impact in order to more accurately guide dosing decisions.
Further to the identification of their high allelic frequency, Patil et al illustrated increased toxicity in a small cohort of 34 Indian patients carrying novel variants c.85T>C, c.1627A>G and c.2194G>A undertaking neoadjuvant FP-containing regimens for head and neck cancer.37 Patients who developed significant toxicity after cycle 1 of therapy were selected for targeted DPYD genotyping and were dose-reduced if a variant was detected. Of the 12 (35%) patients who developed grade ≥2 toxicity, 7 carried c.2194G>A, 5 carried c.85T>C, 2 carried c.1627G>A, 2 carried c.496A>G and 1 carried c.1601G>A. One patient did not carry any of these DPYD variants. Importantly, 5 patients were compound heterozygote carriers of multiple variants. There were no carriers of c.1905+1G>A or c.2846A>T. Variant carriers underwent dose adjustment for subsequent cycles, although Patil et al acknowledges that the guidelines were inaccurate for these specific variants, and 2/10 variant carriers (20%) went on to develop severe toxicity even after dose adjustment. Though this is a small study, it highlights several issues. First, the prevalence of DPYD variants is uniquely different to that of Caucasian populations, both in frequency and clinical effect. Second, there is a high incidence of compound heterozygotes in this selected population, which further complicates dosing decisions. While single variant carriers of c.2194G>A, c.85T>C, and c.1627G>A all experienced significant toxicity, no cases of isolated c.1601G>A or c.496A>G were documented, making phenotypic correlation of genotype effect difficult to confirm. It is unclear if these variants are independently significant or if they exist in linkage with other variants without clinical implication. Additionally, there may also have been more toxicity overall in this cohort given the use of triplet therapy, Overall toxicity in wild-type carriers was not included and therefore no conclusions can be drawn about relative toxicity of treatment in carriers of variants versus those who are wild type.
There are several important points to consider when interpreting DPYD genotype results and their impact on clinical tolerance of FP agents. Firstly, a wild-type result on a limited variant panel does not confer wild-type status for all known clinically significant variants that could be present within DPYD. Second, the absolute impact of compound heterozygote and homozygote carriers are more challenging to predict, and ideally DPD deficiency should be quantified using DPD activity measurement. Third, DPYD variants can have differing phenotypic expression between patients with different ethnic origins. The reason for this is probably multifactorial, but includes differences in linkage disequilibrium, genetic penetrance of variants across ethnic populations and differing genetic traits associated with 5-FP metabolism.
Genotype/Phenotype Correlation of DPYD Variants
The pursuit of genotype/phenotype correlation should extend across a diverse range of ethnic populations. The importance of this is illustrated by Negarandeh et al who conducted a prospective observational study of 73 Iranian colorectal cancer patients undergoing FP chemotherapies.38 They found 4/73 (5.5%) patients were heterozygous for c.1905+1G>A, tested via RFLP PCR. Both c.2846A>T and c.2194G>A were also investigated, and both were absent in the study population. FP dosing was not altered in light of DPYD variant carrier status. There were no differences in grade 3–4 toxicity between c.1905+1G>A carriers and wild-type comparators, with an overall incidence of 18.2%. Although this is a small study, it illustrates that c.1905+1G>A variant prevalence is high within this Iranian cohort compared with other Caucasian and non-Caucasian data. It also demonstrates a limited clinical impact of the c.1905+1G>A variant on FP tolerability and toxicity. This is in contrast to the significant metabolic impact that c.1905+1G>A imparts on Caucasian carriers. Quantification of DPD activity in this cohort would help to clarify the genotype/phenotypic correlation between variant and wild-type carriers.
Khushman et al conducted a genotype/phenotype correlative evaluation in 67 patients, 76% Caucasian, 22% African-American and 2% Hispanic from the United States.39 This retrospective analysis collated retrospective DPYD genotyping including DPYD *2A (tested in 100% of patients), c.2846A>T (82%) c.1679T>G (82%) c.85T>C (42%) and c.1590T>C (42%). Variants were found in 17/67 (25%) of patients; 13/17 (76%) were carriers of c.85T>C. The remaining 4 patients carried c.1905+1G>A and c.2846A>T, and c.1679T>G. Eight of the 13 c.85T>C carriers were treated with full dose FP chemotherapy and developed grade 3–4 diarrhoea. Three developed skin toxicity. It is unclear what percentage of variant carriers within this small cohort were African-American or Hispanic. Maharjan et al set out to reproduce the genotype/phenotype correlation from Khushman et al on a larger scale using retrospective data from 113 patients who underwent DPYD genotyping for c.1905+1G>A, c.1679T>G, and c.2846A>T.40 31% were African-American, 66% were Caucasian and 3% were other unspecified ethnicities. Of the total 113 patient cohort, DPYD c.85T>C was identified in 29 (25.7%) Caucasian, 26 (23%) of African-American patients and 2 (1.8%) of unspecified ethnicity. Grade 3–4 toxicity occurred in 46% of c.85T>C carriers, including grade 3–4 diarrhoea in 19% of carriers and grade 3–4 skin toxicity in 11%. This is compared to grade 3–4 toxicity in 38% of wild-type carriers (p=0.44). Of those who received full dose FP chemotherapy, 22/46 (48%) developed severe toxicity (p=0.83). This larger study, while still evaluating only a relatively small cohort size, showed similar prevalence of c.85T>C in Caucasian and African-American patients. The increase in toxicity incurred by carriers treated with full dose FP, while not statistically significant, could be argued to carry important clinical significance. This again highlights that different DPYD variants confer different phenotypic impact between ethnic populations and reaffirms the importance of considering genotyping for variants outside of the four typical Caucasian variants in patients of non-Caucasian ethnicity.
There is further evidence in support of a phenotypic effect of c.85T>C outside Caucasian patient populations. Varma et al conducted a focussed evaluation of c.85T>C genotype/phenotype correlation in 145 South Indian colorectal cancer patients being treated with capecitabine 1000mg/m2 twice daily for 14 days in 21-day cycles.41 They conducted a pharmacokinetic evaluation of drug level at 2 and 3 hours after administration. c.85T>G was found in 38/145 (26%), similar to that described by Khushman et al and Maharjan.39,40 Carriers had increased 5-FU concentrations at 3 hrs after administration of capecitabine compared to wild-type patients, Clinically, c.85T>C carriers developed increased incidence of HFS and thrombocytopaenia. There was limited data about other clinical toxicities such as mucositis and neutropaenia/myelosuppression, which Varma et al accepts as a limitation of this study and a goal for future exploration.
Another controversial DPYD variant, c.1601G>A, shows conflicting functional impact across various studies. Offer et al conducted an in silico study that concluded that c.1601G>A (along with c.85T>C) was not linked to decreased DPD activity and actually increased FP metabolic activity.42 Conversely, Loganayagam et al reported high incidence of grade 3–4 toxicity in 10/14 patients heterozygous for c.1601G>A in a study of 430 patients.43 Additionally, 2/24 variant carriers were compound heterozygotes with c.1601G>A and another significant DPYD variant allele. Both patients were hospitalised with grade 4 toxicity. And, 15.3% of this cohort were of African or Asian ancestry, however the ethnicity of the variant carriers is not reported. Nor was a quantitative measure of DPD activity obtained in variant carriers. Seck et al also confirmed decreased DPD function in c.1601G>A carriers, but normal function in c.85T>C carriers in their correlative study of genotype with DPD mRNA expression and DPD enzyme activity in 157 Caucasian patients.44 Both Naushad and Hariprakash identified increased toxicity in c.1601G>A carriers in their large-scale studies of South Asian (including South Indian) populations.45,46 Interestingly, Maekawa’s study in a Japanese cohort shows the absence of c.1601G>A, along with the Caucasian variants, again illustrating variability between various non-Caucasian populations.31 Variant c.1601G>A is not included in the 2018 CPIC guidelines and is marked as “fully functional” in the DPWG guidelines, implying that more evidence is needed to confirm clinical effect outside of Caucasian populations.4,11 Nevertheless, c.1601G>A cannot be ignored outside of Caucasian populations, given that the data is suggestive of clinical effect in certain populations.
Whole-Genome Sequencing and Ethnic Variability
Whole-genome sequencing allows for comprehensive genomic screening, which can be applied across large population sizes. Both Hariprakash et al and Naushad et al conducted WGS studies of several thousand subjects, both looking at DPYD genetic variants within South Asian ethnic groups, including individuals of Indian, Singaporean, Malaysian, Sri Lankan, Pakistani and Bangladeshi ancestry.45,46 In a pooled analysis, Hariprakash et al combined data from 16 studies of South Asian participants, utilising targeted, whole exome and whole-genome sequencing techniques. They included 3140 participants in this in silico analysis. Additionally, 110 Indian cancer patients undertaking FP chemotherapy were recruited for prospective targeted sequencing of 15 DPYD variants. They compared the calculations from various predictive software tools and found that the total number of calculated deleterious variants in the DPYD gene ranged from 38 (SIFT) and 39 (Polyphen-2) to 65 (PROVEAN). There were 14 variants mutually agreed upon by all software tools. This in silico analysis identified a high prevalence of c.85T>C, c.1627A>G, c.2194G>A, c.496A>G and c.1906–14763G>A within the South Asian population, with particularly high frequency of c.2194A>G and c.1906–14763G>A compared to other ethnic populations. Naushad et al describe genetic clustering of genotypes within a population of 2000 healthy Indian subjects.45 They used WGS combined with clinical guidelines and bioinformatics platforms to perform an in silico analysis of 11 variants. Classical variants 1236G>A and c.1905+1G>A were found at low MAFs, compared to more prevalent novel variants c.85T>C (MAF 25%), c.1627A>G (9%), c.496A>G (9%) and c.2194G>A (8.4%). Of the novel variants, c.496A>G, c.1601A>G and c.2194G>A were reported to display a deleterious effect based upon in silico analyses.
Both studies concluded that there is marked heterogeneity of variant distribution between global populations. In the Hariprakash et al patient cohort, 19/110 patients were carriers of c.496A>G. Carriers were at increased risk of serious FP toxicity, in particular mucositis (OR 7.2 heterozygote/OR 151 homozygote, p= 0.04) and diarrhoea (OR 7.9 heterozygote/OR 71 homozygote p=0.01). Two c.1905+1G>A heterozygotes were identified, both suffering grade 3 toxicities. There were three variants identified (including c.2194G>A and two rarer novel variants) that showed no toxicity correlation.46 This data is supported by the in silico results from Naushad et al but is in contrast to real-world data from Patil (2016), suggesting significant toxicity in 7 c.2194G>A carriers also in an Indian cohort of 34 head and neck cancer patients.37,45 It is possible that the differences in the FP containing regimens (mono and doublet therapy in Hariprakash’s cohort, versus neoadjuvant triplet therapy in Patil’s cohort) could have confounded the toxicity data, leading to a clinical correlation of DPYD variants to toxicity in Patil’s patients. DPD activity correlation may have offered more conclusive evidence of effect but was not included in this study.
Discussion
Differences across ethnic groups regarding both the genotypic and phenotypic characteristics of FP metabolism have long been recognised but not well understood. It is clear that there are inter-ethnic differences in DPYD variant frequency, DPYD variant expression, and the clinical impact of these variants, as well as differences in measured DPD activity. Mean DPD activity levels differ significantly in non-Caucasian populations compared to Caucasian levels.22,24,25 More data is needed to discern whether and how this ethnic variation contributes to population-based phenotypic differences in metabolism of FP and increased toxicity within non-Caucasian groups. Measurement of DPD activity is not standardized, making a universal threshold of normal activity difficult to define. Reasons for this, as described by Knickman et al could include failure to account for the circadian fluctuation of DPD and differences in testing equipment and protocols across laboratories.47 There is a role for DPD activity testing as a confirmatory test to determine the degree of deficiency in DPYD variant carriers (compared to local controls) once identified on genotyping, rather than its utilisation in mass-screening at this stage. Standardization of DPD activity testing, including an acceptable turnaround time and median values of significance (normal, partial and profound deficiency) is yet to be achieved and remains a barrier to the incorporation of DPD activity testing into service laboratories.
DPYD variants that have little or no effect in Caucasian populations can have profound implications for DPD activity and FP tolerability and toxicity in non-Caucasian groups. It is clear that a number of “novel” variants as labelled within the guidelines, including c.557A>G, c.85T>C, c.2194G>A, c.496A>G and c.1601G>A have a limited effect in Caucasian carriers but illustrate a significant impact on both DPD function and FP tolerability and toxicity when evaluated in non-Caucasian populations. The opposite is also true; the prevalence of typical Caucasian variants c.1905+1G>A, c.2846A>T, c.1236G>A, and c.1679T>G is diverse and even non-existent in some non-Caucasian populations.29–31 The limited recognition of novel variants in the guidelines is likely reflective of the abundant data describing DPYD variants in Caucasian populations compared to the limited but important data from non-Caucasian populations. With low frequency of each variant, substantial data is needed to verify clinical significance. It is foreseeable that our understanding of novel variants will improve as more studies are undertaken that focus on non-Caucasian patient populations. Ideally, dose decision support for variants of significance in non-Caucasian populations will eventually be included in consortia guidelines, and consideration of ethnicity built into screening tools and interpretation of results.
The diversity of DPYD variants across different ethnic populations complicates the current concept of DPYD genotyping as a screening tool to identify those at risk of increased toxicity. In many centres, DPYD genotyping incorporates a targeted selection of variants, leaving gaps where patients of non-Caucasian ancestry may not be as extensively or appropriately genotyped for variants relevant to their genetic lineage.48–50 This limitation suggests that a preferable strategy may be DPYD genotype screening using a DNA sequencing approach that would identify all DPYD-related variants and allow for cross-referencing against genomic databases which are ethnically diverse, such as gnomAD51 (Genome Aggregation Database https://gnomad.broadinstitute.org/). Currently, clinicians rely upon patients self-identifying their ethnic origins, which may not include historical ancestral lineage, as this may not be known. DNA sequencing may help to identify ethnic ancestry pertinent to DPD activity and DPYD gene variant expression that could impact clinical decision-making and guide safer FP dosing.
Ideally, in addition to genotyping, patients intending to receive FP chemotherapy should undergo synchronous phenotyping, either by measuring DPD activity or assessment of the UH2/U ratio. These techniques are varied in methodological approach and interpretation and still have their limitations, and ideally should be standardised if they are to be included in a clinical screening model of care. Synchronous genotype/phenotype assessment would fulfil two purposes. Firstly, DPYD variant carriers should be phenotyped to quantify the DPD enzyme impact and thereby more confidently guide dosing decisions. Furthermore, it would help to determine cumulative DPD deficiency in compound heterozygotes and homozygotes. Second, synchronous DPD testing will help to identify the proportion of patients who are DPD deficient without an identifiable DPYD variant, again allowing for safer dosing decisions. We know that a proportion of patients with DPD deficiency do not appear to carry a known DPYD variant, and suspect that the impairment of enzyme activity and or expression is related to other factors (including down-stream regulators and other genes implicit in DPD production or elimination).52–54 DPD quantification and genotypic correlation can help to clarify the expected effect of both classical and novel variants, which we know differ in both frequency and effect across ethnic populations.
More extensive genomic and phenotype testing obviously incurs a higher cost per patient. This may, however, be financially feasible in regions where the efficacy of targeted screening is lower due to increased ethnic diversity within the target population. Increased efficacy of screening can decrease financial expenditure on variant carriers and thereby reduce the burden on health care systems (hospitalisations, intensive care admissions, medication) in managing severe toxicity. This has proved cost-effective in retrospective and limited prospective analyses in Caucasian populations.55,56 Centres throughout Europe that include the UK are already incorporating pre-treatment screening for DPYD gene variants and DPD activity. This clinical implementation of testing is effective in reducing FP-related toxicity in patients by introducing genotype-guided fluoropyrimidine dosing but has not yet been extensively studied outside Caucasian populations.
There is a paucity of data describing the prevalence of DPD deficiency and DPYD variant carriers in Australia, as well as the incidence of severe toxicity related to FP administration. A single study has been conducted which illustrated a lower identification rate of DPYD variants c.1905+1G>A, c.1679T>G, c.2846A>T, c.1236G>A and c.557A>G; 7/176 (4%) compared to European data.57 The multicultural ethnic variability within Australia may partially account for the lower incidence of Caucasian variants, although this was not explored in the Lee study.
There are currently no data describing the ethnic distribution of patients receiving FP chemotherapy in Australia. Furthermore, there is no data to describe DPYD variants in Aboriginal Australians. This is in part due to both historical and cultural apprehension toward genetic testing, embedded in the historical mismanagement of genetic material and poor attention to ethical conduct and consent.58,59 This has necessitated important improvements in the consensual collection and management of biological material collected from patients within Australia, and identification of DPYD variants in Aboriginal Australians may be possible in the future.
The clinical implications of this DPYD genotyping and intervention currently focus upon safety of FP chemotherapy, by minimization of toxicities which develop during treatment. This has probable follow-on implications for cancer-related outcomes, disease free survival, progression-free survival and overall survival of cancer patients who have impaired tolerance and compliance with FP regimens. As we understand through limited data, adjusting chemotherapy dose based on the DPYD genotypic and DPD phenotypic characteristics of the individual does not have a detrimental impact of the anticancer effect of treatment. Limited studies have concluded equal efficacy of FPs in genotype/phenotype-guided dose-reduced patients compared to wild-type patients who are not dose-reduced, but further research is needed to confirm these findings from small studies/impressions.2,60,61
Linkage disequilibrium of DPYD variants may help to clarify the diverse functional impact of certain variants, such as those described by Offer (2013) and Maekawa.31,36 Ethnic heterogeneity of individual variants may extend to linkage patterns and help to explain the variation of effect of specific variants between ethnically diverse people and warrants further investigation. Furthermore, the impact of other pharmacogenomic drivers within the FP metabolic pathway (such as dihydropyrimidinase) is somewhat elusive at present and is one of several directions of further enquiry to understand the aetiology of patients who do not carry known functionally significant DPYD variant(s).
Conclusion
Screening for deficiency of DPD and associated DPYD variants is not a “one-size-fits-all” exercise. Consideration must be given to ethnic ancestry and diversity incurred in both DPD activity and causative DPYD variants within non-Caucasian populations. Single modality results should be interpreted with caution and efforts should be made to correlate DPYD variants with their phenotypic effect. Furthermore, current targeted screening may need to become more comprehensive to incorporate variants of significance in non-Caucasians in order to be truly predictive of FP toxicity risk in ethnically diverse societies.
Abbreviations
FP, fluoropyrimidine; 5-FU, 5-fluorouracil; DPD, dihydropyrimidine dehydrogenase (enzyme); DPYD, dihydropyrimidine dehydrogenase (gene); PBMC, peripheral blood mononuclear cells; HPLC, high performance liquid chromatography; PCR, polymerase chain reaction; MAF, minor allele frequency; LD, linkage disequilibrium gnomAD, Genome Aggregation Database; HGVS, Human Genome Variation Society; SNP, single nucleotide polymorphism.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Henricks LM, Opdam FL, Beijnen JH, Cats A, Schellens JHM. DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: call for a drug label update. Ann Oncol. 2017;28(12):2915–2922.
2. Henricks LM, van Merendonk LN, Meulendijks D, et al. Effectiveness and safety of reduced-dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: a matched pair analysis. Int J Cancer. 2019;144(9):2347–2354.
3. Diasio RB. Clinical implications of dihydropyrimidine dehydrogenase on 5-FU pharmacology. Oncology (Williston Park). 2001;15(1Suppl 2):
4. Lunenburg C, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28(4):508–517.
5. Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16(4):215–237.
6. Braakhekke JP, Renier WO, Gabreels FJ, De Abreu RA, Bakkeren JA, Sengers RC. Dihydropyrimidine dehydrogenase deficiency. Neurological aspects. J Neurol Sci. 1987;78(1):71–77.
7. Meta-Analysis Group In C; Levy E, Piedbois P, et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol. 1998;16(11):3537–3541.
8. Loganayagam A, Arenas-Hernandez M, Fairbanks L, Ross P, Sanderson JD, Marinaki AM. The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer Chemother Pharmacol. 2010;65(2):403–406.
9. Boisdron-Celle M, Capitain O, Faroux R, et al. Prevention of 5-fluorouracil-induced early severe toxicity by pre-therapeutic dihydropyrimidine dehydrogenase deficiency screening: assessment of a multiparametric approach. Semin Oncol. 2017;44(1):13–23.
10. Varughese LA, Lau-Min KS, Cambareri C, et al. DPYD and UGT1A1 pharmacogenetic testing in patients with gastrointestinal malignancies: an overview of the evidence and considerations for clinical implementation. Pharmacotherapy. 2020;40(11):1108.
11. Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210–216.
12. Henricks LM, Lunenburg C, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459–1467.
13. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(16):1639–1650.
14. Deenen MJ, Tol J, Burylo AM, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17(10):3455–3468.
15. Henricks LM, Lunenburg CA, Meulendijks D, et al. Translating DPYD genotype into DPD phenotype: using the DPYD gene activity score. Pharmacogenomics. 2015;16(11):1277–1286.
16. Stavraka C, Pouptsis A, Okonta L, et al. Clinical implementation of pre-treatment DPYD genotyping in capecitabine-treated metastatic breast cancer patients. Breast Cancer Res Treat. 2019;175(2):511–517.
17. Saif MW. Dihydropyrimidine dehydrogenase gene (DPYD) polymorphism among Caucasian and non-Caucasian patients with 5-FU- and capecitabine-related toxicity using full sequencing of DPYD. Cancer Genomics Proteomics. 2013;10(2):89–92.
18. Lu Z, Zhang R, Diasio RB. Dihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapy. Cancer Res. 1993;53(22):5433–5438.
19. Harris BE, Carpenter JT, Diasio RB. Severe 5-fluorouracil toxicity secondary to dihydropyrimidine dehydrogenase deficiency. A potentially more common pharmacogenetic syndrome. Cancer. 1991;68(3):499–501.
20. Fleming RA, Milano G, Thyss A, et al. Correlation between dihydropyrimidine dehydrogenase activity in peripheral mononuclear cells and systemic clearance of fluorouracil in cancer patients. Cancer Res. 1992;52(10):2899–2902.
21. Ackland SP, Garg MB, Dunstan RH. Simultaneous determination of dihydrofluorouracil and 5-fluorouracil in plasma by high-performance liquid chromatography. Anal Biochem. 1997;246(1):79–85.
22. Morsman JM, Sludden J, Ameyaw MM, et al. Evaluation of dihydropyrimidine dehydrogenase activity in South-west Asian, Kenyan and Ghanaian populations. Br J Clin Pharmacol. 2000;50(3):269–272.
23. Etienne MC, Lagrange JL, Dassonville O, et al. Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol. 1994;12(11):2248–2253.
24. Sohn DR, Cho MS, Chung PJ. Dihydropyrimidine dehydrogenase activity in a Korean population. Ther Drug Monit. 1999;21(2):152–154.
25. Mattison LK, Fourie J, Desmond RA, Modak A, Saif MW, Diasio RB. Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin Cancer Res. 2006;12(18):5491–5495.
26. Meinsma R, Fernandez-Salguero P, Van Kuilenburg AB, Van Gennip AH, Gonzalez FJ. Human polymorphism in drug metabolism: mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol. 1995;14(1):1–6.
27. Terrazzino S, Cargnin S, Del Re M, Danesi R, Canonico PL, Genazzani AA. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: a meta-analysis. Pharmacogenomics. 2013;14(11):1255–1272.
28. Uzunkoy A, Dilmec F, Ozgonul A, van Kuilenburg AB, Akkafa F. Investigation of IVS14+ 1G > A polymorphism of DPYD gene in a group of Turkish patients with colorectal cancer. Anticancer Res. 2007;27(6B):3899–3902.
29. He YF, Wei W, Zhang X, et al. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in Chinese cancer patients. J Clin Pharm Ther. 2008;33(3):307–314.
30. Elraiyah T, Jerde CR, Shrestha S, et al. Novel deleterious dihydropyrimidine dehydrogenase variants may contribute to 5-fluorouracil sensitivity in an East African population. Clin Pharmacol Ther. 2017;101(3):382–390.
31. Maekawa K, Saeki M, Saito Y, et al. Genetic variations and haplotype structures of the DPYD gene encoding dihydropyrimidine dehydrogenase in Japanese and their ethnic differences. J Hum Genet. 2007;52(10):804–819.
32. Hishinuma E, Narita Y, Saito S, et al. Functional characterization of 21 allelic variants of dihydropyrimidine dehydrogenase identified in 1070 Japanese individuals. Drug Metab Dispos. 2018;46(8):1083–1090.
33. Yokoi K, Nakajima Y, Matsuoka H, et al. Impact of DPYD, DPYS, and UPB1 gene variations on severe drug-related toxicity in patients with cancer. Cancer Sci. 2020;111(9):3359–3366.
34. Slatkin M. Linkage disequilibrium–understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008;9(6):477–485.
35. Zaanan A, Dumont LM, Loriot MA, Taieb J, Narjoz C. A case of 5-FU-related severe toxicity associated with the p.Y186C DPYD variant. Clin Pharmacol Ther. 2014;95(2):136.
36. Offer SM, Lee AM, Mattison LK, Fossum C, Wegner NJ, Diasio RB. A DPYD variant (Y186C) in individuals of African ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther. 2013;94(1):158–166.
37. Patil VM, Noronha V, Joshi A, et al. Dihydropyrimidine dehydrogenase mutation in neoadjuvant chemotherapy in head and neck cancers: myth or reality? South Asian J Cancer. 2016;5(4):182–185.
38. Negarandeh R, Salehifar E, Saghafi F, et al. Evaluation of adverse effects of chemotherapy regimens of 5-fluoropyrimidines derivatives and their association with DPYD polymorphisms in colorectal cancer patients. BMC Cancer. 2020;20(1):560.
39. Khushman M, Patel GK, Hosein PJ, et al. Germline pharmacogenomics of DPYD*9A (c.85T>C) variant in patients with gastrointestinal malignancies treated with fluoropyrimidines. J Gastrointest Oncol. 2018;9(3):416–424.
40. Maharjan AS, McMillin GA, Patel GK, et al. The prevalence of DPYD*9A(c.85T>C) genotype and the genotype-phenotype correlation in patients with gastrointestinal malignancies treated with fluoropyrimidines: updated analysis. Clin Colorectal Cancer. 2019;18(3):e280–e286.
41. Varma A, Jayanthi M, Dubashi B, Shewade DG, Sundaram R. Genetic influence of DPYD*9A polymorphism on plasma levels of 5-fluorouracil and subsequent toxicity after oral administration of capecitabine in colorectal cancer patients of South Indian origin. Drug Metab Pers Ther. 2020;35:4.
42. Offer SM, Fossum CC, Wegner NJ, Stuflesser AJ, Butterfield GL, Diasio RB. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74(9):2545–2554.
43. Loganayagam A, Arenas Hernandez M, Corrigan A, et al. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer. 2013;108(12):2505–2515.
44. Seck K, Riemer S, Kates R, et al. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin Cancer Res. 2005;11(16):5886–5892.
45. Naushad SM, Hussain T, Alrokayan SA, Kutala VK. Pharmacogenetic profiling of dihydropyrimidine dehydrogenase (DPYD) variants in the Indian population. J Gene Med. 2020;23:e3289.
46. Hariprakash JM, Vellarikkal SK, Keechilat P, et al. Pharmacogenetic landscape of DPYD variants in south Asian populations by integration of genome-scale data. Pharmacogenomics. 2018;19(3):227–241.
47. Knikman JE, Gelderblom H, Beijnen JH, Cats A, Guchelaar HJ, Henricks LM. Individualized dosing of fluoropyrimidine-based chemotherapy to prevent severe fluoropyrimidine-related toxicity - What are the options? Clin Pharmacol Ther. 2020;23(1):e3289.
48. Loriot MA, Ciccolini J, Thomas F, et al. [Dihydropyrimidine déhydrogenase (DPD) deficiency screening and securing of fluoropyrimidine-based chemotherapies: update and recommendations of the French GPCO-Unicancer and RNPGx networks]. Bull Cancer. 2018;105(4):397–407. Swedish.
49. Martens FK, Huntjens DW, Rigter T, Bartels M, Bet PM, Cornel MC. DPD testing before treatment with fluoropyrimidines in the Amsterdam UMCs: an evaluation of current pharmacogenetic practice. Front Pharmacol. 2019;10:1609.
50. National Health Service E. Clinical Commissioning Urgent Policy Statement Pharmacogenomic Testing for DPYD Polymorphisms with Fluoropyrimidine Therapies [URN 1869] (200603P); 2020.
51. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443.
52. Coenen MJH, Paulussen ADC, Breuer M, et al. Evolution of dihydropyrimidine dehydrogenase diagnostic testing in a single center during an 8-year period of time. Curr Ther Res Clin Exp. 2019;90:1–7.
53. Schwab M, Zanger UM, Marx C, et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26(13):2131–2138.
54. Rosmarin D, Palles C, Church D, et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: investigation in the QUASAR2 study, systematic review, and meta-analysis. J Clin Oncol. 2014;32(10):1031–1039.
55. Henricks LM, Lunenburg C, de Man FM, et al. A cost analysis of upfront DPYD genotype-guided dose individualisation in fluoropyrimidine-based anticancer therapy. Eur J Cancer. 2019;107:60–67.
56. Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2016;34(3):227–234.
57. Lee B, Lingaratnam S, Alexander M, et al. Feasibility of Implementation of a pharmacist-Led DPYD gene testing for patients newly commencing 5-Fluorouracil or Capecitabine. Asia Pac J Clin Oncol. 2020;16(S8):108–207.
58. Elsum I, McEwan C, Kowal EE, Cadet-James Y, Kelaher M, Woodward L.Inclusion of Indigenous Australians in biobanks: a step to reducing inequity in health care. Med J Aust. 2019;211(1):7–9.
59. Kowal EE. Genetic research in Indigenous health: significant progress, substantial challenges. Comment. Med J Aust. 2012;197(7):384.
60. Mercier C, Brunet C, Yang C, et al. Pharmacoeconomic study in head and neck cancer patients: impact of prospective DPD deficiency screening with 5-fluorouracil (5-FU) dose tailoring on toxicities-related costs. J Clin Oncol. 2009;27(15_suppl):6515.
61. Launay M, Dahan L, Duval M, et al. Beating the odds: efficacy and toxicity of dihydropyrimidine dehydrogenase-driven adaptive dosing of 5-FU in patients with digestive cancer. Br J Clin Pharmacol. 2016;81(1):124–130.
62. Zhao C, Li S, Liu Q. Meta-analysis of molecular targeted agents in the treatment of elderly patients with metastatic colorectal cancer: does the age matter? J Chemother. 2016;28(4):321–327.
63. Froehlich TK, Amstutz U, Aebi S, Joerger M, Largiader CR. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int J Cancer. 2015;136(3):730–739.
64. Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73(6):1958–1968.
65. Saif MW, Lee AM, Offer SM, McConnell K, Relias V, Diasio RB. A DPYD variant (Y186C) specific to individuals of African descent in a patient with life-threatening 5-FU toxic effects: potential for an individualized medicine approach. Mayo Clin Proc. 2014;89(1):131–136.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.