")
Back to Journals » Cancer Management and Research » Volume 12
Estrogen-Induced Stromal FGF18 Promotes Proliferation and Invasion of Endometrial Carcinoma Cells Through ERK and Akt Signaling
Authors Wu J , Tao X , Zhang H, Yi XH, Yu YH
Received 17 March 2020
Accepted for publication 7 July 2020
Published 4 August 2020 Volume 2020:12 Pages 6767—6777
DOI https://doi.org/10.2147/CMAR.S254242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Jian Wu,1,2 Xiang Tao,3,4 Hong Zhang,5 Xiang-Hua Yi,1 Yin-Hua Yu4
1Department of Pathology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, People’s Republic of China; 2Department of Pathology, Gongli Hospital, Second Military Medical University, Shanghai 200135, People’s Republic of China; 3Department of Pathology, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200032, People’s Republic of China; 4Department of Gynecology, Obstetrics and Gynecology Hospital, Fudan University, Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, Shanghai, 200011, People’s Republic of China; 5Department of Pharmacy, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, People’s Republic of China
Correspondence: Xiang Tao Email [email protected]
Hong Zhang Email [email protected]
Objective: The aim of this study was to evaluate whether estrogen promoted the proliferation and invasion of endometrial carcinoma (EC) cells through paracrine FGFs in endometrial stromal cells (ESCs).
Patients and Methods: We screened gene alterations in a primary ESC culture after 10 nM estrogen treatment using an Agilent mRNA microarray. We knocked down stromal FGF18 expression in a co-culture system and aimed to explore the contribution of E2-induced stromal FGF18 to the proliferation and invasion of EC cells. To determine the effective receptors and detailed downstream signaling of FGF18, we co-cultured estrogen-treated hESCs with FGFR1-, FGFR2-, FGFR3- or FGFR4-knockdown Ishikawa cells. Finally, we detected FGF18 expression in clinical samples, including several primary cultures of different ESCs and a series of tissue microarrays (TMAs) of 90 patients with EC.
Results: A few genes altered significantly in estrogen-treated primary ESCs, but only FGF18 was noticeably enhanced among the FGF family genes. Knockdown of FGF18 expression in hESCs inhibited the promoting effect of FGF18 on the proliferation and invasion of EC cells. FGF18 bound FGFR2 and FGFR3 in Ishikawa cells to activate downstream ERK and Akt pathways and to promote the viability of EC cells. The FGF18-FGFR2 and FGF18-FGFR3 pathways had close correlations with Survivin and CD44V6 expression but not with P53. Primary ESCs of endometrioid EC (EEC, type I EC) had higher FGF18 expression than ESCs of normal endometrium (NE), endometrial atypical hyperplasia (EAH) and type II EC.
Conclusion: Estrogen induced FGF18 in ESCs to promote the proliferation and invasion of EC cells, and FGFR inhibitors should be considered as promising candidate targets for EC treatment.
Keywords: FGF18, paracrine, proliferation, invasion, endometrial stromal cells, ESCs, endometrial carcinoma, EC
Introduction
Worldwide, endometrial cancer (EC) is the most prevalent invasive gynecologic malignancy.1 Despite recent therapeutic advances, in many cases, treatment failure results in cancer recurrence, metastasis, and death.1,2 The 5-year survival rate is only 84% for white women and 62% for black women in the United States.3 Imbalances in sex steroid hormones—excess stimulation of endometrial epithelium by estrogen relative to progesterone—are often conceptualized as a leading paradigm to account for the etiology of endometrial carcinomas.4 Most studies are entirely focused on neoplastic cells themselves and do not pay sufficient attention to the microenvironment in which the neoplastic cells survive.5 The tumor microenvironment is a heterogeneous population of cells mainly composed of epithelial cells, stromal fibroblasts, and inflammatory and endothelial cells. In fact, the crosstalk between the tumor and stroma promoting tumor growth and invasion has clearly indicated the dual role of stromal cells in the normal and cancerous states.2 Unfortunately, the details of the related mechanism of the correlation between estrogen and stroma remain largely unclear, even though accumulated data have shown that E2 might have no direct effect on E2-induced uterine epithelial proliferation.6
It is known that after dispersed form blood vessels, estrogen directly acts on nearby stromal cells instead of “distant” epithelial cells. Furthermore, in addition to the direct effects on epithelial cells, estrogen can regulate epithelial cells in a paracrine fashion through stromal cells. In the female reproductive tract, it also appears that steroid receptors in ESCs, but not in the epithelium, may be required for the action of E2, thus demonstrating the paracrine role of stromal cells in endometrial function.6 Therefore, genetic alterations at the molecular level in ESCs might precede this alteration in epithelial cells and essentially act as the initial driver of EC pathogenesis. Accumulating data indicate that estrogen mediates the biological behaviors of epithelial and endothelial cells by regulating paracrine activities in ESCs through signaling pathways such as the SDF-1α/CXCR4, TNF-α/HGF/C-met and VEGF/VEGFR1 pathways.7–9 Interestingly, estrogen also induces ESCs of the mouse uterus to upregulate stromal FGF10 and Bmp8a expression, which promotes epithelial proliferation.10 However, we do not know whether estrogen indirectly controls the proliferation and invasion of human epithelial cells by paracrine FGFs in ESCs.
The present study demonstrated that estrogen-induced FGF18 was elevated in stromal cells and stimulated the growth and invasion of EC cells. FGF18 bound FGFR2 or FGFR3 to activate downstream ERK and AKt pathways and to promote EC cell viability with a close correlation with Survivin and CD44V6 but not with P53 expression. Patients with endometrioid endometrial carcinoma (EEC) had higher stromal FGF18 expression.
Patients and Methods
Patients
The endometrial samples that were used for primary culture were obtained from patients with normal endometrium (NE), atypical endometrial hyperplasia (AEH) and endometrioid endometrial carcinoma (EEC). A series of tissue microarrays that included 90 patients with EC were performed in this study. All patients were treated with radical surgery for EC and pelvic lymphadenectomy at Gongli Hospital between January 2008 and November 2012. None of the patients had preoperative chemotherapy. The histology subtypes consisted of 75 cases of type I and 15 cases of type II EC. In accordance with the Declaration of Helsinki, the Ethical Committee of Gongli Hospital of Second Military Medical University approved this study. Written informed consent was obtained from all patients.
Drugs and Antibodies
17β-Estradiol (E2, Sigma-Aldrich, Missouri, USA) was dissolved in DMSO at a concentration of 1 μM and stored at −20 °C. The Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) reagent was purchased from Lizhi Biological Company. Antibodies against Vimentin (catalog no. ab92547), FGF18 (catalog no. ab86571), GAPDH (catalog no. ab8245), ER (catalog no. ab108398), FGFR1 (catalog no. ab824), FGFR2 (catalog no. ab109372), FGFR3 (catalog no. ab155960), FGFR4 (catalog no. ab240205), P53 (ab26), Survivin (ab76424) and CD44V6 (ab78960) were purchased from Abcam. Antibody against CKpan (catalog no. RAB-0050) was purchased from MXB Biotechnology (China). Antibodies against AKt (catalog no. 9272S), phosphorylated (p)-Akt (Ser473) (catalog no. 9271), P44/42 MAPK (ERK1/2) (catalog no. 4695) and phosphorylated (p)-p44/42 MAPK (ERK1/2) (catalog no. 9488) were purchased from Cell Signaling Technology, Inc.
Cell Culture
The EC cell lines Ishikawa and Hec-1A were obtained from Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, Obstetrics and Gynecology Hospital of Fudan University. The immortalized human ESC line hESCs, established from a patient with uterine leiomyoma at Obstetrics and Gynecology Hospital of Fudan University, were obtained from Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases. Primary ESCs were isolated from patients with proliferative endometrium at the Obstetrics and Gynecology Hospital, Fudan University. Briefly, the endometrial samples were washed with PBS three times, cut into 1–3 mm3 pieces, and digested for 30 min at 37 °C in PBS containing 2 mg/mL collagen IV (Invitrogen, USA). Stromal cells were separated from epithelial cells by sequential sieving through 100 µm, 70 µm and 40 µm nylon sieves, with ESCs passing into the filtrate. All cell lines and primary-culture ESCs were cultured in DMEM/F-12 medium (Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA) and antibiotics, including streptomycin (100 µg/mL), penicillin (100 µg/mL) and amphotericin B (0.5 µg/mL) (all Sigma, USA). Some epithelial cells were mixed with ESCs during the process of primary culture. However, epithelial cells are difficult to passage, so the mixed epithelial cells disappeared after passaging. Therefore, we only used ESCs that had been passaged 2–3 times for the following tests. To identify the origin of primary culture cells, we detected CK and Vimentin expression in primary culture cells with a WB assay. ESCs were used for subsequent experiments only when the purity exceeded 95%. All cell lines in our experiment were authenticated by STR profiling.
Estrogen Treatment
Primary ESCs, which can be reseeded and cultured for approximately 10 generations without genetic changes, were treated with 10 nM E2 for 48 h to identify the changes in FGF family genes. Both hESCs and primary ESCs were evaluated for FGF18 expression after 0, 10 and 100 nM E2 treatments. The hESCs were co-cultured with EC cells at a concentration of 10 nM E2. After centrifugation and sterile filtration, the cell medium (CM) of hESCs treated with E2 for 72 h was collected and stored at −20 °C.
Agilent Microarray
After primary ESCs were treated with 0 nM (the control group) or 10 nM E2 for 3 d and 7 d, 1×106 ESCs were suspended in 1.0 mL of TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The mRNA was reverse-transcribed into cDNA (Takara), and the quality was verified before sending the samples to KangCheng Bio-Tech (Shanghai, China) for Agilent Whole Human Genome Array GeneChip microarray analysis. Data analysis was performed using Agilent Feature Extraction Software. Differential expression of genes was expressed as the fold change, and compared with gene expression in the control group. Genes with a 2-fold difference simultaneously in both the 3 d and 7 d groups were considered significant. To focus our research on certain fundamental and crucial genes, we added the fold value of the differentially expressed genes of the 3 d and 7 d groups and selected those genes with a total sum of fold value ≥ 5 as genes of interest in our study.
Western Blotting
Primary cultured ESCs and the ESC line hESCs were lysed with RIPA buffer supplemented with protease inhibitors, and proteins were quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime, China). Whole-cell lysates (20 μg of protein per lane) were subjected to 4–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking with 5% BSA for 2 h, PVDF membranes were incubated with antibodies at 4 °C. Afterwards, antigen-antibody complexes were incubated with goat anti-rabbit HRP-conjugated secondary antibody for 1 h at room temperature. Finally, detection was performed with electrochemiluminescence (ECL, Thermo, USA). All WBs were repeated three times, and the average values were used for subsequent statistical analyses.
Co-Culture Experiment
After digestion and suspension, 5×105 EC cells and 1×105 hESCs were seeded into the lower chambers and upper inserts of 24-well plates, respectively, for the co-culture experiment. Tumor cells were cultured with MEM/F-12 medium and 10% FBS and E2 at a concentration of 10 nM. The proliferation of EC cells was detected by the CCK-8 assay. Each experiment was repeated at least three times in triplicate.
Cell Proliferation
After co-culture of hESC and EC cells for 0, 24, 48 and 72 h, Ishikawa or Hec-1A cells were washed three times with PBS, and 100 μL of CCK-8 (Dojindo, Kumamoto, Japan) solution was added to every well of a 24-well plate. After incubation for 1 h, the absorbance of cells was measured at 450 nm using an ELISA reader (BioTek, Winooski, VT, USA) according to the manufacturer’s instructions. Each experiment measured six samples at a time, and each experiment was performed for a minimum of three times.
Transwell Assay
A Transwell assay (Costar; Corning, Inc.) was performed to evaluate the invasive ability of Ishikawa and Hec-1A cells in vitro. The upper inserts were coated with growth factor-reduced Matrigel for the invasive assay. Ishikawa or Hec-1A cells were seeded into the upper inserts at a concentration of 1×105 cells. The medium in the upper inserts contained 50% serum-free DMEM-F12 medium and 50% CM of estrogen-treated hESCs a total volume of 500 μL. Medium containing 10% FBS was added to the lower chambers as a chemoattractant. Following incubation at 37 °C for 24 h, the medium of the upper insert was discarded, and cells on the upper surface, especially the edge, of the membrane were removed by a Q-tip. The invaded cells were fixed with formaldehyde and stained using 0.5% crystal violet (Sigma-Aldrich; Merck KGaA). The number of invaded cells was counted in five randomly selected fields.
Cell Transfection
We chose the immortalized ESC line hESCs for our transfection assay because the rate of cell infection in the ESC line hESCs was higher than that in primary-culture ESCs. FAM-siRNAs against FGF18, FGFR1, FGFR2, FGFR3 and FGFR4 were designed and synthesized by GenePharma Inc. (Shanghai, China). The transfection of siRNA-FGF18 in hESCs and the transfection of siRNA-FGFR1, -FGFR2, -FGFR3 and -FGFR4 in Ishikawa cells were performed using Lipofectamine 3000 (Invitrogen, Life Technologies, USA) in a 6-well plate with 105 cells. The transfection efficiency of antibodies was detected by WB 48 h after transfection. SiRNA-FGF18-transfected hESCs were co-cultured with EC cells at a concentration of 10 nM E2 for the subsequent evaluation of cell proliferation and invasion. Ishikawa cells transfected with siRNA-FGFR1, siRNA-FGFR2, siRNA-FGFR3 and siRNA-FGFR4 were used to identify the downstream signaling pathways of FGF18.
Immunohistochemistry
Formalin-fixed, paraffin-embedded archival tissue samples were retrieved from 90 sampled cases. TMA was constructed according to the method by Matysiak et al.11 Immunohistochemical staining with FGF18 antibody was performed on the TMA sections using the Bond Polymer Intense Detection System (Leica Microsystems, Inc., Germany) according to the manufacturer’s protocol with minor modifications. The intensity of immunostaining was scored as 0, negative; 1+, weak; 2+, moderate; or 3+, intense. The proportion of positive-stained cells was assigned to one of four categories: 0, ≤ 5%; 1, 6–25%; 2, 26–50% and 3, 50%-75%; or 4, >75%. The staining intensity and the proportion of positively stained cells were summed to calculate the cumulative score. According to the immunostaining score, the 90 EC cases were divided into a low expression group (0–6 score) and a high FGF18 expression group (8–12 score). FGF18 was positively expressed in stromal and epithelial cells, but in this study, we only scored stromal FGF18 expression.
Statistics Methods
Statistical analyses were performed using SPSS Statistics software version 20.0 (IBM Corporation, USA), and the data are presented as the mean ± standard deviation (SD). The results of the protein expression, cell proliferation and invasion assays were analyzed using Student’s t-tests. Associations between stromal FGF18 expression levels and clinicopathological variables were assessed using Chi-square tests. The results with a P value ≤ 0.05 were considered significant (* P≤0.05; ** P≤0.01; *** P≤0.001).
Results
Identification of ESCs and E2 Treatment
Primary culture ESCs were derived from patients that were diagnosed with normal endometrium (NE), endometrial atypical hyperplasia (EAH) and endometrioid endometrial carcinoma (EEC). To identify the purity of the different ESCs, we detected Vimentin and CKpan expression in different kinds of ESCs and hESCs by WB. The WB results indicated that all ESCs were positive for Vimentin but negative for CKpan expression (Figure 1A). The present study aimed to understand the paracrine effects of estrogen-treated ESCs to ensure that ESCs express estrogen receptors (ERs). The results showed that primary culture ESCs and hESCs expressed both ERα and ERβ proteins. After treatment with 10 nM E2 for 72 h, primary ESCs and hESCs had morphological alterations, such as oval- and short fusiform shapes becoming slim- and fishbone-like arranged shapes (Figure 1B).
Estrogen Promoted ESCs to Increase Paracrine FGF18 Expression
We analyzed the results of the Agilent microarray and found 25 upregulated and 20 downregulated genes with significant differential expression. These differentially expressed genes were involved in signaling pathways, transcription factor function, energy metabolism and RNA regulation. We show 11 genes that were identified to be involved in signaling pathways in Table 1. We summarize the characteristics of mRNA expression for the FGF-related family genes after estrogen treatment for 3 d in Figure 2A. The results revealed 11 upregulated and 10 downregulated genes of FGF-related family members (including ligands and receptors). However, only the expression of FGF18 mRNA was upregulated 3.46-fold compared with the control group, which was not treated with estrogen. To verify the results of the Agilent microarray, we examined FGF18 expression in primary ESCs and the ESC line hESCs after treatment with 0, 10 and 100 nM E2 by WB. The expression of FGF18 protein in ESCs was increased after E2 treatment. Compared with that in the control groups, which were only cultured with DMEM-F12 medium, stromal FGF18 expression in the 10 nM and 100 nM E2 treatment groups was elevated (Figure 2B). Furthermore, upregulated FGF18 expression was found in hESCs after 10 nM and 100 nM E2 treatments (Figure 2C). ELISA results also demonstrated that paracrine FGF18 proteins in the CM from ESCs and hESCs were enhanced after treatments with 10 and 100 nM E2 (Figure 2D).
 |
Table 1 The mRNA Expression of Genes with Significant Difference in ESCs That Being Involved in Signaling Pathways After 10 nM E2 Treatment |
E2 Promoted the Proliferation and Invasion of EC Lines via Paracrine FGF18 Expression
The EC cell lines Ishikawa and Hec-1A were co-cultured with hESCs to observe paracrine growth promotion in adjacent epithelial cells (Figure 3A). Estrogen-treated hESCs promoted the proliferation of Ishikawa and Hec-1A cells through a paracrine mechanism. Compared with the control group, the estrogen-treated experimental group had more strongly increased proliferation in both Ishikawa and Hec-1A cells after 48 and 72 h of culture (Figure 3B and C). As the model (Figure 3D) showed, the numbers of invading Ishikawa and Hec-1A cells increased after co-culture with CM from estrogen-treated hESCs for 24 h (Figure 3E and F). WB demonstrated that FGF18 expression in FGF18-knockdown hESCs was obviously less than that in the negative control cells (Figure 3G). ELISA results also revealed that despite treatment with 10 nM E2 for 48 h, the paracrine FGF18 protein level in the CM of siRNA FGF18-transfected hESCs decreased markedly (Figure 3H). When co-cultured with si-FGF18-treated hESCs, Ishikawa and Hec-1A cells had decreased proliferation compared to the negative control groups of si-NC-treated hESCs (Figure 3I and J). The number of invading Ishikawa and Hec-1A cells was decreased after culture with CM from estrogen-treated FGF18-knockdown hESCs for 24 h (Figure 3K and L).
FGF18 Bound FGFR2 and FGFR3 to Activate Downstream ERK and Akt Signaling
To ascertain the effective receptors and detailed downstream pathways of stromal paracrine FGF18, we knocked down FGFR1-4 expression in Ishikawa cells (Figure 4A). We co-cultured hESCs with FGFR1-, FGFR2-, FGFR3- or FGFR4-knockdown Ishikawa cells treated with E2 at a concentration of 10 nM and explored the expression of a few downstream target genes (Figure 4B). WB results showed that FGFR2- and FGFR3-knockdown Ishikawa cells had reduced expression of phospho-ERK, phospho-Akt, Survivin and CD44V6 (Figure 4C). In contrast, FGFR1- and FGFR4- knockdown cells had the same expression of phospho-ERK, phospho-AKT, Survivin and CD44V6 as the control group of si-NC Ishikawa cells. Our results demonstrated that FGF18 bound FGFR2 and FGFR3 to activate downstream ERK and Akt signaling and had close correlations with Survivin and CD44V6 expression. The expression of ERK, Akt, and P53 was not different among all FGFR-knockdown groups, which indicated that these genes were not downstream target genes of FGF18. FGFR2- and FGFR4-knockdown Ishikawa cells were co-cultured with hESCs treated with 10 nM E2 for 1 d (cell invasion) and 3 d (cell proliferation). The results showed that FGF18/si-FGFR2 signaling inhibited the proliferation and invasion of Ishikawa cells compared to FGF18/si-NC and FGF18/si-FGFR4 signaling (Figure 4D and E). FGF18-FGFR2 or FGFR3 signaling should play major roles in the proliferation and invasion of EC cells.
Clinical Patients with Type I EC Had Higher Stromal FGF18 Expression
Five cases of primary culture ESCs derived from NE, EAH and EEC patients were assayed for FGF18 expression by WB (Figure 5A). The results demonstrated that ESCs in EEC had higher FGF18 expression than ESCs in NE and EAH (Figure 5B). In contrast, FGF18 expression in ESCs of the NE and EAH groups was not different. We also measured FGF18 expression in EC TMAs by using immunohistochemistry staining. Stromal (* shown) and epithelial (arrow shown) cells of type I EC had stronger expression of FGF18, whereas only the epithelium of type II EC had increased FGF18 expression (Figure 5C). The analysis of the FGF18 score in EC TMA revealed that patients with type I EC had higher stromal FGF18 scores than patients with type II EC or with proliferative phase (Figure 5D).
Discussion
The tumor microenvironment plays an essential role in the malignant transformation of epithelial cells and invasion of tumor cells. The FGF-FGFR pathway plays important physiopathological roles in controlling the cells of the extracellular matrix, mainly fibroblasts and endothelial cells, and mediates the cross-talk between tumor cells and the surrounding stroma.3,12,13 The FGF family contains 23 members, namely, 18 ligands and 5 FGF receptors (FGFRs).14 Located on chromosome 14p11, FGF18 promotes the proliferation of normal fibroblasts, liver and intestinal cells.15,16 Moreover, FGF18 also plays a major role in manipulating the growth and invasion of tumor cells. Autocrine FGF18, produced by a mutation in tumor cells, is overexpressed in gastric, breast and lung cancers and stimulates tumor cell growth.17–19 Simultaneously, tumor cells stimulate stromal cells to produce and release paracrine FGFs, which activate FGFR expression and constitute the paracrine loop of tumor cells.14 It has been reported that autocrine and paracrine FGF18 is clearly involved in regulating the behaviors of colon and liver cancers.20,21
There have been a few attempts to clarify the role of FGF18 in EC. It has been reported that bazedoxifene, an estrogen receptor modulator, can reduce stromal FGF18 expression by inhibiting the inducing effects of estrogen on ESCs.22 Another finding indicated that progesterone inhibits epithelial proliferation by inducing Stromal Heart and neural crest derivatives expressed transcript 2 (HAND2) to repress FGF18 expression in ESCs.23 These data are consistent with our findings that estrogen-induced stromal FGF18 stimulated the viability of EC cells. Hence, we can conclude that paracrine FGF18 in estrogen-treated ESCs might play major roles in EC carcinogenesis. The fact that stromal FGF18 was overexpressed in patients with type I EC further supported our conclusion because type I EC is closely associated with estrogen. In contrast, Flannery et al found that endometrial adenocarcinoma elevates FGF18 expression compared with normal endometrial epithelium.22 This finding was consistent with our observation that epithelial FGF18 was overexpressed in TMA, including cases with type I and type II EC. According to the notion that epithelial FGF18 plays a major role in the stimulation of epithelium in most solid cancers, it was reasonable to presume that epithelial FGF18 might exert some crucial functions in EC carcinogenesis. Autocrine epithelial FGF18 might have effects on both type I and type II EC, whereas paracrine stromal FGF18 only has effects on type I EC. However, further studies are needed to determine the extent to which autocrine FGF18 of epithelial cells is involved.
Our data revealed that FGF18 activated FGFR2 and FGFR3 to transmit proliferative signaling through the ERK and AKt pathways. In recent years, there has been increasing interest in the downstream signaling of FGF18, which has proven that the ERK and/or AKt pathways are major downstream pathways of FGF18 in lung, gastric, liver and breast cancers.17,19,24,25 Survivin and CD44V6 are downstream targets of ERK and/or AKt signaling in several malignancies, such as colorectal, breast and oral cancers.26–28 Some FGFs of FGF-family genes, such as FGF8 and FGF2, can promote Survivin expression in rectal and small cell lung cancers.29,30 Blocking FGFR expression in pancreatic cancer cell lines also inhibits Survivin expression.31 Our experiment suggested that the FGF18-FGFR2 and FGF18-FGFR3 pathways had a close correlation with Survivin and CD44V6 in EC through the ERK and Akt pathways. The enhanced FGF18-induced Survivin and CD44V6 protein levels are helpful in understanding the promotion of EC cell proliferation and invasion.
Inhibition of the FGF/FGFR pathway as a therapeutic approach in various cancers has been applied for several years, with its consequent effects on both parenchymal and stromal tumor compartments.32 Moreover, approximately 10% of EC cases have an FGFR2 mutation that is associated with shorter patient survival. It seems that we should consider inhibitors of FGF-FGFR signaling as a candidate approach for the targeted therapy of EC.
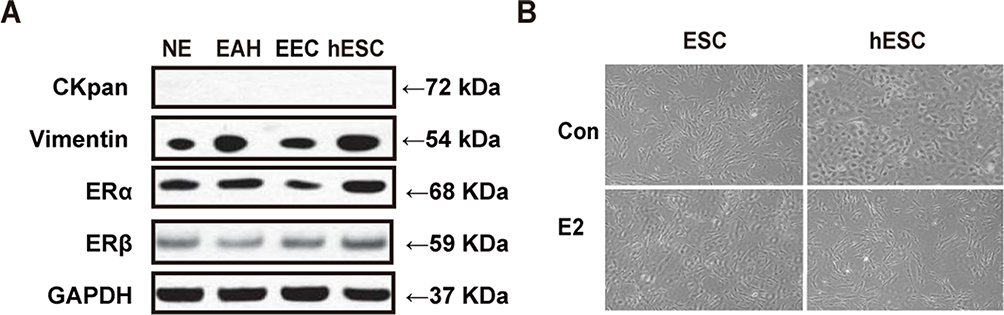 |
Figure 1 Identification of ESCs and E2 treatment. (A) The WB results indicated that of the different ESCs (NE, EAH and EEC) were positive for Vimentin, ERα and ERβ, but negative for CKpan expression. (B) Primary culture ESCs and hESCs had morphological alterations from oval- and short fusiform-shaped to slim- and fishbone-like arranged shapes after E2 treatment. |
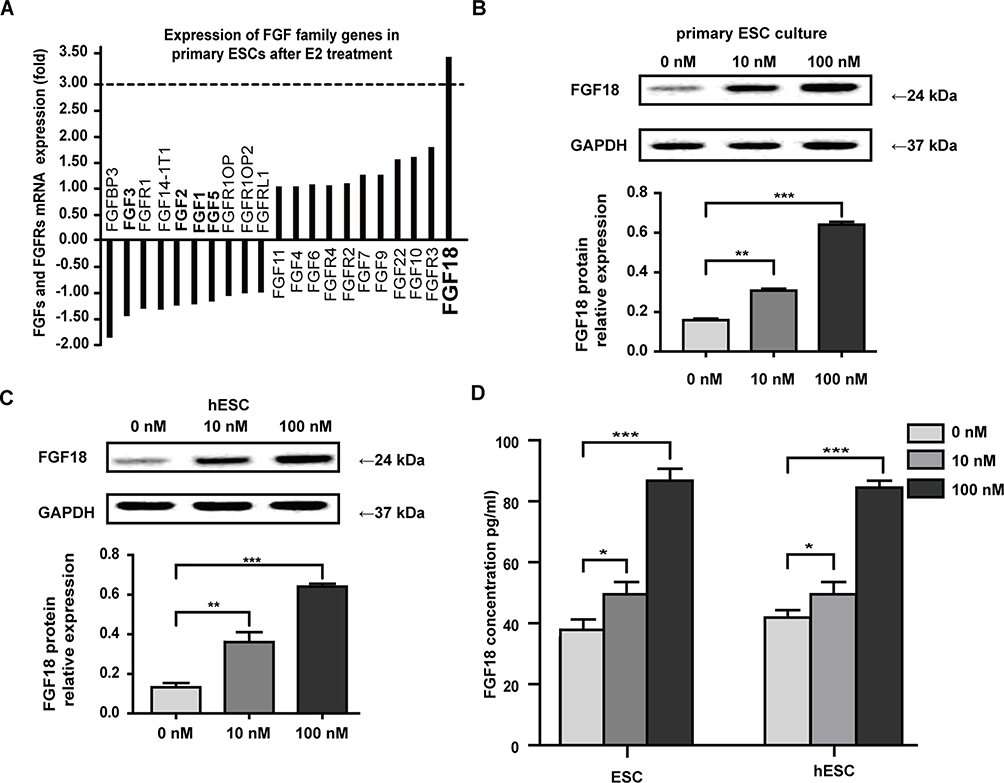 |
Figure 2 Estrogen promoted ESCs to increase paracrine FGF18 expression. (A) The results of the Agilent microarray revealed 11 upregulated and 10 downregulated genes of FGF-related family members after 10 nM E2 treatment for 3 d, but only FGF18 expression was upregulated 3.46-fold. (B and C) FGF18 expression was elevated in primary ESCs and hESCs after 10 and 100 nM E2 treatment. (D) ELISA results demonstrated that paracrine FGF18 protein levels in CM from ESCs and hESCs were enhanced after 10 and 100 nM E2 treatments for 48 h (*P≤0.05; **P≤0.01; ***P≤0.001). |
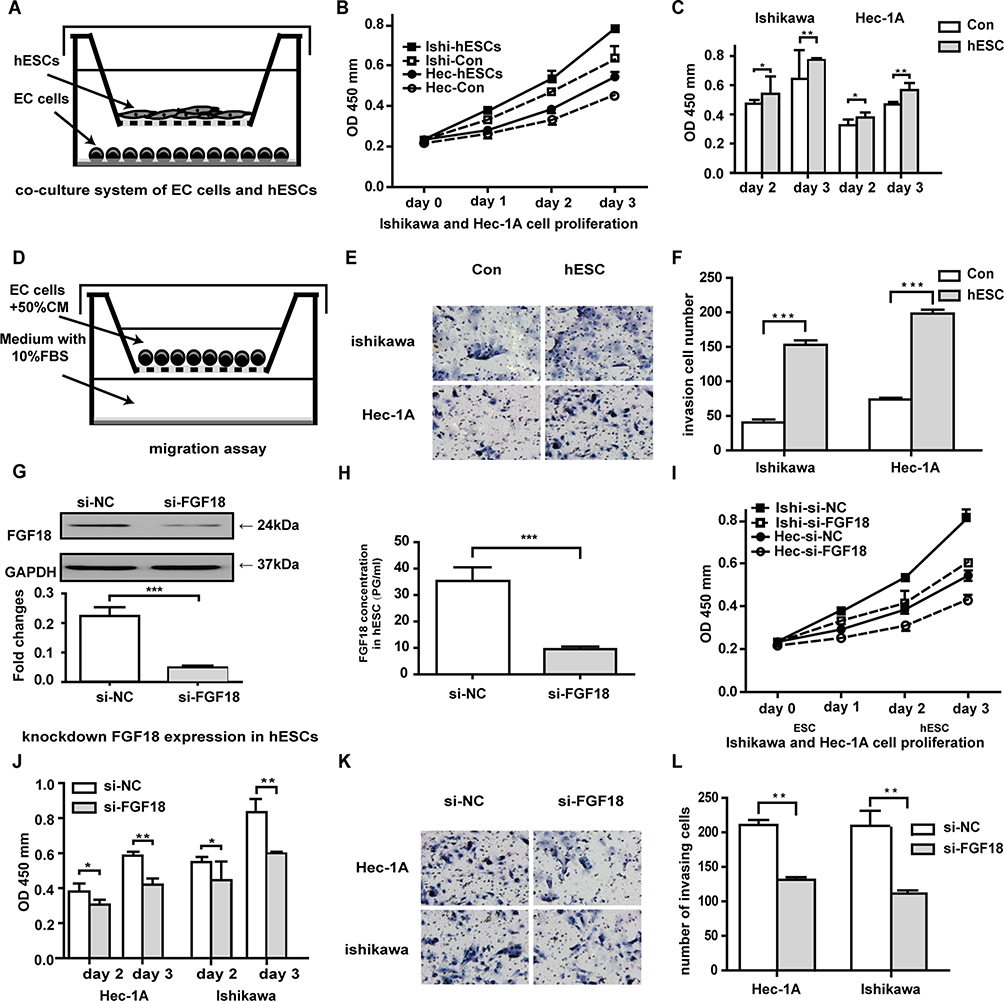 |
Figure 3 E2 promotes the proliferation and invasion of EC lines via paracrine FGF18 expression. (A) The model of co-cultured hESCs and ECs. (B and C) Estrogen-treated hESCs promoted the proliferation of Ishikawa and Hec-1A cells through a paracrine mechanism. (D) The model for the cell invasion assay. (E and F) The numbers of invading Ishikawa and Hec-1A cells increased after co-culture with CM from estrogen-treated FGF18-knockdown cells for 24 h. (G) FGF18 expression in FGF18-knockdown hESCs was obviously less than that in the negative control cells. (H) In spite of treatment with 10 nM E2 for 48 h, ELISA results revealed that the paracrine FGF18 protein levels in CM of siRNA FGF18-transfected hESCs decreased markedly. (I and J) After co-culture with si-FGF18 hESCs, Ishikawa and Hec-1A cells had decreased proliferation compared to the negative control groups of si-NC hESCs. (K and L) The number of invading Ishikawa and Hec-1A cells was decreased after culture with CM from estrogen-treated FGF18-knockdown cells for 24 h (*P≤0.05; **P≤0.01; ***P≤0.001). |
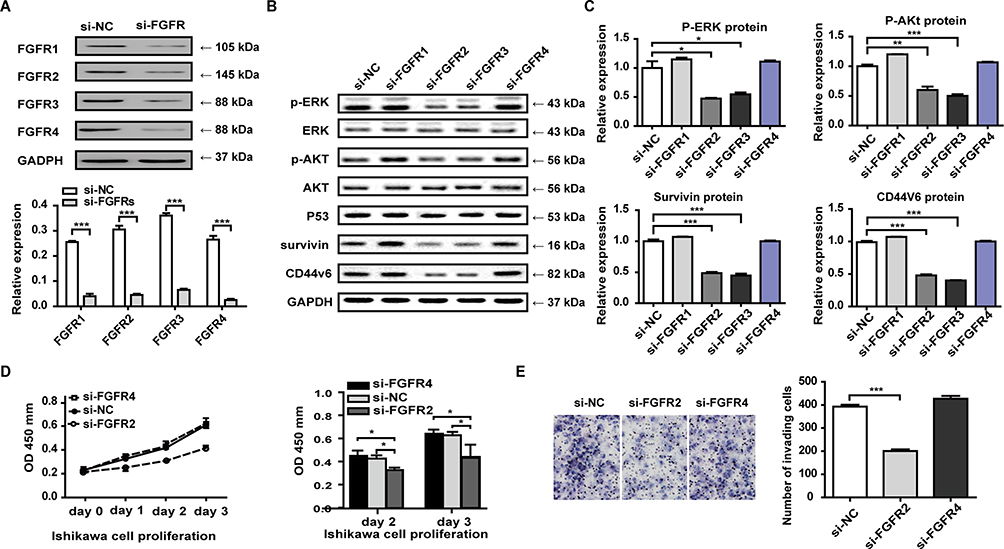 |
Figure 4 FGF18 bound FGFR2 and FGFR3 to activate downstream ERK and Akt signaling. After knockdown of FGFR expression in Ishikawa cells (A), a few downstream target genes of FGF18-FGFR signaling were detected by WB assay (B). (C) FGFR2- and FGFR3-knockdown Ishikawa cells had reduced expression of phospho-ERK, phospho-Akt, Survivin and CD44V6; FGFR1- and FGFR4- knockdown cells had the same expression of phospho-ERK, phospho-AKT, Survivin and CD44V6 as the control group of si-NC Ishikawa cells; and the expression of ERK, Akt, and P53 was not different among all FGFR-knockdown groups. (D and E) After co-culturing FGFR2/4-knockdown Ishikawa cells and hESCs, the results showed that FGF18/si-FGFR2 signaling inhibited the proliferation and invasion of Ishikawa cells compared to FGF18/si-NC or FGF18/si-FGFR4 signaling (*P≤0.05; **P≤0.01; ***P≤0.001). |
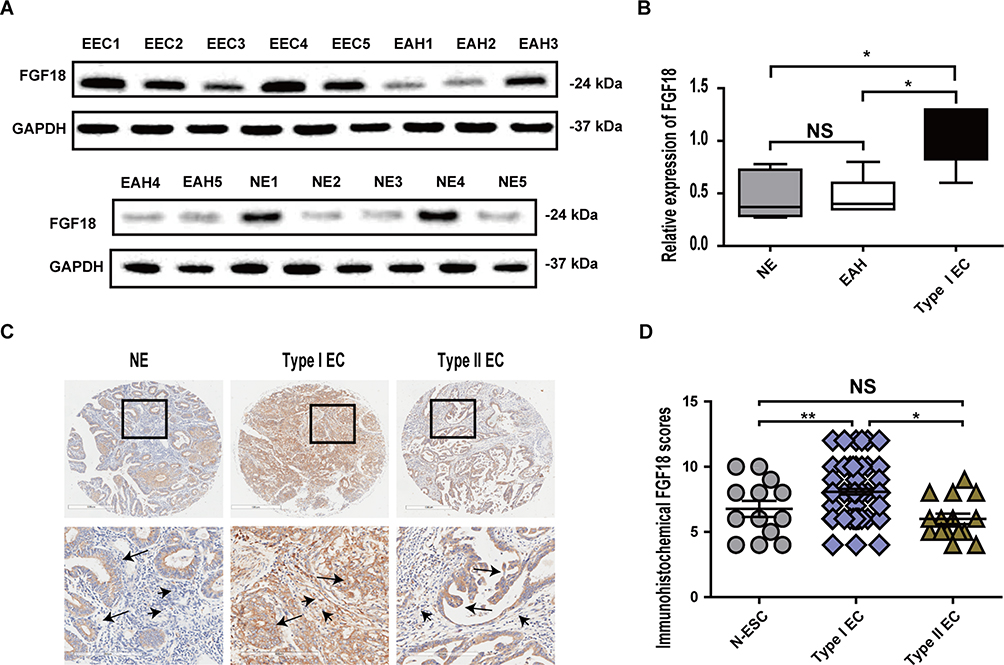 |
Figure 5 Clinical patients with type I EC had higher stromal FGF18 expression. (A) FGF18 expression was detected in 5 primary culture ESCs derived from NE, EAH and EEC patients using WB. (B) The results demonstrated that ESCs in EEC had higher FGF18 expression than ESCs in NE and EAH, but FGF18 expression levels in ESCs of the NE and EAH groups were not different. (C) Stromal and epithelial FGF18 expression in EC TMAs was determined by immunohistochemistry staining, and the results showed that stromal (short arrow shown) and epithelial (long arrow shown) cells of type I EC had strong expression of FGF18, whereas only the epithelium of type II EC had increased FGF18 expression. (D) TMA revealed that patients with type I EC had higher stromal FGF18 scores than patients with type II EC or with proliferative phase endometrium (*P≤0.05; **P≤0.01). |
Acknowledgments
This study was funded by the Weak Subject Construction Foundation of Pudong New Area Health Planning Commission, Shanghai, China (No. PWZbr2017-21), National Natural Science Foundation of China Grant (N0. 81402151) and the New Teacher Fund of Ministry of Education, Shanghai’s Fudan University (No. 20120071120084).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
2. Sahoo SS, Zhang XD, Hondermarck H, Tanwar PS. The emerging role of the microenvironment in endometrial cancer. Cancers. 2018;10:408.
3. Bell DW, Ellenson LH. Molecular genetics of endometrial carcinoma. Annu Rev Pathol. 2019;14:339–367.
4. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591. doi:10.1038/nrc1408
5. West RB, van de Rijn M. Experimental approaches to the study of cancer-stroma interactions: recent findings suggest a pivotal role for stroma in carcinogenesis. Lab Invest. 2007;87(10):967–970. doi:10.1038/labinvest.3700666
6. Cooke PS, Buchanan DL, Young P, et al. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci U S A. 1997;94(12):6535–6540. doi:10.1073/pnas.94.12.6535
7. Tsutsumi A, Okada H, Nakamoto T, Okamoto R, Yasuda K, Kanzaki H. Estrogen induces stromal cell-derived factor 1 (SDF-1/CXCL12) production in human endometrial stromal cells: a possible role of endometrial epithelial cell growth. Fertil Steril. 2011;95(1):444–447. doi:10.1016/j.fertnstert.2010.08.037
8. Li M, Xin X, Wu T, Hua T, Wang H, Wang H. Stromal cells of endometrial carcinoma promotes proliferation of epithelial cells through the HGF/c-Met/Akt signaling pathway. Tumour Biol. 2015;36(8):6239–6248. doi:10.1007/s13277-015-3309-2
9. Okada H, Tsutsumi A, Imai M, Nakajima T, Yasuda K, Kanzaki H. Estrogen and selective estrogen receptor modulators regulate vascular endothelial growth factor and soluble vascular endothelial growth factor receptor 1 in human endometrial stromal cells. Fertil Steril. 2010;93(8):2680–2686. doi:10.1016/j.fertnstert.2009.08.056
10. Chung D, Gao F, Jegga AG, Das SK. Estrogen mediated epithelial proliferation in the uterus is directed by stromal Fgf10 and Bmp8a. Mol Cell Endocrinol. 2015;400:48–60. doi:10.1016/j.mce.2014.11.002
11. Matysiak BE, Brodzeller T, Buck S, et al. Simple, inexpensive method for automating tissue microarray production provides enhanced microarray reproducibility. Appl Immunohistochem Mol Morphol. 2003;11:269–273. doi:10.1097/00129039-200309000-00011
12. Sakai S, Iwata C, Tanaka HY, et al. Increased fibrosis and impaired intratumoral accumulation of macromolecules in a murine model of pancreatic cancer co-administered with FGF-2. J Control Release. 2016;230:109–115. doi:10.1016/j.jconrel.2016.04.007
13. Guzy RD, Li L, Smith C, et al. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J Biol Chem. 2017;292(25):10364–10378. doi:10.1074/jbc.M117.791764
14. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi:10.1038/nrc2780
15. Hu MC, Qiu WR, Wang YP, et al. FGF-18, a novel member of the fibroblast growth factor family, stimulates hepatic and intestinal proliferation. Mol Cell Biol. 1998;18(10):6063–6074. doi:10.1128/MCB.18.10.6063
16. Hu MC-T, Wang Y-P, Qiu WR. Human fibroblast growth factor-18 stimulates fibroblast cell proliferation and is mapped to chromosome 14p11. Oncogene. 1999;18(16):2635–2642. doi:10.1038/sj.onc.1202616
17. Zhang J, Zhou Y, Huang T, et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene. 2019;38(1):33–46. doi:10.1038/s41388-018-0430-x
18. Yu Z, Lou L, Zhao Y. Fibroblast growth factor 18 promotes the growth, migration and invasion of MDA‑MB‑231 cells. Oncol Rep. 2018;40(2):704–714. doi:10.3892/or.2018.6482
19. Chen T, Gong W, Tian H, et al. Fibroblast growth factor 18 promotes proliferation and migration of H460 cells via the ERK and p38 signaling pathways. Oncol Rep. 2017;37(2):1235–1242. doi:10.3892/or.2016.5301
20. Sonvilla G, Allerstorfer S, Stättner S, et al. FGF18 in colorectal tumour cells: autocrine and paracrine effects. Carcinogenesis. 2007;29(1):15–24. doi:10.1093/carcin/bgm202
21. Gauglhofer C, Sagmeister S, Schrottmaier W, et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology. 2011;53(3):854–864. doi:10.1002/hep.24099
22. Flannery CA, Fleming AG, Choe GH, et al. Endometrial Cancer-Associated FGF18 expression is reduced by bazedoxifene in human endometrial stromal cells in vitro and in murine endometrium. Endocrinology. 2016;157(10):3699–3708. doi:10.1210/en.2016-1233
23. Li Q, Kannan A, DeMayo FJ, et al. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011;331(6019):912–916. doi:10.1126/science.1197454
24. Guo P, Wang Y, Dai C, et al. Ribosomal protein S15a promotes tumor angiogenesis via enhancing Wnt/β-catenin-induced FGF18 expression in hepatocellular carcinoma. Oncogene. 2018;37(9):1220–1236. doi:10.1038/s41388-017-0017-y
25. Song N, Zhong J, Hu Q, et al. FGF18 enhances migration and the epithelial-mesenchymal transition in breast cancer by regulating Akt/GSK3β/Β-catenin signaling. Cell Physiol Biochem. 2018;49(3):1019–1032. doi:10.1159/000493286
26. Ye Q, Cai W, Zheng Y, Evers BM, She Q-B. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene. 2014;33(14):1828–1839. doi:10.1038/onc.2013.122
27. Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8(1):129. doi:10.1186/1471-2407-8-129
28. Kashyap T, Pramanik KK, Nath N, et al. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. 2018;86:234–243. doi:10.1016/j.oraloncology.2018.09.028
29. Harpain F, Ahmed MA, Hudec X, et al. FGF8 induces therapy resistance in neoadjuvantly radiated rectal cancer. J Cancer Res Clin Oncol. 2010;10(2):77–86. doi:10.1007/s00432-018-2757-7
30. Xiao D, Wang K, Zhou J, et al. Inhibition of fibroblast growth factor 2-induced apoptosis involves survivin expression, protein kinase C alpha activation and subcellular translocation of Smac in human small cell lung cancer cells. Acta Biochim Biophys Sin (Shanghai). 2008;40(4):297–303. doi:10.1111/j.1745-7270.2008.00401.x
31. Taeger J, Moser C, Hellerbrand C, et al. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther. 2011;10(11):2157–2167. doi:10.1158/1535-7163.MCT-11-0312
32. Giacomini A, Chiodelli P, Matarazzo S, Rusnati M, Presta M, Ronca R. Blocking the FGF/FGFR system as a “two-compartment” antiangiogenic/antitumor approach in cancer therapy. Pharmacol Res. 2016;107:172–185. doi:10.1016/j.phrs.2016.03.024
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.