")
Back to Journals » OncoTargets and Therapy » Volume 14
Establishment of a Risk Signature Based on m6A RNA Methylation Regulators That Predicts Poor Prognosis in Renal Cell Carcinoma
Authors Wei J, Qian Y, Tang Y, Ge X, Jiang K, Fang Y, Fu D, Kong X, Xiao Q, Ding K
Received 25 October 2020
Accepted for publication 23 December 2020
Published 14 January 2021 Volume 2021:14 Pages 413—426
DOI https://doi.org/10.2147/OTT.S288663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Jingsun Wei,1,2,* Yucheng Qian,1,2,* Yang Tang,1,2 Xiaoxu Ge,1,2 Kai Jiang,1,2 Yimin Fang,1,2 Dongliang Fu,1,2 Xiangxing Kong,1 Qian Xiao,1 Kefeng Ding1,2
1Department of Colorectal Surgery and Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China; 2Cancer Institute, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kefeng Ding
Department of Colorectal Surgery and Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, Zhejiang, People’s Republic of China
Tel/Fax +86-571-87784820
Email [email protected]
Purpose: N6-methyladenosine (m6A) modifications represent one of the most common methylation modifications, and they are mediated by m6A RNA methylation regulators. However, their functions in renal cell carcinoma (RCC) are not completely understood. The aim of this study was to investigate the effects of the regulators in RCC.
Materials and Methods: The expression levels of the 13 main m6A RNA methylation regulators in RCC were detected and consensus clustering was performed to explore their relationships with RCC. Thereafter, a risk signature based on the regulators was established. This risk model was fully verified by conducting prognostic analyses using two datasets (The Cancer Genome Atlas [TCGA] and Gene Expression Omnibus [GEO] datasets) and a ROC curve analysis.
Results: Of the 13 main m6A regulators, six were significantly upregulated and four were significantly downregulated in 893 RCC cases compared to 128 normal controls in the TCGA database. Consensus clustering based on the regulators identified two clusters of RCC cases, which were significantly associated with a pathological characteristic (T status). Thus, these results indicated that m6A RNA methylation regulators were associated with RCC. Thereafter, a risk model involving two of the regulators (METTL14 and WTAP) was established. The alterations in the mRNA and protein expression levels of these two regulators were further confirmed based on Human Protein Atlas data and real-time PCR in RCC and normal cell lines. The results indicated that the risk model may serve as an independent prognostic marker of overall survival, and it was also associated with clinicopathological characteristics (T status, M status, pathological stage, and gender) in RCC.
Conclusion: Collectively, the results of this study indicated that the risk model (based on two m6A RNA methylation regulators) may serve as an independent prognostic indicator of RCC, which may aid further investigation into m6A RNA modification in RCC.
Keywords: renal cell carcinoma, m6A methylation, TCGA, prognostic signature
Introduction
Renal carcinoma, also known as renal cell carcinoma (RCC), is one of the most common malignant tumors of the urinary system, and it accounts for 2–3% of all malignant tumors.1 Each year worldwide, ~295,000 individuals are diagnosed with RCC, resulting in 134,000 deaths.2 The pathological types of RCC comprise clear cell, papillary, and chromophobe RCC, with >75% of cases being clear cell RCC.3 Although >50% of RCC patients are diagnosed with localized RCC, 30% have metastasis at initial diagnosis.4,5 RCC is highly angiogenic, with a hypoxic tumor environment, and it is not sensitive to chemotherapy, so surgical resection is the only effective treatment.6 However, 20% of patients who have surgery experience local recurrence or distant metastases.7 Due to the lack of obvious symptoms in early RCC, early accurate diagnosis is a challenge, with one-third of patients displaying distant metastases at diagnosis, along with local recurrence being common after early surgical resection.8,9 Therefore, identifying a risk signature to evaluate the prognosis of RCC patients at the diagnosis stage is of significance.
Epigenetic modification is highly associated with the occurrence and progression of tumors. It involves the regulation of gene function and expression levels via DNA methylation, thus affecting tumor progression. N6-methyladenosine (m6A) modification of RNA is the most common post-transcriptional modification in eukaryotic cells, accounting for 80% of RNA methylation modifications.10 m6A methylation is involved in the intricate regulation of important genes in many cellular activities. Additionally, several studies have demonstrated that m6A methylation has an important role during the occurrence and progression of tumors.11–13
It has been reported that m6A modification is a dynamic reversible process.14 m6A-related proteins can be divided into three categories based on function: methyltransferases (writers), demethylases (erasers), and m6A-binding proteins/effectors (readers). Writers mediate RNA methylation, and they include METTL3, METTL14, WTAP, ZC3H13, RBM15, and KIAA1429 (also known as VIRMA). The first two are the most common writers, and they can catalyze m6A methylation of mRNA in vitro and in vivo15 Some writers, such as WTAP, do not display methyltransferase activity. However, WTAP interacts with the METTL3/METTL14 complex to influence m6A methyltransferase activity,16 ZC3H13 promotes m6A methylation by anchoring WTAP,17 and RBM15 and KIAA1429 facilitate the recruitment of the METTL3/METTL14 complex.18 Moreover, erasers are able to “erase” RNA methylation modification signals, mediating RNA demethylation. FTO, an eraser, uses ferrous iron as a cofactor along with α-ketoglutarate to oxidize the N-methyl at the m6A site to hydroxymethyl, thereby altering the cellular m6A level.14 Lastly, readers are responsible for “reading” the methylation information. They can be involved in downstream RNA translation and degradation. There are two “reading” modes: direct reading, where the readers (such as the YTH protein family, YTHDC1, YTHDC2, YTHDF1, and YTHDF2) selectively interact with the m6A site of RNA,19 and indirect reading. This involves the m6A methylation changing the secondary structure of the RNA and thereby rendering it accessible to certain “readers” such as heterogeneous nuclear ribonucleoprotein C (HNRNPC), which can bind to other proteins and promote mRNA maturation.20
The interaction between m6A mRNA methylation and tumors has received increasing attention. Increasing evidence indicates that m6A mRNA methylation is closely associated with tumorigenesis and development, and m6A-related proteins are important regulators of these processes.10 In this study, RNA sequencing data from The Cancer Genome Atlas (TCGA) database were used to analyze the expression of 13 major m6A RNA methylation regulators in 893 RCC patients. In addition, a risk signature, based on two of the m6A methylation regulators, was established to predict the prognosis of the RCC patients. This risk signature was used to divide the RCC cases into the high- and low-risk groups (based on the median risk score). Thereafter, the associations between the high/low-risk groups and both overall survival and various clinicopathological characteristics were assessed.
Methods
Datasets
Transcriptome (RNA sequencing) data and relevant clinical information on RCC patients (including those with clear cell, papillary, and chromophobe RCC) were acquired from the TCGA database (cancergenome.nih.gov). The TCGA dataset included information on 893 RCC patients and 128 normal controls. Additionally, a Gene Expression Omnibus (GEO) dataset (GSE29609; ncbi.nlm.nih.gov/geo), involving 39 RCC patients, was used to validate the prognostic value of the risk model constructed based on TCGA data.
Selection of m6A RNA Methylation Regulators
Thirteen major m6A regulators were analyzed: ZC3H13, WTAP, METTL3, METTL14, RBM15, KIAA1429, HNRNPC, YTHDC1, YTHDC2, YTHDF1, YTHDF2, ALKBH5, and FTO. An expression matrix of the 13 genes was established, and data on the clinicopathological characteristics were also obtained and utilized in the bioinformatics analyses.
Cell Culture
RCC cell lines (786–0 and 769-P) were cultured in Roswell Park Memorial Institute (RPMI)‐1640 medium (Gibco, CA, USA) with 10% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin. Another RCC cell line (Caki-1) was cultured in McCoy’s 5A (Gibco, USA) with 10% FBS (Life Technologies), 100 U/mL penicillin, and 100 mg/mL streptomycin. The 293T cell line (the control kidney cell line) was cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) with 10% FBS (Life Technologies), 100 U/mL penicillin, and 100 mg/mL streptomycin. The cells were cultured at 37°C in 5% CO2. All cells were acquired from the American Type Culture Collection (ATCC; Rockville, MD, USA).
Real-Time PCR
The RNA was extracted from the cell lines using TRIzol Reagent (Invitrogen, CA, USA) and reverse transcribed (PrimeScript™ RT Master Mix; Takara, Kyoto, Japan) to acquire cDNAs. Real-time PCR was performed using SYBR® Premix Ex Taq™ GC (Takara). β-actin was used as the internal control. The primers for METTL14 and WTAP (m6A RNA methylation regulators) are listed in Table S1. The 2−ΔΔCt method was used to calculate the relative expression of these genes.
Bioinformatics Analyses
The limma package (version 3.22.7) in R was used to analyze the expression of the 13 m6A regulators in the TCGA dataset.21 Subsequently, the vioplot package (version 3.4) was used to visualize the expression of these genes in the RCC and normal tissues. A consensus clustering algorithm in the ConsensusClusterPlus package (version 1.52.0) inR22 was used to evaluate the stability of clustering and determine the optimum number of clusters. Least absolute shrinkage and selection operator (LASSO) Cox regression was used to establish a risk signature for predicting overall survival based on the m6A regulators. The coefficients of the selected genes were determined, with penalty parameter (λ) tuning based minimum criteria. The glmnet package (version 3.0) and the survival package were used to execute this program.23 The following formula was used to calculate the risk score for each patient: Risk score = β1×ExprGene1 + β2×ExprGene2 + … + βn×ExprGenen, where β represents the coefficients (1 to n). The median risk score was used as the cut-off value to determine the high- and low-risk groups. The predictive performance of the risk model was assessed using a receiver operating characteristic (ROC) curve analysis.24 Furthermore, Kaplan–Meier analysis was used to compare overall survival between the high- and low-risk groups, and univariate and multivariate Cox regression analyses of the association between the risk score and overall survival were conducted. The clinicopathological characteristics of the high/low-risk groups (based on the median risk score) were also investigated.
Statistical Analysis
One-way analysis of variance (ANOVA) was used to compare the expression levels of the 13 m6A regulators in the TCGA dataset. Student’s t-test was used to explore the associations between the high/low-risk groups of RCC patients (based on the median risk score) and clinicopathological characteristics, such as age, gender, tumor stage, and tumor, node, and metastasis (TNM) status. Kaplan–Meier analysis and the log rank test were used to analyze the overall survival of RCC patients in the two clusters in the TCGA dataset and in the high- and low-risk groups (based on the median risk score) in the TCGA and GEO datasets. Furthermore, univariate and multivariate Cox regression analyses were used to assess the associations of the m6A RNA methylation regulator expression levels, the risk score, and other clinicopathological characteristics with overall survival.
Cases with missing data in the TCGA dataset were excluded from the Kaplan–Meier and Cox regression analyses of overall survival, leaving 634 cases for these analyses. P<0.05 was considered to indicate statistical significance. The statistical analyses were performed using R software (version 3.6; www.r-project.org).
Results
Expression of m6A RNA Methylation Regulators in RCC
The m6A RNA methylation regulators play key roles in the formation and development of various tumors. First, the expression levels of these regulators (Table 1) in 893 RCC and 128 normal tissues obtained from the TCGA database were compared. Of the 13 genes, ten genes displayed significant differences. Six genes (METTL3, RBM15, YTHDF1, YTHDC2, ALKBH5, and FTO) were significantly upregulated and four genes (METTL14, ZC3H13, HNRNPC, and YTHDF2) were significantly downregulated in RCC patients compared to normal controls (Figure 1A and B). Furthermore, the expression levels of the 13 regulators were positively correlated (Figure 1C).
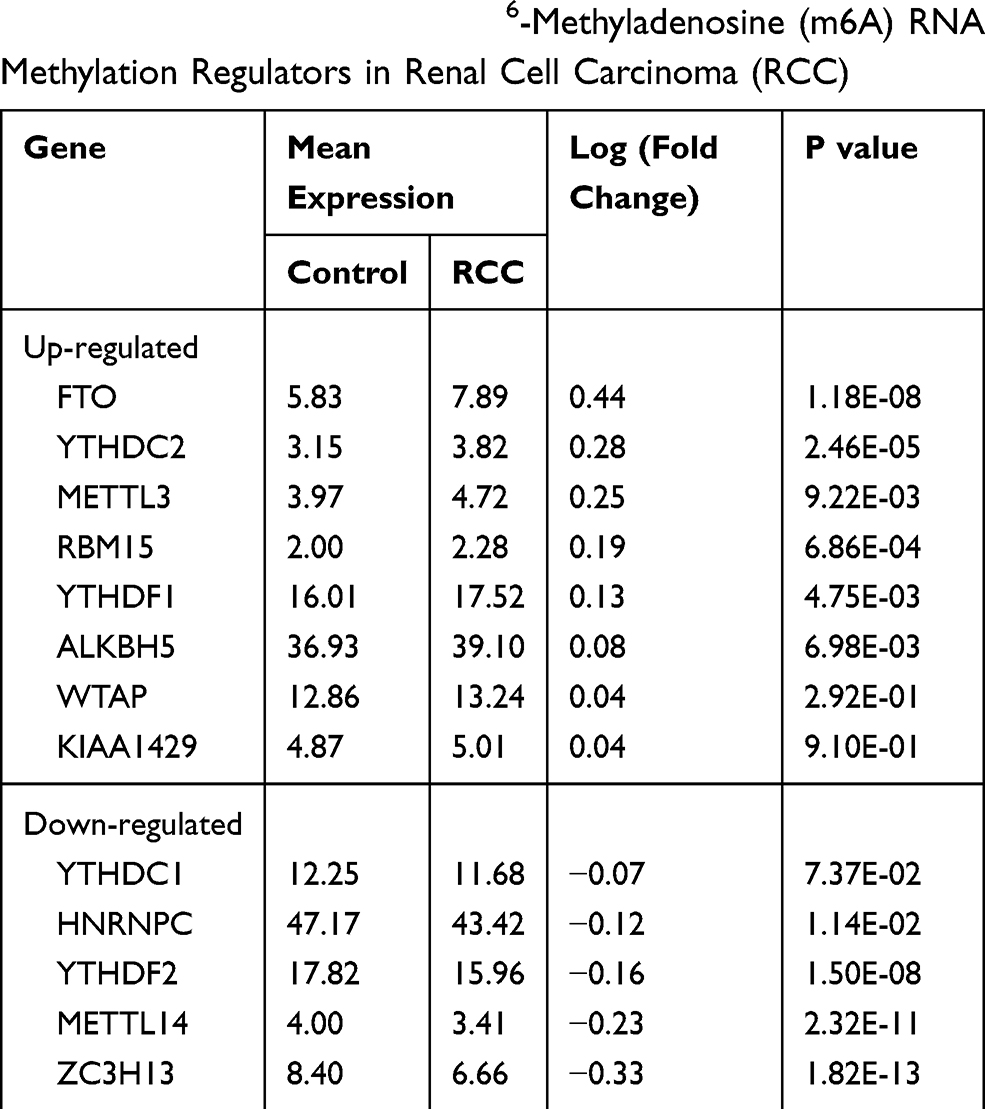 |
Table 1 Expression of the 13 N6-Methyladenosine (m6A) RNA Methylation Regulators in Renal Cell Carcinoma (RCC) |
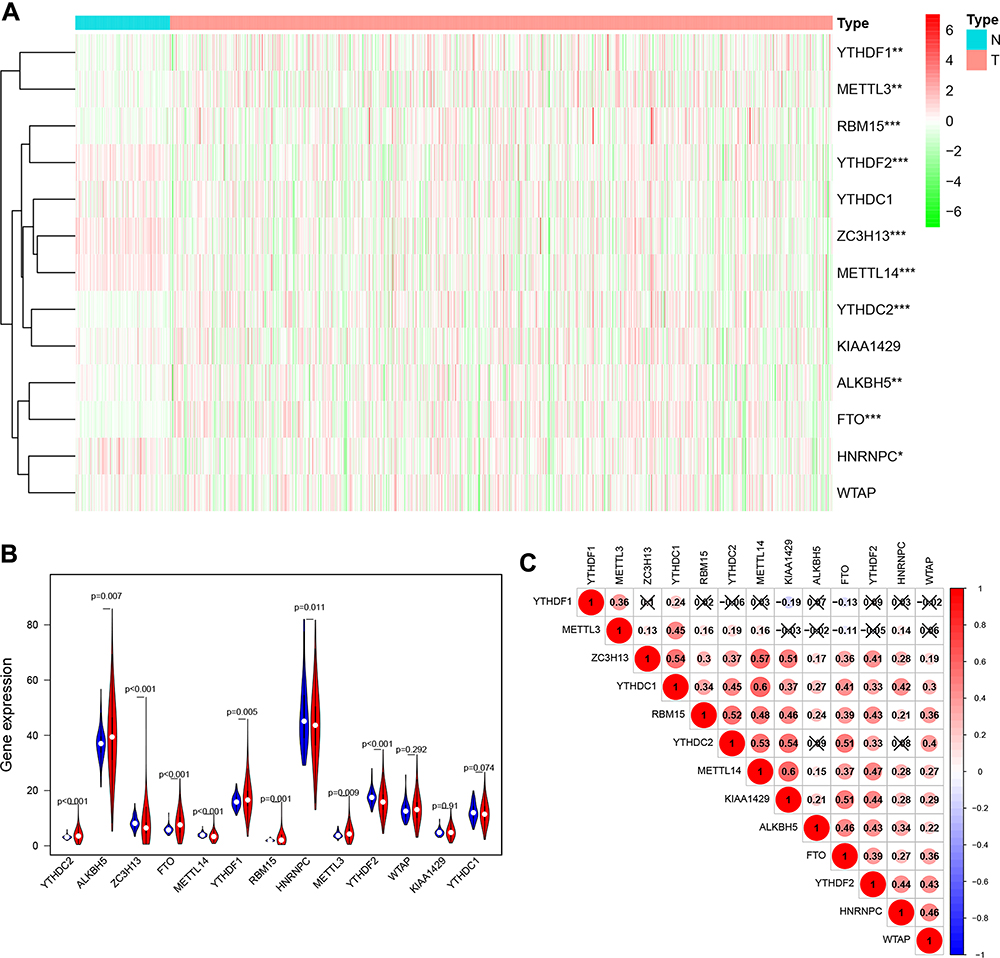 |
Figure 1 Expression levels of m6A RNA methylation regulators in RCC. (A) Heatmap of the expression of 13 m6A RNA methylation regulators in RCC tissues compared to normal tissues. (B) Violin plots of the expression of the 13 regulators in RCC. (C) Spearman correlation analysis of the 13 regulators in RCC. *P<0.05, **P<0.01, and ***P<0.001. Abbreviations: RCC, renal cell carcinoma; m6A, N6-methyladenosine. |
Identification of Two Clusters of RCC Cases by Consensus Clustering Analysis
The ConsensusClusterPlus package was used to perform a consensus clustering analysis based on the expression levels of the 13 m6A RNA methylation regulators in the 893 RCC tissues in the TCGA database. The results indicated that the relative change in the area under the cumulative distribution function (CDF) curve obviously decreased when k=3 (Figure 2A and B). However, the consensus matrix showed that there was considerable classification overlap among the clusters when the cases were divided into three clusters. Therefore, the cases were divided into two clusters (Figure 2C). For k=2–9, the distributions of the RCC cases are presented in Figure 2D. Principal component analysis of the two clusters was used to assess the performance of the classification. The results indicated that the cluster 1 cases clustered together, as did the cluster 2 cases (Figure 2E).
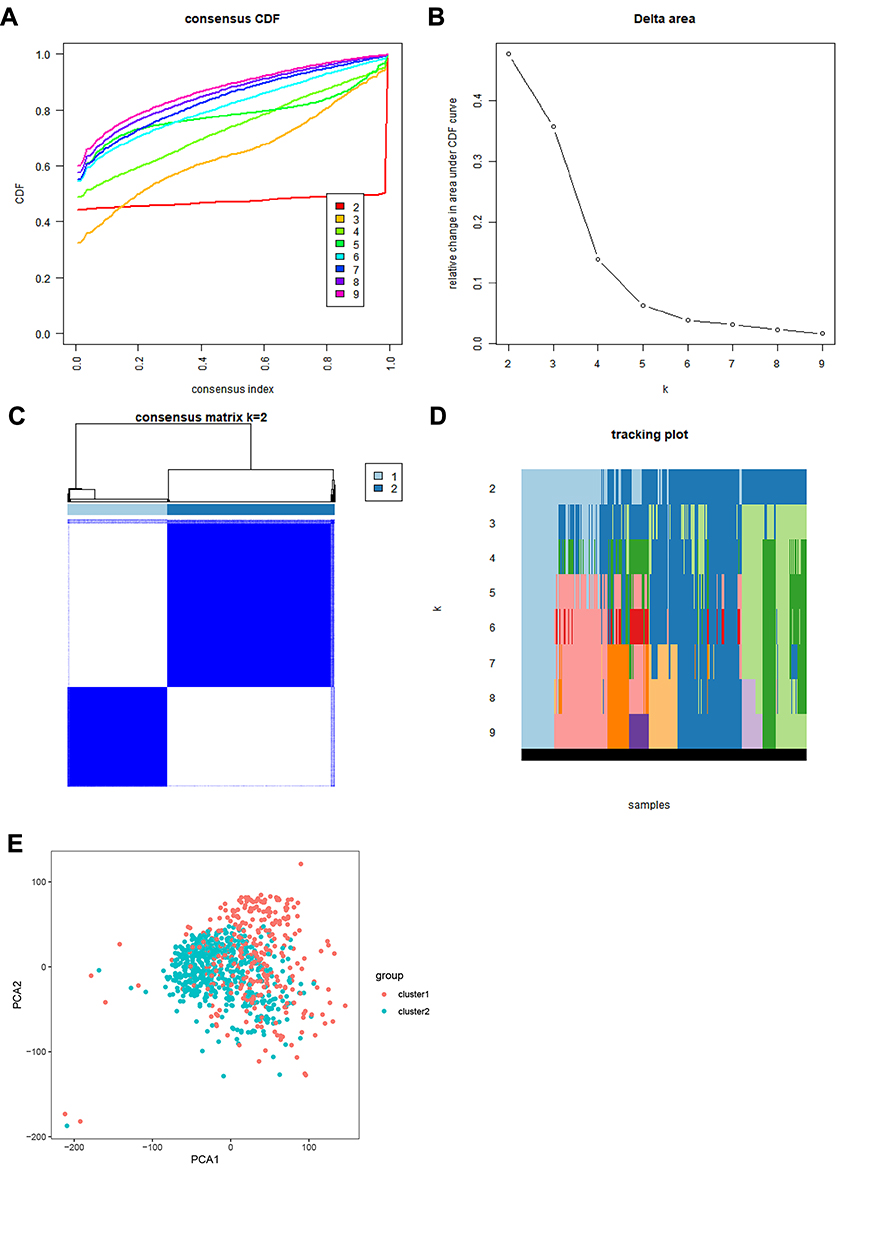 |
Figure 2 Consensus clustering analysis of RCC cases. (A) CDF curves for k=2–9. (B) Relative change in the area under the CDF curves for k=2–9. (C) Consensus matrix for k=2. (D). Tracking plot of cases for k=2–9. (E). Principal component analysis plot based on the RNA expression profiles of the 13 major m6A regulators, which grouped the cases into two clusters. Red: cluster 1; blue: cluster 2. Abbreviations: CDF, cumulative distribution function; RCC, renal cell carcinoma. |
Cluster 1 and 2 are Associated with a Clinicopathological Characteristics (T Status) in RCC
To further understand the clustering results, a Kaplan–Meier analysis of the overall survival of 634 RCC patients in the two clusters was conducted. The results indicated that cluster 2 had a worse prognosis compared to cluster 1, but the difference was not significant (P=0.084; Figure 3A). Moreover, the clusters were significantly associated with T status (Figure 3B). Collectively, the results indicated that the clusters were closely associated with RCC.
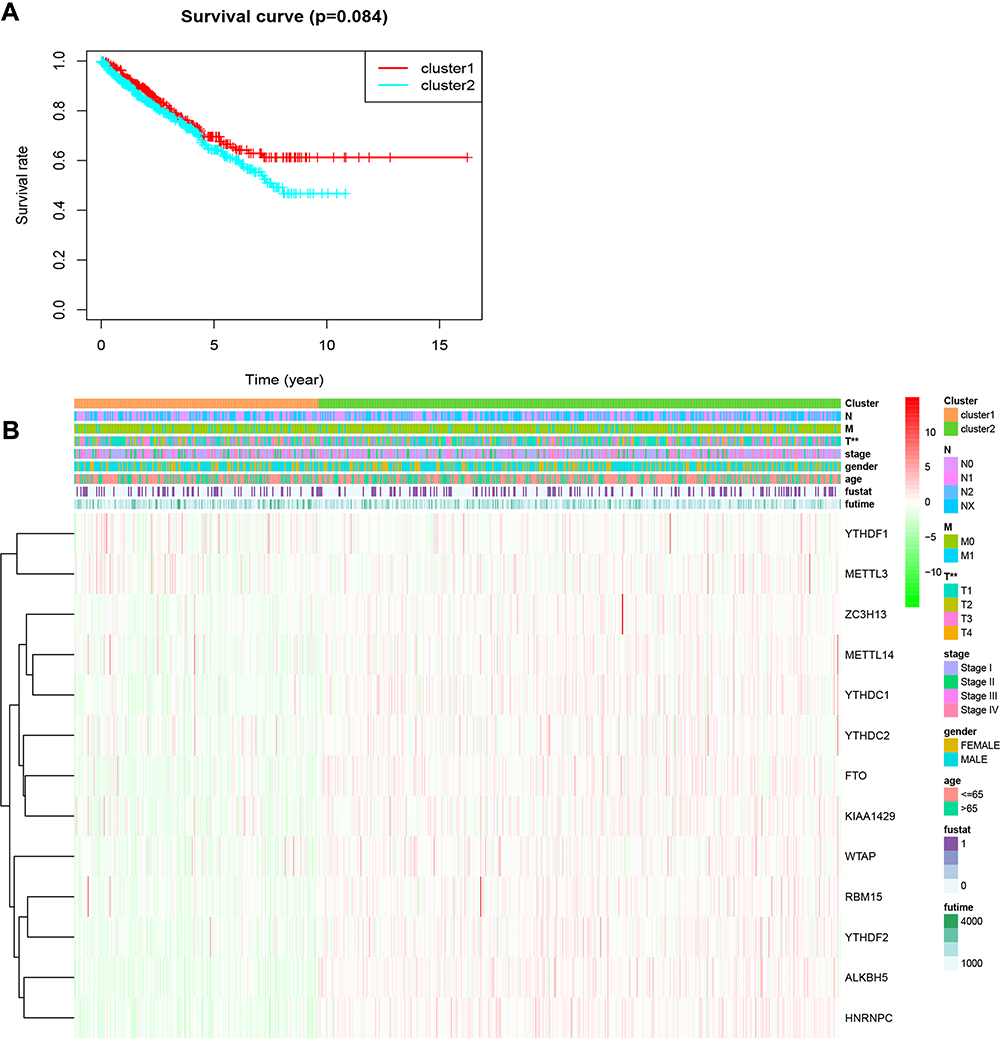 |
Figure 3 Overall survival and clinicopathological characteristics of RCC patients in the two clusters. (A) Kaplan–Meier curves of overall survival of RCC patients in clusters 1 (red) and 2 (blue). (B) Heatmap showing the associations between clinicopathological characteristics and the expression of each of the 13 m6A RNA methylation regulators in clusters 1 and 2. Red: upregulated; green: downregulated. **P<0.01. Abbreviations: m6A, N6-methyladenosine; RCC, renal cell carcinoma. |
Prognostic Risk Model Based on the m6A RNA Methylation Regulators
Subsequently, the associations between the expression levels of the m6A RNA methylation regulators and prognosis in RCC patients were assessed. Univariate Cox regression analyses were conducted according to the expression levels of the regulators in the TCGA database. The results indicated that WTAP (hazard ratio [HR], 1.051; 95% confidence interval [CI], 1.021–1.082), HNRNPC (HR, 1.016; 95% CI, 1.003–1.028), METTL3 (HR, 1.086; 95% CI, 1.017–1.161), YTHDC1 (HR, 0.955; 95% CI, 0.90–0.98), and METTL14 (HR, 0.730; 95% CI, 0.638–0.836) were significantly associated with overall survival (Figure 4A).
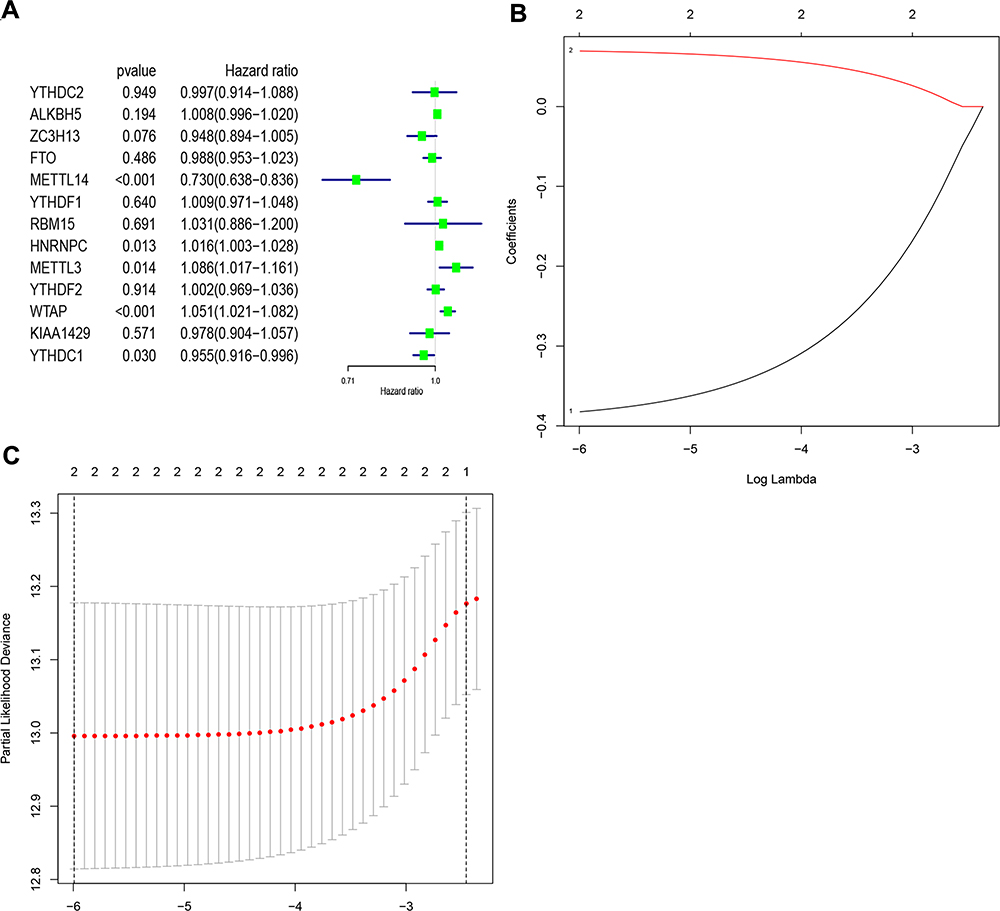 |
Figure 4 Identification of risk signature based on m6A RNA methylation regulators. (A) Univariate Cox regression results (hazard ratio, 95% confidence interval) for the 13 m6A RNA methylation regulators. (B and C) Coefficients from the multivariate LASSO Cox regression. LASSO: least absolute shrinkage and selection operator. Abbreviation: m6A, N6-methyladenosine. |
Thereafter, two of the genes with P<0.001 in the univariate analyses of TCGA data (METTL14 and WTAP) were selected, and the LASSO Cox regression algorithm was subsequently applied. These two genes were selected for the risk signature via the minimum criteria. The risk scores were then determined using the coefficients acquired from the LASSO algorithm (Figure 4B and C). The patients were then divided into high- and low-risk groups based on the median risk score.
Risk Scores are Associated with Clinicopathological Characteristics in RCC
To further analyze the associations between the risk score and the clinical traits of RCC patients, the associations between the high/low-risk groups and both overall survival and various clinicopathological characteristics were assessed. First, the results indicated that the high-risk group had a worse overall survival compared to the low-risk group (Figure 5A). Second, the associations between the high/low-risk groups and clinicopathological characteristics, including TNM status, stage and gender, were determined (Table 2), and strong associations between the high/low-risk groups and M status, T status, stage, and gender were identified (Figure 5C). The results also indicated that WTAP was downregulated while METTL14 was upregulated in the high-risk group compared to the low-risk group.
 |
Table 2 Clinical Characteristics of the Patients in High- and Low-Risk Groups (Based on the Median Risk Score) |
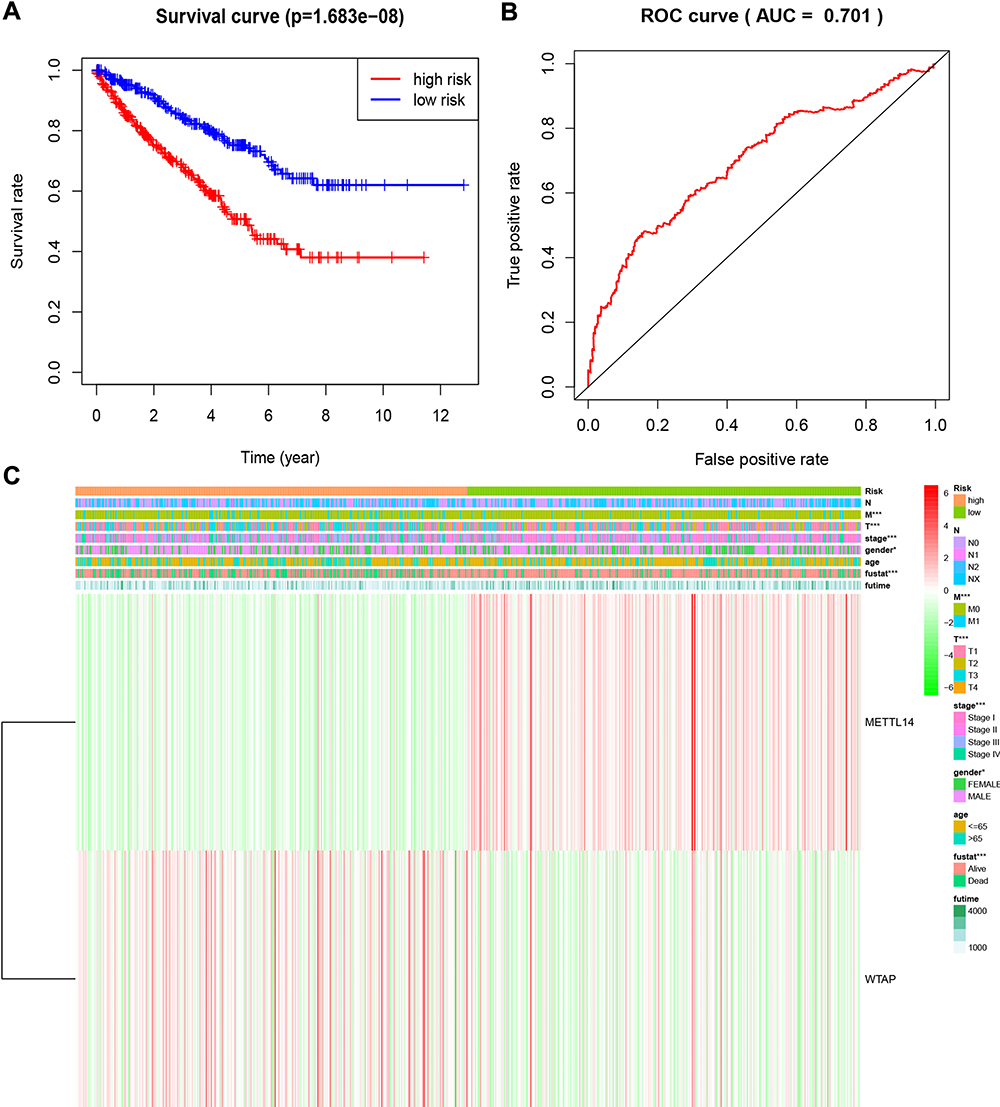 |
Figure 5 Overall survival and clinicopathological characteristics of RCC patients in the high- and low-risk groups. (A). Kaplan–Meier curves of overall survival of patients in the high- and low-risk groups (based on the median risk score involving two m6A RNA methylation regulators). (B) Receiver operating characteristic curve showing the predictive performance of the risk model. (C) Heatmap showing the associations between clinicopathological characteristics and the expression of two m6A RNA methylation regulators (METTL14 and WTAP) in the high- and low-risk groups. Red: upregulated; green: downregulated. *P<0.05 and ***P<0.001. Abbreviations: RCC, renal cell carcinoma; m6A, N6-methyladenosine. |
A ROC curve analysis was used to further verify the predictive performance of the risk signature. The area under the ROC curve was 0.701 (Figure 5B), which indicated that the model had high predictive performance.
Validation in the Gene Expression Omnibus (GEO) Dataset
The GSE29609 GEO dataset (involving 39 RCC patients) was used to further validate the prognostic value of the risk signature in RCC patients. Based on the median risk score, the 39 patients were divided into high- and low-risk groups. The results indicated that the patients in the high-risk group had a significantly worse prognosis than the patients in the low-risk group (P<0.05; Figure 6). The result was consistent with the TCGA results, indicating that the risk model was accurate and effective.
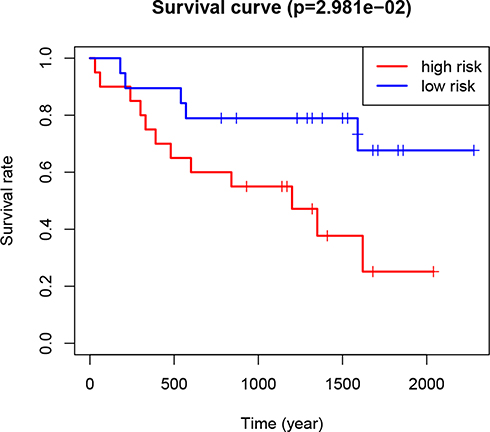 |
Figure 6 Kaplan–Meier curves of overall survival of RCC patients in the high- and low-risk groups (based on the median risk score involving two m6A RNA methylation regulators) in the Gene Expression Omnibus dataset. Abbreviations: m6A, N6-methyladenosine; RCC, renal cell carcinoma. |
Cox Regression Analysis Indicates That the Risk Signature is an Independent Prognostic Indicator
Univariate (Figure 7A) and multivariate (Figure 7B) Cox regression analyses indicating the associations between the clinicopathological characteristics (including risk score) and overall survival were performed. The results indicated that age, stage, and risk score were significantly associated with overall survival in RCC (Figure 7A and B). Collectively, these results indicated that the risk score obtained from the risk signature may serve as an independent prognostic factor for overall survival in RCC patients.
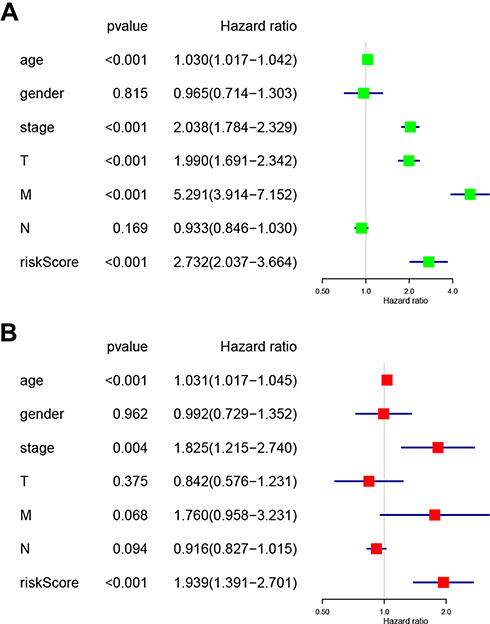 |
Figure 7 Associations between the clinicopathological characteristics and overall survival of RCC patients. (A) Univariate and (B) multivariate Cox regression results (hazard ratio, 95% confidence interval) indicating the associations between the clinicopathological characteristics and the overall survival of RCC patients. Abbreviation: RCC, renal cell carcinoma. |
Validation of mRNA and Protein Expression Levels of the Two m6A RNA Methylation Regulators
To further verify our results, information from the Human Protein Atlas (HPA) database was used to analyze the protein expression of the two m6A RNA methylation regulators: METTL14 and WTAP. The protein expression of METTL14 was higher in RCC tissues than normal tissues (Figure 8A), while the protein expression of WTAP was higher in normal tissues than RCC tissues (Figure 8B). These results were consistent with the expression of METTL14 and WTAP at the mRNA level (Figure 8C and D); our results showed that compared to the control kidney cell line (293T), METTL14 mRNA was significantly decreased in the RCC cell lines (786–0 and 769-P), while WTAP mRNA was significantly increased in RCC cell lines (786–0 and Caki-1). These results demonstrate that METTL14 and WTAP showed significant changes in RCC at both the mRNA and protein levels, indicating that they have important roles in RCC.
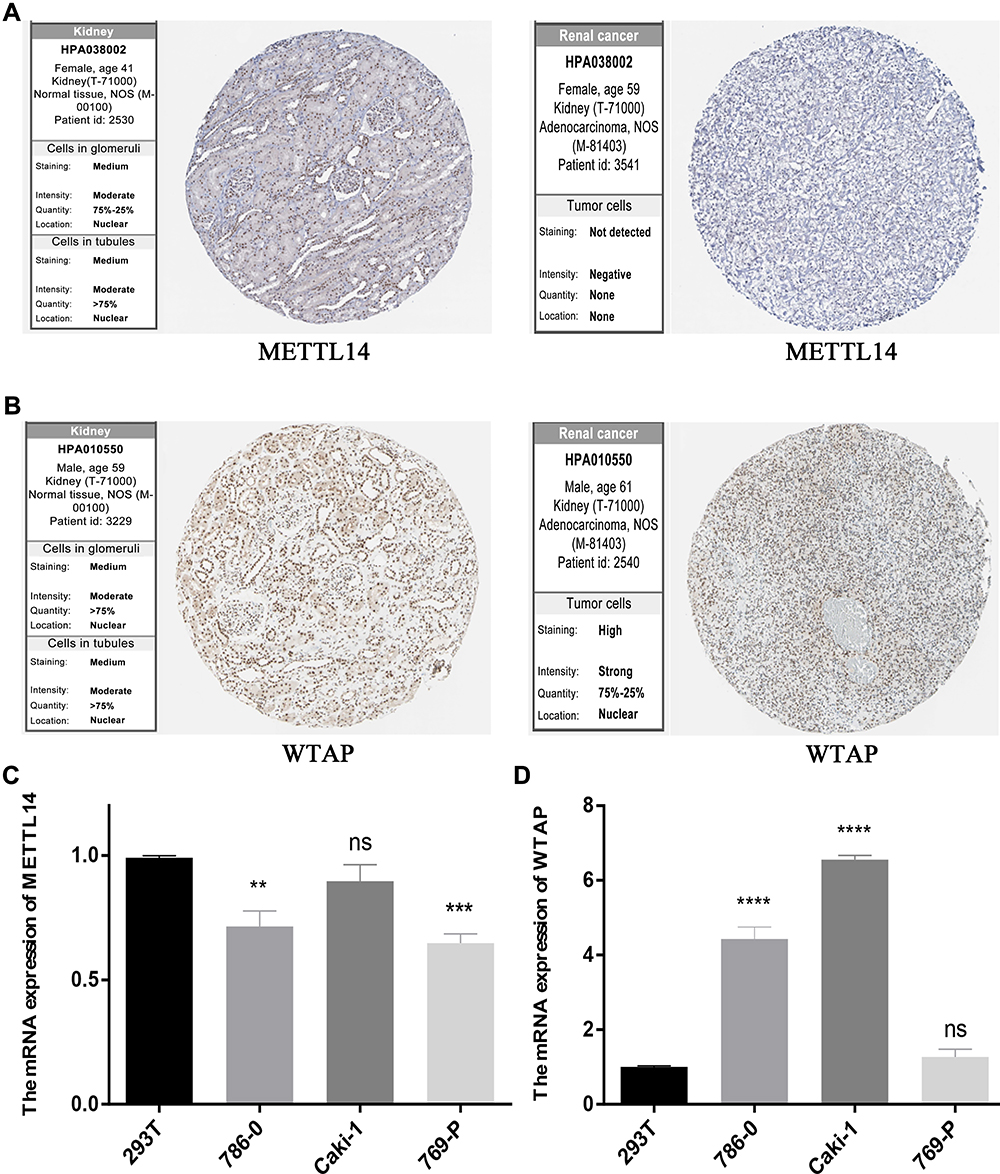 |
Figure 8 METTL14 and WTAP levels. (A) METTL14 and (B) WTAP protein expression in normal and RCC tissues from the Human Protein Atlas, as detected by immunohistochemical analysis. (C) METTL14 and (D) WTAP mRNA expression in normal (293T) and RCC cell lines. **P<0.01, ***P<0.001, and ****P<0.0001. Abbreviation: RCC, renal cell carcinoma. |
Discussion
The majority of RCC cases are clear cell RCC.3 Patients with early RCC have no obvious symptoms, so some of the patients already have metastases at pathological diagnosis. Molecularly targeted therapies are prone to drug resistance. RCC patients typically have a poor prognosis and the 5-year survival rate is <40%.25 Interferon and interleukin-2-based immunotherapy, as the first-line treatment, displays 15% efficacy.26 The occurrence and development of RCC is a complicated process involving numerous factors, such as smoking, obesity, genetics, and cell cycle changes.27 Thus, the precise mechanisms underlying RCC are complex. Developing novel therapeutic strategies based on these mechanisms is critical.
m6A modification of RNA refers to methylation of the nitrogen atom at position 6 of adenine (A), catalyzed by methyltransferase.28 Each mRNA has 3–5 m6A methylation sites that regulate RNA stability, localization, transport, splicing, and translation at the post-transcriptional level.29 Increasing evidence has demonstrated the role of m6A modification in acute myeloid leukemia (AML), hepatocellular carcinoma (HCC), breast cancer, and other types of cancer. In malignant tumors, m6A modification can induce a carcinogenic or anticancer effect. However, little is known about the roles of m6A methylation regulators in RCC. The risk model involving m6A RNA methylation regulators in RCC in this study provides new insights for further research into RCC.
According to the 893 RCC and 128 normal tissues analyzed in this study, the expression of the 13 m6A RNA methylation genes was significantly altered in RCC patients compared to normal controls, which indicated that m6A RNA modification may serve a role in RCC. Among the 13 regulators, the change in FTO expression was the most significant. FTO is a member of the non-heme Fe (II) and α-ketoglutarate-dependent dioxygenase ALKB protein family.30 Recent studies have suggested that FTO expression may be associated with several tumors, such as breast, thyroid, endometrial, and gastric cancer.31,32 Li et al reported that FTO affects the occurrence and resistance of leukemia by reducing m6A levels in the ASB2 and RARA target genes.33 Zhou et al demonstrated that FTO expression was significantly increased in patients with cervical squamous cell carcinoma (CSCC), and FTO and β-catenin expression have prognostic value in CSCC.34 These studies above have investigated the biomolecular mechanism underlying FTO-induced m6A modification during cancer development, but further investigation is required.
To further study the effect of the 13 m6A regulators on the clinical characteristics and prognosis of RCC, clinical data were downloaded from the TCGA database for analysis. Consensus clustering was performed to divide the RCC cases into two clusters. The results indicated that the clusters were associated with a clinicopathological characteristic (T status) in RCC. The results also indicated that the expression of the m6A RNA methylation regulators differed between the two clusters. Therefore, this analysis suggested that the m6A RNA methylation regulators were strongly associated with RCC.
The associations between the m6A RNA methylation regulators and the prognosis of RCC were further investigated by performing univariate Cox regression analyses. Based on the results, two m6A RNA methylation regulators with P<0.01 (METTL14 and WTAP) were selected for LASSO Cox regression analysis to establish a risk signature. Additionally, based on the heatmap, METTL14 was upregulated and WTAP was downregulated in the high-risk group compared to the low-risk group. Although METTL14 and WTAP are both writers, their expression levels and roles in tumors are not the same. WTAP is upregulated in AML, which promotes tumorigenesis and development. In contrast, METTL14 is downregulated in HCC, where it has a suppressive role.35,36 Each m6A RNA methylation regulator works differently in different tumors. For example, METTL3 is downregulated in glioblastoma and has an inhibitory role, but it is upregulated in lung cancer and HCC.37–39 The roles of METTL14 and WTAP in RCC are not completely understood and require further investigation.
Our results demonstrated that the high/low-risk groups (based on the median risk score) were significantly associated with RCC prognosis. Moreover, the area under the ROC curve for the risk signature was 0.701, which indicated that the predictive performance of the model was good. In addition, the high/low-risk groups were associated with clinicopathological characteristics (P<0.05; T status, M status, stage, and gender). Finally, univariate and multivariate Cox regression analyses were performed to assess the associations between the risk score (and other clinicopathological characteristics) and overall survival. The results indicated that the risk signature may serve as an independent prognostic factor in RCC. Therefore, WTAP and METTL14 may serve as independent prognostic factors in RCC.
WTAP is an essential protein in m6A methylation modification40 as it coordinates METTL3-METTL14 heterodimer localization and promotes m6A methylation.16 This indicates that the carcinogenic effect of WTAP may be closely related to the m6A methyltransferase complex. In addition, WTAP is linked to the development of other cancers. WTAP expression was found to be significantly increased in pancreatic ductal adenocarcinoma (PDAC) and was an independent prognostic factor in PDAC.41 WTAP may affect RCC cell proliferation by regulating the stability of CDK2 mRNA, leading to the occurrence and development of cancer.41 Therefore, WTAP may serve as a novel target for the diagnosis and treatment of RCC.
METTL14 is an important component of the m6A methyltransferase complex.15 Recent studies have reported its role in the malignant phenotype of tumors. One study demonstrated that METTL14 knockout in AML cell lines effectively inhibited proliferation.42 Moreover, METTL14 and FTO were downregulated in HCC,36 which indicated that m6A modification involved a complex feedback regulation mechanism involving both the writer and the eraser. To date, few studies have investigated the role of METTL14 in RCC. This study indicated that METTL14 was upregulated in the high-risk group compared to the low-risk group, which suggested that METTL14 upregulation is associated with poor RCC prognosis.
The specific regulation of the m6A methylation regulators and their potential biological functions require further investigation, especially regarding the two m6A genes (which encode writers) included in the risk signature in this study.
Conclusions
In conclusion, the results indicated that the risk score may serve as an independent prognostic factor and may also predict clinicopathological characteristics of RCC. However, the roles of m6A RNA methylation regulators in RCC are not completely understood, so further investigation of the precise underlying mechanisms is required. The results of this study may aid in investigating prognosis, diagnosis, and therapeutic strategies for RCC.
Abbreviations
m6A, N6-methyladenosine; RCC, renal cell carcinoma; TCGA, The Cancer Genome Atlas; LASSO, least absolute shrinkage and selection operator; GEO, Gene Expression Omnibus; CSCC, cervical squamous cell carcinoma; HCC, hepatocellular carcinoma; PDAC, pancreatic ductal adenocarcinoma; HPA, Human Protein Atlas.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This article does not contain any studies of human participants performed by any of the authors.
Consent for Publication
Not applicable.
Acknowledgments
We thank Wei Lu and Maxwell Hwang for technical help. Thanks to urologist Dr. Yi Li for reviewing the article.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was funded by the National Key R&D Program of China (2017YFC0908200), the Key Technology Research and Development Program of Zhejiang Province (2017C03017), and the National Natural Science Foundation of China (81772545).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Rossi SH, Klatte T, Usher-Smith J, Stewart GD. Epidemiology and screening for renal cancer. Review. World J Urol. 2018;36(9):1341–1353. doi:10.1007/s00345-018-2286-7
2. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Ficarra V. Renal cell carcinoma. Nat Rev Disease Primers. 2017;3:17009.
3. Srinivasan R, Ricketts CJ, Sourbier C, Linehan WM. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res. 2015;21(1):10–17. doi:10.1158/1078-0432.CCR-13-2993
4. Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. research support, N.I.H., extramural. N Engl J Med. 2016;374(2):135–145. doi:10.1056/NEJMoa1505917
5. Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Research Support, N.I.H., Intramural Review. Nat Rev Urol. 2010;7(5):245–257. doi:10.1038/nrurol.2010.46
6. Ljungberg B, Bensalah K, Canfield S, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol. 2015;67(5):913–924. doi:10.1016/j.eururo.2015.01.005
7. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Research Support, Non-U.S. Gov’t Review. Cancer Treat Rev. 2008;34(3):193–205. doi:10.1016/j.ctrv.2007.12.001
8. Dogan B, Canda AE, Akbulut Z, Balbay MD. Re: histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J Urol. 2010;184(5):2211–2212. doi:10.1016/j.juro.2010.06.125
9. Pantuck AJ, Zisman A, Belldegrun AS. Belldegrun as. the changing natural history of renal cell carcinoma. J Urol. 2001;166(5):1611–1623. doi:10.1016/S0022-5347(05)65640-6
10. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Review. Cell Res. 2018;28(5):507–517. doi:10.1038/s41422-018-0034-6
11. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Research Support, Non-U.S. Gov’t Review. Mol Cancer. 2019;18(1):176. doi:10.1186/s12943-019-1109-9
12. Gong D, Zhang J, Chen Y, et al. The m(6)A-suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP-induced Ca(2+) influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J Exp Clin Cancer Res. 2019;38(1):233. doi:10.1186/s13046-019-1223-y
13. Li X, Tang J, Huang W, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):96103–96116. doi:10.18632/oncotarget.21726
14. Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Nat Chem Biol. 2011;7(12):885–887. doi:10.1038/nchembio.687
15. Liu J, Yue Y, Han D, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 10(2):93–95. doi:10.1038/nchembio.1432
16. Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Research Support, Non-U.S. Gov’t. Cell Res. 2014;24(2):177–189. doi:10.1038/cr.2014.3
17. Wen J, Lv R, Ma H, et al. Zc3h13 regulates nuclear rna m 6 a methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028–1038.e6. doi:10.1016/j.molcel.2018.02.015
18. Yue Y, Liu J, Cui X, Cao J, Liu J. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discovery. 2018;4(1):10. doi:10.1038/s41421-018-0019-0
19. Hsu PJ, Zhu Y, Ma H, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–1127. doi:10.1038/cr.2017.99
20. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Nature. 2015;518(7540):560–564. doi:10.1038/nature14234
21. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Research Support, Non-U.S. Gov’t. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
22. Wilkerson MD, Neil HD. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;12:12.
23. Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for cox’s proportional hazards model via coordinate descent. J Stat Softw. 2011;39(5):1–13. doi:10.18637/jss.v039.i05
24. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143(1):29–36. doi:10.1148/radiology.143.1.7063747
25. Kaplan S, Ekici S, Dogan R, Demircin M, Ozen H, Pasaoglu I. Surgical management of renal cell carcinoma with inferior vena cava tumor thrombus. Am J Surg. 2002;183(3):292–299. doi:10.1016/s0002-9610(02)00782-1
26. Winkler GS. The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol. 2010;222(1):66–72. doi:10.1002/jcp.21919
27. Hunt JD, van der Hel OL, McMillan GP, Boffetta P, Brennan P. Renal cell carcinoma in relation to cigarette smoking: meta-analysis of 24 studies. Meta-Analysis Research Support, U.S. Gov’t, P.H.S. Int J Cancer. 2005;114(1):101–108. doi:10.1002/ijc.20618
28. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3ʹ UTRs and near stop codons. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Cell. 2012;149(7):1635–1646. doi:10.1016/j.cell.2012.05.003
29. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293–306. doi:10.1038/nrg3724
30. Gerken T, Girard CA, Tung YCL, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–1472. doi:10.1126/science.1151710
31. Sigurdson AJ, Brenner AV, Roach JA, et al. Selected single-nucleotide polymorphisms in FOXE1, SERPINA5, FTO, EVPL, TICAM1 and SCARB1 are associated with papillary and follicular thyroid cancer risk: replication study in a German population. Carcinogenesis. 2016;37(7):677–684. doi:10.1093/carcin/bgw047
32. Tan A, Dang Y, Chen G, Mo Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int J Clin Exp Pathol. 2015;8(10):13405–13410.
33. Li Z, Weng H, Su R, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA demethylase. research support, Non-U.S. Gov’t Research Support, N.I.H., Extramural. Cancer Cell. 2017;31(1):127–141. doi:10.1016/j.ccell.2016.11.017
34. Zhou S, Bai ZL, Xia D, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Research Support, Non-U.S. Gov’t. Mol Carcinog. 2018;57(5):590–597. doi:10.1002/mc.22782
35. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Bansal S. WTAP is a Novel Oncogenic Protein in Acute Myeloid Leukemia. Leukemia. 2014;28(12):1171–1174. doi:10.1038/leu.2014.16
36. Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Research Support, Non-U.S. Gov’t. Hepatology. 2017;65(2):529–543. doi:10.1002/hep.28885
37. Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Research Support, Non-U.S. Gov’t. Hepatology. 2018;67(6):2254–2270. doi:10.1002/hep.29683
38. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62:3. doi:10.1016/j.molcel.2016.03.021
39. Cui Q, Shi H, Ye P, et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Research Support, Non-U.S. Gov’t Research Support, N.I.H., Extramural. Cell Rep. 2017;18(11):2622–2634. doi:10.1016/j.celrep.2017.02.059
40. Schwartz S, MR M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5ʹ sites. Cell Rep. 2014;8(1):284. doi:10.1016/j.celrep.2014.05.048
41. Li BQ, Huang S, Shao QQ, et al. WT1-associated protein is a novel prognostic factor in pancreatic ductal adenocarcinoma. Oncol Lett. 2017;13(4):2531–2538. doi:10.3892/ol.2017.5784
42. Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA mA Modification. Cell Stem Cell. 2018;22(2):191–205.e9. doi:10.1016/j.stem.2017.11.016
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.