")
Back to Journals » Infection and Drug Resistance » Volume 16
Establishment and Methodological Evaluation of a Method for Rapid Detection of Helicobacter pylori and Virulence Genes Based on CRISPR-Cas12a
Authors Zhu Y , Lin C, Xu H, Xia Z, Yang W, Tang H, Hu X, Jiang T, Liu Z, Shen J
Received 17 November 2022
Accepted for publication 18 January 2023
Published 25 January 2023 Volume 2023:16 Pages 435—443
DOI https://doi.org/10.2147/IDR.S398098
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Yi Zhu,1,2 Chunhui Lin,1,2 Huaming Xu,3 Zhaoxin Xia,1,2 Wensu Yang,1,2 Hao Tang,4 Xinyi Hu,1,2 Tong Jiang,1,2 Zhen Liu,1,2 Jilu Shen1,2
1The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China; 2Anhui Public Health Clinical Center, Hefei, People’s Republic of China; 3The First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, People’s Republic of China; 4The Second Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China
Correspondence: Jilu Shen, Tel +86 151 5515 2963, Email [email protected]; [email protected]
Introduction: More than half of the world’s people are infected or have been infected with Helicobacter pylori. This infection is related to many diseases, with its pathogenicity related to virulence factors. Therefore, the rapid diagnosis of H. pylori and genotyping of virulence genes play an extremely important role in the clinical treatment and control of transmission.
Methods: To this end, we developed a molecular detection method based on RPA- CRISPR-Cas12a technology for the specific genes 16S rDNA gene, cytotoxin associated gene A(cagA), and vacuolating cytotoxin A (vacA) of H. pylori.
Results: The results of which were displayed by lateral flow strips. Macroscopic observation takes only about 25 minutes and the sensitivity is 2ng/microliter.
Discussion: The method is simple, convenient to operate and has low costs, and can therefore be applied widely to the detection and typing of H. pylori in various environments such as primary hospitals, community clinics, outdoors, and large medical institutions.
Keywords: Helicobacter pylori, virulence genes, recombinase polymerase amplification, CRISPR-Cas12a, lateral flow immunochromatographic strip
Introduction
Helicobacter pylori (H. pylori) is a Gram-negative, spiral microaerophilic bacterium that infects more than half of the world’s population.1,2 Studies have shown that this bacterium is the main pathogenic factor in chronic gastritis and peptic ulcers, and is related to the occurrence and development of gastric cancer.3,4 For several decades, H. pylori is classified as a class I carcinogen by the International Agency for Research on Cancer (IARC).5
The cytotoxin-associated gene A(cagA) and vacuolating cytotoxin gene (vacA) of H. pylori are the main pathogenic factors.6 Clinically, these strains are generally divided into type I or type II, with type I being positive for cagA and/or vacA and type II negative for both cagA and vacA in H. pylori serum antibody test.7 Type II strains are almost non-toxic and generally cause only chronic superficial gastritis or no clinical symptoms after infection. Eradication may not be a blind option for people with type II and no clinical symptoms. Therefore, cagA and vacA virulence genotyping of H. pylori have become the key judgment indicators for clinical eradication treatment of patients with a H. pylori infection.
At present, the commonly used detection methods for H. pylori include a 13C or 14C urea breath test (UBT), H. pylori fecal antigen test, and H. pylori serum antibody test.8 However, these methods do not meet the needs of clinical detection and treatment due to several disadvantages such as radioactive contamination and inability to either genotyping the virulence of the strains or distinguish between previous and ongoing infections. In order to compensate for these shortcomings, we attempted to achieve rapid, accurate, and convenient detection method of H. pylori by combining recombinase polymerase amplification (RPA) with CRISPR-Cas12a technology.
RPA is a novel isothermal acid amplification technique first proposed by Piepenburg et al in the UK in 2006.9 This amplification can be completed under constant temperature conditions of 37–42 °C for 10–20 min and does not need a thermal cycle, water bath, or metal bath to meet the reaction conditions. This greatly reduces the cost, while the portable equipment can be deployed for field diagnosis, especially in areas with limited resources, making the method very suitable for instant detection. The CRISPR-Cas system is an adaptive immune system formed during the long-term evolution of prokaryotes. The system is distinguished from other systems by the complementary pairing of lead RNA (gRNA) and target sequence nucleic acid, as well as by the presence of PAM sites on the target sequence nucleic acid.10 When Cas12a is activated by a specific target sequence, any single-stranded DNA can be subjected to non-specific cleavage.11 Isothermal amplification techniques including RPA and LAMP have been used in combination with CRISPR-Cas12a for a variety of in vitro diagnostic and biological assays, including SARS-CoV-2,12 methicillin-resistant Staphylococcus aureus,13 leptospirosis,14 and HCV.15
Based on these findings we attempted to develop a novel method based on a RPA-Cas12a-lateral flow immunochromatographic strip, which only required 25 min for the reaction process and achieved a sensitivity of 2 ng/microliter. Because large equipment and instruments were not needed, only one water bath or metal bath was required, and therefore the method could be used for testing in places other than a clinical central laboratory, such as primary hospitals, community clinics, and outdoor locations.
Materials and Methods
Reagents and Chemicals
All the primers (Table 1) used in this study were designed independently by our laboratory and synthesized by Shanghai Sangon Biotech Company (Shanghai, China). The standard strain of H. pylori ATCC700392 was purchased from BeNa Culture Collection (Beijing, China). The FAM-BHQ1 double-labeled single-stranded DNA probe, FAM-biotin double-labeled single-stranded DNA probe, and crRNA were all purchased from GeneBiogist (Shanghai, China). The RPA kit and lateral flow immunochromatographic strip were obtained from TwistDx (Cambridge, UK). LbaCas12a and NEB buffer 3.1 were obtained from New England Biolabs (Beijing, China). PCR Master Mix was purchased from Takara (Dalian, China). The clinical saliva samples were collected in the expiratory room of the Department of Gastroenterology, the First Affiliated Hospital of Anhui Medical University.
 |
Table 1 Oligonucleotide Sequences Used in the Study |
Design and Screen of the RPA Primers and crRNA
RPA Primer Design
We used dry-lab16 technologies to design RPA primers. At first, the H. pilori 16S rDNA gene (EU544199) and cagA (JF798703) and vacA gene (GQ331984) sequences were retrieved from the GenBank database of NCBI. The RPA primers were then designed using Primer Premier 5.0, with the primer having the highest score selected for screening. The results showed that the RPA primers were superior to the other primers for band brightness in agarose gel electrophoresis after RPA amplification.
crRNA Design
The crRNA was designed for the TTTN PAM site of the target sequence of the target gene.17 First, a FAM-BHQ1 double-labeled single-stranded DNA probe was added to the system and used in the CRISPR-Cas12a cleavage system to screen for crRNA using fluorescence intensity measured by a fluorescent quantitative PCR instrument. The FAM-biotin double-labeled single-stranded DNA probe was then used in the CRISPR-Cas12a cleavage system for methodological validation of the RPA-Cas12a- lateral flow immunochromatographic strip.
Culture Collection of the Bacterial Strain
The standard strain ATCC700392 was used for the solid state culture, using the method for H. pylori described by reference Fiorini, G.18 The culture results are shown in Figure 1A.
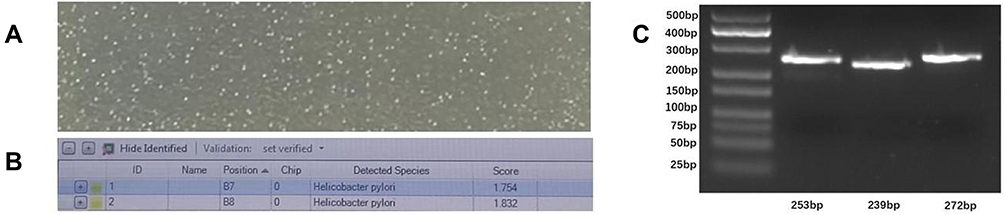 |
Figure 1 (A) Needle-like colonies appeared in the culture medium of H. pylori. (B) The colonies in Figure A were picked and identified as H. pylori by mass spectrometry. (C) Agarose gel electrophoresis of the RPA amplification of the three target genes. From left to right, they respectively expressed 16S rDNA, the cagA and vacA genes, and the lengths of the corresponding target fragments were 253 bp, 239 bp, and 272 bp, respectively. |
Bacterial DNA Extraction
The target bacterial strain was selected and DNA extraction was carried out using the nucleic acid extraction kit. The concentration and OD value of the extracted nucleic acid were then detected using a Nanodrop one spectrophotometer. The OD260/OD280 ration was required to be between 1.8 and 2.0.
Target Fragment Amplification
The extracted nucleic acids were used as templates for RPA and PCR amplification and amplified according to the instructions of the kit. The reaction was carried out in a 1.5 mL Eppendorf tube. After the reaction system was mixed evenly, an equal amount of paraffin oil was added to the surface to prevent aerosol pollution.
- The reaction system of RPA (50 microliters) consisted of 29.5 microliters of primer-free rehydration buffer, 2.4 microliters of forward primer (10 μM) and reverse primer (10 μM), making a total of 13.2 microliters of template and DEPC water, and 2.5 microliters of MgOAc (280 mM). The reaction was carried out at 39°C for 15 min.
- The PCR reaction system (30 microliters) consisted of 1×SYBR qPCR mix, 0.6 microliters of forward primer and reverse primer (final concentration 0.2 uM), 0.6 microliters of ROX reference solution, 2 microliters of DNA, and complement of DEPC water to 30 microliters. The reaction conditions were as follows: pre-denaturation at 94°C for 3 min, denaturation at 94°C for 20s, annealing at 63°C for 30s, and extension at 72°C for 30s, with 40 cycles in total.
Establishment of the RPA-Cas12a- Immunochromatographic Assay
The reaction cleavage system (20 microliters) consisted of 5 microliters of RPA amplification product, 3 microliters of crRNA (10uM), 2 microliters of NEB buffer 3.1, 1 microliter of Cas12a protein, 1 microliter of FAM-Biotin single-stranded DNA probe, and 8 microliters of DEPC H2O. The mixtures were then incubated in a metal bath at 37°C for 10 min, followed by aspiration of 1uL of product into a 1.5 mL Eppendorf tube. A 99 microliters aliquot of the incubation solution was then added and mixed for application to the immunochromatographic test strips, with the results observed within 5 min.
Interpretation of the results: When the target gene was amplified by RPA, Cas12a could not only recognize and cleave the target gene, but also non-specifically cleave the probes labeled with FAM-biotin.
The cleaved probes were captured by the biotin ligand bound on the test line. As the biotin ligand could not capture the gold-labeled antibody, no band was displayed, and the result was positive. Conversely, the biotinylated ligand on the test line captures the gold-labeled antibody and when the probe is not cleaved, this displays the corresponding band indicating a negative result (Figure 2).
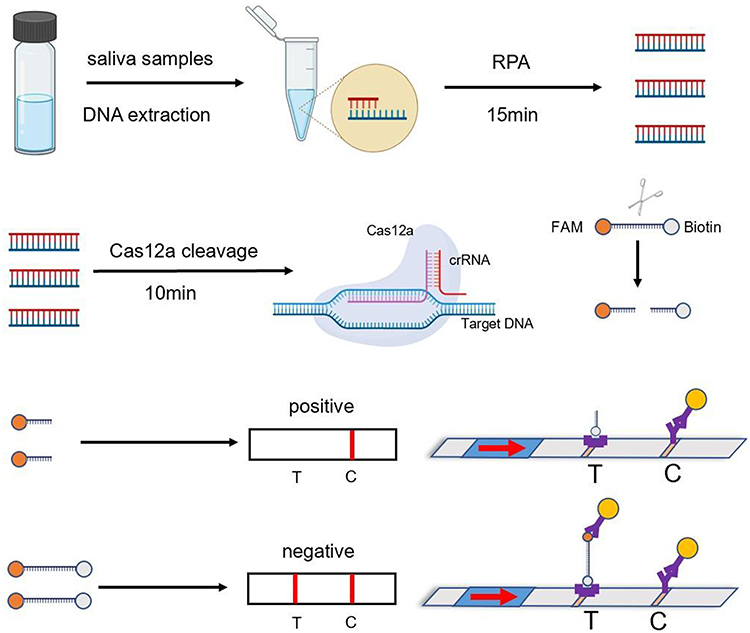 |
Figure 2 Detection method based on a RPA-Cas12a- transverse flow immunochromatographic strip. |
Minimum Detection Limit
The extracted template DNA was subjected to a series of dilutions at 40, 20, 10, 5, 2, and 1 ng/ microliter, respectively. The 16S rDNA gene was amplified by RPA and PCR, and the brightness of the corresponding bands was observed to determine the lowest detected concentration of the two detection methods. To interpret the results with as few cuts as possible, we also diluted the probes with an initial concentration of 100 μM to 10 and 1 μM, and 800, 600, 400, 200, and 100 nM, respectively. DEPC-treated water was then used as a template to simulate the reaction process of the RPA-Cas12a-lateral flow immunochromatographic test strip, with the coloration of the test line observed to clearly show the lowest probe concentration of the strip for subsequent experiments.
Evaluation of the Clinical Application
Using the detection method of RPA-Cas12a-immunochromatography established above, 28 positive samples of a 14C expiratory test, 3 positive samples of a 13C expiratory test, and 20 negative samples of a 14C expiratory test were tested respectively. The results were compared with UBT, and the positive and negative coincidence rates were calculated by the Chi-square test.
Results
Screening Results of Primers for the RPA Amplification System
The nucleic acid extracted from the H. pylori ATCC700392 strain was used as the experimental sample for RPA amplification, with the products then subjected to agarose gel electrophoresis. As shown in Figure 1C, three pairs of RPA primers for three genes of H. pylori were shown to meet the requirements.
Screening Results of crRNA in the H. pylori CRISPR-Cas12a Cleavage System
Two crRNAs were designed for each of the RPA-amplified regions of the 16S rDNA, cagA, and vacA genes of H. pylori, while a single-stranded DNA probe double-labeled with FAM and BHQ1 was used in the CRISPR-Cas12a cleavage system. The fluorescence values detected by the fluorescent quantitative PCR instrument showed that crRNA No.1, 3, and 5 were superior to those of No.2, 4, and 6, so the crRNA No.1, 3, and 5 were selected for the subsequent cleavage experiments of 16S rDNA, and cagA, and vacA genes, respectively (Figure 3).
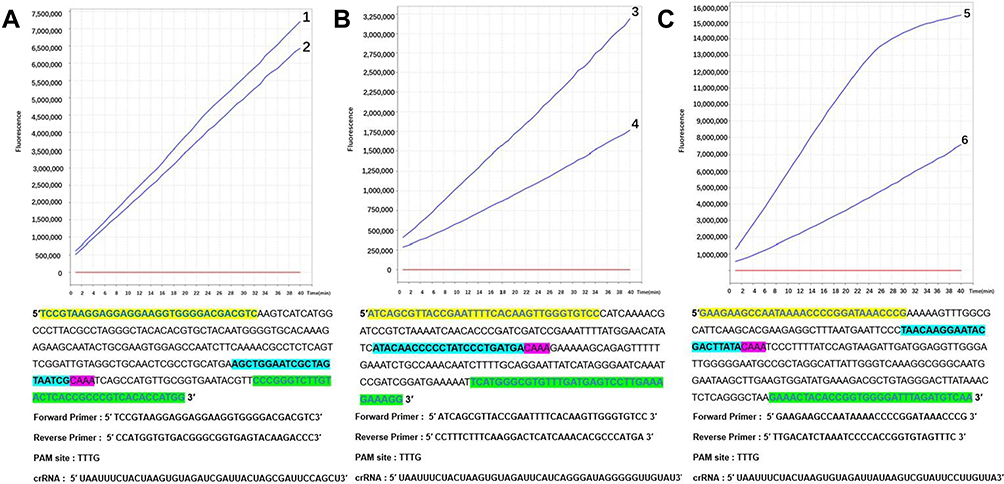 |
Figure 3 (A–C) show the monitoring of fluorescence values of the CRISPR-Cas12a cleavage system targeting the target sequences of H. pylori 16S rDNA, cagA, and vacA genes, respectively, with two crRNAs designed per gene. Finally, the crRNA corresponding to No.1, No.3, and No.5 with good peak initiation was selected for the subsequent cleavage experiment. The corresponding RPA primers and the positions of the crRNA and PAM sites are shown below each selected crRNA. |
Establishment of the RPA-Cas12a- Lateral Flow Immunochromatographic Strip Assay and Its Sensitivity Detection
The screened crRNA was used in the probe concentration dilution experiment and the sensitivity test of the RPA-Cas12a- lateral flow immunochromatographic strip. The results are shown as follows:
- Dilution of the probe concentration to 200 nM revealed distinct bands (Figure 4), so an initial probe concentration of 200 nM was selected for the subsequent experiments.
- The minimum detection concentrations for agarose gel electrophoresis of RPA and PCR amplification products were both 1 ng/microliter (Figure 5A and B).
- RPA-Cas12a-lateral flow immunochromatographic strip test results showed a minimum detectable concentration of 2 ng/microliter (Figure 5C).
 |
Figure 4 (A) No.1, 2, 3, 4, and 5 indicate that the concentrations of FAM-biotin double-labeled single-stranded DNA probes were 0, 100, 10, 1, and 0.1 μM, respectively. (B) No. 6, 7, 8, 9, 10, and 11 indicate probe concentrations of 1 μM, 800 nM, 600 nM, 400 nM, 200 nM, and 100 nM, respectively. |
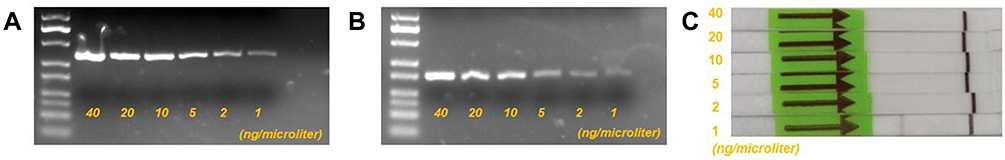 |
Figure 5 (A) RPA agarose gel electrophoresis with template concentrations of 40, 20, 10, 5, 2, and 1 ng/microliter, respectively. (B) PCR agarose gel electrophoresis with template concentrations of 40, 20, 10, 5, 2, and 1 ng/microliter, respectively. (C) The results of the strips are shown after cleavage by the CRISPR-Cas12a system and correspond to Figure A in sequence from top to bottom. |
Clinical Sample Detection Evaluation
A total of 51 saliva samples (31 positives and 20 negatives) were detected with a RPA-Cas12a-lateral flow immunochromatographic strip for the 16S rDNA, cagA, and vacA genes (Figure 6). The Chi-square test showed that the positive and negative coincidence rate of the 16S rDNA positive test results and UBT were both 100%. In addition, for samples with positive UBT and 16S rDNA, the detection rates of the cagA and vacA genes were 64.5% and 48.4%, respectively (Figures 6 and 7).
 |
Figure 6 DETECT validation for detection of H. pylori in clinical samples. (A) A total of 31 UBT positive samples were all positive for the 16SrDNA gene test with only the control line. (B) The 16S rDNA gene assay of 20 UBT negative samples was negative for both the test and control lines because the probe was not cleaved. (C) 20 of the 31 UBT positive samples tested positive for the cagA gene. (D) 15 of the 31 UBT positive samples were found to be positive for the vacA gene. |
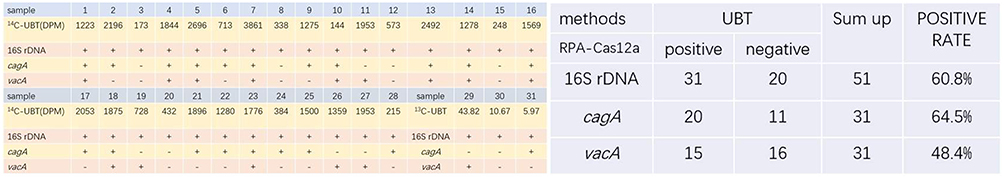 |
Figure 7 Detection results of the clinical samples. |
Discussion
H. pylori gastritis is associated etiologically with peptic ulcer and gastric cancer and is highly infectious.19 The infection is usually acquired in childhood, especially through intra-family transmission.20 There is evidence that timely diagnosis and eradication of H. pylori can improve gastric mucosal inflammation, promote ulcer healing, and reduce the incidence of gastric cancer.21 The virulence of the strain plays an important role in the clinical consequences of a H. pylori infection.22 H. pylori has two important toxins, cagA and vacA, which are the main pathogenic factors of the organism.23 Studies have shown that cagA toxin has a causal relationship with the occurrence of gastropathy, especially gastric cancer.24 VacA has also been reported to induce apoptosis in gastric epithelial cells.25 As mentioned previously, for patients with cagA, or vacA-negative, the infection may only be associated with the occurrence of chronic superficial gastritis, or no clinical symptoms, and cannot be treated. Therefore, we also genotyped the cagA and vacA virulence genes when detecting H. pylori.
The current commonly methods used to detect H. pylori have many limitations. The 13C urea breath test (13C-UBT) is the gold standard for detecting H. pylori infections,26 but cannot type the organism. Serological tests can also not distinguish between an active infection and previous exposure to H. pylori.27 Although fluorescent quantitative PCR method can be used for typing, this requires large-scale advanced equipment and professional technical personnel to operate and is therefore not suitable for areas with limited resources.28 In addition, traditional endoscopy and other methods can cause invasive damage to the patient. Based on the above limitations, we aimed to design a simple, fast, and accurate method for detecting and typing H. pylori, in order to meet the needs of clinical detection and treatment. Studies have reported that the genetic material (DNA) of H. pylori can be detected in gastric biopsies, saliva, feces, or dental plaque samples.29 Compared with other sample types, the collection and processing of saliva is simpler, and therefore we selected saliva samples as the research material.
We designed a new technology based on a RPA-Cas12a-lateral flow immunochromatographic strip.30 In order to meet the requirements of rapid and accurate detection, we greatly reduced the dependence on large-scale advanced equipment, with the whole reaction process only requiring a water bath or metal bath. Through 15 min of RPA amplification and 10 min of CRISPR-Cas12a trans-cutting, the products could be read using a lateral flow immunochromatographic strip, making it very suitable for Point of Care Technology (POCT), which greatly reduced the turnaround time.31 In order to solve the problem of aerosol pollution caused by uncapping after RPA reaction, we added the activator of the RPA reaction and immediately added an equal amount of paraffin oil to isolate the air after mixing.32 For the verification process of the clinical specimens, saliva samples from 31 positive patients and 20 negative patients were collected. By using a RPA-Cas12a- lateral flow immunochromatographic test strip, the positive and negative coincidence rates of the obtained results with the clinical UBT test results were both shown to be 100%, confirming the specificity of the method. Using agarose gel electrophoresis we found that the bands could be observed by both RPA amplification and PCR methods at a template concentration of 1ng/microliter, indicating that RPA amplification was comparable to PCR sensitivity. The sensitivity of RPA is up to 2 ng/microliter when cleaved after amplification to show the results on the strips. Compared with PCR, our method was more rapid. More importantly, our method does not depend on large-scale instruments, and can be used in places with poor medical conditions, such as clinics and primary hospitals. The combination of CRISPR-Cas12a technology and RPA technology has two significant advantages. On the one hand, the specificity of binding of crRNA to target fragments in the CRISPR-Cas12a reaction system effectively prevents the influence of RPA false-positive results. On the other hand, in this experiment, based on the uniqueness of the test strip detection principle, in order to achieve interpretation of the results with as few amplifications and cleavage as possible, we performed a series of dilutions of the probe concentration, and finally set the probe concentration at 200 nM. This method effectively utilizes the high-efficiency amplification of RPA and the high-efficiency cleavage ability of Cas12a protein, and greatly shortens the time required to observe the results. Despite these advantages, the technique has some limitations. For example, crRNA is easily degraded and the storage conditions are relatively strict, requiring it to be placed at −20°C or below.33 Therefore, storage methods and conditions need to be improved to ensure a longer shelf-life at room temperature for large-scale and extensive clinical use. We consider that not being dependent on instruments will facilitate the universal detection of various pathogens, including H. pylori.
Ethics Approval and Consent to Participate
This study involved the use of the patient’s saliva, which had been given with the informed consent of the subjects, and all specimens were numerically numbered and did not affect the patient’s privacy. All procedures performed in studies involving human participants were in accordance with the ethical standards of the medical ethics committee of the First Affiliated Hospital of Anhui Medical University (Anhui Public Health Clinical Center)(reference number: LISC20210802) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Acknowledgments
The authors would like to express their gratitude to EditSprings for the expert linguistic services provided.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded through a grant from Anhui Provincial Department of Education for university cooperative research and public health collaborative innovation project in Anhui Province in 2020 (Grant No. GXXT-2020-016) and a grant from Anhui Provincial Health Commission for key scientific research projects in 2021 (Grant No. AHWJ2021a011).
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Salama NR, Hartung ML, Müller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11(6):385–399. doi:10.1038/nrmicro3016
2. Reshetnyak VI, Burmistrov AI, Maev IV. Helicobacter pylori: commensal, symbiont or pathogen? World J Gastroenterol. 2021;27(7):545–560. doi:10.3748/wjg.v27.i7.545
3. Jonaitis L, Pellicano R, Kupcinskas L. Helicobacter pylori and nonmalignant upper gastrointestinal diseases. Helicobacter. 2018;23(Suppl 1):e12522. doi:10.1111/hel.12522
4. Wu JY, Lee YC, Graham DY. The eradication of Helicobacter pylori to prevent gastric cancer: a critical appraisal. Expert Rev Gastroenterol Hepatol. 2019;13(1):17–24. doi:10.1080/17474124.2019.1542299
5. Testerman TL, Morris J. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol. 2014;20(36):12781–12808. doi:10.3748/wjg.v20.i36.12781
6. El Khadir M, Boukhris Alaoui S, Benajah DA, et al. VacA genotypes and cagA-EPIYA-C motifs of Helicobacter pylori and gastric histopathological lesions. Int J Cancer. 2020;147(11):3206–3214. doi:10.1002/ijc.33158
7. Basso D, Navaglia F, Brigato L, et al. Analysis of Helicobacter pylori vacA and cagA genotypes and serum antibody profile in benign and malignant gastroduodenal diseases. Gut. 1998;43(2):182–186. doi:10.1136/gut.43.2.182
8. Ranjbar R, Behzadi P, Farshad S. Advances in diagnosis and treatment of Helicobacter pylori infection. Acta Microbiol Immunol Hung. 2017;64(3):273–292. doi:10.1556/030.64.2017.008
9. Piepenburg O, Williams CH, Stemple DL, Armes NA, Haber J. DNA detection using recombination proteins. PLoS Biol. 2006;4(7):e204. doi:10.1371/journal.pbio.0040204
10. Liu G, Lin Q, Jin S, Gao C. The CRISPR-Cas toolbox and gene editing technologies. Mol Cell. 2022;82(2):333–347. doi:10.1016/j.molcel.2021.12.002
11. Chen JS, Ma E, Harrington LB, et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360(6387):436–439. doi:10.1126/science.aar6245
12. Broughton JP, Deng X, Yu G, et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat Biotechnol. 2020;38(7):870–874. doi:10.1038/s41587-020-0513-4
13. Li Y, Shi Z, Hu A, et al. Rapid one-tube RPA-CRISPR/Cas12 detection platform for methicillin-resistant Staphylococcus aureus. Diagnostics. 2022;12(4). doi:10.3390/diagnostics12040829
14. Jirawannaporn S, Limothai U, Tachaboon S, et al. Rapid and sensitive point-of-care detection of Leptospira by RPA-CRISPR/Cas12a targeting lipL32. PLoS Negl Trop Dis. 2022;16(1):e0010112. doi:10.1371/journal.pntd.0010112
15. Kham-Kjing N, Ngo-Giang-Huong N, Tragoolpua K, Khamduang W, Hongjaisee S. Highly specific and rapid detection of hepatitis C virus using RT-LAMP-coupled CRISPR-Cas12 assay. Diagnostics. 2022;12(7):1524. doi:10.3390/diagnostics12071524
16. Behzadi P, Ranjbar R. DNA microarray technology and bioinformatic web services. Acta Microbiol Immunol Hung. 2019;66(1):19–30. doi:10.1556/030.65.2018.028
17. Vanegas KG, Jarczynska ZD, Strucko T, Mortensen UH. Cpf1 enables fast and efficient genome editing in Aspergilli. Fungal Biol Biotechnol. 2019;6:6. doi:10.1186/s40694-019-0069-6
18. Fiorini G, Vakil N, Zullo A, et al. Culture-based selection therapy for patients who did not respond to previous treatment for Helicobacter pylori infection. Clin Gastroenterol Hepatol. 2013;11(5):507–510. doi:10.1016/j.cgh.2012.12.007
19. Salavati S, Ahmadi Hedayati M, Ahmadi A, Fakhari S, Jalili A. Relationship between Helicobacter pylori cagA genotypes infection and IL-10 and TGFβ1 genes’ expression in gastric epithelial cells. Int J Prev Med. 2020;11:20. doi:10.4103/ijpvm.IJPVM_536_18
20. Lee YC, Dore MP, Graham DY. Diagnosis and treatment of Helicobacter pylori infection. Annu Rev Med. 2022;73:183–195. doi:10.1146/annurev-med-042220-020814
21. Suzuki S, Kusano C, Horii T, Ichijima R, Ikehara H. The ideal Helicobacter pylori treatment for the present and the future. Digestion. 2022;103(1):62–68. doi:10.1159/000519413
22. Hedayati MA, Salavati S, Dell Agli M. Transcriptional profile of Helicobacter pylori virulence genes in patients with gastritis and gastric cancer. Can J Infect Dis Med Microbiol. 2021;2021:1309519. doi:10.1155/2021/1309519
23. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196–202. doi:10.1016/j.canlet.2013.08.016
24. Fujii Y, Murata-Kamiya N, Hatakeyama M. Helicobacter pylori CagA oncoprotein interacts with SHIP2 to increase its delivery into gastric epithelial cells. Cancer Sci. 2020;111(5):1596–1606. doi:10.1111/cas.14391
25. Yahiro K, Akazawa Y, Nakano M, et al. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov. 2015;1:15035. doi:10.1038/cddiscovery.2015.35
26. Huang J, Cui J. Evaluation of Helicobacter pylori infection in patients with chronic hepatic disease. Chin Med J. 2017;130(2):149–154. doi:10.4103/0366-6999.197980
27. Malfertheiner P, Megraud F, O’Morain CA, et al. Management of Helicobacter pylori infection--the Maastricht IV/ Florence consensus report. Gut. 2012;61(5):646–664. doi:10.1136/gutjnl-2012-302084
28. Babu B, Ochoa-Corona FM, Paret ML. Recombinase polymerase amplification applied to plant virus detection and potential implications. Anal Biochem. 2018;546:72–77. doi:10.1016/j.ab.2018.01.021
29. Bordin DS, Voynovan IN, Andreev DN, Maev IV. Current Helicobacter pylori diagnostics. Diagnostics. 2021;11:8. doi:10.3390/diagnostics11081458
30. Sun Y, Yu L, Liu C, et al. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J Transl Med. 2021;19(1):74. doi:10.1186/s12967-021-02741-5
31. Patel SK, Pratap CB, Jain AK, Gulati AK, Nath G. Diagnosis of Helicobacter pylori: what should be the gold standard? World J Gastroenterol. 2014;20(36):12847–12859. doi:10.3748/wjg.v20.i36.12847
32. Zen LPY, Lai MY, Lau YL. Elimination of contamination in loop-mediated isothermal amplification assay for detection of human malaria. Trop Biomed. 2020;37(4):1124–1128. doi:10.47665/tb.37.4.1124
33. Xu H, Tang H, Li R, et al. A new method based on LAMP-CRISPR-Cas12a-lateral flow immunochromatographic strip for detection. Infect Drug Resist. 2022;15:685–696. doi:10.2147/idr.S348456
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.