")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Esomeprazole alleviates the damage to stress ulcer in rats through not only its antisecretory effect but its antioxidant effect by inactivating the p38 MAPK and NF-κB signaling pathways
Authors Xie W , Huang X , Chen R , Chen R , Li T , Wu W, Huang Z
Received 6 November 2018
Accepted for publication 16 May 2019
Published 22 August 2019 Volume 2019:13 Pages 2969—2984
DOI https://doi.org/10.2147/DDDT.S193641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Wei Xie,*,1 Xielin Huang,*,2 Renpin Chen,1 Ruru Chen,1 Tang Li,1 Wei Wu,1 Zhiming Huang1
1Department of Gastroenterology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Department of Gastrointestinal Surgery, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Wu; Zhiming Huang
Department of Gastroenterology, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang Street, Wenzhou 325000, People’s Republic of China
Tel +86 135 6629 0340
Fax +86 137 0665 8620
Email [email protected]; [email protected]
Background: Stress ulcer is a severe complication in critically ill patients and causes a high mortality. The proton pump inhibitor esomeprazole is widely applied in the treatment of stress ulcers because of its powerful acid suppression ability. However, the mechanism of stress ulcer and the precise gastroprotective effect of esomeprazole in stress ulcer remain unclear.
Purpose: In the present study, the rats with water-immersed and restraint (WIR)-induced stress ulcer were used to further elucidate the anti-ulcerogenic capacity of esomeprazole in stress ulcer in addition to its anti-acid secreting ability.
Methods and results: The rats were randomly divided into 5 groups: control group (NS), water-immersed and restraint group (WIR), high-dose application of esomeprazole plus stress ulcer-induced group (HE+WIR), low-dose application of esomeprazole plus stress ulcer-induced group (LE+WIR), and high-dose application of esomeprazole without stress ulcer-induced group (HE). Our study showed that the pretreatment of esomeprazole alleviated gastric tissue damage in both macroscopic and histopathological manifestations. Pretreatment of esomeprazole elevated the decline in PEG2 level affected by WIR; and it inhibited the secretion of gastric acid, gastrin and pepsin. Moreover, esomeprazole exerted its antioxidant effects by reducing malondialdehyde levels, enhancing the expressions of antioxidant factors like glutathione and superoxide dismutase (SOD) and reducing the compensatory transcriptional elevation of SOD1 gene. Esomeprazole also reduced the levels of MPO (myeloperoxidase), tumor necrosis factor (TNF)-α and interleukin (IL)-1β according to its anti-inflammatory effects. We further explored the possible mechanism of esomeprazole pretreatment on stress ulcer and demonstrated that esomeprazole attenuated the high phosphorylation levels of nuclear factor kappa B (NF-κB) p65 and p38 MAPK, and decreased the NF-κB p65 nuclear translocation induced by WIR related stress ulcer.
Conclusion: Our study provides some evidence that the esomeprazole pretreatment exerts gastroprotective effects in WIR-induced stress ulcer through not only its antisecretory effect but also its antioxidant effect by inactivating the p38 MAPK and NF-κB signaling pathways.
Keywords: stress ulcer, esomeprazole, oxidative stress, NF-κB, MAPK-p38
Introduction
Stress ulcer, sometimes called stress-related mucosal disease (SRMD), generally refers to an acute mucosal lesion of the stomach or even the duodenum that occurs during shock, trauma, post-operation and severe bacterial infections.1 It is an intense threat to many critically ill patients because it could cause a high mortality.1–4 Imbalance between aggressive factors (such as gastric acid, pepsin) and defense factors (such as mucosal blood flow and prostaglandin E2, PGE2) accounts for the pathophysiology of stress ulcer.1,5 However, the precise mechanism still remains unclear.
ROS are atoms or molecules which can cause oxidative stress that promote the development of epithelial necrosis and mucosal ulceration.7,12 Malondialdehyde (MDA) is one of the most important products of membrane lipid peroxidation and is regarded as an advisable marker of ROS-mediated tissue damage.7,13–15 Superoxide dismutase (SOD) and glutathione (GSH) are two free radical scavengers which play a critical antioxidative role. Changes in these three indicators can be used to reflect the severity of tissue oxidative damage.
The mitogen-activated protein kinases (MAPKs) are major intracellular signal transduction pathways and include several subgroups, and p38 MAPK is one of the most representative members. As a redox-sensitive kinase, it is activated by extracellular stress stimuli, such as ROS overproduction or inflammation.7,16 Oxidative stress increases the phosphorylation of p38 MAPK and activates the p38 MAPK pathway.17 The activated p38 MAPK pathway is thought to be closely related to the activation of the nuclear factor kappa B (NF-κB) pathway,7 which is shown to respond directly to stress and inflammatory reaction and plays a vital role in the pathogenesis of stress ulcer.6,8 RELA (p65) is a subunit of NF-κB that is responsible for activating specific gene transcription. In general, p65 is bound to IκB proteins located in the cytoplasm as an inactive form. Various pro-inflammatory factors and ROS can either directly18,19 or via the activated p38-MAPK signaling pathway exit the NF-κB pathway through the modified IκBs degradation and the p65 translocation into the nucleus. Tumor necrosis factor (TNF)-α and IL-1β, as the downstream molecules of NF-κB pathway, have a central role in mediating neutrophil infiltration and mucosal damage involved in stress ulcer.6–11,20,24 They can also act as the external stimuli for the formation of ROS and lead to the activation of NF-κB by promoting the nuclear translocation of the p65 subunit.21–23 This positive feedback loop may exacerbate the damage to the gastric mucosa.25,26 Myeloperoxidase (MPO) is the specific markers of neutrophils and the activity of MPO can reflect the severity of inflammation in tissues.27,28
Esomeprazole belongs to proton pump inhibitors (PPIs) and is widely used in clinical practice because of a satisfactory acid inhibition effect.29 Recently, many studies found that, besides inhibiting acid secretion, PPIs such as esomeprazole also protected gastric mucosa through mechanisms connected with the mitigation of oxidative damage, the inhibition of inflammation and the suppression of NF-κB and p38 MAPK pathways.14,28,30–32 Most of these studies focused on diseases such as NSAID-induced gastric-intestinal ulcers or gastro-esophagitis reflux disease. However, the detailed mechanisms underlying the relief of stress ulcers by esomeprazole are still unclear.
Water-immersed and restraint (WIR) of rat is recognized as the most reproducible and clinically relevant animal model of stress ulcer and frequently used in medical research.7 Therefore, by using such animal model, this study aims to further verify the function of esomeprazole in suppressing gastric secretion and to explore the mechanisms involved in the regulation of ROS, inflammatory factors TNF-α and IL-1β, and NF-κB and p38 MAPK pathways. Our study provides a better understanding of esomeprazole’s gastric protective effects in the WIR-induced experimental stress ulcer, thus supporting the application of esomeprazole in stress ulcers.
Materials and methods
Animals
Fifty male Sprague Dawley (SD) rats, weighing 200±20 g, were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China) and raised in individual cages with a relatively constant temperature. The animals were in a controlled environment and had free access to food and water. All the operations were conducted ethically following the Guide for the Care and Use of the Administration Committee of Experimental Animals of Wenzhou Medical University (Permit no. wydw2017-0074). All animals were acclimated in the above environment for 1 week before the experiment.
Preventive use of esomeprazole and establishment of stress ulcer models
The esomeprazole was provided by CHIA TAI TIANQING (CTTQ) Pharmaceutical (Lianyungang, Jiangsu, China). All substances administered to animals were dissolved in the normal saline. The rats were randomly divided into five groups of ten rats each as follows: control group (NS), water-immersed and restraint group (WIR), high-dose application of esomeprazole plus stress ulcer-induced group (HE+WIR), low-dose application of esomeprazole plus stress ulcer-induced group (LE+WIR) and high-dose application of esomeprazole without stress ulcer-induced group (HE). In HE+WIR group and HE group, animals were subjected to daily intraperitoneal injection of esomeprazole at a high dose (50 mg/kg/day), animals in LE+WIR group were subjected to the administration of esomeprazole at a low dose (10 mg/kg/day). The rats in NS group and WIR group received the same amount of normal saline. The doses of esomeprazole were selected based on preliminary experiments and drug instructions. The rats in the above groups received prophylactic administration for 7 days.
The rat stress ulcer model was achieved with a method called water-immersed and restraint (WIR).6 Briefly, the rats were fasted for 24 hrs but allowed free access to water before the experiment. Then, the rats for the WIR group, the WIR+HE group and the WIR+LE group were restrained and immersed in water to the depth of the xiphoid process at 21°C for 12 hrs to induce the stress ulcer. Then, these rats were taken out, unrestrained and gathered together with other groups (NS and HE) for the next step.
Assessment of gastric acid secretion
Determination of gastric acid was carried out based on the methods used in previous studies.14,33,34 After the WIR treatment for 12 hrs, all rats from five groups were rounded up and anesthetized with isoflurane, and then the rat’s abdomen was opened by midline laparotomy and the stomach was fully exposed, the pylorus was then ligated and the abdominal incision was temporarily closed. Two hours later, the incision was opened again, the lower end of the esophagus was ligated and the entire stomach was then separated. The gastric content was emptied, transferred into graduated centrifuge tubes and centrifuged for 10 mins (4°C, 3000× g). The pH of the supernatant obtained after centrifugation was measured and recorded with a pH meter (METTLER TOLEDO, Zurich, Switzerland)
Macroscopic assessment of the gastric mucosa
The assessment of gastric mucosal damage was scored according to previous research carried out by Guth.35 Briefly, the stomach contents and extravagated blood were rapidly washed out to obtain a legible view. Mucosal lesions were scored by an unaware investigator. Petechial lesions were scored as 1, injuries less than 1 mm were scored as 2, injuries ranged from 1 to 2 mm were scored as 3, injuries ranged from 2 to 4 mm were scored as 4 and injuries beyond 4 mm were scored as the actual length in mm.
Pathological examination of the gastric mucosa
The stomach tissues were fixed in 4% paraformaldehyde solution (Solarbio, Beijing, China) for 24 hrs, then dehydrated through ethanol (75–100%), followed by clearing with xylenes. Then, the tissues were embedded, sectioned and stained with H&E (Solarbio, Beijing, China). Specimen slices were observed under a light microscope at a magnification of either x100 or x400. Microscopic evaluation was carried out by a pathologist according to a previous research.36 Briefly, the pathological scores were evaluated in three aspects: mucosal necrosis, inflammatory cells infiltration and congestion. Every pathologic change was divided into six ranks (0–5 scores) independently based on the ratio of changed area relative to that of whole gastric mucosa (less than 5%, between 10% and 20%, between 20% and 40%, between 40% and 60%, between 60% and 80%, and more than 80%). The scores of mucosal necrosis, inflammatory cells infiltration and congestion were added up to total scores.
Evaluation of oxidative markers and MPO activity in the gastric tissue
Gastric tissues were weighed accurately and homogenized. After a centrifugation, the MDA, GSH, SOD and MPO in the supernatants were measured with the commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). The results were expressed as nmol/mgprot of tissue for MDA; μmol/gprot of tissue for GSH, U/mgprot of tissue for SOD and U/gprot of tissue for MPO.
ELISA assessment
Blood samples were allowed to stand at room temperature for 20 mins until they were naturally coagulated, and then they were centrifuged. Gastric tissues mixed with PBS were fully homogenized and centrifuged again. The supernatant of each sample was transferred to new and labeled centrifuge tubes. The ELISA kits (Shanghai Boyun Biotech, Shanghai, China) were used following the manufacturer’s instructions to detect the concentrations of TNF-α, IL-1β, PGE2, gastrin and pepsin in the serums or tissue homogenates.
Reverse-transcription quantitative polymerase chain reaction analysis (RT-qPCR)
Total RNA was extracted from the gastric tissues which were homogenized with TRIzol reagent (Sigma, Santa Clara, CA, USA) according to the instructions provided by the manufacturer. The levels of RNA were measured using Nanodrop 2000 ultraviolet-visible spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and all specimens were appropriately diluted to the identical concentration. Total RNA of each sample (2 µg) was reverse-transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). The mRNA expression was quantified by RT-qPCR on the ABI 7500 Sequence-Detection System using SYBR Green Real-time PCR Master Mix Plus (Applied Biosystems; Thermo Fisher scientific, Waltham, MA, USA). The cycling condition of RT-qPCR was set as follows: 1 cycle of pre-degeneration for 10 mins at 95°C, 40 amplification cycles of 15 s at 95°C and 60 s at 60°C. The expression levels of the samples were normalized to that of the internal control GAPDH gene as the reference gene. Results were calculated via 2−∆∆Cq analysis. All primers used in the experiment were synthesized by Sangon Biotech Company (Shanghai, China) and listed in Table 1.
 |
Table 1 Nucleotide sequence of the primers used for the real-time PCR assays |
Western blotting analysis
The gastric sample was first mixed in the phosphatase inhibitor cocktail (Beyotime, Shanghai, China), PMSF (Beyotime, Shanghai, China) and RIPA lysis buffer (Beyotime, Shanghai, China), and then placed in a homogenizer for homogenization under an ice-cold environment. After standing for 30 mins, all samples were centrifuged with a setting of 12,000 rpm for 15 mins at 4°C. The protein concentration was detected by the bicinchoninic acid protein assay kit (Beyotime, Shanghai, China). About 30 μg of the supernatant was loaded on the 12% protein gel and adequately separated with SDS-PAGE. The proteins were transferred to the PVDF membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked in the 5% skim milk for 2 hrs and washed in TBST for 3 times and then incubated individually overnight at 4°C, with primary rabbit antibodies against NF-κB p65 (1:1000, Abcam, Cambridge, UK); rabbit antibodies against phosphor-NF-κB p65 (1:500, Abcam, Cambridge, UK); rabbit antibodies against p38-MAPK (1:1000, CST, Boston, MA, USA); rabbit antibodies against phosphor-p38 MAPK (1:1000, CST, Boston, MA, USA); rabbit antibodies against GADPH (1:1000, Abcam, Cambridge, UK). After cleaning with the TBST, the membranes were incubated with a secondary antibody for 1 hr at room temperature. Immunoreactive bands were visualized using an enhanced chemiluminescence detection kit (Bio-Rad Laboratories, Inc. Hercules, CA, USA), and Image Lab software version 4.1 (Bio-Rad Laboratories, Inc. Hercules, CA, USA) was used for signal collection and densitometric image analysis.
Immunohistochemistry
Gastric samples were fixed in 4% paraformaldehyde and embedded in paraffin. The embedded samples were then deparaffinated and rehydrated. Antigens were carefully retrieved in a citrate buffer for 15 mins at 100°C. Endogenous peroxidases of the samples were quenched by incubation in 3% hydrogen peroxidase solution for 15 mins, and then the sample sections were blocked at 37°C for about 1 hr using 5% goat serum. Thereafter, the sample sections were rinsed in PBS buffer and incubated overnight at 4°C with primary antibodies of NF-κB p65 (1:1,000, Abcam, Cambridge, UK), phosphor-NF-κB p65 (1:50, Abcam, Cambridge, UK) and phosphor-p38 MAPK (1:100 CST, Boston, MA, USA). Then, all sample sections were incubated with the specific secondary antibody at ambient temperature for 30 mins. The sections were then rewashed, dyed by DAB-substrate-chromagen, counterstained with hematoxylin and dehydrated. The sections were observed and photographed with a biological imaging microscope (Olympus Corporation, Tokyo, Japan).
Statistical methods
Statistical analysis was performed using GraphPad Prism version 7.00 (GraphPad Software, Inc., San Diego, CA, USA). Data were presented as mean ± standard error of the mean (SEM). According to our pre-experiment and clinical experiences of treatment of peptic ulcer, the sample size was calculated using the formula: N=2[(a+b)2σ2]/(μ1–μ2).237 Data distribution was checked by the Shapiro– Wilk test. Independent sample t-test was used to compare the difference between the HE+WIR group and the LE+WIR group. The data of macroscopic assessment (lesion score) and pathological examination (pathological scores) among multiple groups were analyzed using Kruskal–Wallis nonparametric test followed by the Dunn’s multiple comparisons test. A one-way ANOVA followed by the Dunnett’s multiple comparisons test was used to compare the differences among multiple groups that data were normally distributed. The level of significance was set at P<0.05.
Results
Pretreatment with esomeprazole mitigates the gastric mucosal injury on macroscopic evaluations
As shown in Figure 1, the rats that received normal saline (the NS group) showed no visible gastric mucosal damage (Figure 1A); by contrast, the rats induced by WIR (the WIR group) had a wide range of mucosal damage (massive hemorrhage, severe edema, mucosal lesion) (Figure 1B). However, the pretreatment with esomeprazole at the dose of 50 mg/kg (the HE+WIR group) decreased the rats’ gastric lesions to an extent that was almost indistinguishable from that of the control group (Figure 1C). The rats treated with esomeprazole at the dose of 10 mg/kg (the LE+WIR group) also revived the mucosal lesion, but the mucosal edema, congestion and mucosal damage could still be found (Figure 1D). The gastric lesions of the rats that received the high dose of esomeprazole without stress ulcer (the HE group) displayed no difference compared with that of the normal group (Figure 1E). Guth’s lesion score was used to further assess the extent of mucosal damage (Figure 1F). WIR significantly increased the lesions with a score of 59.7±4.3 in the WIR group and 0.6±0.2 in the NS group (P<0.001). The pretreatment with esomeprazole at both high (50 mg/kg) and low (10 mg/kg) concentrations significantly decreased the mucosal lesions to lower scores of 5.7±0.8 for the HE+WIR group and 31.9±3.1 for the LE+WIR group from the score of 59.7±4.3 for the WIR group (P=0.0001<0.001).
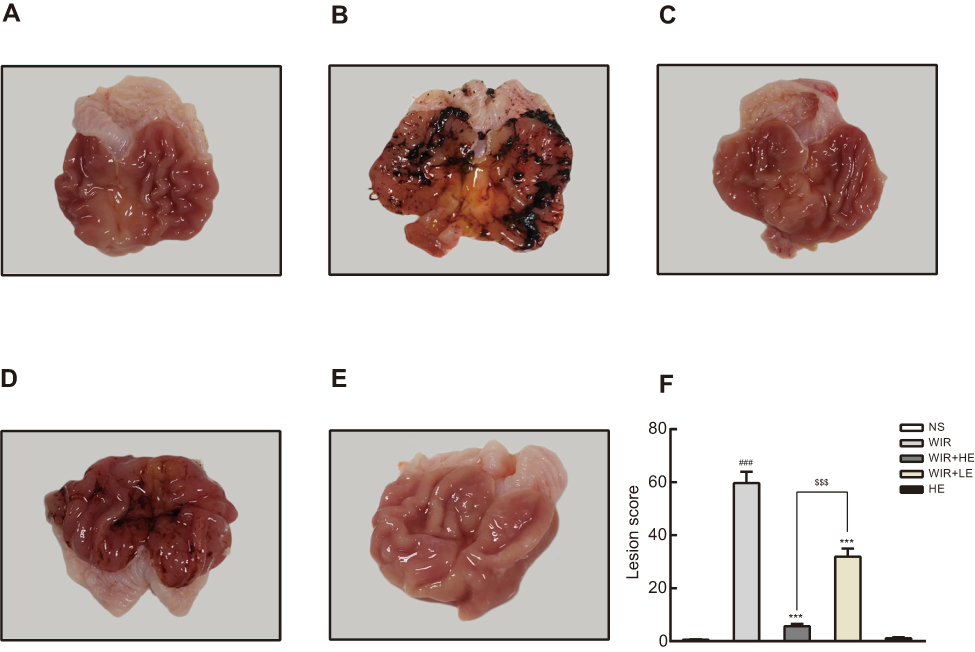 |
Figure 1 Macroscopic appearance of rat stomach. Notes: (A–E) Macroscopic images of the stomach. (F) Lesion score. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. ###P<0.001 was considered significantly different compared with the NS group; ***P<0.001 was considered significantly different compared with the WIR group; $$$P<0.001 was considered significantly different compared with the LE+WIR group. |
Pretreatment with esomeprazole mitigates the gastric mucosal injury on microscopic evaluations
More detailed examination under microscopic level showed that the WIR group displayed an extensive destruction of the gastric mucosa. According to the pathological score mentioned above, no obvious pathological changes were observed in the NS group (Figure 2A and D), while severe necrosis of mucosal epithelial cells, congestion and inflammatory cells infiltration were observed in the gastric mucosa epithelium in the WIR group, dilatating and congesting vessels and edema also existed in the submucosa of the WIR group, the WIR group and the NS group had the scores of 9.2±0.8 and 0.1±0.1, respectively (P=0.0001<0.001) (Figure 2B, E and K). Esomeprazole (50 mg/kg) markedly alleviated the gastric mucosal lesion, congestion and inflammatory cells infiltration with the sores of 1.2±0.3 and 9.2±0.8 for the HE+WIR group and the WIR group, respectively (P=0.0001<0.001) (Figure 2C, F and K). Esomeprazole (10 mg/kg) also alleviated these pathologic changes with the scores of 5.4±0.5 for the LE+WIR group and 9.2±0.8 for the WIR group (P=0.0001<0.001) (Figure 2G, I and K). Again, the high-dose pretreatment of esomeprazole was more effective than the low-dose pretreatment (1.2±0.3 in the HE+WIR versus 5.4±0.5 in the LE+WIR)(P=0.017<0.05) (Figure 2K). Using esomeprazole (50 mg/kg) in the HE group did not cause any changes in the mucosa (0.2±0.1 of the HE group versus 0.1±0.1 of the NS group) (P>0.99) (Figure 2H, J and K).
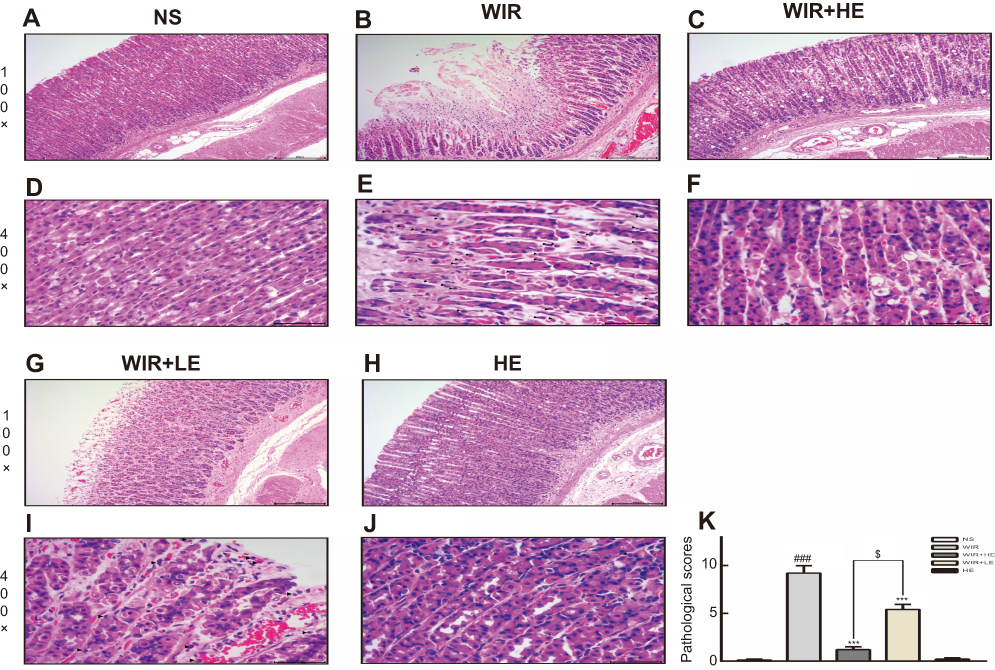 |
Figure 2 H&E-stained gastric cross-sections and microscopic changes of rat. Notes: (A–C, G, H) Microscopic images of the gastric mucosa, 100×. (D–F, I, J) Inflammatory cells among the gastric epithelium, 400×. ▲ labels inflammatory cells. (K) Pathological score. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. ###P<0.001 was considered significantly different between the WIR group and the NS group; ***P<0.001 was considered significantly different between the WIR group and either the HE-WIR group or the LE-WIR group; $P<0.05 was considered significantly different between the HE-WIR group and the LE+WIR group. |
Esomeprazole inhibits gastric secretion in stress ulcer
We further examined the effect of esomeprazole on acid secretion and the levels of gastrin and pepsin. As expected, the pH value of the NS group was relatively low compared with that of the WIR group (P=0.007<0.01). The pretreatment with esomeprazole significantly increased pH value in a dose-dependent manner (P=0.03<0.05) (Figure 3A; Table 2). Interestingly, the pH value of the HE group was higher than that of the NS group, suggesting that esomeprazole used at the high dose without stress ulcer could cause a significant inhibition of acid secretion (P=0.001<0.01) (Figure 3A; Table 2).
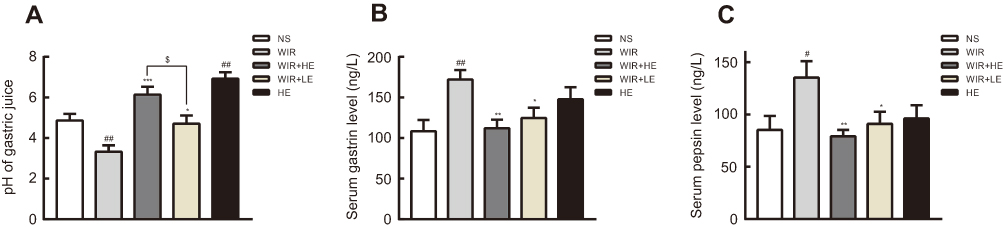 |
Figure 3 Effect of esomeprazole on pH, gastrin and pepsin in stress ulcer induced by WIR. Notes: (A) pH value of the gastric juice measured by the pH meter; (B) levels of serum gastrin analyzed by ELISA; (C) levels of serum pepsin analyzed by ELISA. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. #P<0.05, ##P<0.01 were considered significantly different compared with the NS group; *P<0.05, **P<0.01, ***P<0.001 were considered significantly different compared with the WIR group; $P<0.05 was considered significantly different. |
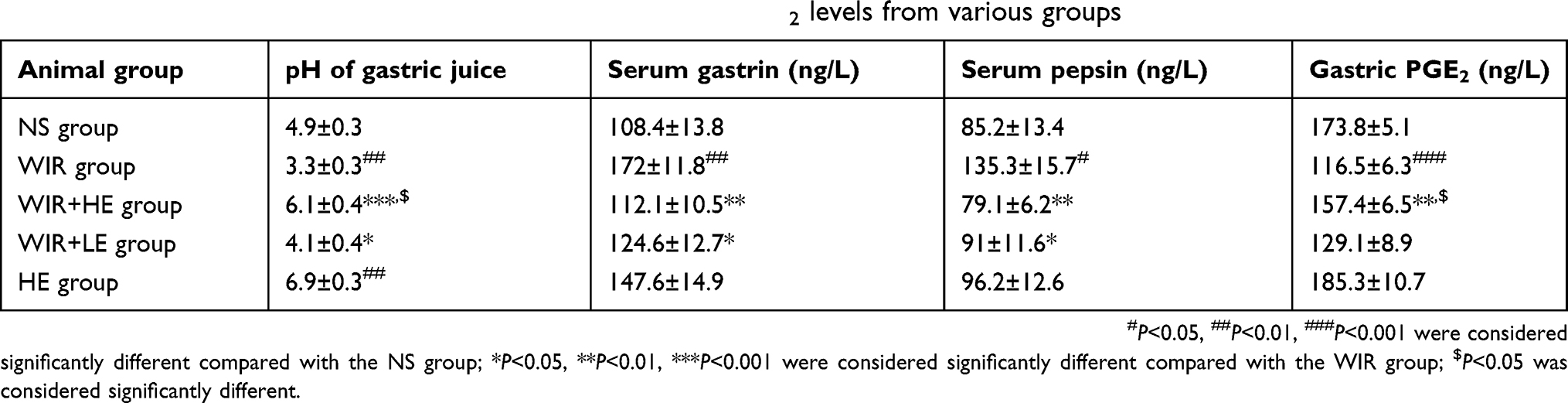 |
Table 2 Data of pH, serum gastrin, serum pepsin and gastric PGE2 levels from various groups |
The serum gastrin levels in the WIR group were elevated compared with that in the NS group (P=0.003<0.01) and decreased in the HE+WIR group (P=0.003<0.01) and in the LE+WIR group (P=0.047<0.05) by the administration of esomeprazole (Figure 3B; Table 2). Despite not statistically significant, it could be observed that the rats accepted the prophylactic administration of high-dose esomeprazole for 7 days in the HE group which had a tendency of increasing serum gastrin compared with that in the rats of the NS group (P=0.121>0.05). Analogously, serum pepsin was also enhanced in the WIR group compared to that in the NS group (P=0.043<0.05), and significantly reduced as a result of prophylactic administration of esomeprazole at high dose (P=0.009<0.01) and at low dose (P=0.042<0.05) (Figure 3C; Table 2).
Pretreatment with esomeprazole ameliorates the oxidative damage to the gastric tissue
We further detected the levels of MDA, GSH and SOD to assess the degree of oxidative stress damage in gastric tissue. Gastric mucosal levels of MDA in the WIR group were significantly elevated compared with that in the NS group (P=0.0001<0.001). The administration of esomeprazole in different doses significantly reduced the MDA levels in the HE+WIR group (P=0.0001<0.001) and the LE+WIR group (P=0.0001<0.001) (Figure 4A; Table 3). Compared with the NS group, the level of GSH in the WIR group was declined (P=0.012<0.05), which was rescued by the high-dose esomeprazole treatment (P=0.036<0.05) (Figure 4B; Table 3). The assessment revealed that the WIR treatment significantly reduced the levels of SOD relative to that of the NS group (P=0.025<0.05), and this condition was reversed by the prophylactic administration of the high-dose esomeprazole (P=0.048<0.05) (Figure 4C; Table 3). Interestingly, the expression of the SOD1 (Cu/Zn superoxide dismutase) gene displayed an opposite effect and was significantly upregulated by the WIR treatment (P=0.0001<0.001), and it was significantly downregulated following the high-dose esomeprazole treatment (P=0.0001<0.001) (Figure 4D). The rats in the HE group did not show any significant changes in the levels of MDA (P=0.963), GSH (P=0.980), SOD (P=0.875) and SOD1 (P=0.809) in the stomach tissue compared with the NS group rats (Figure 4A–D).
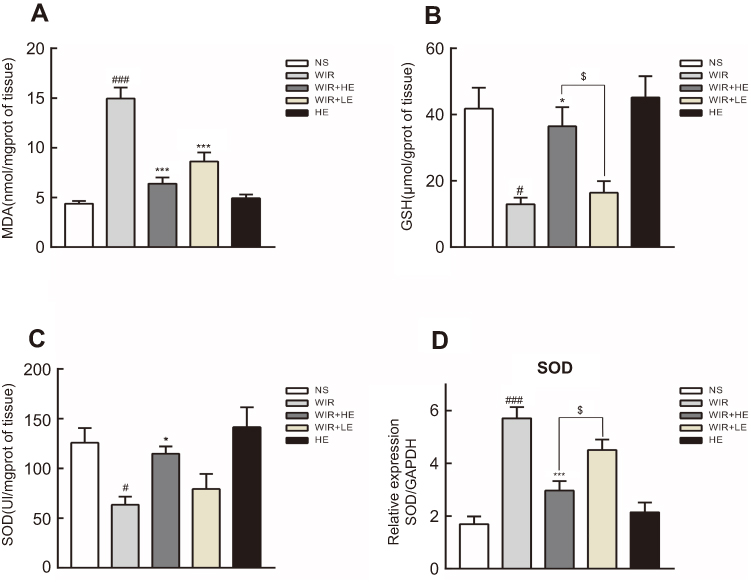 |
Figure 4 Effect of esomeprazole on oxidative stress markers in stress ulcer induced by WIR. Notes: (A–C) Levels of MDA, GSH and SOD in the gastric tissue were measured. (D) Total mRNA was extracted from colonic tissue and reversely transcribed into cDNA. The mRNA expressions of SOD1 gene were detected by RT-qPCR and normalized with GAPDH mRNA levels. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. #P<0.05, ###P<0.001 were considered significantly different compared with the NS group; *P<0.05, ***P<0.001 were considered significantly different compared with the WIR group; $P<0.05 was considered significantly different compared with the LE+WIR group. Abbreviations: MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. |
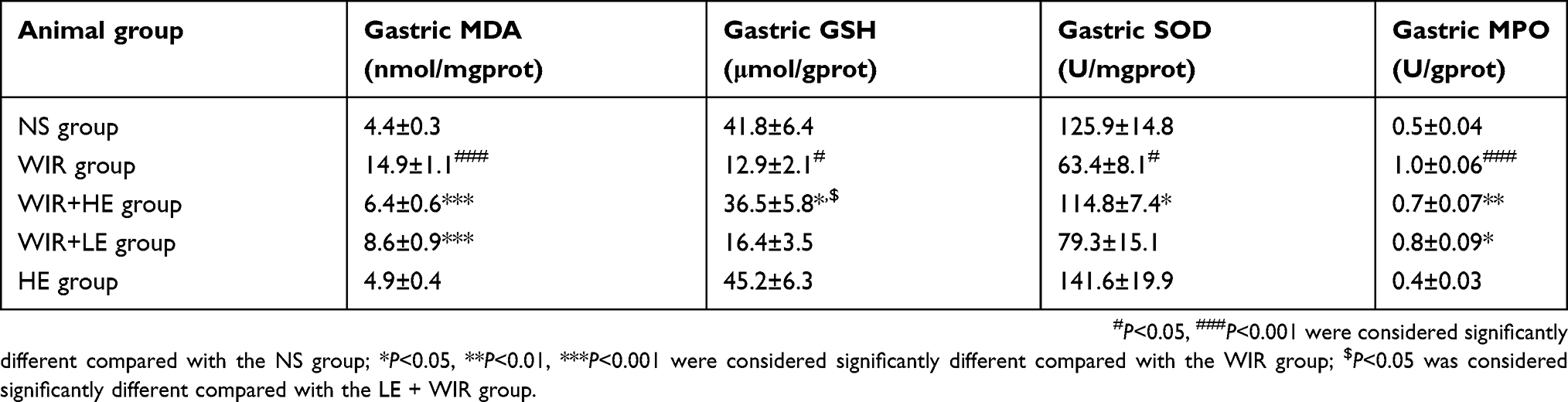 |
Table 3 Data of gastric MDA, gastric GSH, gastric SOD and gastric MPO levels from various groups |
Prophylactic application of esomeprazole alleviates the inflammatory damage in stress ulcer
Our ELISA results showed that the expressions of TNF-α and IL-1β in serums (Figure 5A and B; Table 4) and in gastric tissues (Figure 5C and D; Table 4) were elevated following WIR stress and significantly suppressed with the prophylactic application of the high-dose esomeprazole (Figure 5C and D; Table 4). Although not as effective as the high-dose esomeprazole, the low-dose esomeprazole also partially inhibited the TNF-α and IL-1β expression in gastric tissues (Figure 5C and D; Table 4). To confirm our results further, we assessed the mRNA expression levels of the pro-inflammatory factor in gastric homogenates by RT-qPCR. The expressions of TNF-α and IL-1β genes in gastric tissues were significantly upregulated due to the treatment of WIR (P<0.001) relative to those of the NS group and downregulated following the esomeprazole treatments at different doses compared with those of the WIR group (Figure 5E and F). MPO in gastric tissues was elevated following WIR stress (P=0.0001<0.001), and significantly suppressed with the prophylactic application of the high-dose esomeprazole (P=0.0012<0.01) and the low-dose esomeprazole (P=0.042<0.05) (Figure 6A, Table 3). Application of esomeprazole to the rats of the HE group did not significantly affect inflammatory factor levels. It was noted that the gastric mucosal PGE2 level was considerably reduced in the WIR group compared with that in the NS group (P=0.0001<0.001), and the high-dose esomeprazole pretreatment significantly increased the PGE2 level (P=0.002<0.01) (Figure 6B, Table 2).
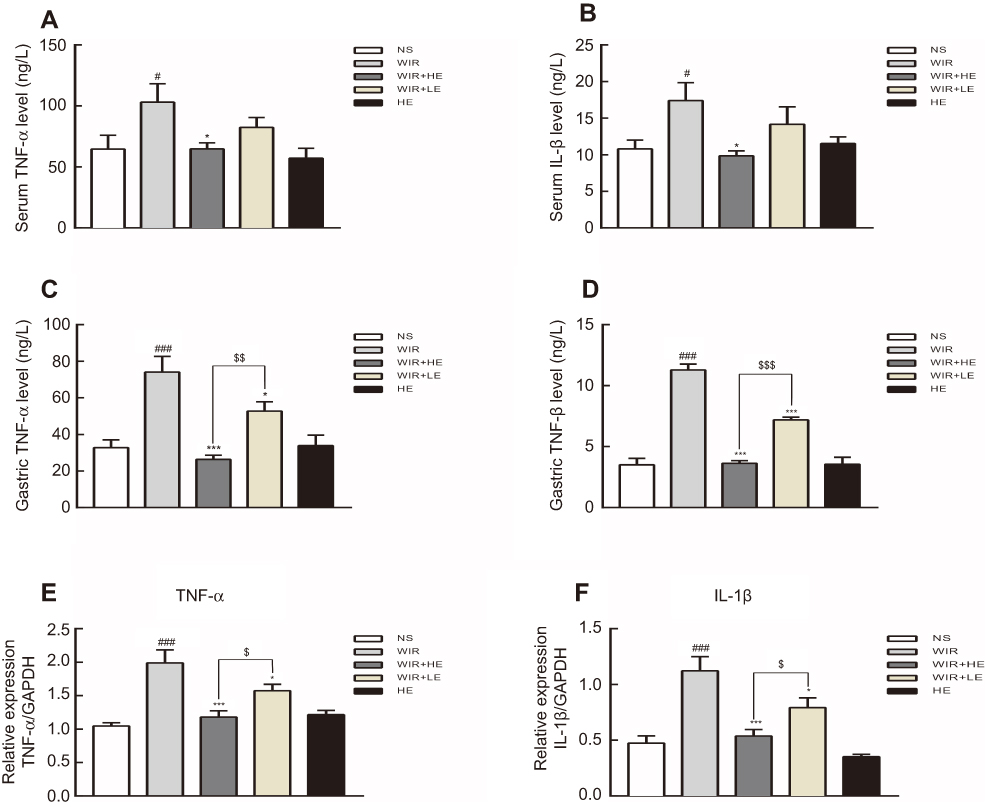 |
Figure 5 Effect of esomeprazole on pro-inflammatory cytokines levels after induction of stress ulcer induced by WIR. Notes: (A and B) Levels of TNF-α and IL-1β in serum detected by ELISA. (C and D) Levels of TNF-α and IL-1β in gastric tissues detected by ELISA. (E and F) Total mRNA was extracted from gastric tissue and reversely transcribed into cDNA. The mRNA expressions of TNF-α and IL-1β genes were detected by RT-qPCR and normalized with GAPDH mRNA levels. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. #P<0.05, ###P<0.001 were considered significantly different compared with NS group; *P<0.05, ***P<0.001 were considered significantly different compared with WIR group; $P<0.05, $$P<0.01, $$$P<0.001 were considered significantly different compared with LE+WIR group.Abbreviations: TNF-α, tumor necrosis factor α; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. |
 |
Table 4 Data of pro-inflammatory cytokines levels detected by ELISA from various groups |
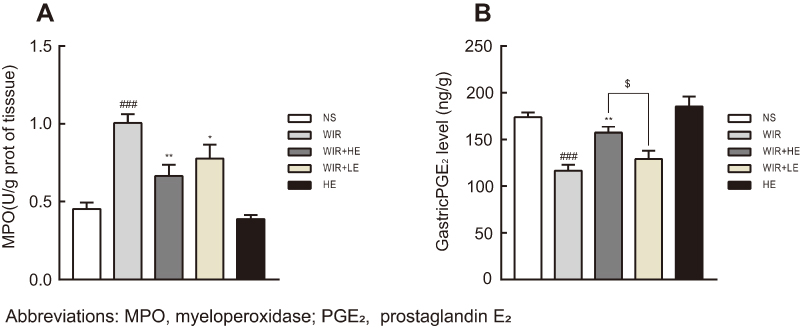 |
Figure 6 Effect of esomeprazole pre-treatment on MPO and PGE2 levels after induction of stress ulcer induced by WIR. Notes: (A) Level of MPO in the gastric tissue. (B) Level of PGE2 in the gastric tissue. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. ###P<0.001 was considered significantly different compared with the NS group; *P<0.05, **P<0.01 were considered significantly different compared with the WIR group; $P<0.05 was considered significantly different compared with the LE+WIR group. |
Pretreatment of esomeprazole inhibits NF-κB and p38 MAPK signaling pathways in stress ulcer
To explore antioxidative and anti-inflammatory benefits of esomeprazole pretreatment in signaling pathways, we measured the phosphorylation of NF-κB p65 and p38 MAPK by the Western blotting (Figure 7A). The phosphorylation of NF-κB p65 was dramatically increased in the WIR-induced stress ulcer and significantly decreased with the pretreatment of esomeprazole in a dose-dependent way (WIR versus NS, P=0.0002<0.001, HE+WIR versus WIR, P=0.0001<0.001, HE+WIR versus LE+WIR, P=0.012<0.05, Figure 7B). The phosphorylation levels of p38 MAPK were significantly elevated in the WIR group and dose-dependently suppressed by esomeprazole (WIR versus NS, P=0.0001<0.001, HE+WIR versus WIR, P=0.0001<0.001, LE+WIR versus WIR, P=0.0001<0.001, HE+WIR versus LE+WIR, P=0.046<0.05, Figure 7C). Immunohistochemistry was consistent with Western blotting results, and the phosphorylation of p38 MAPK and NF-κB p65 in the NS group and the HE group showed little or no positive expression, while they were dramatically increased in the gastric epithelial cells in the WIR group. Pretreatment with esomeprazole dose-dependently reduced the phosphorylation of p38 MAPK and NF-κB p65 in a gastric epithelial cell in stress ulcer models (Figure 8A and B). In addition, the immunohistochemistry results on the nuclear translocation of NF-κB p65 showed that NF-κB p65 proteins were expressed at a low level and equally localized in the cytoplasm of gastric epithelial cells in the NS and HE groups (Figure 8C). The nuclear translocation of NF-κB p65 was observed in some gastric epithelial cells in the WIR group, and the prophylactic usage of esomeprazole dose-dependently blocked the nuclear translocation.
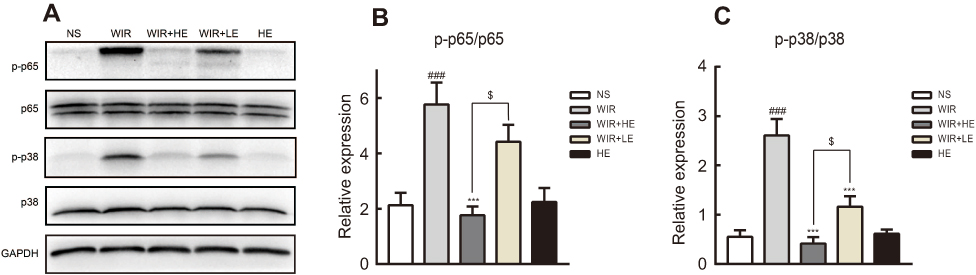 |
Figure 7 Effect of esomeprazole pretreatment on p38-MAPK and NF-κB activity after induction of stress ulcer induced by WIR. Notes: (A) The proteins were extracted from gastric tissue and analyzed by Western blotting. GAPDH levels were used as internal controls. The mean density values of targeted protein were expressed as a ratio relative to that of GAPDH. (B) Relative protein level of p-p65/p65. (C) Relative protein level of p-p38/p38. The data are expressed as the mean ± SEM of ten animals per group and at least three independent experiments. ###P<0.001 was considered significantly different compared with the NS group; ***P<0.001 was considered significantly different compared with the WIR group; $P<0.05 was considered significantly different compared with the LE+WIR group.Abbreviations: p38-MAPK, mitogen-activated protein kinase p38; NF-κB, nuclear factor kappa B; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. |
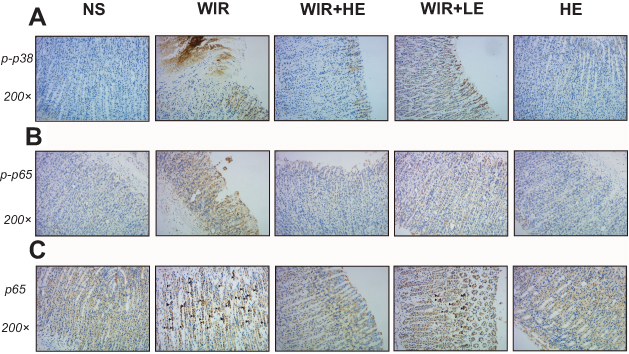 |
Figure 8 Representative pictures showing the immunohistochemical analysis of p-p38, p-p65 and NF-κB p65 in sections of gastric mucosa obtained from rats. Notes: (A and B) The immunohistochemical staining of p-p38 and p-p65 in the gastric tissues (magnification, 200×) in different groups, brown staining denotes positive expression. (C) The immunohistochemical detection of the nuclear translocation of p65 subunit in gastric tissues, ▲ labels epithelial cells with hyperchromatic area around nucleus (magnification, 200×) in different groups. Abbreviations: p-p38, phosphorylated p38; p-p65, phosphorylated p65; NF-κB, nuclear factor kappa B. |
Discussion
The development of stress ulcers in rats subjected to WIR stress is related to vagal stimulation of gastrin release, followed by increased acid secretion of gastric juice.38 The prophylactic use of PPIs is an indispensable treatment in the clinical practice because of their puissant ability of acid inhibition3,39–41 by the back diffusion of hydrogen ions into the mucosa.42
In our study, the pH of the rat stomach in the WIR group was significantly reduced during the progression of stress ulcers and was dose-dependently reversed by the pretreatments with esomeprazole due to its vigorous acid suppression ability (Figure 3A). Our study also showed that prophylactic application of esomeprazole decreased the secretion of pepsin (Figure 3C) in agreement with previous studies.43 We speculate that the decrease of pepsin levels may be partially associated with the inhibition of acid caused by esomeprazole.
Some studies pointed out that esomeprazole can be used to suppress gastric acid output which can reflectively elevate the level of gastrin, and that hypergastrinemia can be considered as a risk factor for gastric carcinoid and cancer.44–46 However, it needs a very long time of administration with a large dose of PPI. In our study, we found that the level of gastrin was significantly elevated after stress and inhibited by pretreatment of esomeprazole. Nevertheless, we noticed that, although it was not significant, the gastrin level in the HE group mildly increased, consistent with the observations of previous research.47–50 Thus, it is possible that esomeprazole could increase gastrin slightly when it is used alone, but it may cause an opposite effect to reduce gastrin level under stress ulcers. Besides, esomeprazole is given acutely in our study, so that it may not bring any visible increase in gastrin levels in the given time. The increase of gastrin level that is feedback-induced by the low acid environment may not surpass the potent inhibitory effect of esomeprazole. It has also been reported that the elevated gastrin level caused by PPIs will return to normal soon after stopping the drug,49 and so a short-term use of esomeprazole is relatively safe. As the secretion of gastrin is inhibited, the amount of gastric acid secretion is also reduced, and the degree of damage in gastric mucosa is relieved too. PGs like PGE2 can increase mucosal blood flow and even inhibit acid secretion.5 Our research showed that pretreatment of esomeprazole dose-dependently elevated the decline of PEG2 level affected by WIR, which indicates that esomeprazole may be able to achieve protective effects by increasing mucosal blood flow and inhibiting acid secretion through PGE2. In short, our study demonstrates that esomeprazole exerts an anti-ulcer effect by inhibiting several damage factors in gastric juice.
Accumulated evidence indicate that ROS plays a very important role in the development of stress ulcers6 and may directly damage tissues or induce some downstream signaling pathways to mediate inflammatory damage to the tissues. Various studies show that esomeprazole has the capacity of antioxidation which is independent of acid inhibition.14,34,51,52 Our results demonstrated that esomeprazole can have an antioxidant effect. Indeed, we observed that stress ulcer elevated MDA production and decreased SOD and GSH levels in gastric mucosa. These changes were notably reversed by prophylactic use of esomeprazole, and the high dose of esomeprazole had a more evident effect. The antioxidant mechanism of esomeprazole may be explained by its conversion into a tetracyclic sulfenamide with the help of gastric acid and its sulfhydryl compounds performed as antioxidants.53 Unexpectedly, from the results of our RT-qPCR, the expression of SOD1 gene increased in the WIR groups but decreased in the groups that were given esomeprazole. It is possible that stress ulcers produce many ROS and consume endogenous antioxidants such as SOD and GSH, which enhances the transcription of SOD1 gene for compensation, and the application of esomeprazole could minimize the need for ROS detoxification by reducing the gene expression of antioxidant, including SOD1.
It has been indicated that PPIs have anti-inflammatory effects, which is independent of the inhibition of gastric acid production.31,54 Consistent with these studies, our study showed that pretreatment with esomeprazole reduced the expression of TNF-α and IL-1β at both transcription and protein levels, and esomeprazole also reduced the gastric MPO level induced by WIR treatment. These results were further confirmed by the reduction of inflammatory cell infiltration and gastric mucosal damage observed under the microscope, indicating that esomeprazole also exerts an anti-inflammatory effect. Our results also showed a dose-dependent effect of esomeprazole in gastric tissue but not in serum, suggesting that the changes of inflammatory factors in gastric tissues under stress ulcers are more representative than in serum in agreement of the view that the gastric epithelium itself also participates in the inflammatory response.7 Besides, the pretreatments of esomeprazole in normal individuals did not affect the inflammatory response.
Although it is traditionally believed that the activation of NF-κB is dependent on the degradation of IκB and the nuclear translocation of NF-κB p65, a growing number of recent studies indicates that post-translational modifications of NF-κB, especially the phosphorylation of NF-κB p65 in serine 276 (Ser 276), are also an essential step in maximizing NF-κB transcriptional activity.17,19,55,56 Research also revealed that ROS can enhance the Ser 276 phosphorylation of NF-κB p65 through cAMP-dependent protein kinase A.57 PPIs blocking NF-κB pathway have been found in some in vitro studies and other disease models.32,58,59 In the present study, NF-κB pathway in the WIR group was significantly activated as expected, and the pretreatment of esomeprazole not only reduced the nuclear translocation of NF-κB p65 subunit but also decreased the formation of the phosphorylated NF-κB p65 subunit. The high-dose pretreatment of esomeprazole enhanced these effects. In short, our study demonstrated for the first time that esomeprazole can dose-dependently inhibit the nuclear translocation and phosphorylation of the NF-κB p65 subunit to reduce the damage caused by stress ulcer. The antioxidation effects of esomeprazole either directly or indirectly inhibit the NF-κB pathway, leading to the reduction of the TNF-α and IL-1β levels and the inhibition of their feedback-induced activations of ROS and NF-κB pathways, and finally reduce the damage caused by the stress ulcers.
It is worth mentioning that some in vitro research showed that low pH can activate the NF-κB pathway.60 So we presume that, under normal circumstances, the mucus bicarbonate barrier works well to prevent gastric epithelial cells from the interference of gastric acid. When stress ulcers occur, ischemia and hypoxia make the barrier weaker, along with the acid back-diffusion, thus causing a dramatic decrease in pH around the gastric epithelial cells, thereby activating the NF-κB pathway in epithelial cells. The high pH environment caused by the prophylactic use of esomeprazole can limit the activation of NF-κB pathway.
Recently, p38 MAPK has been shown to be closely related to the NF-κB pathway activation as a vital upstream event. For instance, phosphorylation of p38 MAPK is responsible for the phosphorylation of IκBα, resulting in its dissociation from the NF-κB p65 subunit, thus facilitating the nuclear translocation of NF-κB p65 subunit.61 Jia Yitao7 pretreated the rats with a selective p38 MAPK inhibitor (SB 239063) and found the phosphorylation in both IκB and p65 subunits decreased, suggesting that p38 MAPK may be involved in the duration of ROS-dependent NF-κB activation under the development of stress ulcer. A study in skeletal myoblasts also revealed that under oxidative stress, activated p38 MAPK can phosphorylate the serine 276 residue (Ser 276) of NF-κB p65, thereby participating in post-transcriptional modification of NF-κB p65 and maximally activating the NF-κB pathway.56 There were some in vitro experiments showing that PPIs could reduce phosphorylation of p38 MAPK, thus inhibiting the p38 MAPK pathway.62 In the present study, the Western blotting and immunohistochemistry experiments revealed that the esomeprazole pretreatments significantly attenuated the high phosphorylation levels of p38 MAPK in gastric epithelium induced by WIR, implying that esomeprazole could dose-dependently suppress p38 MAPK signaling pathways in stress ulcer, thereby interfering the activation of downstream NF-κB signaling pathway and the expression of pro-inflammatory factors. Taken together, we concluded that the inhibition of the NF-κB pathway by esomeprazole is multifaceted through the p38 MAPK pathway.
In summary, we presume that the antioxidative effect of esomeprazole may inactivate the p38 MAPK pathway, which weakens the phosphorylation of NF-κB p65, leading to a downregulated expression of TNF-α and IL-1β, simultaneously interfering the positive feedback of TNF-α and IL-1β to the ROS signaling pathways with the production of ROS and the cross-regulation of upstream signaling pathways.
Conclusion
Our results indicate that, in addition to invasive factors such as gastric acid and pepsin, inflammation damages caused by ROS relate to a cascade of activations among p38 MAPK and NF-κB signaling pathways which play a very important role in the pathophysiological insults of stress ulcer. The positive feedback loop between the ROS signaling pathways and proinflammatory factors aggravates mucosal lesions. The pretreatments with esomeprazole exert a protective effect on the rats with WIR-induced stress ulcer not only through an anti-secretory mechanism but also through an anti-inflammatory and antioxidative effects by dose-dependently inactivating the p38 MAPK and NF-κB signaling pathways. This will contribute to the application of esomeprazole in stress ulcer. However, a better understanding of the mechanisms involved in the regulatory network of oxidative inflammatory damage in stress ulcer and the more effective using of esomeprazole in patients with stress ulcer remains to be achieved.
Acknowledgments
CHIA TAI TIANQING (CTTQ) Pharmaceutical provided esomeprazole. This work was supported by Grants from National Natural Science Foundation of China (grant no. 81570495).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bardou M, Quenot JP, Barkun A. Stress-related mucosal disease in the critically ill patient. Nat Rev Gastroenterol Hepatol. 2015;12(2):98–107. doi:10.1038/nrgastro.2014.235
2. Monnig AA, Prittie JE. A review of stress-related mucosal disease. J Vet Emerg Crit Car. 2011;21(5):484–495. doi:10.1111/j.1476-4431.2011.00680.x
3. Spirt MJ. Stress-related mucosal disease: risk factors and prophylactic therapy. Clin Ther. 2004;26(2):197–213.
4. Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med. 2013;41(3):693–705. doi:10.1097/CCM.0b013e3182758734
5. Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135(1):41–60. doi:10.1053/j.gastro.2008.05.030
6. Jia YT, Ma B, Wei W, et al. Sustained activation of nuclear factor-kappaB by reactive oxygen species is involved in the pathogenesis of stress-induced gastric damage in rats. Crit Care Med. 2007;35(6):1582–1591. doi:10.1097/01.CCM.0000266824.82280.17
7. Jia YT, Wei W, Ma B, et al. Activation of p38 MAPK by reactive oxygen species is essential in a rat model of stress-induced gastric mucosal injury. J Immunol. 2007;179(11):7808–7819. doi:10.4049/jimmunol.179.11.7808
8. Liu X, Chen Z, Mao N, Xie Y. The protective of hydrogen on stress-induced gastric ulceration. Int Immunopharmacol. 2012;13(2):197–203. doi:10.1016/j.intimp.2012.04.004
9. Akanda MR, Park BY. Involvement of MAPK/NF-kappaB signal transduction pathways: camellia japonica mitigates inflammation and gastric ulcer. Biomed Pharmacother. 2017;95:1139–1146. doi:10.1016/j.biopha.2017.09.031
10. Morsy MA, Heeba GH, Abdelwahab SA, Rofaeil RR. Protective effects of nebivolol against cold restraint stress-induced gastric ulcer in rats: role of NO, HO-1, and COX-1,2. Nitric Oxide. 2012;27(2):117–122. doi:10.1016/j.niox.2012.06.001
11. Elshazly SM, Abd El Motteleb DM, Ibrahim I. Hesperidin protects against stress induced gastric ulcer through regulation of peroxisome proliferator activator receptor gamma in diabetic rats. Chem Biol Interact. 2018;291:153–161. doi:10.1016/j.cbi.2018.06.027
12. Shian WM, Sasaki I, Kamiyama Y, Naito H, Matsuno S, Miyazawa T. The role of lipid peroxidation on gastric mucosal lesions induced by water-immersion-restraint stress in rats. Surg Today. 2000;30(1):49–53. doi:10.1007/PL00010046
13. Adamczyk-Sowa M, Sowa P, Pierzchala K, Polaniak R, Labuz-Roszak B. Antioxidative enzymes activity and malondialdehyde concentration during mitoxantrone therapy in multiple sclerosis patients. J Physiol Pharmacol. 2012;63(6):683–690.
14. Fornai M, Colucci R, Antonioli L, et al. Effects of esomeprazole on healing of nonsteroidal anti-inflammatory drug (NSAID)-induced gastric ulcers in the presence of a continued NSAID treatment: characterization of molecular mechanisms. Pharmacol Res. 2011;63(1):59–67. doi:10.1016/j.phrs.2010.10.013
15. Jia YT, Wei D, Chen XL, Xia ZF. [Activation of nuclear factor-kappaB signaling pathway in rat gastric mucosa during stress ulceration]. Zhonghua Shao Shang Za Zhi. 2006;22(5):351–354.
16. Ramiro-Puig E, Casadesus G, Lee HG, et al. Neuroprotective effect of cocoa flavonoids on in vitro oxidative stress. Eur J Nutr. 2009;48(1):54–61. doi:10.1007/s00394-008-0761-4
17. Zhang J, Wang X, Vikash V, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. 2016;2016:4350965. doi:10.1155/2016/4350965
18. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. doi:10.1038/nri3423
19. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
20. Yeo D, Hwang SJ, Kim WJ, Youn HJ, Lee HJ. The aqueous extract from Artemisia capillaris inhibits acute gastric mucosal injury by inhibition of ROS and NF-kB. Biomed Pharmacother. 2018;99:681–687. doi:10.1016/j.biopha.2018.01.118
21. Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10(4):693–695.
22. Oeckinghaus A, Hayden M, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi:10.1038/ni.2065
23. Ciuffa R, Caron E, Leitner A, Uliana F, Gstaiger M, Aebersold R. Contribution of mass spectrometry-based proteomics to the understanding of TNF-alpha signaling. J Proteome Res. 2017;16(1):14–33. doi:10.1021/acs.jproteome.6b00728
24. Hamaguchi M, Watanabe T, Higuchi K, Tominaga K, Fujiwara Y, Arakawa T. Mechanisms and roles of neutrophil infiltration in stress-induced gastric injury in rats. Dig Dis Sci. 2001;46(12):2708–2715. doi:10.1023/A:1012779530004
25. Roberge S, Roussel J, Andersson D, et al. TNF-α-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc Res. 2014;103(1):90–99. doi:10.1093/cvr/cvu112
26. Clauzure M, Valdivieso AG, Massip Copiz MM, Schulman G, Teiber ML, Santa-Coloma TA. Disruption of interleukin-1beta autocrine signaling rescues complex I activity and improves ROS levels in immortalized epithelial cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PLoS One. 2014;9(6):e99257. doi:10.1371/journal.pone.0099257
27. Li W, Wang X, Zhi W, et al. The gastroprotective effect of nobiletin against ethanol-induced acute gastric lesions in mice: impact on oxidative stress and inflammation. Immunopharmacol Immunotoxicol. 2017;39(6):354–363. doi:10.1080/08923973.2017.1379088
28. Al-Quraishy S, Othman MS, Dkhil MA, Abdel Moneim AE. Olive (Olea europaea) leaf methanolic extract prevents HCl/ethanol-induced gastritis in rats by attenuating inflammation and augmenting antioxidant enzyme activities. Biomed Pharmacother. 2017;91:338–349. doi:10.1016/j.biopha.2017.04.069
29. Saccar CL. The pharmacology of esomeprazole and its role in gastric acid related diseases. Expert Opin Drug Metab Toxicol. 2009;5(9):1113–1124. doi:10.1517/17425250903124363
30. Peng YC, Huang LR, Shyu CL, Cheng CC, Ho SP. Interaction of omeprazole and Helicobacter pylori-induced nuclear factor-kappaB activation and mediators in gastric epithelial cells. J Chin Med Assoc. 2014;77(11):567–572. doi:10.1016/j.jcma.2014.07.006
31. Tanigawa T, Watanabe T, Higuchi K, et al. Lansoprazole, a proton pump inhibitor, suppresses production of tumor necrosis factor-alpha and interleukin-1beta induced by lipopolysaccharide and helicobacter pylori bacterial components in human monocytic cells via inhibition of activation of nuclear factor-kappaB and extracellular signal-regulated kinase. J Clin Biochem Nutr. 2009;45(1):86–92. doi:10.3164/jcbn.08-267
32. Huo X, Zhang X, Yu C, et al. In oesophageal squamous cells exposed to acidic bile salt medium, omeprazole inhibits IL-8 expression through effects on nuclear factor-kappaB and activator protein-1. Gut. 2014;63(7):1042–1052. doi:10.1136/gutjnl-2013-305533
33. Fornai M, Natale G, Colucci R, et al. Mechanisms of protection by pantoprazole against NSAID-induced gastric mucosal damage. Naunyn Schmiedebergs Arch Pharmacol. 2005;372(1):79–87. doi:10.1007/s00210-005-1075-1
34. Colucci R, Fornai M, Antonioli L, et al. Characterization of mechanisms underlying the effects of esomeprazole on the impairment of gastric ulcer healing with addition of NSAID treatment. Dig Liver Dis. 2009;41(6):395–405. doi:10.1016/j.dld.2008.10.004
35. Guth PH, Paulsen G. Aspirin-induced gastric injury in the rat: histologic changes and sucralfate cytoprotection. Proc Soc Exp Biol Med. 1987;184(4):423–428.
36. Fu Y, Wu HQ, Cui HL, Li YY, Li CZ. Gastroprotective and anti-ulcer effects of oxymatrine against several gastric ulcer models in rats: possible roles of antioxidant, antiinflammatory, and prosurvival mechanisms. Phytother Res. 2018. doi:10.1002/ptr.6148
37. Noordzij M, Dekker FW, Zoccali C, Jager KJ. Sample size calculations. Nephron Clin Pract. 2011;118(4):c319–323. doi:10.1159/000322830
38. Debas HT, Carvajal SH. Vagal regulation of acid secretion and gastrin release. Yale J Biol Med. 1994;67(3–4):145–151.
39. Schubert ML. Physiologic, pathophysiologic, and pharmacologic regulation of gastric acid secretion. Curr Opin Gastroenterol. 2017;33(6):430-438.
40. Jedynak M, Siemiatkowski A, Jasiewicz P. [Stress related mucosal disease prophylaxis in intensive care unit patients]. Pol Merkur Lekarski. 2008;24(139):48–53.
41. Sesler JM. Stress-related mucosal disease in the intensive care unit: an update on prophylaxis. AACN Adv Crit Care. 2007;18(2):119–126; quiz 127-118. doi:10.1097/01.AACN.0000269254.39967.8e
42. Frank WO. Gastric acid secretion and mucosal defense mechanisms with special reference to the role of cimetidine in critically ill patients. Clin Ther. 1986;8 Suppl A:2–13.
43. Thippeswamy A, Sajjan M, Palkar M, Koti B, Viswanathaswamy A. Comparative study of proton pump inhibitors on dexamethasone plus pylorus ligation induced ulcer model in rats. Indian J Pharm Sci. 2010;72(3):367–371. doi:10.4103/0250-474X.70486
44. Nørsett K, Laegreid A, Kusnierczyk W, et al. Changes in gene expression of gastric mucosa during therapeutic acid inhibition. Eur J Gastroenterol Hepatol. 2008;20(7):613–623. doi:10.1097/MEG.0b013e3282f5dc19
45. Havu N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion. 1986;35(Suppl 1):42–55. doi:10.1159/000199381
46. Larsson H, Håkanson R, Mattsson H, Ryberg B, Sundler F, Carlsson E. Omeprazole: its influence on gastric acid secretion, gastrin and ECL cells. Toxicol Pathol. 1988;16(2):267–272. doi:10.1177/019262338801600220
47. Zhang SL, Li H, He X, et al. Alkaloids from Mahonia bealei posses anti-H(+)/K(+)-ATPase and anti-gastrin effects on pyloric ligation-induced gastric ulcer in rats. Phytomedicine. 2014;21(11):1356–1363. doi:10.1016/j.phymed.2014.07.007
48. de Almeida AB, Luiz-Ferreira A, Cola M, et al. Anti-ulcerogenic mechanisms of the sesquiterpene lactone onopordopicrin-enriched fraction from Arctium lappa L. (Asteraceae): role of somatostatin, gastrin, and endogenous sulfhydryls and nitric oxide. J Med Food. 2012;15(4):378–383. doi:10.1089/jmf.2011.0025
49. Parente N, Bari Olivier N, Refsal K, Johnson C. Serum concentrations of gastrin after famotidine and omeprazole administration to dogs. J Vet Intern Med. 2014;28(5):1465–1470. doi:10.1111/jvim.12408
50. Singh N, Singh P, Shrivastva S, et al. Gastroprotective effect of anti-cancer compound rohitukine: possible role of gastrin antagonism and H(+) K (+)-ATPase inhibition. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(3):277–286. doi:10.1007/s00210-011-0711-1
51. Koch TR, Petro A, Darrabie M, Opara EC. Effect of the H, K-ATPase inhibitor, esomeprazole magnesium, on gut total antioxidant capacity in mice. J Nutr Biochem. 2004;15(9):522–526. doi:10.1016/j.jnutbio.2004.03.003
52. Biswas K, Bandyopadhyay U, Chattopadhyay I, Varadaraj A, Ali E, Banerjee RK. A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J Biol Chem. 2003;278(13):10993–11001. doi:10.1074/jbc.M210328200
53. Pastoris O, Verri M, Boschi F, et al. Effects of esomeprazole on glutathione levels and mitochondrial oxidative phosphorylation in the gastric mucosa of rats treated with indomethacin. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(4):421–429. doi:10.1007/s00210-008-0314-7
54. Balza E, Piccioli P, Carta S, et al. Proton pump inhibitors protect mice from acute systemic inflammation and induce long-term cross-tolerance. Cell Death Dis. 2016;7:e2304. doi:10.1038/cddis.2016.218
55. Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5(5):392–401. doi:10.1038/nrm1368
56. Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18(12):2238–2251. doi:10.1016/j.cellsig.2006.05.004
57. Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal. 2007;19(7):1419–1433. doi:10.1016/j.cellsig.2007.01.020
58. Handa O, Yoshida N, Fujita N, et al. Molecular mechanisms involved in anti-inflammatory effects of proton pump inhibitors. Inflamm Res. 2006;55(11):476–480. doi:10.1007/s00011-006-6056-4
59. Song J, Seo C, Kim T, et al. Protective effects of manassantin A against ethanol-induced gastric injury in rats. Biol Pharm Bull. 2016;39(2):221–229. doi:10.1248/bpb.b15-00642
60. O’Toole D, Abdel-Latif MM, Long A, et al. Low pH and Helicobacter pylori increase nuclear factor kappa B binding in gastric epithelial cells: a common pathway for epithelial cell injury? J Cell Biochem. 2005;96(3):589–598. doi:10.1002/jcb.20539
61. Calleros L, Lasa M, Toro MJ, Chiloeches A. Low cell cholesterol levels increase NFkappaB activity through a p38 MAPK-dependent mechanism. Cell Signal. 2006;18(12):2292–2301. doi:10.1016/j.cellsig.2006.05.012
62. Martins de Oliveira R, Antunes E, Pedrazzoli J
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.