")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Eremomycin pyrrolidide: a novel semisynthetic glycopeptide with improved chemotherapeutic properties
Authors Olsufyeva EN, Shchekotikhin AE , Bychkova EN, Pereverzeva ER , Treshalin ID, Mirchink EP, Isakova EB, Chernobrovkin MG, Kozlov RS, Dekhnich AV, Preobrazhenskaya MN
Received 12 May 2018
Accepted for publication 2 July 2018
Published 10 September 2018 Volume 2018:12 Pages 2875—2885
DOI https://doi.org/10.2147/DDDT.S173923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Evgenia N Olsufyeva,1 Andrey E Shchekotikhin,1,2 Elena N Bychkova,1 Eleonora R Pereverzeva,1 Ivan D Treshalin,1 Elena P Mirchink,1 Elena B Isakova,1 Mikhail G Chernobrovkin,3 Roman S Kozlov,4 Andrey V Dekhnich,4 Maria N Preobrazhenskaya1,†
1Gause Institute of New Antibiotics, Moscow, Russia; 2Mendeleyev University of Chemical Technology, Moscow, Russia; 3Drugs Technology, Khimki, Russia; 4Institute of Antimicrobial Chemotherapy, Smolensk State Medical University, Smolensk, Russia
†This author passed away on December 25, 2014
Purpose: Development of new semisynthetic glycopeptides with improved antibacterial efficacy and reduced pseudoallergic reactions.
Methods: Semisynthetic glycopeptides 3–6 were synthesized from vancomycin (1) or eremomycin (2) by the condensation with pyrrolidine or piperidine. The minimum inhibitory concentration (MIC) for the new derivatives was measured by the broth micro-dilution method on a panel of clinical isolates of Staphylococcus and Enterococcus. Acute toxicity (50% lethal dose, maximum tolerated doses), antibacterial efficacy on model of systemic bacterial infection with S. aureus and pseudoallergic inflammatory reaction (on concanavalin A) of eremomycin pyrrolidide (5) were evaluated in mice according to standard procedures.
Results: The eremomycin pyrrolidide (5) was the most active compound and showed a high activity against Gram-positive bacteria: vancomycin-susceptible staphylococci and enterococci (minimum inhibitory concentrations [MICs] 0.13–0.25 mg/L), as well as vancomycin-intermediate resistant Staphylococcus aureus (MICs 1 mg/L). Antimicrobial susceptibility tested on a panel of 676 isolates showed that 5 had similar activity for the genera Staphylococcus and Enterococcus with MIC90=0.5 mg/L, while vancomycin had MIC90=1–2 mg/L. The number of resistant strains of Enterococcus faecium (vancomycin-resistant enterococci) (MIC =64 mg/L) with this value was 7 (8%) for vancomycin (1) and 0 for the compound 5. In vivo comparative studies in a mouse model of systemic bacterial infection with S. aureus demonstrated that the efficacy of 5 was notably higher than that of the original antibiotics 1 and 2. In contrast to 1, compound 5 did not induce pseudoallergic inflammatory reaction (on concanavalin A).
Conclusion: The new semisynthetic derivative eremomycin pyrrolidide (5) has high activity against staphylococci and enterococci including vancomycin-resistant strains. Compound 5 has a higher efficacy in a model of staphylococcal sepsis than vancomycin (1) or eremomycin (2). In striking contrast to natural antibiotics, the novel derivative 5 does not induce a pseudoallergic inflammatory reaction to concanavalin A and therefore has no histamine release activity. These results indicate the advantages of a new semisynthetic glycopeptide antibiotic eremomycin pyrrolidide (5) which may be a prospective antimicrobial agent for further pre-clinical and clinical evaluations.
Keywords: semisynthetic glycopeptides, antibacterial activity, pseudoallergic reaction, antibacterial efficacy
Introduction
Polycyclic glycopeptide antibiotics are highly active against Gram-positive bacteria of Staphylococcus, Streptococcus, Enterococcus, and Clostridium genera, including strains resistant to β-lactams, fluoroquinolones, and tetracyclines. Vancomycin (1) (Figure 1) and teicoplanin are the drugs of last resort for treatment of life-threatening infections caused by Gram-positive human pathogens.1 These antibiotics are especially effective for treatment of staphylococci infections caused by multi-resistant strains, especially methicillin-resistant Staphylococcus aureus (MRSA).2,3 The antibiotics of this class do not have any alternative thus far, but their application is restricted by the emergence of resistance to glycopeptides, and by severe side effects.4 Due to its short half-life, rapid excretion, and a low accumulation in tissues, vancomycin is inefficient for treatment of pneumonia and some infections caused by Gram-positive bacteria.5–8
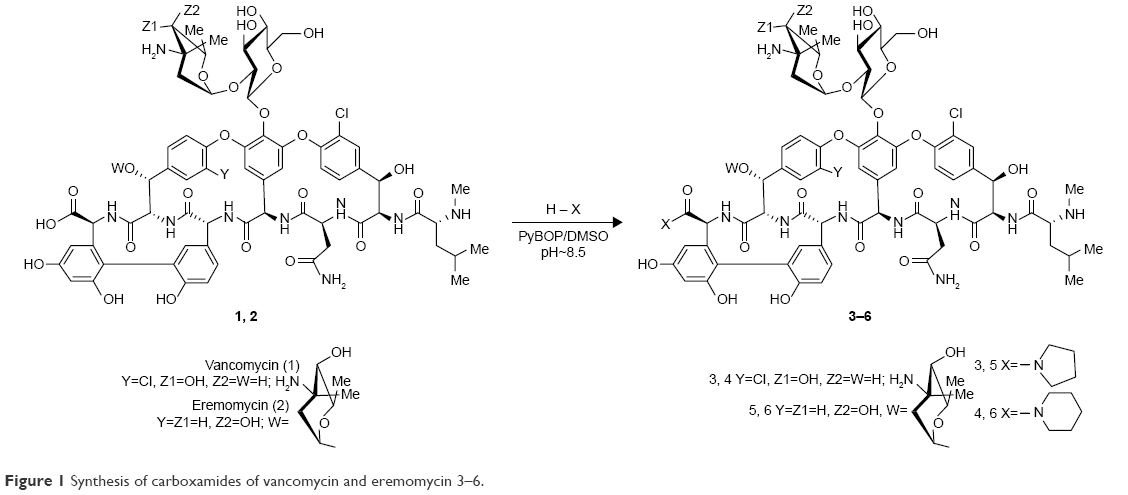 | Figure 1 Synthesis of carboxamides of vancomycin and eremomycin 3–6. |
Glycopeptide antibiotics selectively interfere with the formation of bacterial cell walls without significant effects on eukaryotic cells. These antibiotics bind to D-Ala-D-Ala dipeptide terminus of growing bacterial peptidoglycan, sequestering the substrate from transpeptidation and transglycosylation reactions of peptidoglycan synthesis.9 Due to the unique mechanism of action of glycopeptides, the resistance of microorganisms to vancomycin did not emerge for a long time.
However, the use of vancomycin (1) can be associated with adverse effects, largely nephro- and ear toxicity. Furthermore, vancomycin causes several types of hypersensitivity reactions, such as “red man syndrome” (RMS). RMS is a form of pseudoallergic drug reaction with symptoms that mimic immunological drug allergies but lack immunological mechanisms.5,10 Vancomycin rarely causes anaphylactic reactions; however, >50% of patients exhibit RMS as a result of histamine release from mast cells and basophils directly activated by vancomycin.11
The wide usage of vancomycin (1) in the clinic and avoparcin in veterinary practice has resulted in the emergence of strains resistant to glycopeptides, such as vancomycin-resistant enterococci (VRE)12 and vancomycin-intermediate resistant S. aureus (VISA) strains.13 The isolates showed a modestly increased minimum inhibitory concentration (MIC) value for vancomycin (1) (MIC ≥8 mg/L). Nevertheless, vancomycin treatment of infections caused by VISA isolates often led to therapeutic failure. The spread of resistance to glycopeptides in enterococci and S. aureus became a serious reason for the decreased efficacy of vancomycin against Gram-positive bacterial infections.3,14,15
Thus, the resistance, adverse effects, and limitations in the application of vancomycin have stimulated the efforts to develop glycopeptides with improved pharmacological properties. Some second-generation glycopeptides have been identified in an extensive search for new agents active against poly-resistant strains of Gram-positive pathogens.3,16,17 Three most promising semisynthetic glycopeptides have been obtained by the modification of natural antibiotics: oritavancin (LY 333328; Eli Lilly, Indianapolis, IN, USA)18 was prepared by modifying chloroeremomycin, telavancin (TD-6424; Theravance Biopharma US, Inc., South San Francisco, CA, USA)19 was obtained from vancomycin, and dalbavancin (BI-397; Actavis, Edison, NJ, USA)20 from A-40926. They were approved in the USA and European Union (EU) for treatment of skin infections and hospital-acquired bacterial pneumonia caused by resistant Gram-positive bacteria.16,21 According to preliminary data, the adverse-effect profile of these glycopeptides was similar to that of drugs currently used to treat severe Gram-positive infections.22 Further clinical evaluation revealed some safety problems, such as an increased risk of developing osteomyelitis with oritavancin; nephrotoxicity, a risk of QT interval prolongation with telavancin, and an elevation of hepatic enzymes after dalbavancin.4
We have synthesized and studied the biological properties of new semi-synthetic glycopeptides for selection of new drug candidates with improved chemotherapeutic properties.23,25 Special attention is given to the derivatives of an original antibiotic of this group, eremomycin (2) (Figure 1), discovered by Gause et al.26 This glycopeptide antibiotic is highly active against Gram-positive bacteria, including the strains resistant to β-lactams, fluoroquinolons, and tetracyclines. It is 3–7 times more potent than one in vitro and in vivo toward many pathogenic strains, while the acute toxicity in animal tests is several times lower that of vancomycin (1).27,28 Investigation of eremomycin (2) in immunological tests in vivo (reaction of generalized anaphylaxis [anaphylactic shock], active skin anaphylaxis, and delayed hypersensitivity reaction) showed that this antibiotic does not have allergenic properties but causes pseudoallergic reactions in rats.27,29,30
Like vancomycin (1), eremomycin (2) is inactive against VRE but moderately potent against VISA (MIC =8 mg/L). Semisynthetic eremomycin carboxamides (C-terminus derivatives) showed a high activity against Gram-positive bacteria, including resistant strains for which the parent antibiotic was inert.23,24,30–32 Eremomycin N-adamantyl-2-amide had a promising activity against a range of clinical isolates, including methicillin-resistant staphylococci and VRE. This agent was equally active against ciprofloxacin-susceptible and ciprofloxacin-resistant Bacillus anthracis strains. The results of in vivo tests demonstrated that this compound efficiently prevented animal death caused by S. aureus or B. anthracis, showing better pharmacokinetic properties compared with 1 or 2.24
Another promising semisynthetic derivative of eremomycin amide was active against vancomycin-susceptible staphylococci and enterococci, as well as against VISA (MIC =0.5–0.25 mg/L).33 In the rotational-echo double solid state nuclear magnetic resonance experiments with a labeled analog (Kim et al) was also shown that this compound has an additional binding site to the main site-D-Ala-D-Ala with fragments of the structure of cell-wall peptidoglycan of S. aureus.34,35 Like several semisynthetic eremomycin derivatives modified at the C-terminus of the peptide core, eremomycin amides exhibited less pronounced pseudoallergic reactions compared with the parent antibiotic 2 in animal tests.30
Due to the increasing prevalence of diseases caused by resistant strains and some adverse effects of glycopeptides, the preparation and development of novel antibacterial antibiotics effective against poly-resistant bacteria (including glycopeptide-resistant strains) with diminishing of side effects remains a problem. The goal of this study was to synthesize and assess the antibacterial activity of new carboxamides of vancomycin and eremomycin (3–6). Among this series, the derivative 5 demonstrated an improved activity against vancomycin-susceptible staphylococci and enterococci, including VISA and VRE strains. Thus, a new semisynthetic eremomycin pyrrolidide (5) showed advantages compared with natural antibiotics 1 and 2.
Materials and methods
Reagents, instruments used and other general information; several properties of the compounds 3–6 and NMR data for eremomycin pyrrolidide 5 (Figures S1–S3) are presented in Supplementary materials.
Glycopeptide antibiotics
Vancomycin hydrochloride (1) was purchased from TEVA (Pharmaceutical Industries Ltd., Debrecen, Hungary). Sulfate of eremomycin (2) was produced at the pilot plant of the Gause Institute of New Antibiotics (Moscow, Russia).
General procedure for the preparation of carboxamides 3–6
Semisynthetic derivatives 3–6 were synthesized from vancomycin (1) or eremomycin (2) by previously described methods23–25,31–33 for glycopeptide carboxamides (Figure 1). Compounds 3–6 were synthesized by condensation of 1 or 2 (1 eq.) with pyrrolidine or piperidine hydrochloride (5 eq.) using benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) as a condensing reagent (1.5 eq.) in dimethyl sulfoxide (DMSO) in the presence of Et3N (pH~8.5). After 1 hour of stirring at 22°C, Et2O was added to the reaction mixture and shaken intensively to partly extract DMSO. The upper ethereal layer was separated, and the oil residue was dissolved in aqueous (0.1 n) H2SO4. The pH value was adjusted to 4.5, and the solution was poured into a stirred acetone to precipitate the product. The latter was filtered off, washed with acetone, and dried in vacuum to obtain a white powder of crude salts 3–6. Purification of compounds was performed using column chromatography on silanized silica gel pre-equilibrated with water (1 g of solid for 70 cm3 of silica gel). The yields of 3–6 were ~48%–66%, and high-performance liquid chromatography (HPLC) purity was 96%–98%. Properties (yield, melting temperature, UV-, IR, HPLC, high-resolution mass spectrometry) and 1H- and 13C-NMR-spectra of 3–6 are presented in Supplementary materials.
Microorganisms
The pre-screening of antibacterial activity of new derivatives 3–6 and starting glycopeptides 1 and 2 was performed on a panel of clinical isolates of Gram-positive microorganisms, including Staphylococcus epidermidis 533, Staphylococcus haemolyticus 602, S. aureus 3797 (VISA HIP-5836; New Jersey), S. aureus 3798 (VISA HIP-5827; Michigan), Enterococcus faecalis 559 (vancomycin susceptible enterococci [VSE]), and Enterococcus faecium 568 (VSE). The strains were kindly provided by colleagues from the Lepetit Research Center (LePetit Group, Biosearch S.p.A., Varese, Italy). The results were usually identical and always within 2-fold of each other. Antimicrobial susceptibility testing of 5 and 1 was performed on the collection of clinical isolates from the Institute of Antimicrobial Chemotherapy (Smolensk, Russia). A total of 676 non-duplicate isolates were evaluated as follows: 255 strains of MRSA, 179 strains of methicillin-sensitive S. aureus, 45 strains of coagulase-negative Staphylococcus, 122 strains of E. faecalis, and 75 strains of E. faecium. We tested the material from hospitalized patients (adults and children) with clinical and laboratory symptoms of infection (out-of-hospital and nosocomial) of the skin and soft tissues, respiratory tract, bones, joints, urinary tract, and abdominal cavity. The in vitro study was performed according to the recommendations of the European Committee on Antimicrobial Susceptibility testing. The MIC values were determined by the broth micro-dilution method using Mueller Hinton broth and National Committee for Clinical Laboratory Standards procedures.24,36 Data are presented in Tables 1 and 2.
 | Table 1 Antibacterial activities (MIC, mg/L) of derivatives 3–6 compared with the antibiotics 1 and 2 |
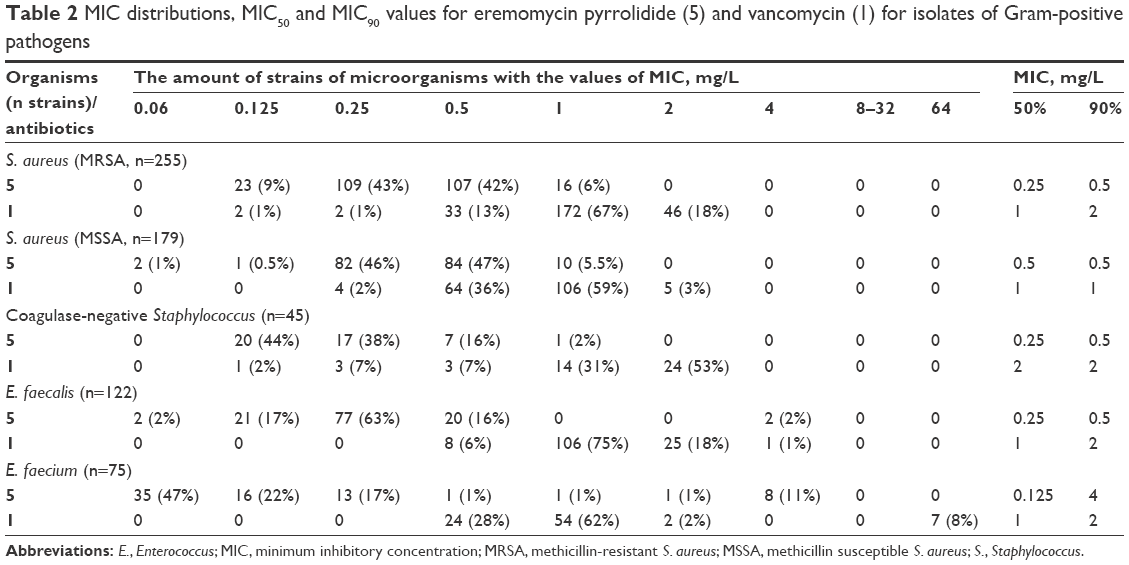 | Table 2 MIC distributions, MIC50 and MIC90 values for eremomycin pyrrolidide (5) and vancomycin (1) for isolates of Gram-positive pathogens |
Animals
Animals were obtained from the facility of the Russian Academy of Sciences (Moscow, Russia). The in vivo experiments were performed in accordance with the European Convention for the Protection of Vertebrate Animals, Directives 86/609/EEC,37 the National Standard of the Russian Federation R 53434–2009 “Good Laboratory Practice”38 and approved by Ethics of Animal Experimentation of Gause Institute of New Antibiotics.
Mouse model of staphylococcal sepsis
A comparative study of the efficacy of sulfate of eremomycin pyrrolidide (5), vancomycin hydrochloride (1), and sulfate of eremomycin (2) was performed using a model of staphylococcal sepsis in mice. We used female SHK colony mice weighing 22–25 g. After a 2-week quarantine, healthy animals were used in experiments. S. aureus (strain 10 sensitive to antibiotics, adapted to growth in mice) was used as an infectious agent. Initially, a 100% lethal dose (LD100) of S. aureus was determined for a given strain of mice in the intravenous (IV) route of the infection. Death of mice was registered daily for 10 days. The LD100 was calculated as 3 × 108 CFU/mouse.
To determine the efficacy of tested drugs, the doses were determined at which 50% and 100% of animals survived (effective dose of compound required to cure 50% and 100% of animals [ED50] and [ED100]). Mice were housed in 10-head cages and infected IV with S. aureus at a lethal dose. Thirty minutes after infection, mice were injected with vancomycin hydrochloride (1), sulfate of eremomycin (2), and sulfate of eremomycin pyrrolidide (5) IV (6 doses in 5% glucose solution). As a control, a cohort of animals infected with S. aureus (at a lethal dose) but not treated with antibiotics was examined. Animals were monitored for 10 days, mortality was recorded daily.
To determine ED50 in the model of staphylococcal sepsis in mice, we used vancomycin hydrochloride (1) at 2.5–7.5 mg/kg; sulfate of eremomycin (2) at 0.5–5.0 mg/kg; and sulfate of eremomycin pyrrolidide (5) at 0.25–4.5 mg/kg. ED50 of the test preparations was determined by death of animals in Barents method (frequency accumulation).
Acute toxicity
The CBA mice (18–22 g) were randomized into groups (n=6–10) and received 1, 2, and 5 as single bolus IV injections. The substances were dissolved in 5% glucose solution (concentration of 1 and 5: 10 mg/mL; concentration of 2: 100 mg/mL) and administered at 200–700 mg/kg (1), 500–3,000 mg/kg (2), and 100–400 mg/kg (5). Acute toxicity was evaluated on the basis of mortality, survival time, and clinical manifestations of intoxication. The 50% lethal dose (LD50) values and maximum tolerated doses (MTD = 10% lethal dose [LD10]) were calculated by the method of Litchfield and Wilcoxon using StatPlus 2006 AnalystSoft StatPlus software.
Assessment of pseudoallergic reaction
The potency of the semisynthetic derivative 5 to induce pseudoallergic reaction was assessed by non-immune activation of histamine release (inflammatory reaction to concanavalin A [Con A]) according to the “Guidelines on the assessment of allergic properties of drugs.”39 The experiment was carried out in male CBA mice (n=40) weighing 26–28 g. After a 2-week quarantine, healthy animals were caged in groups of 10 and fed ad libitum with standard laboratory food and tap water.
Compounds 1, 2, and 5 in 5% glucose solution were injected IV at single doses of MTD. Similarly, 10 mL/kg of 5% glucose solution was administered to the control group (n=10). One hour post injection, 0.5% Con A in isotonic sodium chloride solution (0.01 mL/10 g body mass) was injected into the pad of the hind legs, and the same volume of isotonic solution of sodium chloride was injected into the contralateral extremity of mice in the experimental and control groups. One hour later, the mice were decapitated, the extremities were amputated at the limit of the hock joint, and their mass was determined. The reaction index of inflammation (RI) was calculated using the formula RI = (Me−Mc)/Mc × 100%, where Me is the mass of the hind leg foot injected with Con A and Mc is the mass of the hind leg foot injected with isotonic sodium chloride solution.
Results and discussion
Chemistry
Modification of the C-terminal carboxyl group of the peptide core is an effective synthetic method of chemical transformation of glycopeptides.16,17,40 We synthesized new vancomycin and eremomycin carboxamides 3–6 (Figure 1). The reaction of 1 or 2 with pyrrolidine or piperidine in the presence of the coupling reagent resulted in pyrrolidides or piperidides of vancomycin 3 and 4 or eremomycin 5 and 6. Several appropriate reagents (PyBOP, O-(benzotriazol-1-yl)-N,N,N″,N″-tetramethyluronium tetrafluoroborate, 1-[Bis(dimethylamino)methylene]–1 H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, 1-propanephosphonic anhydride (), diphenylphosphoryl azide, or [1-cyano-2-ethoxy-2-oxoethylidenaminooxy]dimethylamino-morpholino-carbenium hexafluorophosphate) were tested for the amidation of glycopeptides.40 The best reagent in this reaction was PyBOP, which gave the final carboxamides 3–6 in 48%–66% yields in optimal conditions. The purity of final derivatives 3–6 was >95% according to HPLC analysis. The analytical and spectroscopic data of new compounds were in full accordance with the assigned structures (Supplementary materials).
In vitro antibacterial testing
Screening of the antibacterial activity of new antibiotics was carried out using a panel of clinical isolates of Gram-positive microorganisms using the micro-dilution method to determine MIC. Generally, the semisynthetic vancomycin derivatives 3 and 4 were less active against the tested strains (MICs ~0.5–8 mg/L) compared with the corresponding eremomycin derivatives 5 and 6 (Table 1). The activity of 3 and 4 or 5 and 6 were close to the potency of the corresponding starting antibiotics 1 or 2. However, it should be noted that carboxamides 4, 5, and 6 were more active against resistant strains VISA (MICs =0.5–2 mg/L) than the starting antibiotics 1 or 2 (MICs =4–8 mg/L).
The derivative 5 showed the highest activity against all tested strains and better MICs values for VSE (S. epidermidis, S. haemolyticus; MICs =0.13–0.25 mg/L) and vancomycin-resistant strains (VISA MICs =1 mg/L) than the carboxamides 3, 4, and 6. Based on results of pre-screening of antimicrobial properties, eremomycin pyrrolidide (5) was selected for a broad assessment of activity compared with vancomycin (1) as the gold standard for treatment Gram-positive human pathogens. The MIC distribution, MIC50, MIC90, and the percent susceptibility rates are summarized in Table 2.
Eremomycin pyrrolidide (5) revealed a high potency against S. aureus, coagulase-negative Staphylococcus, and Enterococcus strains, exceeding that of vancomycin (1) by 2–4 dilutions. Of note, eremomycin pyrrolidide (5) had similar activity for the genera Staphylococcus and Enterococcus (except for E. faecium) with MIC90=0.5 mg/L while vancomycin had MIC90=1–2 mg/L. The activity of both antibiotics against Staphylococcus and Enterococcus strains had no dependence on resistance to beta-lactams, fluoroquinolones, tetracyclines, and aminoglycosides. The distribution of MICs was monomodal for Staphylococcus and E. faecalis. Strains of vancomycin-resistant E. faecium (VRE, MIC >64 mg/L) were susceptible to 5, but an increased MIC value for these isolates led to the increase of MIC90 to 4 mg/L, which was slightly higher than that of 1 (MIC90=2 mg/L).
The derivative 5 was significantly more active than vancomycin (1) for E. faecium (MIC50 0.125 mg/L vs 1 mg/L, respectively). It is important to mention that the number of resistant strains of E. faecium (MIC >64 mg/L) with this value was 7 (8%) for vancomycin (1) and zero for 5 (Table 2). Given that eremomycin pyrrolidide (5) demonstrated an obvious advantage over vancomycin (1), we compared their antibacterial efficacy in vivo and their acute toxicity in mice.
Staphylococcal sepsis in mice
Figure 2 indicates that the survival rate of mice infected with S. aureus increased after IV single-dose injections of 5 in a dose-dependent manner. Moreover, the efficacy of 5 in this model was notably superb compared with vancomycin (1) and eremomycin (2). Thus, the calculated ED50 values for 5 (0.8 mg/kg) were 2.5 and 5 times lower than those for 2 or 1 (2.0 and 4.1 mg/kg, respectively). Survival of 100% animals was achieved with eremomycin pyrrolidide (5) at a noticeably lower dose (2.5 mg/kg) than after the administration of natural antibiotics 2 or 1 (4.5 and 6.5 mg/kg, respectively).
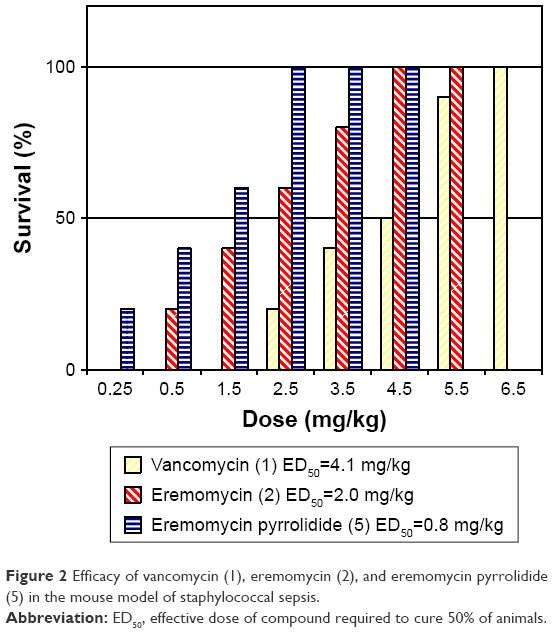 | Figure 2 Efficacy of vancomycin (1), eremomycin (2), and eremomycin pyrrolidide (5) in the mouse model of staphylococcal sepsis. |
Acute toxicity
The signs of acute toxicity caused by a single IV administration of 1, 2, and 5 were generally similar. After the injection of the biggest doses, the animals in all groups died within 1–3 hour exhibiting neurotoxicity. Lower doses were lethal 1–3 days post treatment due to cardiovascular failure. Quantitative parameters of acute toxicity of 1, 2, and 5 are presented in Table 3. The LD50 and MTD (LD10) values for 1 correlated with the results reported by US Pharmacopeia.41,42 The acute toxicity of 2 (LD50 ~1,500 mg/kg) was ~ 3-fold lower than that of 1 (LD50 ~500 mg/kg), but the acute toxicity of 5 (LD50 ~250 mg/kg) was higher about 2-fold. Nevertheless, the ratio between therapeutic (ED50) and toxic doses (MTD) for compound 5 was 1/150 compared with 1/90 for compound 1.
 | Table 3 Acute toxicity LD50 and MTD (LD10) of vancomycin (1), eremomycin (2), and eremomycin pyrrolidide (5) in mice |
Pseudoallergic reaction
Finally, we compared the potency to induce a non-immune activation of histamine release for the selected derivative 5 and natural antibiotics 1 and 2. The inflammatory reaction to Con A is based on the capacity of this lectin to directly act on mast cell and basophil receptors, thereby liberating the inflammatory mediators. The results of this test demonstrated that the IV injection of vancomycin (1) and eremomycin (2) produced statistically significant elevations of Me with respect to Mc (Table 4). The RI values in these groups were 52.1% and 42.7%, respectively. In contrast, a non-significant difference was found in Me and Mc in mice treated with 5. The RI value in this group exceeded the respective value in the control cohort by only 5.5%.
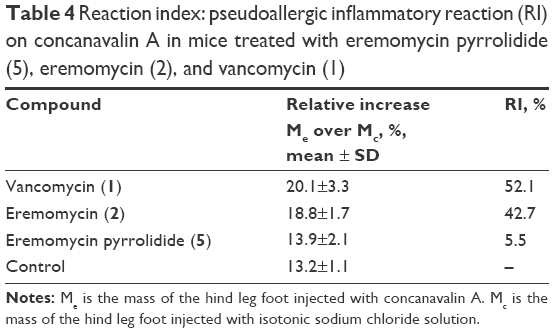 | Table 4 Reaction index: pseudoallergic inflammatory reaction (RI) on concanavalin A in mice treated with eremomycin pyrrolidide (5), eremomycin (2), and vancomycin (1) |
Conclusion
Four new glycopeptides carboxamides of 3–6 were obtained by amidation of natural antibiotics vancomycin (1) or eremomycin (2) with pyrrolidine or piperidine in the presence of the coupling reagent PyBOP. Evaluation of biological properties of eremomycin pyrrolidide (5) demonstrated a pronounced antibacterial activity in vitro against a panel of clinical isolates of Gram-positive bacteria, including strains resistant to glycopeptides (VISA and VRE). The highest in vivo efficacy was also achieved in a murine model of staphylococcal sepsis. Eremomycin pyrrolidide (5) showed advantages over vancomycin (1) in all in vitro tests against Gram-positive bacteria. The response to Con A showed that 5 had no histamine release activity and, therefore, did not induce a pseudoallergic reaction. In addition, compound 5 demonstrated a favorable toxicology profile because its therapeutically efficient dose was 150-fold smaller than MTD. Thus, eremomycin pyrrolidide (5) is advantageous over the natural antibiotics vancomycin (1) and eremomycin (2) and is perspective for further investigations.
Acknowledgments
We are grateful to Natalia M Malyutina for HPLC analysis, Alexandr M Korolev for electrospray ionization mass spectra, and Yurii N Luzikov for NMR-spectra (all of whom are from the Gause Institute of New Antibiotics, Moscow). †Professor Maria N Preobrazhenskaya passed away on December 25, 2014.
Disclosure
The authors report no conflicts of interest in this work.
References
Sievert DM, Rudrik JT, Patel JB, Mcdonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis. 2008;46(5):668–674. | ||
Nicolaou KC, Boddy CNC, Brase S, Winssinger N. Chemistry biology, and medicine of glycopeptide antibiotics. Angew Chem Int Ed. 1999;38(15):2096–2152. | ||
Binda E, Marinelli F, Marcone GL. Old and New Glycopeptide Antibiotics: Action and Resistance. Antibiotics. 2014;3(4):572–594. | ||
van Bambeke F, van Laethem Y, Courvalin P, Tulkens PM. Glycopeptide antibiotics: from conventional molecules to new derivatives. Drugs. 2004;64(9):913–936. | ||
Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68(9):1781–1790. | ||
Li Z, Willke RJ, Pinto LA, et al. Comparison of Length of Hospital Stay for Patients with Known or Suspected Methicillin-Resistant Staphylococcus Species Infections Treated with Linezolid or Vancomycin: A Randomized, Multicenter Trial. Pharmacotherapy. 2001;21(3):263–274. | ||
Bailie GR, Neal D. Vancomycin ototoxicity and nephrotoxicity. A review. Med Toxicol Adverse Drug Exp. 1988;3(5):376–386. | ||
Polk RE, Red man syndrome. Ann Pharmacother. 1998;32(7–8):840. | ||
Barna JC, Williams DH. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu Rev Microbiol. 1984;38:339–357. | ||
Farnam K, Chang C, Teuber S, Gershwin ME. Nonallergic drug hypersensitivity reactions. Int Arch Allergy Immunol. 2012;159(4):327–345. | ||
Kupstaité R, Baranauskaité A, Pileckyté M, Sveikata A, Kadusevicius E, Muckiené G. Severe vancomycin-induced anaphylactic reaction. Medicina. 2010;46(1):30–33. | ||
Aarestrup FM, Seyfarth AM, Emborg HD, Pedersen K, Hendriksen RS, Bager F. Effect of abolishment of the use of antimicrobial agents for growth promotion on occurrence of antimicrobial resistance in fecal enterococci from food animals in Denmark. Antimicrob Agents Chemother. 2001;45(7):2054–2059. | ||
Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124(7):2836–2840. | ||
Chen CY, Tien FM, Sheng WH, et al. Clinical and microbiological characteristics of bloodstream infections among patients with haematological malignancies with and without neutropenia at a medical centre in northern Taiwan, 2008–2013. Int J Antimicrob Agents. 2017;49(3):272–281. | ||
Flokas ME, Karageorgos SA, Detsis M, Alevizakos M, Mylonakis E. Vancomycin-resistant enterococci colonisation, risk factors and risk for infection among hospitalised paediatric patients: a systematic review and meta-analysis. Int J Antimicrob Agents. 2017;49(5):565–572. | ||
Ashford PA, Bew SP. Recent advances in the synthesis of new glycopeptide antibiotics. Chem Soc Rev. 2012;41(3):957–978. | ||
Olsufyeva EN, Tevyashova AN. Synthesis, Properties, and Mechanism of Action of New Generation of Polycyclic Glycopeptide Antibiotics. Curr Top Med Chem. 2017;17(19):2166–2198. | ||
Corey GR, Arhin FF, Wikler MA, et al. Pooled analysis of single-dose oritavancin in the treatment of acute bacterial skin and skin-structure infections caused by Gram-positive pathogens, including a large patient subset with methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2016;48(5):528–534. | ||
Masterton R, Cornaglia G, Courvalin P, Lode HM, Rello J, Torres A. The clinical positioning of telavancin in Europe. Int J Antimicrob Agents. 2015;45(3):213–220. | ||
Huband MD, Castanheira M, Farrell DJ, et al. In vitro activity of dalbavancin against multidrug-resistant Staphylococcus aureus and streptococci from patients with documented infections in Europe and surrounding regions (2011–2013). Int J Antimicrob Agents. 2016;47(6):495–499. | ||
O’Driscoll T, Crank CW. Vancomycin-resistant enterococcal infections: epidemiology, clinical manifestations, and optimal management. Infect Drug Resist. 2015;8:217–230. | ||
Guskey MT, Tsuji BT. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy. 2010;30(1):80–94. | ||
Miroshnikova OV, Printsevskaya SS, Olsufyeva EN, et al. Structure-activity relationships in the series of eremomycin carboxamides. J Antibiot. 2000;53(3):286–293. | ||
Maples KR, Wheeler C, Ip E, et al. Novel semisynthetic derivative of antibiotic Eremomycin active against drug-resistant gram-positive pathogens including Bacillus anthracis. J Med Chem. 2007;50(15):3681–3685. | ||
Plattner J, Chu D, Mirchink EP, Isakova EB, Preobrazhenskaya MN, Olsufyeva EN. N′-(alpha-aminoacyl)- and N′-alpha-(N-alkylamino)acyl derivatives of vancomycin and eremomycin II. Antibacterial activity of N′-(alpha-aminoacyl)- and N′-alpha-(N-alkylamino)acyl derivatives of Vancomycin and Eremomycin. J Antibiot. 2007;60(4):245–250. | ||
Gause GF, Brazhnikova MG, Lomakina NN, et al. Eremomycin-new glycopeptide antibiotic: chemical properties and structure. J Antibiot. 1989;42(12):1790–1799. | ||
Gol’dberg LE, Stepanova ES, Vertogradova TP, Shevniuk LA, Shepelevtseva NG. Preclinical toxicological study of the new antibiotic eremomycin. Its acute toxicity for laboratory animals. Antibiot Med Biotekhnol. 1987;32(12):910–915. | ||
Malkova IV. [Experimental study of the antibacterial activity and chemotherapeutic efficacy of the novel glycopeptide eremomycin]. Antibiot Chemother. 1989;34(1):52–56. Russian. | ||
Filippos’iants ST, Shevniuk LA. Experimental study of histamine-liberating properties of eremomycin. Antibiot Khimioter. 1989;34(3):209–212. | ||
Pavlov AY, Berdnikova TF, Olsufyeva EN, et al. Carboxamides and hydrazide of eremomycin. J Antibiot. 1996;49(2):194–198. | ||
Printsevskaya SS, Reznikova MI, Korolev AM, et al. Synthesis and study of antibacterial activities of antibacterial glycopeptide antibiotics conjugated with benzoxaboroles. Future Med Chem. 2013;5(6):641–652. | ||
Olsufyeya EN, Berdnikova TF, Miroshnikova OV, Reznikova MI, Preobrazhenskaya MN. Chemical Modification of Antibiotic Eremomycin at the Asparagine Side Chain. J Antibiot. 1999;52(3):319–324. | ||
Solov’eva SE, Printsevskaya SS, Olsuf’eva EN, Batta G, Preobrazhenskaya MN. New derivatives of eremomycin containing 15N or F atoms for NMR study. Bioorg Chem. 2008;34(6):831–839. | ||
Kim SJ, Cegelski L, Preobrazhenskaya M, Schaefer J. Structures of Staphylococcus aureus cell-wall complexes with vancomycin, eremomycin, and chloroeremomycin derivatives by 13C{19F} and 15N{19F} rotational-echo double resonance. Biochemistry. 2006;45(16):5235–5250. | ||
Chang J, Zhou H, Preobrazhenskaya M, Tao P, Kim SJ. The Carboxyl Terminus of Eremomycin Facilitates Binding to the Non-d-Ala-d-Ala Segment of the Peptidoglycan Pentapeptide Stem. Biochemistry. 2016;55(28):4003. | ||
Pavlov AY, Lazhko EI, Preobrazhenskaya MN. A new type of chemical modification of glycopeptides antibiotics: aminomethylated derivatives of eremomycin and their antibacterial activity. J Antibiot. 1997;50(6):509–513. | ||
Council of Europe European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes. Strasbourg, 18.III.1986. Council of Europe, ETS No. 123. 1986. Available from: https://rm.coe.int/168007a67b. Accessed August 28, 2018. | ||
National state standard GOST P 53434-2009 the Russian Federation standard “The principles of Good Laboratory Practice” (approved and put into effect by the Order of the Federal Agency for Technical Regulation and Metrology of December 2, 2009), No 544. | ||
Supotnitskiy МV, Gorbunova EV, Korsun LV, Lebedinskaya EV. Aspects of preclinical studies at the stage of developing non-viral vector constructions designed for gene therapy. Scientific Centre for Expert Evaluation of Medicinal Products Bulletin. 2014;3:30–8. Available from: http://supotnitskiy.ru/stat/stat118.htm. Accessed August 28, 2018. Russian. | ||
Sigma-Aldrich. Peptide synthesis, coupling reagents. ChemFiles 2007;7:2. https://www.sigmaaldrich.com/technical-documents/articles/chemfiles/coupling-reagents.html | ||
Product monograph - Vancomycin hydrochloride for injection, USP, 2017:21. Available from: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m87760.html. Accessed August 28, 2018. | ||
USP, Inc., Rockville, MD, USA. USPDI Drug information for the health care professional; 1995; 1:2755-2758. Available from: http://www.worldcat.org/title/usp-di-drug-information-for-the-health-care-provider/oclc/606251784/. Accessed August 28, 2018. |
Supplementary materials
Reagents, instruments used, and other general information
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA), Fluka (Munich, Germany), and Merck (Kenilworth, NJ, USA). The evaluation of the reactions, column eluates, and all final samples were analyzed by thin layer chromatography (TLC) using Merck Silica Gel 60F254 plates (Merck) in systems containing EtOAc-PrOH-25%-NH4OH, 30:30:22 or 1:1:1. In addition, purity of the final compounds was demonstrated by HPLC (column Kromasil 100-5C8, 4.6 × 250 mm, size 5 μm; Ekzo Nobel, Sweden) with loop 20 μL using 2 systems: A – 0.2% HCOONH4 (pH 4.2) and MeCN 5→60%, 30 min; B – 0.6% HCOONH4, pH 7.8 and MeCN 8→70%, 40 min. Reaction products were purified by reversed-phase column chromatography on Merck Silanized Silica Gel (0.063–0.2 mm). Melting points were determined using a Buchi SMP-20 apparatus and are uncorrected. Optical rotation was measured by AA-55 series polarimeter (Optical Activity Ltd., Huntingdon, UK). UV-spectra were determined by UNICO 2804 single-beam infrared scanning spectrophotometer (Unico, Inc., Franksville, WI, USA). The IR spectra were obtained on a Nicolet-iS10 Fourier transform IR spectrometer (DTGS detector, splitter – KBr) (Thermo Fisher Scientific, Waltham, MA, USA) with a Smart Performer module equipped with a ZnSe-crystal. The spectra were run on the range of 3,000–650 cm−1 with a resolution of 4 cm−1. The spectra were preceded using the OMNIC-7.0 program package. High resolution mass spectra were recorded with electron spray ionization (ESI) on a Bruker Daltonics microOTOF-QII instrument (Bruker Daltonics GmbH Life Sciences, Bremen, Germany). NMR spectra were recorded on a Varian VXR-400 instrument (Varian Inc., Palo Alto, CA, USA) operated at 400 and 100 MHz for 1H and 13C, respectively. Chemical shifts were measured in D2O using 3-(trimethylsilyl)-1-propanesulfonic acid, sodium salt (DSS) as an internal standard or MeOH as an internal standard.
Several properties of the compounds 3–6
Vancomycin pyrrolidide (3). Yield 48%. White powder. Melting temperature 215°C–220°C (decomposition). TLC on Merck Silica Gel 60F254 plates in system EtOAc-PrOH-25%-NH4OH, 30:30:22, Rf = 0.51. HPLC: Rt = 19.07 min (A system). [α]D22 = −16°, c = 0.25, H2O. UV-spectra, H2O: λmax = 279 nm, E1%1 cm = 25.0. IR-spectra, strong bands: 3,323, 1,644, 1,504, 1,229, 1,045 cm−1. ESI MS: found, 1,501.49, calculated for C70H82Cl2N10O23, 1,501.50.
Vancomycin piperidide (4). Yield 65%. White powder. Melting temperature >220°C (decomposition); TLC on Merck Silica Gel 60F254 plates in system EtOAc-PrOH-25%-NH4OH, 30:30:22, Rf = 0.58. HPLC: Rt = 20.73 min (A system). [α]D22 = −24°, c = 0.25, H2O. UV-spectra, H2O: λmax = 279 nm, E1%1 cm = 23.0. IR-spectra, strong bands: 3,289, 1,645, 1,504, 1,228, 1,045 cm−1. ESI MS: found 1,515.46, calculated for C71H84Cl2N10O23, 1,515.52.
Eremomycin pyrrolidide (5). Yield 66%. White powder. Melting temperature >220°C (decomposition). TLC on Merck Silica Gel 60F254 plates in system EtOAc-PrOH-25%-NH4OH, 30:30:22. Rf = 0.36. HPLC: Rt = 27.31 min (B system). [α]D°C = −64°, c = 0.25, H2O. UV-spectra, H2O: λmax = 282.5 nm, E1%1 cm= 29.4. IR-spectra, strong bands: 3,286, 1,644, 1,504, 1,212, 1,063 cm−1. ESI MS: found ESI MS: found 1,610.62, calculated for C77H96ClN11O25, 1,610.63.
Eremomycin piperidide (6). Yield 52%. White powder. Melting temperature >220°C (decomposition). TLC on Merck Silica Gel 60F254 plates in system EtOAc-PrOH-25%-NH4OH, 30:30:22, Rf = 0.46. HPLC: Rt = 28.04 min (B system). [α]D22 = −64°, c = 0.25, H2O; UV-spectra, H2O: λmax = 282 nm, E1%1 cm = 31.2. IR-spectra, strong bands: 2,949, 1,683, 1,652, 1,515, 1,102, 1,028 cm−1. ESI MS: found ESI MS: found 1,624.65, calculated for C78H98ClN11O25, 1,624.65.
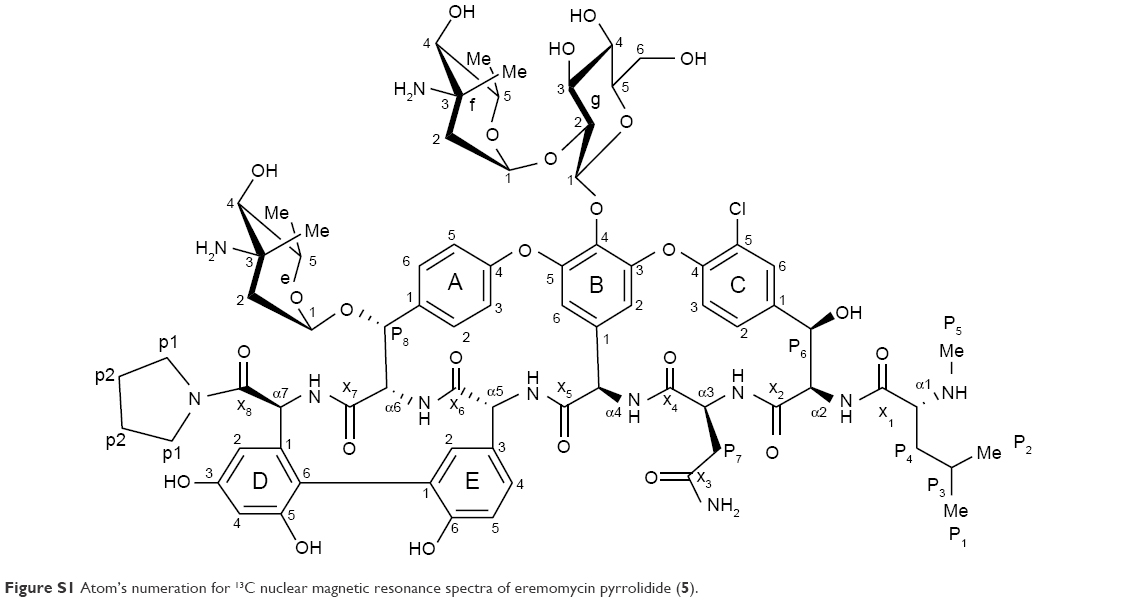 | Figure S1 Atom’s numeration for 13C nuclear magnetic resonance spectra of eremomycin pyrrolidide (5). |
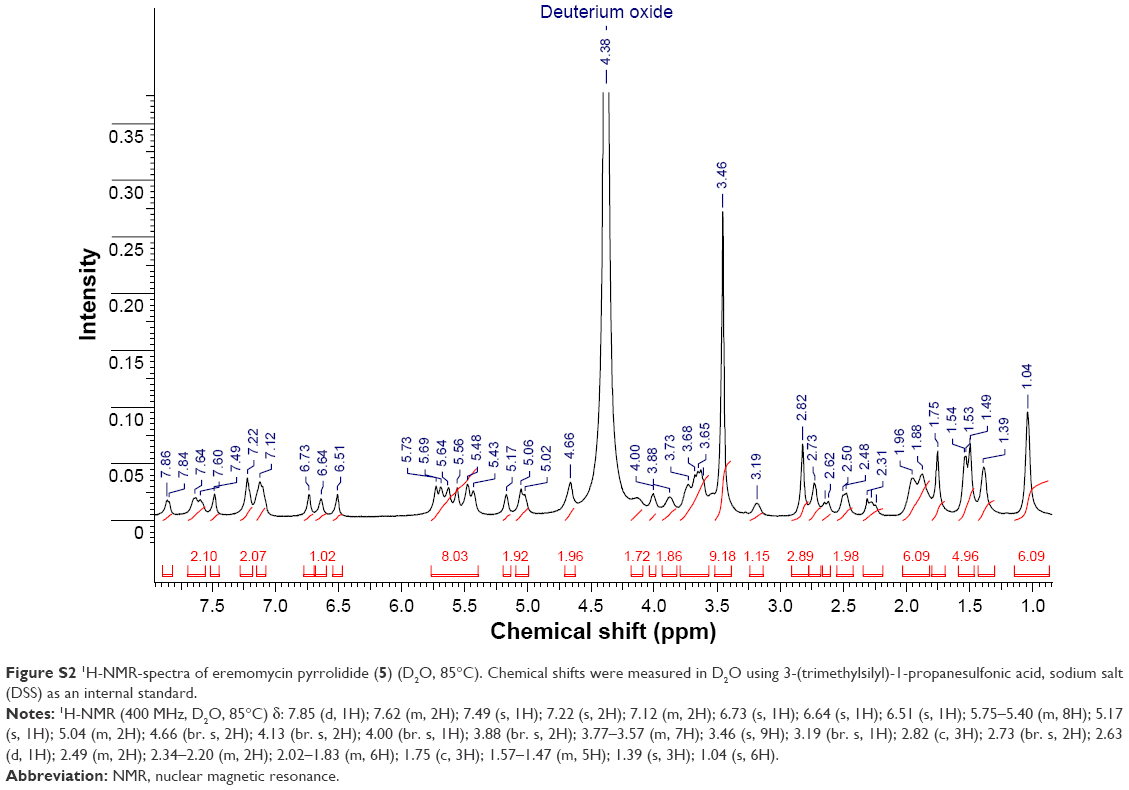 | Figure S2 1H-NMR-spectra of eremomycin pyrrolidide (5) (D2O, 85°C). Chemical shifts were measured in D2O using 3-(trimethylsilyl)-1-propanesulfonic acid, sodium salt (DSS) as an internal standard. |
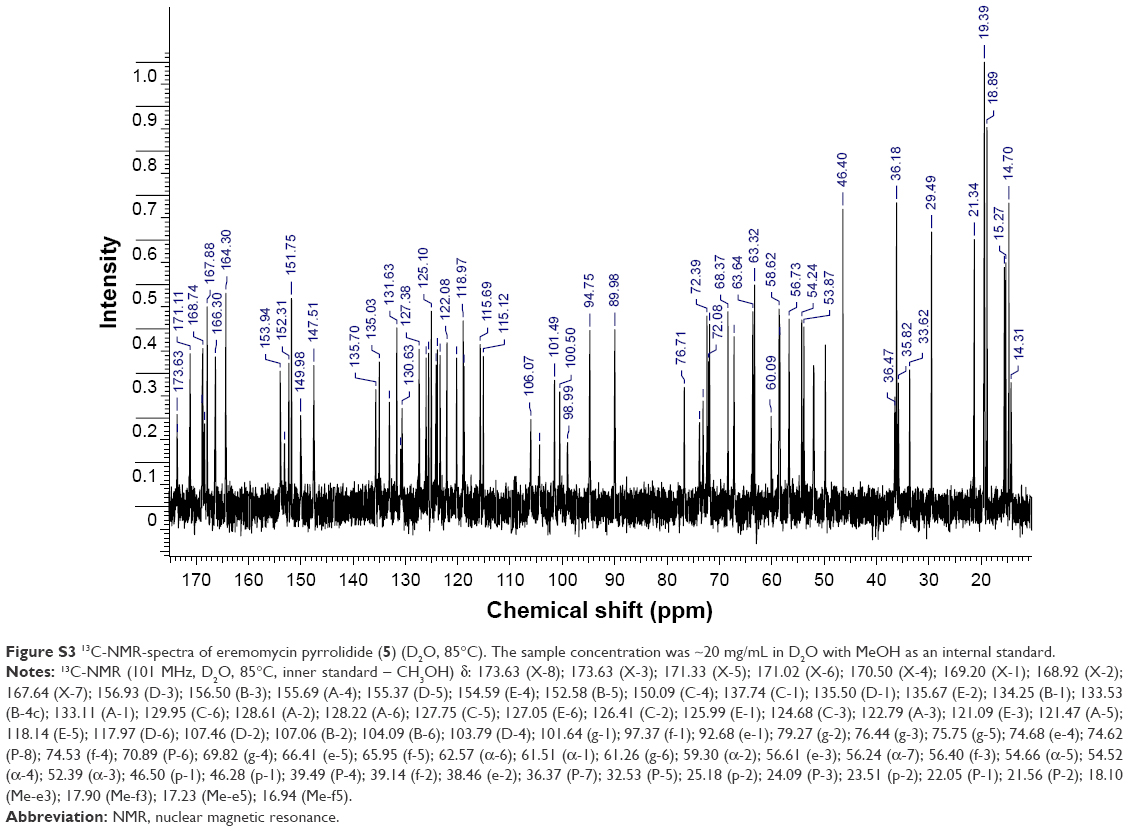 | Figure S3 13C-NMR-spectra of eremomycin pyrrolidide (5) (D2O, 85°C). The sample concentration was ~20 mg/mL in D2O with MeOH as an internal standard. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.