")
Back to Journals » OncoTargets and Therapy » Volume 12
Epstein-Barr Virus-Encoded Products Promote Circulating Tumor Cell Generation: A Novel Mechanism of Nasopharyngeal Carcinoma Metastasis
Authors Yang Z, Wang J, Zhang Z, Tang F
Received 24 October 2019
Accepted for publication 10 December 2019
Published 6 January 2020 Volume 2019:12 Pages 11793—11804
DOI https://doi.org/10.2147/OTT.S235948
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Zongbei Yang,1,2 Jing Wang,2 Zhenlin Zhang,1 Faqing Tang2
1Zhuhai People’s Hospital, Zhuhai Hospital of Jinan University, Zhuhai, People’s Republic of China; 2Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, People’s Republic of China
Correspondence: Faqing Tang
Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha 410013, People’s Republic of China
Tel/Fax +86-731-89762688
Email [email protected]
Abstract: Epstein–Barr virus (EBV) is a specific tumorigenic factor in the pathogenesis of nasopharyngeal carcinoma (NPC). Viral products encoded by EBV (LMP1, LMP2A, EBNA1, and miRNAs) have been shown to promote NPC metastasis. EBV-encoded oncoproteins and miRNAs have been shown to induce epithelial–mesenchymal transition (EMT) indirectly by inducing EMT transcription factors (EMT-TFs). These EBV-encoded products also promote the expression of EMT-TFs through post-transcriptional regulation. EMT contributes to generation of circulating tumor cells (CTCs) in epithelial cancers. CTCs exhibit stem cell characteristics, including increased invasiveness, enhanced cell intravasation, and improved cell survival in the peripheral system. EBV may contribute NPC metastasis through promoting generation of CTCs. Furthermore, CTC karyotypes are associated with NPC staging, therapeutic sensitivity, and resistance. We summarized studies showing that EBV-encoded virus-proteins and miRNAs promote generation of NPC CTCs, and highlighted the associated mechanism. This synthesis indicated that EBV mediates NPC metastasis through generation of CTCs.
Keywords: nasopharyngeal carcinoma, circulating tumor cell, metastasis, Epstein-Barr virus
Introduction
Nasopharyngeal carcinoma (NPC) is a cancer generated in nasopharynx epithelium. The tumor epicenter is frequently observed at the fossa of Rosenmüller, which is the location from which the tumor invades adjacent anatomical spaces or organs.1 According to the International Agency for Research on Cancer (IARC), there were about 129 000 new cases of NPC in 2018, which accounted for only 0.7% of all newly diagnosed cancers.2 The geographical distribution of NPC is extremely unbalanced, with >70% of new cases in east and southeast Asia. NPC is one of the leading malignancies in Southeast Asia, particularly in Southern China. Epidemiological trends in the past few decades have shown that lifestyle and environment may be the main contributors to NPC pathogenesis.1,2 These risk factors include Epstein-Barr virus (EBV) infection, environmental carcinogen,3 high-risk dietary habits and high-risk genotypes.2,4–6 Although the diagnostic techniques for NPC have improved and individualized comprehensive chemoradiotherapy strategies have been developed, treatment of NPC remains a significant challenge. The main barriers to effective treatment of NPC are tumor metastasis and recurrence. NPC is a highly malignant and metastatic tumor, and symptoms are often absent until the cancer has metastasized to other sites, such as the neck. More than 70% of newly diagnosed cases are classified as locoregionally advanced disease, with 5-year overall survival (OS) rate of 30–50%.1,7 Treatment failure due to distant metastasis, relapse, or both, results in 20 to 30% of patient deaths.8
Studies have shown that EBV is a major contributor to NPC pathogenesis. Furthermore, EBV contributes to NPC metastasis. The EBV-encoded proteins, EBV nuclear antigen 1 (EBNA1), latent membrane protein (LMP) 1, LMP2A and LMP2B can promote cellular transformation, resulting in altered gene expression and increased growth, survival, and invasion of these transformed cells.9,10 In addition, EBV-encoded miRNAs are involved in NPC development11 and metastasis.12 Recent reports showed that EBV-DNA levels were associated with numbers of circulating tumor cells (CTCs) in patients with NPC,13 and EBV-DNA levels and CTC number have been shown to correlate with clinical outcomes of patients with NPC undergoing treatment.14 CTCs and EBV DNA are predictive markers of NPC metastasis,15 and EBV activation is associated with the number of NPC CTC.16 Therefore, we speculate that EBV may participate in NPC metastasis through metastasis.
CTCs are cells derived from primary or metastatic tumors that reach the blood circulation, and are the progenitors of distant metastasis.17 CTCs play an important role in metastasis of various malignancies. Moreover, circulating tumor microemboli (CTM, a cluster of 2 or more CTCs) have also been shown to contribute to tumor metastasis of various cancer types, and are more malignant and aggressive than CTCs.18–22 Analysis of CTCs has provided significant insight into the metastatic process.23 In clinical cases of NPC, CTCs were closely associated with NPC stage, with later clinical stages associated with higher CTC-positive rates.24 Therefore, CTCs may serve as biomarkers for monitoring the therapeutic efficacy of treatments for NPC.25 Studies have shown that EBV proteins participate in NPC metastasis,26,27 and recent reports suggested that EBV may be involved in CTC generation.14,16 Based on these reports, we hypothesized that EBV-mediated CTCs are major contributors to NPC metastasis. In this review, we summarized evidence that EBV promotes NPC metastasis and highlighted the associated mechanisms. We also summarized studies that showed that EBV-encoded products participate in generation of CTCs.
EBV Participates in NPC Pathogenesis
According to the World Health Organization (WHO), NPC tissues have been classified as keratinizing squamous, non-keratinizing, or basaloid squamous. Non-keratinizing cancers can be further subdivided into differentiated non-keratinizing and undifferentiated carcinoma. The keratinizing (keratinizing) subtype accounts for less than 20% of cases worldwide, and this tumor type is relatively rare in southern China. The non-keratinizing subtype constitutes most cases in endemic areas (>95%) and is predominantly associated with EBV infection.28,29 Multiple factors including EBV infection, host genetic, and environmental factors contribute to the development of NPC (Figure 1). The presence of monoclonal EBV episomes in NPC indicates that viral infection precedes clonal expansion of malignant cells.30 EBV has been shown to localize in high-grade (severe dysplastic and in situ carcinoma), preinvasive lesions in the nasopharynx, but not in low-grade lesions or normal nasopharyngeal epithelium. Both high-grade and in situ carcinomas have been to carry monoclonal EBV genomes.31,32
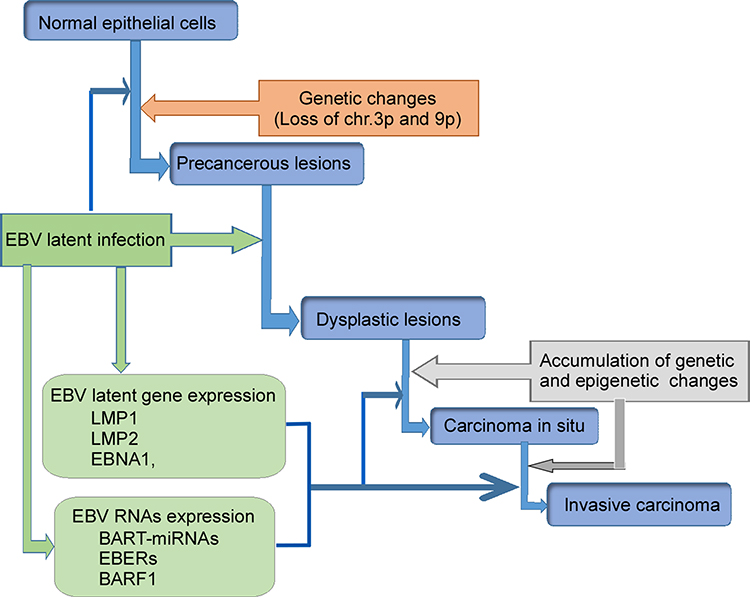 |
Figure 1 Schematic illustration of EBV-encoded products participating in NPC development. Normal epithelial cells being infected with EBV generate genetic changes, and become precancerous lesions. EBV infection produces LMP1, LMP2, EBNA1, BART-miRNA, EBERs, and BARF1. These EBV-products further induce precancerous lesions and dysplastic lesions, and finally result in carcinoma. These virus-products also promote tumor metastasis. Abbreviations: BARF1, BamHI-A rightward frame 1; EBER, Epstein-Barr-Encoded-RNA; EBNA1, EBV nuclear antigen 1; EBV, Epstein–Barr virus; LMP, latent membrane protein. |
EBV can readily infect and transform normal B lymphocytes in vitro.29 Type II EBV latency was originally identified in NPC biopsies.33 Persistent EBV infection and genetically altered epithelial cells are prerequisites for initiation of tumorigenic transformation (Figure 1). Prolonged exposure of the nasopharyngeal mucosa to environmental carcinogens results in DNA damage, and leads to genetic changes in epithelial cells that promote establishment of persistent EBV infection. EBV-DNA encodes type II EBV latency gene products, such as LMP1, LMP2, EBV nuclear antigen (EBNA)-1, BART-miRNAs, EBV-encoded RNAs (EBERs), and BARF1, which disrupt cellular signaling pathways, promote cell proliferation, regulate the host microenvironment, and promote invasive nasopharyngeal EBV infection (Figure 1).33 These findings indicated that EBV infection may be a major pathogenic factor for NPC.
EBV Promotes NPC Invasion and Metastasis
EBV is associated with NPC metastasis.26,27 An EBV-encoded oncoprotein, LMP1 has been shown to trigger a number of signaling pathways, including the NF-κB, PI3K/Akt and mitogen-activated protein kinase (MAPK) pathways, Each of these pathways is actively involved in induction of the epithelial-mesenchymal transition (EMT).34–36 Studies have shown that LMP1 down-regulates E-cadherin expression through promoter methylation.35,36 Furthermore, LMP1 promotes transcriptional inhibition of E-cadherin to promote EMT.37–39 In addition, LMP1 regulates the transcription factors Twist, Snail, and β-catenin (Figure 2).36,38,39 Another important EBV-related oncoprotein, LMP2A has been shown to be over-expressed in most EBV-associated cancers, particularly NPC.40 Immunostaining showed that LMP2A was mainly localized at the tumor invasive front,40 and cell experiments demonstrated that LMP2A induced EMT through activation of the 4EBP1–eIF4E axis, which resulted in enhanced expression of metastatic tumor antigen-1 through targeting of the rapamycin (mTOR) pathway. LMP2A also augments invasive/migratory ability and induces changes in EMT-like biomarkers (Figure 2). EBNA1 up-regulates EMT biomarker expression and induces NPC invasion and metastasis.41 In addition, EBNA1 induces EMT through regulation of transforming growth factor-β (TGF-β), ZEB, Slug, Snail, miR-200, vimentin, occludins-1, and E-cadherin (Figure 2), which are important genes associated with EMT.41,42 Multiple EBV-encoded proteins mediate EMT, and EBV-mediated EMT may be an important factor in NPC metastasis.
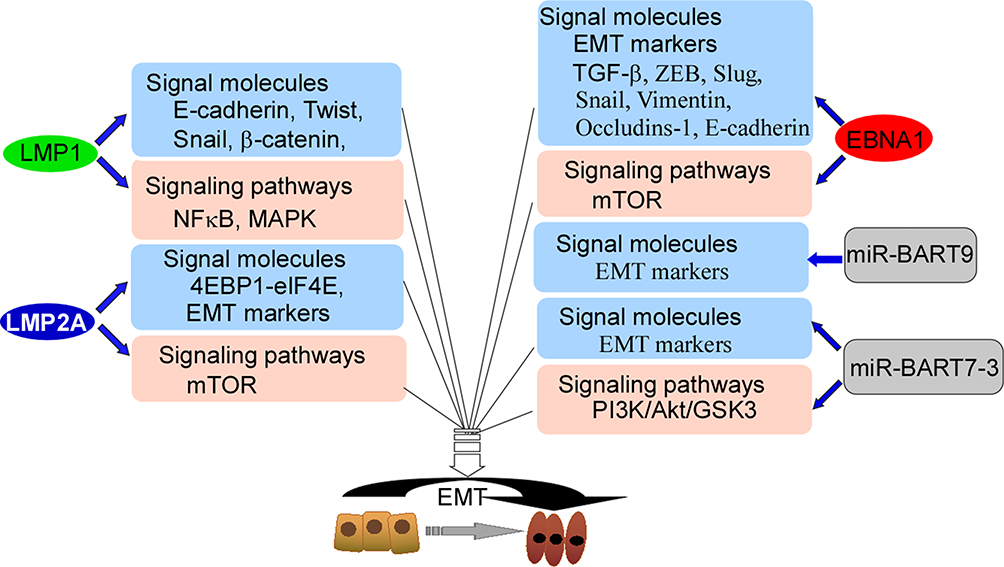 |
Figure 2 The model of EBV-encoded products targeting signal molecules and mediating signal-pathways. LMP1 targets E-cadherin, Twist, Snail, and β-catenin, and also activates NFκB and MAPK, and finally participates in EMT. LMP2A targets 4EBP1-elF4E and EMT markers, and activates mTOR, and finally participates in EMT. EBNA1 regulates EMT markers, TGF-β, ZEB, Slug, Snail, Vimentin, Occludins-1 and E-cadherin, and also activates mTOR, and finally participates in EMT. miR-BART9 and miR-BART7-3 regulate EMT markers, and miR-BART7-3 activates PI3K/Akt/GSK3 pathway, and finally participates in EMT. Abbreviations: MAPK, mitogen-activated protein kinase; EMT, epithelial–mesenchymal transition; miR, microRNA; NFκB, nuclear factor kappa B; TGF-β, transforming growth factor-β; ZEB, zinc finger E-box-binding. |
miRNAs encoded by EBV promote NPC metastasis through promotion of EMT.34,43 Twenty-five EBV-associated miRNAs precursors and 44 mature miRNAs have been identified, and maps at the BHRF1 (4 miRNA) and BART regions (40 miRNA) of EBV genome.44 miR-BART9 has been shown to be over-expressed in all EBV-positive NPC tissues and to promote NPC cell metastasis by targeting E-cadherin and inducing a mesenchymal phenotype (Figure 2).45 A recent study showed that miR-BART7-3p down-regulated epithelial markers, which resulted in mesenchymal features through modulation of the PI3-K/Akt/GSK-3 signaling pathway. These effects resulted in increased expression and nuclear accumulation of Snail and β-catenin in NPC, which correlated positively with lymph node metastasis.46 In EBV-positive gastric carcinoma, EBNA1 mediates EMT by inhibiting the miR-200 family, resulting in up-regulation of ZEB1 and ZEB2. Other latency types I genes (BARF0, LMP2A, EBERs) have a synergistic effect on down-regulation of the miR-200 family (Figure 2).47 miRNAs encoded by EBV mediate EMT of NPC cells, resulting in NPC metastasis.
EBV Promotes Generation of NPC CTCs
Clinical studies have shown that EBV-DNA levels were associated with CTC number in patients with NPC.14,16 During tumor development, various types of cells are observed in the peripheral blood, such as CTCs and CTC clusters. In addition, the presence of disseminated tumor cells (DTCs) in the peripheral blood which are normally present in bone marrow is an important factor in metastasis development.48 CTC clusters consist of various types of cells, such as tumor cells, accessory cells, stromal fibroblasts, endothelial cells, platelets, and immune cells. Complexes of these cells are called microemboli.49 The inner microenvironment of CTC clusters may protect them from being lysed by immune attacks and shear stress in the peripheral blood, resulting in facilitation of metastasis.50 Generation of CTCs by EMT results from four steps: 1) detachment from the tumor mass; 2) invasion of the basal membrane and surrounding tissues; 3) entry of vessels; 4) survival in the peripheral system. The EMT process and the associated regulatory networks promote CTC generation, which increases tumor cell invasiveness, promotes cell intravasation, and facilitates cell survival in the peripheral system. Molecular changes during EMT are regulated by EMT-inducible transcription factors (EMT-TFs). These factors include Snail (Snail family zinc finger transcriptional factors), ZEB1 (zinc finger E-box binding homeobox), Twist (Twist family BHLH transcriptional factor), transcription factor 4 (TCF4), and forkhead box C2 (FOXC2).51–53 In addition to EMT-TFs, some extracellular molecules (TGF-β, FGF, EGF, HGF, Wnt, Notch, and Hedgehog), and related pathways (MAPK, PI3K, NF-κB, Wnt/β-catenin, and Notch) in the tumor microenvironment may also be involved in tumor cell EMT (Figure 3).52,54–56
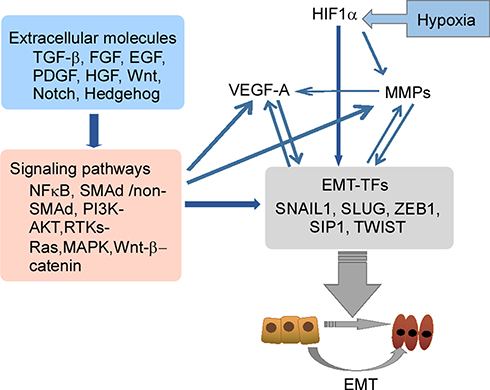 |
Figure 3 The EMT-related regulatory networks. EMT-TFs including Snail 1, Snail 2 (Slug), ZEB1 and Twist play a central role in the networks, and regulate molecular changes during EMT. The EMT regulatory network is an interactive, integrated and precisely regulated network where some important extracellular molecules in the tumor microenvironment, such as TGFβ, HGF, FGF, Wnt and Notch, bind to their respective receptors to induce EMT. Abbreviations: EGF, epidermal growth factor; EMT, epithelial–mesenchymal transition; HGF, hepatocyte growth factor; FGF, fibroblast growth factor; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor kappa B; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β. |
A hallmark of EMT is functional loss of E-cadherin (encoded by CDH1), which is believed to be a suppressor of invasion during carcinoma progression.57 Down-regulation of E-cadherin is an important step in tumor invasion. LMP-1 down-regulates E-cadherin gene expression and induces cell migration activity through cellular DNA methylation.47 miR-BART9 has been shown to be expressed in all EBV-positive NPC tissues, and has been shown to increase E-cadherin expression and induce mesenchymal phenotypes to promote NPC metastasis.45 Several TFs that strongly inhibit cadherin-1 (CDH1),58 such as the Snail, ZEB and basic helix-loop-helix (bHLH) family members are believed to be involved in tumor progression. The Snail zinc finger family is comprised of Snail1 and Snail2 (Slug). Each is capable of binding to the E-box sequences of the E-cadherin gene, resulting in transcriptional repression.59,60 Several miRNAs located in EBV BART clusters (BART-miRNA) may be collectively involved in regulation of EMT genes. miR-BART10-3p can suppress the activity of βTrCP E3 ubiquitin ligase through inhibition of BTRC expression, which results in increased expression of β-catenin and Snail.61 The zinc finger E-box-binding family proteins, ZEB1 (δEF1) and ZEB2 (SIP1), can down-regulate E-cadherin expression, and have been implicated as effectors of malignancy in multiple different human tumors.62,63 ZEB1 has been shown to be a key player in maintenance of EBV latency in certain physiologically relevant cell types. In addition, ZEB2 plays an important role in maintenance of EBV latency.64 Furthermore, ZEB1 and ZEB2 can repress the expression of the EBV BZLF1 gene by directly binding to the ZV element of Zp. The product of the BZLF1 gene, BZLF1 protein, is a key regulator in the switch from EBV latency to lytic replication.65–67 Therefore, both ZEB1 and ZEB2 can regulate the switch between latency and lytic replication of EBV.64
Increasing evidence has indicated that some bHLH factors, and the Id HLH subfamily, play important roles in tumor cell invasion and metastasis. Twist, a member of the bHLH family, represses CDH1 repressor and induces EMT. bHLH factors play significant roles in modulation of the cell-cycle, proliferation and angiogenesis in tumor development.68,69 TGF-β is an inducer of EMT, and is a major cytokine. Further, TGF-β promotes metastasis and cancer development. In the early stages of tumor development, TGF-β signaling plays a suppressive role by inhibiting cell cycle progression from G1 to S, and promoting apoptosis, senescence and differentiation.70–72 In contrast, in advanced tumors, TGF-β acts as a tumor promoter by inducing EMT, migration, invasion, metastasis, and immune escape.72–74 EBV-positive and -negative cells respond differently to TGF-β. For example, EBV-negative cells are sensitive to TGF-β-mediated growth inhibition and apoptosis75–77 and these responses do not occur in EBV-positive cells.78–80 The EBV-positive NPC cell line, C666-1, has been shown to be resistant to TGF-β1-mediated growth inhibition and apoptosis. However, the EBV-negative NPC cell line, CNE-2 did not respond to exogenous TGF-β1.81 Loss of TGF-β responsiveness is a critical event in tumorigenesis of EBV-infected cells. Smad 2/3 are bound to Smad 4 to form a complex. When this complex becomes phosphorylated, it is translocated into the nucleus and interacts with transcriptional factors that regulate the expression of EMT-specific genes.82 TGF-β promotes EMT through activation of the PI3K/Akt signaling through non-Smad signaling pathways (Figure 3).83 In addition, TGF-β has been shown to play different roles in different stages of various cancers. TGF-β maintains cell proliferation and differentiation under physiological conditions or early cancer, but can promote invasion and metastasis in advanced cancers.84 Furthermore, TGF-β directly activates core EMT TFs (Snail, ZEB and Twist). TGF-β is critical for Snail1 activation, and Snail1 can also be activated by the Wnt family, Notch proteins and receptor tyrosine kinases (RTKs–Ras). These pathways are activated to varying degrees under physiological or pathological conditions.51
A negative-feedback mechanism was recently described for SNAI1 regulation by scattering factor (such as hepatocyte growth factor, HGF), which involves the MAPK target protein early growth response (EGR)-1.85 Inhibitor of DNA binding (Id) protein can repress the activities of bHLH class I proteins. In mammalian cell systems, Id protein can be induced by several effectors that promote EMT and/or oncogenic pathways, such as TGFβ-BMP, vascular endothelial grown factor (VEGF) or insulin-like growth factor (IGF)-1, which results in activation of Ras, β-catenin, or phosphatidylinositol 3-kinase (PI3K).68,69 LMP1-mediating Id1 is involved in suppression of p16INK4a expression in nasopharyngeal epithelial cells. During EMT progression, the components responsible for intercellular junctions, such as E-cadherin, claudin, occludins, and desmosomes, are directly down-regulated by Snail, Slug, and Smad interacting protein (SIP)-1 (Figure 3).86–89 The expressions of these proteins are involved in cytoskeleton reconstruction, which changes cell morphology to a spindle-like shape for suitable migration.90 Some important extracellular factors, such as TGFβ, FGF and Wnt, participate in the regulation network associated with EMT and/or matrix metalloproteinase (MMP) expression.52,54,55 The mechanisms of activation of this network are similar to those of extracellular factors under hypoxic conditions.91–94 Hypoxia-inducible factor (HIF)-1, a principal oxygen-sensing transcription factor, is comprised of HIF1A and HIF1H subunits.95 The expression of HIF1A is elevated in EBV type II and type III latently infected cells, and is involved in induction of VEGF. LMP1 regulates HIF1A expression through increased production of reactive oxygen species and activation of the p42/p44 MAPK pathway (Figure 3).96 Under physiological conditions, modification of HIF1A by prolyl hydroxylase domain enzyme (PHD)-1/3, results in recruitment of von Hippel-Lindau (VHL) protein97 and components of the ubiquitin ligase system to promote ubiquitination and proteasomal degradation of HIF1A. The expression levels of HIF1A are typically low under normoxic condition. In addition, LMP1 increases Siah1 levels, and high Siah1 induces degradation of PHD1/PHD3 to maintain appropriate levels of PHD1/PHD3.98 When levels of PHD1/PHD3 are low, HIF1A is not subject to ubiquitin-mediated protein degradation, resulting in accumulation.99 HIF1 is a transcription factor that controls the expression of at least 40 genes involved in tumor angiogenesis, invasion and metastasis.100 These findings showed that regulatory factors and pathways activated by EBV infection play important roles in generation of CTCs, which promote cell invasion, angiogenesis, intravasation, therapy resistance, and tumor cell survival.
The Role of CTCs in NPC
CTCs are highly heterogeneous, and the molecular features of CTCs often differ among subpopulations, which may play different roles in cancer progression.101,102 Increasing numbers of studies have begun to evaluate the genotypes and phenotypes of CTCs. Markers of EMT have the potential for use as biomarkers of CTCs in various cancers. EMT has been shown to promote invasion and motility,103,104 and CTC subpopulations with EMT markers may contribute to cancer progression.105,106 Expression of EMT markers in CTCs has important clinical implications, as EMT is believed to promote stemness of CTCs,107 and overexpression of EMT markers in CTCs has been shown to be accompanied by increased expression stem cell markers such as ALDH1 and CD133 in breast cancer.108–110 Markers of EMT can be classified into three categories: epithelial cell markers, mesenchymal markers, and regulatory factors.111 Epithelial markers are molecular biomarkers that are often used to detect CTCs and confirm their epithelial origin. Traditional epithelial-based CTC detection techniques may not detect some invasive and highly metastatic cells in the peripheral circulation, and CTCs with pure mesenchymal or heterozygous EMT phenotypes may be better indicators of risk of tumor metastasis than CTCs with a purely epithelial phenotype.112–115 EMT-TFs and their related pathways regulate molecular elements in EMT progression, and these elements, such as Twist, Snail, Slug, and ZEB1, may also be markers of EMT. Twist, Snail, Slug, and ZEB1 down-regulate E-cadherin and are associated with cancer progression. In addition, these TFs are also good indicators of CTC EMT status.51,116 Studies have shown that PI3K and Akt act as central elements of the PI3K/Akt/mTOR pathway, and can be used as mesenchymal markers in CTCs.108,117,118 Recent studies have attempted to identify specific EMT markers in CTCs. These studies indicated that EMT and stem cell markers are frequently over-expressed in CTCs, regardless of cancer type. These corresponding EMT and stem cell markers were detected in 18% and 5% of CTC-negative group.108 Therefore, increasing numbers of studies have explored genotypes and phenotypes of CTCs, and focused on cancer-specific phenotypes. For example, some reports have evaluated human epidermal growth factor receptor (HER)-2 levels in CTCs of patients with breast cancer.119,120 AR gene status in CTCs of patients with prostate cancer,121 epidermal growth factor receptor (EGFR) mutations in patients with lung cancer, and Kirsten rat sarcoma viral oncogene (KRAS) mutations in patients with colorectal cancer.122,123 A number of studies have reported that CTCs have a close relationship with clinical characteristics in various types of cancer.124–126
Studies that have evaluated CTCs in NPC are lacking. Cyclooxygenase (COX) −2 is an enzyme involved in conversion of arachidonic acid to prostaglandins, and has been to stimulate tumor cell proliferation, angiogenesis, and invasiveness, and to promote resistance apoptosis.127,128 In patients with NPC, the percentage of CTCs that expressed COX-2 at baseline and at the end of treatment were 66.4% and 46.1%, respectively.129 Expression of COX-2 in CTCs was significantly associated with unfavorable treatment response, and patients with high COX-2 levels were at increased risk of local-regional relapse and distant metastasis.129 MMP9 has been shown to participate in degradation of environmental barriers, which results in increased risk of metastasis.130 The positive rate of MMP9 in mesenchymal CTCs was very high (71.2%), and was low in the complex of epithelial and mesenchymal. However, the proportion of cells that exhibited moderate MMP9 expression was highest in hybrid CTCs, and the mechanisms associated with MMP9 expression in these cells has not been characterized.131 Future studies should focus on core molecules in the signaling pathways activated by EBV in NPC.
Conclusion
NPC is significantly different from other epithelial head and neck tumors. NPC has significant regional and etiological characteristics, and EBV infection is specific pathogenic factor for NPC. The non-keratinizing (non-keratinizing) subtype constitutes most cases in endemic areas (>95%), and is predominantly associated with EBV infection.28,29 EBV-DNA encodes type II EBV latency gene products, such as LMP1, LMP2, EBNA1, BART-miRNAs, EBERs, and BARF1. These gene products induce EMT by indirectly inducing EMT-TFs.33 EBV-encoded products also promote EMT-TF expression through post-transcriptional regulation.36,38,39 EMT promotes CTC generation through detachment from the tumor mass, invasion of the basement membrane and surrounding tissues, and survival of CTCs in the periphery. EMT phenotypes are commonly used to distinguish CTC subtypes. Studies have suggested that CTC count and karyotyping may indicate disease severity, and dynamic monitoring of CTC number may allow for assessment of treatment outcomes in real-time. Recent studies have shown that CTC karyotypes in recent years showed that CTC karyotyping may provide a potential method for monitoring chemical resistance and predicting chemical efficacy during treatment of NPC, and evaluation of CTCs may be critical during follow-up of patients with NPC. CTC karyotype is also associated with NPC staging, chemosensitivity, and drug resistance.129 However, the clinical significance of CTC detection in NPC requires further characterization.
Abbreviations
BARF1, BamHI-A rightward frame 1; bHLH, basic helix-loop-helix; CDH1, cadherin-1; COX2, cyclooxygenase2; CTC, circulating tumor cells; DTC, disseminated tumor cell; EBNA1, EBV nuclear antigen 1; EBV, Epstein–Barr virus; EBER, EBV-encoded RNA; EGF, epidermal growth factor; EGR1, early growth response 1; EGFR, epidermal growth factor receptor; EMT, epithelial–mesenchymal transition; EMT-TF, EMT-transcript factor; FGF, fibroblast growth factor; FOXC2, forkhead box C2; HER2, human epidermal growth factor receptor2; HGF, hepatocyte growth factor; HIF, hypoxia-inducible factor; Id, inhibitor of DNA binding; IGF1, insulin-like growth factor 1; KRAS, kirsten rat sarcoma viral oncogene; LMP, latent membrane protein; MAPK, mitogen-activated protein kinase; miRNA, microRNA; MMP, matrix metalloproteinase; NFκB, nuclear factor kappa B; NPC, nasopharyngeal carcinoma; PHD, prolyl hydroxylase domain enzyme; PI3K, phosphatidylinositol3-kinase; SIP1, Smad interacting protein1; TCF4, transcription factor4, TGF-β, transforming growth factor-β; VEGF, vascular endothelial grown factor; VHL, von Hippel-Lindau; ZEB, zinc finger E-box-binding.
Acknowledgments
We thank the members in Clinical Laboratory of Hunan Cancer Hospital and Xiangya Medical School of Central South University for contributions and Dr, Dong Z in Hormel Institute of University of Minnesota helpful discussion.
Author Contributions
All authors contributed towards data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Funding
This work was supported in part by the National Natural Science Foundation of China (81872226, 81502346), Hunan Provincial Natural Science Foundation of China (2018JJ6131, 2019JJ40175), Changsha Science and Technology Project (kg1801107), and Research Projects of Hunan Health Commission (B2019084).
Disclosure
The authors declare no competing financial interests in this work.
References
1. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64–80. doi:10.1016/s0140-6736(19)30956-0
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.v68.6
3. Tang FQ, Duan CJ, Huang DM, et al. HSP70 and mucin 5B: novel protein targets of N,N’-dinitrosopiperazine-induced nasopharyngeal tumorigenesis. Cancer Sci. 2009;100(2):216–224. doi:10.1111/j.1349-7006.2008.01028.x
4. Zong YS, Sham JS, Ng MH, et al. Immunoglobulin A against viral capsid antigen of Epstein-Barr virus and indirect mirror examination of the nasopharynx in the detection of asymptomatic nasopharyngeal carcinoma. Cancer. 1992;69(1):3–7. doi:10.1002/1097-0142(19920101)69:1<3::aid-cncr2820690104>3.0.co;2-7
5. Chen C, Shen LJ, Li BF, Gao J, Xia YF. Smoking is a poor prognostic factor for male nasopharyngeal carcinoma treated with radiotherapy. Radiother Oncol. 2014;110(3):409–415. doi:10.1016/j.radonc.2013.08.003
6. Sousa H, Mesquita L, Ribeiro J, Catarino R, Breda E, Medeiros R. Polymorphisms in host immune response associated genes and risk of nasopharyngeal carcinoma development in Portugal. Immunobiology. 2016;221(2):145–152. doi:10.1016/j.imbio.2015.09.015
7. Rottey S, Madani I, Deron P, Van Belle S. Modern treatment for nasopharyngeal carcinoma: current status and prospects. Curr Opin Oncol. 2011;23(3):254–258. doi:10.1097/CCO.0b013e328344f527
8. Liao XB, Mao YP, Liu LZ, et al. How does magnetic resonance imaging influence staging according to AJCC staging system for nasopharyngeal carcinoma compared with computed tomography? Int J Radiat Oncol Biol Phys. 2008;72(5):1368–1377. doi:10.1016/j.ijrobp.2008.03.017
9. Tsao SW, Tsang CM, To KF, Lo KW. The role of Epstein-Barr virus in epithelial malignancies. J Pathol. 2015;235(2):323–333. doi:10.1002/path.4448
10. Marquitz AR, Mathur A, Shair KH, Raab-Traub N. Infection of Epstein-Barr virus in a gastric carcinoma cell line induces anchorage independence and global changes in gene expression. Proc Natl Acad Sci U S A. 2012;109(24):9593–9598. doi:10.1073/pnas.1202910109
11. Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS One. 2010;5(9). doi:10.1371/journal.pone.0012745
12. Cai L, Ye Y, Jiang Q, et al. Epstein-Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat Commun. 2015;6:7353. doi:10.1038/ncomms8353
13. Xie XQ, Luo Y, Ma XL, et al. Clinical significance of circulating tumor cells and their expression of cyclooxygenase-2 in patients with nasopharyngeal carcinoma. Eur Rev Med Pharmacol Sci. 2019;23(16):6951–6961. doi:10.26355/eurrev_201908_18735
14. Vo JH, Nei WL, Hu M, et al. Comparison of circulating tumour cells and circulating cell-free Epstein-Barr virus DNA in patients with nasopharyngeal carcinoma undergoing radiotherapy. Sci Rep. 2016;6(1):13. doi:10.1038/s41598-016-0006-3
15. You R, Liu YP, Lin M, et al. Relationship of circulating tumor cells and Epstein-Barr virus DNA to progression-free survival and overall survival in metastatic nasopharyngeal carcinoma patients. Int J Cancer. 2019;145(10):2873–2883. doi:10.1002/ijc.32380
16. He C, Huang X, Su X, et al. The association between circulating tumor cells and Epstein-Barr virus activation in patients with nasopharyngeal carcinoma. Cancer Biol Ther. 2017;18(11):888–894. doi:10.1080/15384047.2017.1281493
17. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14(9):623–631. doi:10.1038/nrc3820
18. Bidard FC, Mathiot C, Delaloge S, et al. Single circulating tumor cell detection and overall survival in nonmetastatic breast cancer. Ann Oncol. 2010;21(4):729–733. doi:10.1093/annonc/mdp391
19. Miller MC, Doyle GV, Terstappen LW. Significance of circulating tumor cells detected by the cell search system in patients with metastatic breast colorectal and prostate cancer. J Oncol. 2010;2010:617421. doi:10.1155/2010/617421
20. Wu C, Hao H, Li L, et al. Preliminary investigation of the clinical significance of detecting circulating tumor cells enriched from lung cancer patients. J Thorac Oncol. 2009;4(1):30–36. doi:10.1097/JTO.0b013e3181914125
21. Okabe H, Tsunoda S, Hosogi H, et al. Circulating tumor cells as an independent predictor of survival in advanced gastric cancer. Ann Surg Oncol. 2015;22(12):3954–3961. doi:10.1245/s10434-015-4483-6
22. Zhang D, Zhao L, Zhou P, et al. Circulating tumor microemboli (CTM) and vimentin+ circulating tumor cells (CTCs) detected by a size-based platform predict worse prognosis in advanced colorectal cancer patients during chemotherapy. Cancer Cell Int. 2017;17:6. doi:10.1186/s12935-016-0373-7
23. Pantel K, Alix-Panabieres C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013;73(21):6384–6388. doi:10.1158/0008-5472.CAN-13-2030
24. Chen Z, Xu L, Xu X, Yuan C. The clinical value of detecting circulating tumour cells in the peripheral blood of nasopharyngeal carcinoma patients. Oncol Lett. 2018;15(5):6283–6290. doi:10.3892/ol.2018.8155
25. Wen Z, Li Z, Yong P, et al. Detection and clinical significance of circulating tumor cells in patients with nasopharyngeal carcinoma. Oncol Lett. 2019;18(3):2537–2547. doi:10.3892/ol.2019.10560
26. Lee WY, Hsiao JR, Jin YT, Tsai ST. Epstein-Barr virus detection in neck metastases by in-situ hybridization in fine-needle aspiration cytologic studies: an aid for differentiating the primary site. Head Neck. 2000;22(4):336–340. doi:10.1002/1097-0347(200007)22:4<336::aid-hed4>3.0.co;2-t
27. Lan YY, Hsiao JR, Chang KC, et al. Epstein-Barr virus latent membrane protein 2A promotes invasion of nasopharyngeal carcinoma cells through ERK/Fra-1-mediated induction of matrix metalloproteinase 9. J Virol. 2012;86(12):6656–6667. doi:10.1128/JVI.00174-12
28. Wang HY, Chang YL, To KF, et al. A new prognostic histopathologic classification of nasopharyngeal carcinoma. Chin J Cancer. 2016;35:41. doi:10.1186/s40880-016-0103-5
29. Young LS, Dawson CW. Epstein-Barr virus and nasopharyngeal carcinoma. Chin J Cancer. 2014;33(12):581–590. doi:10.5732/cjc.014.10197
30. Raab-Traub N, Flynn K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell. 1986;47(6):883–889. doi:10.1016/0092-8674(86)90803-2
31. Pathmanathan R, Prasad U, Sadler R, Flynn K, Raab-Traub N. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N Engl J Med. 1995;333(11):693–698. doi:10.1056/NEJM199509143331103
32. Chan AS, To KF, Lo KW, et al. High frequency of chromosome 3p deletion in histologically normal nasopharyngeal epithelia from southern Chinese. Cancer Res. 2000;60(19):5365–5370.
33. Brooks L, Yao QY, Rickinson AB, Young LS. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol. 1992;66(5):2689–2697.
34. Chen X, Bode AM, Dong Z, Cao Y. The epithelial-mesenchymal transition (EMT) is regulated by oncoviruses in cancer. FASEB J. 2016;30(9):3001–3010. doi:10.1096/fj.201600388R
35. Tsai CL, Li HP, Lu YJ, et al. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006;66(24):11668–11676. doi:10.1158/0008-5472.can-06-2194
36. Cyprian FS, Al-Antary N, Al Moustafa AE. HER-2/Epstein-Barr virus crosstalk in human gastric carcinogenesis: a novel concept of oncogene/oncovirus interaction. Cell Adh Migr. 2018;12(1):1–4. doi:10.1080/19336918.2017.1330244
37. Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A. 2002;99(15):10084–10089. doi:10.1073/pnas.152059399
38. Horikawa T, Yang J, Kondo S, et al. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67(5):1970–1978. doi:10.1158/0008-5472.CAN-06-3933
39. Horikawa T, Yoshizaki T, Kondo S, Furukawa M, Kaizaki Y, Pagano JS. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br J Cancer. 2011;104(7):1160–1167. doi:10.1038/bjc.2011.38
40. Kong QL, Hu LJ, Cao JY, et al. Epstein-Barr virus-encoded LMP2A induces an epithelial-mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog. 2010;6(6):e1000940. doi:10.1371/journal.ppat.1000940
41. Wang L, Tian WD, Xu X, et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer. 2014;120(3):363–372. doi:10.1002/cncr.28418
42. Gaur N, Gandhi J, Robertson ES, Verma SC, Kaul R. Epstein-Barr virus latent antigens EBNA3C and EBNA1 modulate epithelial to mesenchymal transition of cancer cells associated with tumor metastasis. Tumour Biol. 2015;36(4):3051–3060. doi:10.1007/s13277-014-2941-6
43. Yoshizaki T, Kondo S, Wakisaka N, et al. Pathogenic role of Epstein-Barr virus latent membrane protein-1 in the development of nasopharyngeal carcinoma. Cancer Lett. 2013;337(1):1–7. doi:10.1016/j.canlet.2013.05.018
44. Skalsky RL, Cullen BR. EBV noncoding RNAs. Curr Top Microbiol Immunol. 2015;391:181–217. doi:10.1007/978-3-319-22834-1_6
45. Hsu CY, Yi YH, Chang KP, Chang YS, Chen SJ, Chen HC. The Epstein-Barr virus-encoded microRNA MiR-BART9 promotes tumor metastasis by targeting E-cadherin in nasopharyngeal carcinoma. PLoS Pathog. 2014;10(2):e1003974. doi:10.1371/journal.ppat.1003974
46. Cai LM, Lyu XM, Luo WR, et al. EBV-miR-BART7-3p promotes the EMT and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene. 2015;34(17):2156–2166. doi:10.1038/onc.2014.341
47. Krishna SM, Kattoor J, Balaram P. Down regulation of adhesion protein E-cadherin in Epstein-Barr virus infected nasopharyngeal carcinomas. Cancer Biomark. 2005;1(6):271–277. doi:10.3233/cbm-2005-1602
48. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458. doi:10.1038/nrc1098
49. Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–1122. doi:10.1016/j.cell.2014.07.013
50. Hong Y, Fang F, Zhang Q. Circulating tumor cell clusters: what we know and what we expect (Review). Int J Oncol. 2016;49(6):2206–2216. doi:10.3892/ijo.2016.3747
51. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. doi:10.1038/nrc2131
52. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. doi:10.1016/j.devcel.2008.05.009
53. Heerboth S, Housman G, Leary M, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4:6. doi:10.1186/s40169-015-0048-3
54. Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med. 2008;22(3):271–275.
55. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi:10.1038/nrc3447
56. Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98(10):1512–1520. doi:10.1111/j.1349-7006.2007.00550.x
57. Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198(1):11–26. doi:10.1016/0304-419x(94)90003-5
58. Peinado H, Cano A. New potential therapeutic targets to combat epithelial tumor invasion. Clin Transl Oncol. 2006;8(12):851–857. doi:10.1007/s12094-006-0148-z
59. Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116(Pt3):499–511. doi:10.1242/jcs.00224
60. Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62(6):1613–1618.
61. Yan Q, Zeng Z, Gong Z, et al. EBV-miR-BART10-3p facilitates epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget. 2015;6(39):41766–41782. doi:10.18632/oncotarget.v6i39
62. Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24(14):2375–2385. doi:10.1038/sj.onc.1208429
63. Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. doi:10.1016/S1097-2765(01)00260-X
64. Ellis AL, Wang Z, Yu X, Mertz JE. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J Virol. 2010;84(12):6139–6152. doi:10.1128/JVI.02706-09
65. Countryman J, Jenson H, Seibl R, Wolf H, Miller G. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J Virol. 1987;61(12):3672–3679.
66. Feederle R, Kost M, Baumann M, et al. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000;19(12):3080–3089. doi:10.1093/emboj/19.12.3080
67. Sinclair AJ, Brimmell M, Shanahan F, Farrell PJ. Pathways of activation of the Epstein-Barr virus productive cycle. J Virol. 1991;65(5):2237–2244.
68. Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5(8):603–614. doi:10.1038/nrc1673
69. Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13(8):410–418. doi:10.1016/S0962-8924(03)00147-8
70. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–821. doi:10.1038/nrc1208
71. Yanagihara K, Tsumuraya M. Transforming growth factor beta 1 induces apoptotic cell death in cultured human gastric carcinoma cells. Cancer Res. 1992;52(14):4042–4045.
72. Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012:381428. doi:10.5402/2012/381428
73. Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8(23):1243–1252. doi:10.1016/s0960-9822(07)00533-7
74. Ueki N, Nakazato M, Ohkawa T, et al. Excessive production of transforming growth-factor beta 1 can play an important role in the development of tumorigenesis by its action for angiogenesis: validity of neutralizing antibodies to block tumor growth. Biochim Biophys Acta. 1992;1137(2):189–196. doi:10.1016/0167-4889(92)90201-l
75. MacDonald I, Wang H, Grand R, et al. Transforming growth factor-beta 1 cooperates with anti-immunoglobulin for the induction of apoptosis in group I (biopsy-like) Burkitt lymphoma cell lines. Blood. 1996;87(3):1147–1154. doi:10.1182/blood.V87.3.1147.bloodjournal8731147
76. Saltzman A, Munro R, Searfoss G, Franks C, Jaye M, Ivashchenko Y. Transforming growth factor-beta-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp Cell Res. 1998;242(1):244–254. doi:10.1006/excr.1998.4096
77. Inman GJ, Allday MJ. Apoptosis induced by TGF-beta 1 in Burkitt’s lymphoma cells is caspase 8 dependent but is death receptor independent. J Immunol. 2000;165(5):2500–2510. doi:10.4049/jimmunol.165.5.2500
78. Blomhoff HK, Smeland E, Mustafa AS, Godal T, Ohlsson R. Epstein-Barr virus mediates a switch in responsiveness to transforming growth factor, type beta, in cells of the B cell lineage. Eur J Immunol. 1987;17(2):299–301. doi:10.1002/(ISSN)1521-4141
79. Arvanitakis L, Yaseen N, Sharma S. Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-beta 1-mediated growth inhibition in EBV-positive B cells. J Immunol. 1995;155(3):1047–1056.
80. Kumar A, Rogers T, Maizel A, Sharma S. Loss of transforming growth factor beta 1 receptors and its effects on the growth of EBV-transformed human B cells. J Immunol. 1991;147(3):998–1006.
81. Xiao J, Xiang Q, Xiao YC, et al. The effect of transforming growth factor-beta1 on nasopharyngeal carcinoma cells: insensitive to cell growth but functional to TGF-beta/Smad pathway. J Exp Clin Cancer Res. 2010;29:35. doi:10.1186/1756-9966-29-35
82. Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–5774. doi:10.1038/sj.onc.1208927
83. Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179(1–2):11–23. doi:10.1159/000084505
84. Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100(15):8621–8623. doi:10.1073/pnas.1633291100
85. Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of snail. EMBO J. 2006;25(15):3534–3545. doi:10.1038/sj.emboj.7601213
86. Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278(23):21113–21123. doi:10.1074/jbc.M211304200
87. Mann JR, Backlund MG, Buchanan FG, et al. Repression of prostaglandin dehydrogenase by epidermal growth factor and snail increases prostaglandin E2 and promotes cancer progression. Cancer Res. 2006;66(13):6649–6656. doi:10.1158/0008-5472.CAN-06-1787
88. Dohadwala M, Yang SC, Luo J, et al. Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006;66(10):5338–5345. doi:10.1158/0008-5472.can-05-3635
89. Yang AD, Camp ER, Fan F, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66(1):46–51. doi:10.1158/0008-5472.CAN-05-3086
90. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28(1–2):15–33. doi:10.1007/s10555-008-9169-0
91. Yang MH, Wu MZ, Chiou SH, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi:10.1038/ncb1691
92. Imai T, Horiuchi A, Wang C, et al. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163(4):1437–1447. doi:10.1016/S0002-9440(10)63501-8
93. Wang L, Yu Y, Guan H, Liu T, Qiao C. 67-kDa Laminin receptor contributes to hypoxia-induced migration and invasion of trophoblast-like cells by mediating matrix metalloproteinase-9. Clin Exp Pharmacol Physiol. 2015;42(5):549–558. doi:10.1111/cep.2015.42.issue-5
94. Nakayama K. cAMP-response element-binding protein (CREB) and NF-kappaB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase MMP1. J Biol Chem. 2013;288(31):22584–22595. doi:10.1074/jbc.M112.421636
95. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi:10.1073/pnas.92.12.5510
96. Wakisaka N, Kondo S, Yoshizaki T, Murono S, Furukawa M, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2004;24(12):5223–5234. doi:10.1128/MCB.24.12.5223-5234.2004
97. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi:10.1038/20459
98. Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. doi:10.1074/jbc.M406026200
99. Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107(1):1–3. doi:10.1016/s0092-8674(01)00518-9
100. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7(14):2090–2096. doi:10.4161/cc.7.14.6324
101. Massard C, Oulhen M, Le Moulec S, et al. Phenotypic and genetic heterogeneity of tumor tissue and circulating tumor cells in patients with metastatic castration-resistant prostate cancer: a report from the PETRUS prospective study. Oncotarget. 2016;7(34):55069–55082. doi:10.18632/oncotarget.v7i34
102. Powell AA, Talasaz AH, Zhang H, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012;7(5):e33788. doi:10.1371/journal.pone.0033788
103. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.007
104. Bonnomet A, Brysse A, Tachsidis A, et al. Epithelial-to-mesenchymal transitions and circulating tumor cells. J Mammary Gland Biol Neoplasia. 2010;15(2):261–273. doi:10.1007/s10911-010-9174-0
105. Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012;79(4):195–208. doi:10.1159/000337106
106. Bednarz-Knoll N, Alix-Panabieres C, Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012;31(3–4):673–687. doi:10.1007/s10555-012-9370-z
107. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi:10.1016/j.cell.2008.03.027
108. Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012;14(1):R15. doi:10.1186/bcr3099
109. Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11(4):R46. doi:10.1186/bcr2333
110. Armstrong AJ, Marengo MS, Oltean S, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9(8):997–1007. doi:10.1158/1541-7786.MCR-10-0490
111. Jie XX, Zhang XY, Xu CJ. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: mechanisms and clinical applications. Oncotarget. 2017;8(46):81558–81571. doi:10.18632/oncotarget.18277
112. Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 2011;8(8):428–439. doi:10.1038/nrurol.2011.85
113. Busch EL, Keku TO, Richardson DB, et al. Evaluating markers of epithelial-mesenchymal transition to identify cancer patients at risk for metastatic disease. Clin Exp Metastasis. 2016;33(1):53–62. doi:10.1007/s10585-015-9757-7
114. Tan TZ, Miow QH, Miki Y, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6(10):1279–1293. doi:10.15252/emmm.201404208
115. Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol. 2016;10(3):374–394. doi:10.1016/j.molonc.2016.01.007
116. Wushou A, Hou J, Zhao YJ, Shao ZM. Twist-1 up-regulation in carcinoma correlates to poor survival. Int J Mol Sci. 2014;15(12):21621–21630. doi:10.3390/ijms151221621
117. Barriere G, Riouallon A, Renaudie J, Tartary M, Rigaud M. Mesenchymal and stemness circulating tumor cells in early breast cancer diagnosis. BMC Cancer. 2012;12:114. doi:10.1186/1471-2407-12-114
118. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi:10.3389/fonc.2014.00064
119. Apostolaki S, Perraki M, Kallergi G, et al. Detection of occult HER2 mRNA-positive tumor cells in the peripheral blood of patients with operable breast cancer: evaluation of their prognostic relevance. Breast Cancer Res Treat. 2009;117(3):525–534. doi:10.1007/s10549-008-0239-3
120. Ligthart ST, Bidard FC, Decraene C, et al. Unbiased quantitative assessment of Her-2 expression of circulating tumor cells in patients with metastatic and non-metastatic breast cancer. Ann Oncol. 2013;24(5):1231–1238. doi:10.1093/annonc/mds625
121. Attard G, Swennenhuis JF, Olmos D, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69(7):2912–2918. doi:10.1158/0008-5472.can-08-3667
122. Yang MJ, Chiu HH, Wang HM, et al. Enhancing detection of circulating tumor cells with activating KRAS oncogene in patients with colorectal cancer by weighted chemiluminescent membrane array method. Ann Surg Oncol. 2010;17(2):624–633. doi:10.1245/s10434-009-0831-8
123. Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359(4):366–377. doi:10.1056/NEJMoa0800668
124. Das M, Riess JW, Frankel P, et al. ERCC1 expression in circulating tumor cells (CTCs) using a novel detection platform correlates with progression-free survival (PFS) in patients with metastatic non-small-cell lung cancer (NSCLC) receiving platinum chemotherapy. Lung Cancer. 2012;77(2):421–426. doi:10.1016/j.lungcan.2012.04.005
125. Ning Y, Zhang W, Hanna DL, et al. Clinical relevance of EMT and stem-like gene expression in circulating tumor cells of metastatic colorectal cancer patients. Pharmacogenomics J. 2018;18(1):29–34. doi:10.1038/tpj.2016.62
126. Kulemann B, Liss AS, Warshaw AL, et al. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol. 2016;37(6):7547–7554. doi:10.1007/s13277-015-4589-2
127. Greenhough A, Smartt HJ, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30(3):377–386. doi:10.1093/carcin/bgp014
128. Simonsson M, Bjorner S, Markkula A, et al. The prognostic impact of COX-2 expression in breast cancer depends on oral contraceptive history, preoperative NSAID use, and tumor size. Int J Cancer. 2017;140(1):163–175. doi:10.1002/ijc.30432
129. Li YJ, Luo Y, Xie XQ, Li P, Wang F. The prognostic value of COX-2 expression on circulating tumor cells in nasopharyngeal carcinoma: a prospective analysis. Radiother Oncol. 2018;129(2):396–402. doi:10.1016/j.radonc.2018.07.022
130. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. doi:10.1038/nrc745
131. Si Y, Lan G, Deng Z, et al. Distribution and clinical significance of circulating tumor cells in nasopharyngeal carcinoma. Jpn J Clin Oncol. 2016;46(7):622–630. doi:10.1093/jjco/hyw046
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.