")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Epigenetic Regulation of PDX-1 in Type 2 Diabetes Mellitus
Received 13 November 2020
Accepted for publication 16 January 2021
Published 2 February 2021 Volume 2021:14 Pages 431—442
DOI https://doi.org/10.2147/DMSO.S291932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Jiangman Liu, Guangping Lang, Jingshan Shi
Key Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi, 563000, People’s Republic of China
Correspondence: Jingshan Shi Tel +86-851-286-436-66
Fax +86-851-286-423-03
Email [email protected]
Abstract: Type 2 diabetes mellitus (T2DM) is a metabolic disease characterized by hyperglycemia which is caused by insufficient insulin secretion or insulin resistance. Interaction of genetic, epigenetic and environmental factors plays a significant role in the development of T2DM. Several environmental factors including diet and lifestyle, as well as age have been associated with an increased risk for T2DM. It has been demonstrated that these environmental factors may affect global epigenetic status, and alter the expression of susceptible genes, thereby contributing to the pathogenesis of T2DM. In recent years, a growing body of molecular and genetic studies in diabetes have been focused on the ways to restore the numbers or function of β-cells in order to reverse a range of metabolic consequences of insulin deficiency. The pancreatic duodenal homeobox 1 (PDX-1) is a transcriptional factor that is essential for the development and function of islet cells. A number of studies have shown that there is a significant increase in the level of DNA methylation of PDX-1 resulting in reduced activity in T2DM islets. The decrease in PDX-1 activity may be a critical mediator causing dysregulation of pancreatic β cells in T2DM. This article reviews the epigenetic mechanisms of PDX-1 involved in T2DM, focusing on diabetes and DNA methylation, and discusses some potential strategies for the application of PDX-1 in the treatment of diabetes.
Keywords: type 2 diabetes mellitus, β-cells, PDX-1, epigenetic, DNA methylation
Introduction
Type 2 diabetes mellitus (T2DM), also known as non-insulin-dependent diabetes mellitus (NIDDM), is a chronic metabolic disorder characterized by hyperglycemia and hyperlipidemia. It is usually caused by overnutrition, insulin resistance, defective insulin secretion, and other endocrine abnormalities.1 Chronic hyperglycemia can cause a variety of complications, including cardiovascular disease, kidney disease, retinal disease, and neurological disease.2–5 Diabetes is currently one of the fastest-growing chronic diseases. The number of patients with T2DM is increasing at alarming rates worldwide, and the current prevalence of 422 million people is expected to rise to 592 million in 2035. According to large-scale data published in BMJ at the end of April 2020, the prevalence of adult diabetes in China reached a new high level, with a total number of 130 million patients and a prevalence rate of 12.8%.6 To date, the pathogenesis of diabetes mellitus has not been fully elucidated, which cannot be solely attributed to the genetic factors. With a rapid increase in the prevalence of diabetes in the world, more studies are required to gain an in-depth understanding of the pathogenesis of diabetes for the development of effective therapies. Some studies have demonstrated that environmental factors such as lifestyle changes and nutrient imbalance may be a more critical role in the development of T2DM.7–9 The epigenetic effects of environmental and genetic factors may lead to alteration in DNA methylation, histone modification, and non-coding RNA in the genes involved, resulting in corresponding changes in the expression of the genes involved in regulation of metabolism, insulin resistance, obesity and other conditions, and leading to the occurrence of T2DM.10–12
Epigenetics plays a major role in mediating environmental factors and gene expression. Epigenetics is defined as heritable changes in gene expression that occurs when the nucleotide sequence of the genome remains unchanged.13 Epigenetic regulation includes DNA methylation, histone modification (methylation, acetylation, etc.) and non-coding RNA (MicroRNA, IncRNA, etc.).11 In recent years, a good number of studies have reported a link between the epigenetics and T2DM.14 It has been demonstrated that DNA methylation is associated with obesity and T2DM.11 On the other hand, drugs based on epigenetic mechanism, MC1568 (histone deacetylase inhibitor), for instance, have been shown to improve insulin secretion in T2DM.15
Epigenetic abnormalities can induce β-cells dedifferentiation, which means that β-cells can change into different cell types and cause insulin secretion dysfunction.16 Dedifferentiation was coined to describe either a reversal of the differentiation trajectory back toward progenitor states or a loss of terminal differentiation markers and phenotypes.17 Previous study found that α-cells lacking DNA methyltransferases 1 (DNMT1) and Aristaless-related homeobox (Arx) were converted into β-cells.18 It seems likely that the reduction in promoter/enhancer methylation within transcriptional control sequences of key β-cell genes such as insulin, PDX-1 and Nkx6.1 combined with loss of Arx permits activation of these genes.19 Dedifferentiation provides opportunities for cell state transition, and the links with epigenetics may provide prospects for cell regeneration.
The formation and maintenance of islet β-cell clusters are related to both intracellular components, such as transcription factors and extracellular components, such as signal growth factors. The PDX-1 (also known as IPF-1, IDX-1, IUF-1, and STF-1) is the most critical intracellular factor in β-cells and known as the “master regulator” of pancreatic development.20,21 The unique feature of PDX-1 protein is its expression solely in the pancreas, and it has received extensive attention in recent years. The islet transcription factor PDX-1 was initially identified as an activator of insulin and somatostatin genes.22 It is a homologous domain protein selectively expressed in the pancreas of adult mice that trans-activates and binds to insulin promoters.23 Deletion of the gene encoding this protein in mice and humans can lead to hypoplasia of the pancreas.23–25 Maintaining the characteristics of pancreatic cells depends on the function of the pancreatic gene regulatory network, which includes numerous interactions between various transcription factors in the pancreas. Transcription factors regulate gene expression by interacting with chromatin DNA. PDX-1 is one of the focal points of this network, and the normal function of the pancreatic cell lineage requires its high concentration (Figure 1).26 For example, Insulin transcript levels and the expression of key transcription factors required for pancreatic development including forkhead box protein a2 (FOXA2), Nkx2.2, and neurogenic differentiation (NeuroD) were significantly increased by exogenous PDX-1 overexpression. PDX-1 also regulates the transcription of other islet genes, such as glucose transporter 2 (GLUT2), glucokinase (GK), and islet amyloid polypeptide (IAPP), etc., suggesting that PDX-1 may play a broad role.
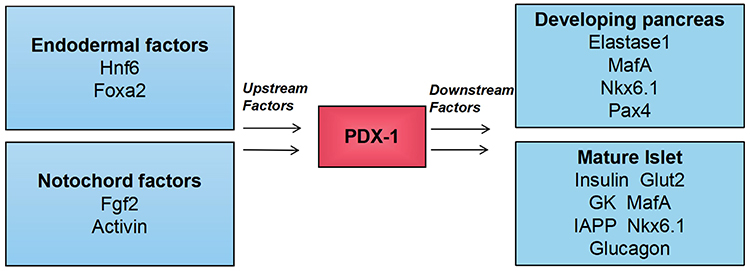 |
Figure 1 Upstream regulator and direct downstream target of PDX-1. The figure highlights the central role of PDX-1 in the pancreas. Extracellular factors (Fgf2 and activin) derived from the notochord and intracellular factors (transcription factors Foxa2 and Hnf6) synergistically activate the expression of PDX-1 in the primitive intestinal endoderm. Abbreviations: Hnf6, transcription factor one cut homeobox 1; Fgf2, fibroblast growth factor 2; Pax4, paired box protein 4. |
PDX-1 is required for normal islet function in adults, and a moderate decrease in PDX-1 impairs normal glucose sensing and insulin secretion.27,28 CpG islands are CG-rich sequences located near coding sequences and they can serve as alternative promoters for their associated genes. Approximately half of mammalian genes have CpG islands. The methylation status of CpG islands within promoter sequences works as an essential regulatory element by modifying the binding affinity of transcription factors to DNA-binding sites. It has been established that the expression of PDX-1 mRNA was negatively related to DNA methylation at the CpG site, and there was a strong negative correlation between DNA methylation at the CpG site in the enhancer region and gene expression.29 Studies have demonstrated that the DNA methylation at multiple CpG sites of PDX-1 promoter and its enhancer in islets of T2DM patients is increased, and the resultant reduction in the expression of PDX-1 mRNA interferes with insulin secretion (Table 1).30 These studies have confirmed the importance of PDX-1 epigenetic regulation in T2DM.
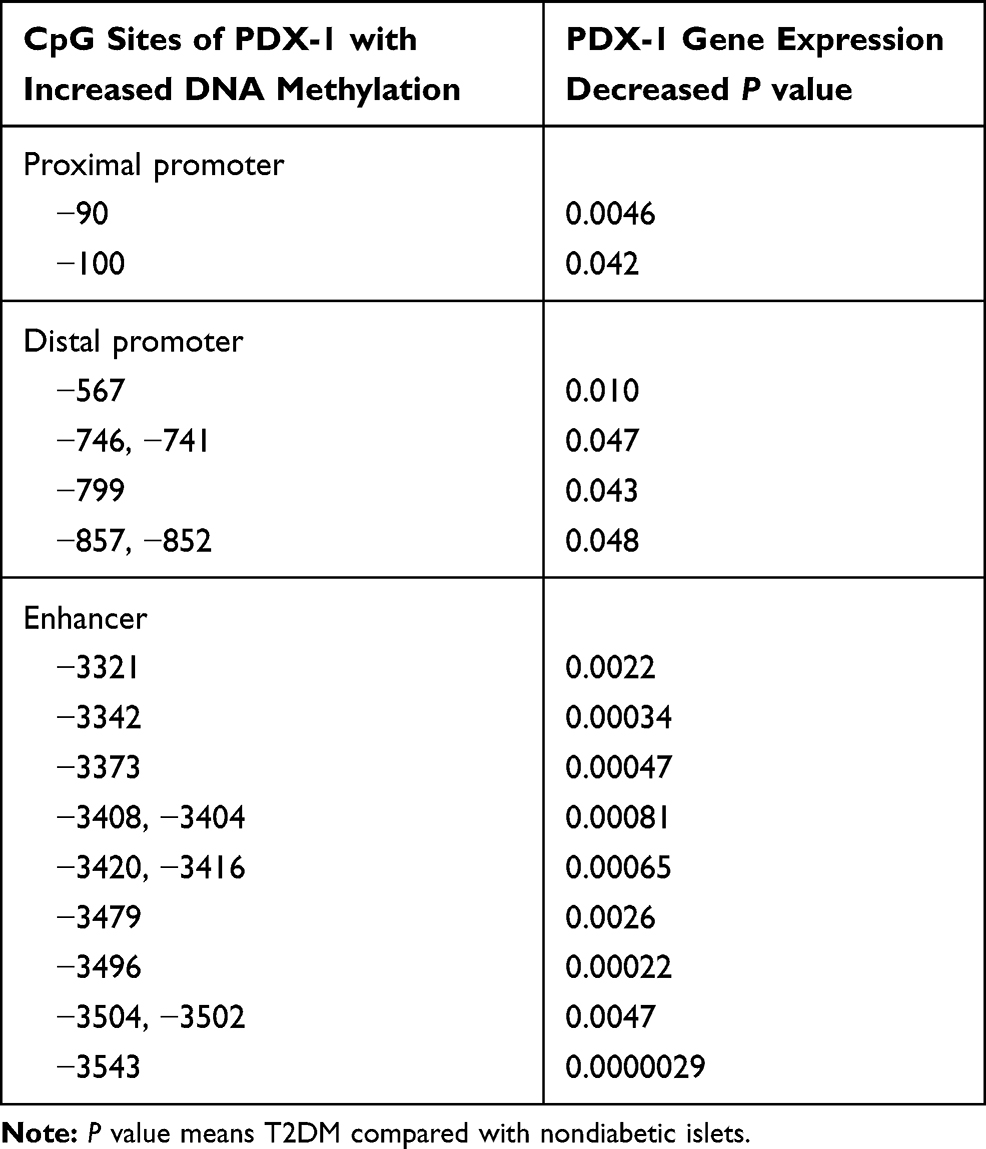 |
Table 1 Correlations Between DNA Methylation of the Proximal and Distal Promoters as Well as Enhancer Regions of PDX-1 and Gene Expression in Human Donors |
Histone Acetylation and Methylation in Diabetes
In cells, DNA wraps around histones to form chromatin structures that act as “on” or “off” switch for gene expression, mainly in the field of epigenetics.16 DNA methylation and histone modification represent two major chromatin remodeling processes that can integrate cellular environmental signals through gene regulation and various transcriptional mechanisms to dynamically regulate gene transcription in response to internal and external stimuli. Consequently, disruption of epigenetic regulation may exert a variety of pathophysiological effects and likely contributes to the etiology of diabetes as well.
Histone methylation usually occurs on lysine or arginine residues of histones H3 and H4. Histone methylation is more stable and durable than histone acetylation. Histone deacetylases (HDACs) deacetylate histones which leads to chromatin compression and inhibits gene transcription. Studies reveal that the epigenetic signature of HDAC3 was significantly elevated in patients with T2DM compared to control subjects.31 HDAC inhibition is observed to have a protective role on β-cell proliferation, function and glucose homeostasis.32 High expression of HDAC7 impairs the secretory ability of rat islet cells, and the expression level of islet HDAC7 is significantly increased in T2DM patients compared with healthy group.33 The HDAC inhibitor MC1568 rescued this secretory impairment caused by increased expression of HDAC7.15
Research has demonstrated that β-cell dysfunction in the pathogenesis of type 2 diabetes is partly due to histone modifications at the PDX-1 promoter.34 Gao et al revealed that pancreatic islet PDX-1 gene proximal promoter histone modifications were pivotal for PDX-1 expression and islet β-cell function in a catch-up growth (CG) model.35 Their animal experiments showed that Liraglutide treatment in the CG rats increased PDX-1 expression with reduced recruitment level of histone H3 lysine 9 dimethylation (H3K9me2) at the PDX-1 proximal promoter. In primary islet culture, the increased PDX-1 mRNA expression after the treatment of HDAC inhibitor (Trichostatin A, TSA) and Liraglutide was found to be negatively correlated with H3K9me2 recruitment at the PDX-1 proximal promoter.
LncRNAs and circRNAs in Diabetes
In addition to classical epigenetic modifications, a variety of noncoding RNAs (ncRNAs) have been found in different cells and organs, many of which are involved in the regulation of metabolic processes. LncRNA is a type of noncoding RNA with a transcription length greater than 200 nucleotides which epigenetically regulate the expression of genes but do not have protein-coding potential. LncRNAs have been found to be involved in the pathophysiological processes of diabetes.36 Studies suggest that lncRNAs H19 interacts with methyl-CPG binding domain protein 1 to recruit H3K9 methyltransferase into its own imprinted gene network, thus controlling gene expression of a given network member.37 LncRNA PDX-1 related lncRNA transcriptional upregulation factor (PLUT) is highly expressed in the nucleus of islet cells, and its enhancer region can bind to the promoter of the key pancreatic cell transcription factor PDX-1 to participate in the regulation of the transcription and differentiation of islet cells.38 The PDX-1, MafA and GLUT2 expression levels were upregulated after lncRNA-p3134 overexpression both in vitro and vivo.39 Studies have shown that lncRNA MALAT1 induces the dysfunction of β cells via the reduction of the H3 histone acetylation of the PDX-1 promoter and subsequently inhibiting the expression of PDX-1, thus suppressing the insulin secretion.40
CircRNAs are closed, circular, non-coding RNAs which have the characteristics of structural stability and tissue specificity.41 Studies have revealed that circRNAs play important roles in the occurrence and development of T2DM.41,42 For instance, circRNA HIPK3 has been shown to contribute to hyperglycemia and insulin resistance via sponging miR-192-5p and upregulating FOXO1.43 Among the selected circRNAs, circRNA ANKRD36 was found to be significantly upregulated in T2DM patients in comparison to the control group.44 More research work is needed to fully understand the functions and mechanisms of circRNAs in the pathophysiology of T2DM.
DNA Methylation and Diabetes
DNA methylation is a basic and stable DNA modification, which not only regulates gene expression, but also plays a significant role in regulating chromosome stability.45–48 It was found that DNA methylation plays a critical role in multiple pathological processes, including metabolic diseases. The role of DNA methylation has been implicated in the progression of T2DM, which may be related to the alteration in mitochondria, cellular proteins, lipids, and nucleic acids caused by inflammation or oxidative stress.49 The strand breaks in DNA can affect DNA methylation and DNA lesions can influence DNA-protein and protein–protein interactions, leading to aberrant patterns of DNA methylation.50 Diet and a sedentary lifestyle may also promote biochemical changes in cells through an epigenetic regulation mechanism. DNA methylation is catalyzed by the DNMTs, and cytosine in CpG dinucleotides is a favorite substrate for DNMTs. Under the catalysis of DNMT, S-adenosine methionine is used as a methyl donor to transfer methyl to the 5th carbon atom of cytosine and becomes 5-methyl cytosine.51 DNMT1 transmits cytosine methylation through replication by recognizing methylated cytosine from existing DNA strands to its new mate, and is mainly responsible for maintaining DNA methylation in cells.52 DNMT3A and DNMT3B are other two catalytically active DNMTs mainly involved in de novo methylation to establish a novel methylation model.53 Notably, DNA methylation is usually associated with gene silencing, and promoter methylation tends to lead to gene silencing, demonstrating a direct link between epigenetic modification and gene activity. There exists accumulating evidence that epigenetic changes caused by methylation may lead to the alteration of metabolic imprinted genes resulting in the up-regulation or down-regulation of the pancreatic development, cell function, peripheral glucose uptake, and insulin resistance.54–56
DNA methylation can modify gene expression in several ways, for example, by altering histone interactions, influencing transcription factor binding, and/or recruitment of methyl-binding proteins.57 However, demethylation occurs with age due to the change in chromatin structure and the presence of binding transcription factors on the distal regulatory elements of the initial β-cells. The cytosine methylation and demethylation system are summarized in Figure 2 and discussed in detail below. Intrauterine growth retardation (IUGR) has been linked to later development of type 2 diabetes in adulthood.58 An abnormal metabolic intrauterine milieu affects the development of the fetus, which may be mediated through permanently modifying gene expression of susceptible cells. Altered gene expression persists after birth, suggesting that an epigenetic mechanism may be responsible for changes in transcription.59 Research found that expression of PDX-1 was permanently reduced in IUGR β-cells and underwent epigenetic modifications throughout the development.60 After the onset of diabetes in adulthood, the CpG island in the proximal promoter was methylated, resulting in permanent silencing of the PDX-1 locus. The molecular mechanism responsible for DNA methylation in IUGR islets is likely to involve histone 3 lysine 9 (H3K9) methylation. As H3K9 methylation occurs during postnatal life in IUGR islets, this would then allow recruitment of the DNMT3A.61 Subsequent to onset of DNA methylation, DNMT1 would then be positioned to maintain the methylated state, locking in PDX-1 silencing in adult IUGR islets.62
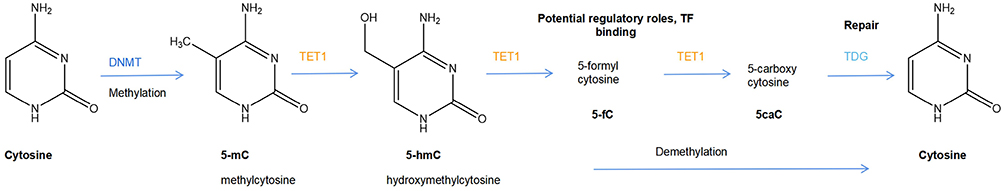 |
Figure 2 The cytosine methylation and demethylation system. 5methyl cytosine (5mC) is generated by the action of DNA methyltransferases (DNMTs). In specific contexts, likely driven by tissue-specific DNA-binding transcription factors, selected 5 mC residues are oxidized by TET enzymes to produce 5hmC (5-hydroxymethyl cytosine), which can be further oxidized to 5 fC (5-formyl cytosine) and 5caC (5-carboxy cytosine). Alternatively, 5fC and 5caC can be excised by thymidine DNA glycosylase (TDG) and repaired by the base excision repair mechanism to cytosine. |
Studies have found that the DNA-binding transcription factors known to maintain mature beta cells such as PDX1 and FOXA2 play a role in the recruitment of Tet enzymes to tissue-specific enhancers in order to initiate active demethylation.63–65 The three enzymes encoded by the Tet loci (“ten-eleven translocation”; Tet 1, Tet 2 and Tet 3) can catalyze the oxidation of 5-methyl cytosine to 5-hydroxy-methyl cytosine (5 hmC), and further to 5-formyl and 5-carboxy-methyl cytosine.66 Therefore, characterization of epigenetic changes (such as DNA methylation), which are closely involved in the regulation of precise β-cells specific gene expression program to maintain the cell’s functional state, may be a key to understanding T2DM islet cell plasticity.
PDX-1 and β-Cells Function
PDX-1 is an essential mediator for the pancreas formation during mammalian development.23,67 In mouse embryos, PDX-1 expression is restricted to the developing pancreatic anlagen and is initiated when the foregut endoderm is committed to a pancreatic fate. PDX-1 is activated during the early phase of embryonic development, followed by other genes associated with glucose-reactive insulin-producing cells (IPCs), such as insulin and GLUT2. In mature pancreas, the function of PDX-1 is primarily confined to regulate genes which are essential the function, proliferation, and survival of the β-cells.68,69 Studies have demonstrated that mice with PDX-1−/- deletion have almost no pancreas formation.23,67 Similarly, studies in animal models of insulin resistance suggested that the down regulation of PDX-1 expression may underlie the pathogenesis of β-cells failure and T2DM.70,71 PDX-1 is usually not present in the islets of gerbils, however, gene transfer experiments have shown that PDX-1 supplementation significantly increases insulin levels and glucose-induced insulin transcription in the islets of these animals.72 And the gradual decrease in PDX-1 protein levels with age may explain the increased incidence of cell dysfunction and impaired glucose tolerance.27,73 Finally, the islet-specific destruction of PDX-1 results in impaired insulin release, glucose intolerance, and diabetes. In humans, the PDX-1 heterozygous mistranslation and frame-shift mutations are linked to reduced insulin secretion, leading to maturity-onset diabetes mellitus of the young (MODY).74–76 Studies have shown that PDX-1 deficiency results in a reduction in cell size and proliferation of β-cells.77,78 The number and volume of islets in mice from the heterozygote PDX-1± decreased and the susceptibility to cell death increased.79,80 Mice in PDX-1± showed hyperglycemia which were associated with increased β-cells death and reduced β-cells mass.81 Notably, PDX-1 expression becomes confined to β-cell over the course of development. The conditional removal of PDX-1 from forming β-cell using insulin-driven Cre transgenic lines results in hyperglycemia, compromised insulin+ (Ins+) cells, and increased glucagon+ (Glu+) cells.82,83 Feltus et al conducted a comprehensive study to identify methylation-sensitive and methylation-resistant genes in order to identify patterns that may elucidate the mechanism underlying methylation-sensitivity.84 Their studies suggested that the continuous high CpG islands methylation limited expression of related genes in fibroblasts, especially the homeobox genes, such as PDX-1. Set7/9 is a lysine methyltransferase which has been shown to interact with PDX-1 and enhance its effects on multiple target genes.85,86 It was demonstrated that methylation mediated by Set7/9 plays an important role in maintaining PDX-1 activity and β-cells function.
It is generally believed that apoptosis is the main cause of beta cell death when the islets or insulin-secreting beta cells are in various pathophysiological states.87 The mechanism of cell apoptosis is complex. It has been suggested that epigenetic mechanism also plays a role in the regulation of apoptosis in the course of nuclear changes experienced by apoptotic cells.88,89 Studies have demonstrated that insulin and/or insulin-like growth factor (IGF) regulate β-cell mass by relieving forkhead transcription factor (Foxo1) inhibition of PDX-1 expression.90 Reduced signaling via the insulin/IGF pathway appears to be an important common mechanism for the sustained reduction of PDX-1 expression in cell failure.91 Epigenetic changes include recruitment of DNMTs, HDAC/msin3 complex and increased miRNA expression, resulting in reduced IGF expression and ultimately a decrease in the transcriptional activity of associated key cellular genes such as PDX-1 and insulin.
Previous studies found that protein level of PDX-1 and mRNA expression of insulin and GLUT2 were significantly reduced in 4 weeks after 90% pancreas excision in rats, suggesting that transcription factors play a vital role in the abnormal development of diabetic cells.92 The presence of transcription factors specificity in the cells may lead to the enhancement or inhibition of the expression of various key proteins. For example, transcription factors PDX-1 and β-cell E-box transactivator 2 (BETA2) are associated with the expression of IAPP, GLUT2, GK, and secretin genes. A decrease in the PDX-1 expression, or activity, resulting in impaired expression of both GLUT2 and insulin may be a more general cause of the development of hyperglycemia that in turn may progress to T2DM.93 Study has found that DNA methylation level may increase abnormally under high glucose concentration, which results in decreased insulin secretion and subsequently leads to diabetes.94 This finding is coincided with reduced binding of the transcription factor PDX-1 to corresponding elements.95 In addition, increased DNA methylation can also reduce PDX-1 mRNA and protein expression. All these suggest a critical role of epigenetic modification of PDX-1 in the regulation of insulin gene expression. Therefore, the studies of PDX-1 may provide insights into the molecular mechanisms that are involved in impaired insulin secretion in humans and the potential therapeutic application of the restoration of β-cells function in the treatment of diabetic patients.
Changes in PDX-1 Methylation Status are Related to Diabetes
The pathogenesis of T2DM is multifactorial, involving β-cells loss and altered β-cells function that may be attributed to the decreased expression of transcription factors such as PDX-1 or Nkx6.1.96 Since DNA methylation can lead to stable alterations in the transcriptional potential, epigenetic mechanisms may partly explain the rapidly increasing prevalence of type 2 diabetes.97 Studies have established that deficiency of PDX-1 in β-cells of the pancreas results in diabetes in rodents.72 In the study of DNA methylation of human samples Yang et al reported that DNA methylation of PDX-1 was negatively correlated with human islet gene expression.30 Of major significance to T2DM is their finding that PDX-1 was one of only 15 genes (of 1749 examined) with CpG islands within the promoter that were methylation-susceptible. Furthermore, comprehensive DNA methylation profiling revealed a significant change in Islet DNA methylation pattern in T2DM compared to no-diabetic donors.98–100 An increase in the methylation at the PDX-1 site reduces the expression of PDX-1 protein in the islets which has been confirmed in the model of T2DM rats.101
Previous studies have shown that an adverse intrauterine environment leads to reduced pancreatic PDX-1 expression and hence impaired insulin secretion and diabetes in adult rats.62 An explanation for this phenotype is that epigenetic modifications of PDX-1, including both increased DNA methylation and histone modifications, are associated with reduced gene transcription in rat islets.62,101–103 Collectively, all these studies evidently support a crucial role of epigenetic modification of PDX-1 in the occurrence and development of T2DM.
The rationale behind demethylation therapies is the ability of DNA methyltransferase inhibitors to revert hypermethylation induced gene silencing. The cytosine analogues 5-azacytosine (azacytidine, Aza) is a drug for epigenetic cancer therapies. This compound function as a DNA methyltransferase inhibitor that has been shown to possess substantial potency in reactivating epigenetically silenced tumor suppressor genes in vitro.104 DNA methylation inhibitor Aza is a cytosine analogue that binds to newly formed DNA in dividing cells, and irreversibly attaches to DNMT1, leading to degradation of DNMT1, resulting in hypomethylation.105 It has been observed that Aza can enhance the expression of the nuclear transcription factor PDX-1 human stem cell lines by immunofluorescence staining.29 The destruction of PDX-1 leads to T2DM and shows increased promoter DNA methylation, histone H3 and H4 deacetylation, and H3K9 methylation in IUGR. These epigenetic changes and the reduction in PDX-1 expression could be reversed by HDAC inhibition.62
Moreover, PDX-1 gene mutation results in neonatal diabetes (NDM), and in NDM screening, even if the pancreas is not diseased, PDX-1 gene mutation can be used as a basis for genetic diagnosis.106 In pancreatic neuroendocrine tumors (PNENs), epigenetic deregulation is considered to be of great significance, and studies have shown that the application of PDX-1 methylation status in the subtyping of PNENs and its prognostic value.107 It is worthy of noting that besides PDX-1 expression, the DNA methylation profile of the PDX-1 gene region is valuable in distinguishing two PNEN subtypes, A and B, which illustrate a link to their respective cell-of-origin, α-cells and β-cells. The two subtypes seem to have different clinicopathological characteristics, a different molecular profile and a different prognosis, with significantly worse outcomes in subtype A PNENs.
The Treatment of T2DM is Related to the Expression of PDX-1
In recent years, preclinical studies have focused on the therapeutic potential of PDX-1 for reversing type 1 diabetes recently. For example, virus-mediated PDX-1 expression in hepatocytes seems to stimulate a range of pancreatic related genes, including insulin, leading to a reversal of chemically induced diabetes in mice.108–111 In addition, the use of protein transduction domains can inspire potential β-cells precursors by in-vivo delivery of PDX-1 proteins, thus opening therapeutic pathways. Studies have demonstrated that the role of glucagon-like peptide 1 (GLP-1) in promoting the growth and formation of adult cells may depend on the activation of PDX-1 in β-cells.112,113 GLP-1 is an insulin secretory promoter, which can increase the expression of PDX-1 and the size of the islets. Current studies have also shown that Exendin-4, a synthetic stable GLP-1 analogue, is a promising drug for the treatment of T2DM,114 which improves cell function by activating PDX-1 transcription.115 Exendin-4 can reverse the inhibitory state of PDX-1, prevent DNMT1 binding to PDX-1, and normalize histone modification and DNA methylation state.116 GLP-1 analogue, liraglutide, promotes insulin synthesis and increases cell viability by restoring the expression of PDX-1 in β-cells cultured with high glucose and high free fatty acids.117 Coxsackievirus B type 4 (CVB4) infection causes unconventional prefoldin RPB5 Interactor (URI) loss in β-cells, leading to DNMT1 expression and PDX-1 silencing, and subsequently inducing β-cell loss and hyperglycemia. Demethylating agent procainamide-mediated DNMT1 inhibition has been shown to reinstate PDX1 expression and protects against diabetes in pancreatic URI-depleted mice.118 Acarbose is a prescribed medicine for the treatment of type 2 diabetes that has been demonstrated to promote the proliferation of islet β-cells and inhibit PDX-1 methylation in islet β-cells from diabetic mice.119 In addition, it has been shown that the expression of PDX-1 can be induced by various approaches as demonstrated in the study of differentiation of mesenchymal stromal cells (MSC) into β-cells.120
Studies reveal that high blood glucose would reduce the expression of PDX-1 gene in human pancreatic islets and increase the DNA methylation of PDX-1.30 Although the expression of PDX-1 was decreased in those cells exposed to high glucose, the expression of PDX-1 methylation and Dnmt1 was markedly increased. The expression level of PDX-1 is related to insulin secretion in human islets. Importantly, islets from T2DM patients with hyperglycemia show increased DNA methylation and reduced PDX-1 expression, leading to insulin secretion suppression and diabetes,30,98,99 which clearly suggests that epigenetic modification of PDX-1 may play a role in the development of T2DM.
Recent research by Xiao et al has found that using gene therapy can reverse diabetes by converting α-cell in the pancreas into fully functioning β-cells.121 They used an adeno-associated virus (AAV) vector technology to secrete into the pancreas two proteins, PDX-1 and macrophage activating factor (Mafa), which reprogrammed α-cells into insulin-producing cells.121 Previous studies by Bramswig et al revealed that treatment of cultured pancreatic islets with a histone methyltransferase inhibitor leads to colocalization of both glucagon and insulin and glucagon and PDX-1 in human islets.122 Their findings suggest that a more targeted manipulation of the histone methylation signature in a histone modification and gene-specific manner might be exploited to promote a more complete human α to β cell fate conversion.
Interestingly, an epigenetic modification approach has recently been employed to promote the differentiation of MSC into glucose-sensitive insulin-secreting cells through epigenetic modification, thus enhancing the regeneration solution of diabetes.29 The results in this study showed that the levels of DNA methylation in cells treated with methyltransferase inhibitor Aza were significantly reduced, and the expression of nuclear transcription factor PDX-1 was enhanced, while the inhibition of methylation enhanced insulin production in differentiated cells.29 The α-cell transcription factor Arx, a DNA-binding protein, is required for the maintenance of α-cells, but it also needs to be repressed in β-cells to prevent their transdifferentiation into glucagon-producing cells.123,124 It seems likely that the reduction in promoter/enhancer methylation within transcriptional control sequences of key β-cell genes such as Insulin, PDX-1 and Nkx6.1 combined with loss of Arx permits activation of these genes.62,125,126 In addition, simultaneous deletions of DNMT1 and the α-cell regulator Arx may induce β-cells reprogramming in vivo.19 All these suggest that additional extrinsic signaling modulation could additively enhance the pace and quality of α-to-β cells conversion achieved after targeted Arx and DNMT1 inactivation. Furthermore, based on the probable effect of DNA methylation on insulin synthesis and secretion, promoting the differentiation of stem cells into insulin-secreting cells under the action of DNA demethylation agents may be a feasible treatment strategy in the future.
Conclusion and Perspectives
Type 2 diabetes is a complex multifactorial disease with both genetic and environmental underpinnings in which epigenetic mechanism is integrated to play a role in the pathological progression. Supplementing classical genetic research with emerging epigenetic tools may be an important approach to explore the molecular causes of complex human disease development. In this review, we elaborate on some of the findings of epigenetic regulation of PDX-1 in diabetes research, and discuss ways to induce the self-reinforcing transcriptional networks that maintain β-cells identity, including PDX-1, which may be a viable treatment strategy for treating T2DM. Further studies of epigenetic regulation of PDX-1 and a more in-depth understanding of PDX-1 in regulating islet-specific genes both in normal and diabetic states will provide insights for identifying potential therapeutic pathways and designing a new generation of epigenetic drugs. In addition, DNA methylation, as a chronic mechanism of long-term gene regulation, is increasingly recognized to be involved in the regulation of chronic diseases. It is possible that the identification of hypermethylated genes that can become demethylated and reactivated by drug treatment will be an important area for future research.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Funds for the Construction of National First-Class Pharmacy Discipline [GNYL(2017)-006].
Disclosure
The authors declared no potential conflicts of interest in this work.
References
1. Galicia-Garcia U, Benito-Vicente A, Jebari S, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. doi:10.3390/ijms21176275
2. Dimova R, Tankova T, Guergueltcheva V, et al. Risk factors for autonomic and somatic nerve dysfunction in different stages of glucose tolerance. J Diabetes Complications. 2017;31(3):537–543. doi:10.1016/j.jdiacomp.2016.11.002
3. Oliveira S, Monteiro-Alfredo T, Silva S, Matafome P. Curcumin derivatives for type 2 diabetes management and prevention of complications. Arch Pharm Res. 2020;43(6):567–581. doi:10.1007/s12272-020-01240-3
4. Low Wang CC, Hess CN, Hiatt WR, et al. Clinical update: cardiovascular disease in diabetes mellitus atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus-mechanisms, management, and clinical considerations. Circulation. 2016;133(24):2459–2502.
5. Ruospo M, Saglimbene VM, Palmer SC, et al. Glucose targets for preventing diabetic kidney disease and its progression. Cochrane Database Syst Rev. 2017;6(6):Cd010137. doi:10.1002/14651858.CD010137.pub2
6. Li Y, Teng D, Shi X, et al. Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: national cross sectional study. BMJ. 2020;369:m997. doi:10.1136/bmj.m997
7. Prasad RB, Groop L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel). 2015;6(1):87–123. doi:10.3390/genes6010087
8. Davegårdh C, García-Calzón S, Bacos K, Ling C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol Metab. 2018;14:12–25. doi:10.1016/j.molmet.2018.01.022
9. Kolb H, Martin S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017;15(1):131. doi:10.1186/s12916-017-0901-x
10. Karachanak-Yankova S, Dimova R, Nikolova D, et al. Epigenetic alterations in patients with type 2 diabetes mellitus. Balkan J Med Genet. 2015;18(2):15–24. doi:10.1515/bjmg-2015-0081
11. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29(5):1028–1044. doi:10.1016/j.cmet.2019.03.009
12. Bansal A, Pinney SE. DNA methylation and its role in the pathogenesis of diabetes. Pediatr Diabetes. 2017;18(3):167–177. doi:10.1111/pedi.12521
13. Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol. 2014;6(1):a018200. doi:10.1101/cshperspect.a018200
14. Dayeh T, Ling C. Does epigenetic dysregulation of pancreatic islets contribute to impaired insulin secretion and type 2 diabetes? Biochem Cell Biol. 2015;93(5):511–521. doi:10.1139/bcb-2015-0057
15. Daneshpajooh M, Eliasson L, Bacos K, Ling C. MC1568 improves insulin secretion in islets from type 2 diabetes patients and rescues β-cell dysfunction caused by Hdac7 upregulation. Acta Diabetol. 2018;55(12):1231–1235. doi:10.1007/s00592-018-1201-4
16. Lu TT, Heyne S, Dror E, et al. The polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018;27(6):1294–1308. doi:10.1016/j.cmet.2018.04.013
17. Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. β-cell dedifferentiation in diabetes is important, but what is it? Islets. 2013;5(5):233–237. doi:10.4161/isl.27494
18. Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20(4):419–429. doi:10.1016/j.devcel.2011.03.012
19. Chakravarthy H, Gu X, Enge M, et al. Converting adult pancreatic islet α cells into β cells by targeting both Dnmt1 and Arx. Cell Metab. 2017;25(3):622–634. doi:10.1016/j.cmet.2017.01.009
20. Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol. 2013;29:81–105. doi:10.1146/annurev-cellbio-101512-122405
21. Arcidiacono B, Iiritano S, Chiefari E, et al. Cooperation between HMGA1, PDX-1, and MafA is essential for glucose-induced insulin transcription in pancreatic beta cells. Front Endocrinol (Lausanne). 2014;5:237. doi:10.3389/fendo.2014.00237
22. Pedica F, Beccari S, Pedron S, et al. PDX-1 (pancreatic/duodenal homeobox-1 protein 1). Pathologica. 2014;106(4):315–321.
23. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi:10.1038/371606a0
24. Puri S, Folias AE, Hebrok M. Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell. 2015;16(1):18–31. doi:10.1016/j.stem.2014.11.001
25. Johnson JD, Ahmed NT, Luciani DS, et al. Increased islet apoptosis in Pdx1± mice. J Clin Invest. 2003;111(8):1147–1160. doi:10.1172/JCI200316537
26. Larsen HL, Grapin-Botton A. The molecular and morphogenetic basis of pancreas organogenesis. Semin Cell Dev Biol. 2017;66:51–68.
27. Brissova M, Shiota M, Nicholson WE, et al. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem. 2002;277(13):11225–11232. doi:10.1074/jbc.M111272200
28. Brissova M, Blaha M, Spear C, et al. Reduced PDX-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. Am J Physiol Endocrinol Metab. 2005;288(4):E707–714. doi:10.1152/ajpendo.00252.2004
29. Elsharkawi I, Parambath D, Saber-Ayad M, Khan AA, El-Serafi AT. Exploring the effect of epigenetic modifiers on developing insulin-secreting cells. Hum Cell. 2020;33(1):1–9. doi:10.1007/s13577-019-00292-y
30. Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol. 2012;26(7):1203–1212. doi:10.1210/me.2012-1004
31. Sathishkumar C, Prabu P, Mohan V, Balasubramanyam M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum Genomics. 2018;12(1):41. doi:10.1186/s40246-018-0173-3
32. Dahllöf MS, Christensen DP, Harving M, Wagner BK, Mandrup-Poulsen T, Lundh M. HDAC inhibitor-mediated beta-cell protection against cytokine-induced toxicity is STAT1 Tyr701 phosphorylation independent. J Interferon Cytokine Res. 2015;35(1):63–70. doi:10.1089/jir.2014.0022
33. Daneshpajooh M, Bacos K, Bysani M, et al. HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia. 2017;60(1):116–125. doi:10.1007/s00125-016-4113-2
34. Gilbert ER, Liu D. Epigenetics: the missing link to understanding β-cell dysfunction in the pathogenesis of type 2 diabetes. Epigenetics. 2012;7(8):841–852. doi:10.4161/epi.21238
35. Gao M, Deng XL, Liu ZH, et al. Liraglutide protects β-cell function by reversing histone modification of Pdx-1 proximal promoter in catch-up growth male rats. J Diabetes Complications. 2018;32(11):985–994. doi:10.1016/j.jdiacomp.2018.08.002
36. Qi M, Zhou Q, Zeng W, et al. Analysis of long non-coding RNA expression of lymphatic endothelial cells in response to type 2 diabetes. Cell Physiol Biochem. 2017;41(2):466–474. doi:10.1159/000456599
37. Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. H19 lncRNA controls gene expression of the imprinted gene network by recruiting MBD1. Proc Natl Acad Sci U S A. 2013;110(51):20693–20698. doi:10.1073/pnas.1310201110
38. Yin DD, Zhang EB, You LH, et al. Downregulation of lncRNA TUG1 affects apoptosis and insulin secretion in mouse pancreatic β cells. Cell Physiol Biochem. 2015;35(5):1892–1904. doi:10.1159/000373999
39. Ruan Y, Lin N, Ma Q, et al. Circulating LncRNAs analysis in patients with type 2 diabetes reveals novel genes influencing glucose metabolism and islet β-cell function. Cell Physiol Biochem. 2018;46(1):335–350. doi:10.1159/000488434
40. Ding H, Wang F, Shi X, et al. LncRNA MALAT1 induces the dysfunction of β cells via reducing the histone acetylation of the PDX-1 promoter in type 1 diabetes. Exp Mol Pathol. 2020;114:104432. doi:10.1016/j.yexmp.2020.104432
41. Stoll L, Sobel J, Rodriguez-Trejo A, et al. Circular RNAs as novel regulators of β-cell functions in normal and disease conditions. Mol Metab. 2018;9:69–83. doi:10.1016/j.molmet.2018.01.010
42. Xu H, Guo S, Li W, Yu P. The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci Rep. 2015;5:12453. doi:10.1038/srep12453
43. Cai H, Jiang Z, Yang X, Lin J, Cai Q, Circular LX. RNA HIPK3 contributes to hyperglycemia and insulin homeostasis by sponging miR-192-5p and upregulating transcription factor forkhead box O1. Endocr J. 2020;67(4):397–408. doi:10.1507/endocrj.EJ19-0271
44. Fang Y, Wang X, Li W, et al. Screening of circular RNAs and validation of circANKRD36 associated with inflammation in patients with type 2 diabetes mellitus. Int J Mol Med. 2018;42(4):1865–1874. doi:10.3892/ijmm.2018.3783
45. Kwak SH, Park KS. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp Mol Med. 2016;48(3):e220. doi:10.1038/emm.2016.7
46. Holliday R. Epigenetics: a historical overview. Epigenetics. 2006;1(2):76–80. doi:10.4161/epi.1.2.2762
47. Xie R, Carrano AC, Sander M. A systems view of epigenetic networks regulating pancreas development and β-cell function. Wiley Interdiscip Rev Syst Biol Med. 2015;7(1):1–11. doi:10.1002/wsbm.1287
48. Quilichini E, Haumaitre C. Implication of epigenetics in pancreas development and disease. Best Pract Res Clin Endocrinol Metab. 2015;29(6):883–898. doi:10.1016/j.beem.2015.10.010
49. Simmons RA. Role of metabolic programming in the pathogenesis of beta-cell failure in postnatal life. Rev Endocr Metab Disord. 2007;8(2):95–104. doi:10.1007/s11154-007-9045-1
50. Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008;266(1):6–11. doi:10.1016/j.canlet.2008.02.026
51. Yin Y, Morgunova E, Jolma A, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337):2239. doi:10.1126/science.aaj2239
52. Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805–811. doi:10.1038/nrg2651
53. Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12(2):206–222. doi:10.1002/cbic.201000195
54. De Jesus DF, Orime K, Kaminska D, et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J Clin Invest. 2020;130(5):2391–2407. doi:10.1172/JCI127502
55. Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes. 2011;60(7):1849–1855. doi:10.2337/db11-0400
56. Chen P, Piaggi P, Traurig M, et al. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia. 2017;60(4):645–655. doi:10.1007/s00125-016-4203-1
57. Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330(6004):622–627. doi:10.1126/science.1190614
58. Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50(10):2279–2286. doi:10.2337/diabetes.50.10.2279
59. Simmons RA. Developmental origins of beta-cell failure in type 2 diabetes: the role of epigenetic mechanisms. Pediatr Res. 2007;61(5 Pt 2):64r–67r. doi:10.1203/pdr.0b013e3180457623
60. Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes. 2003;52(3):734–740. doi:10.2337/diabetes.52.3.734
61. Li H, Rauch T, Chen ZX, Szabó PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281(28):19489–19500. doi:10.1074/jbc.M513249200
62. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118(6):2316–2324. doi:10.1172/JCI33655
63. Burtscher I, Barkey W, Lickert H. Foxa2-venus fusion reporter mouse line allows live-cell analysis of endoderm-derived organ formation. Genesis. 2013;51(8):596–604. doi:10.1002/dvg.22404
64. Gao T, McKenna B, Li C, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014;19(2):259–271. doi:10.1016/j.cmet.2013.12.002
65. Doyle MJ, Sussel L. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes. 2007;56(8):1999–2007. doi:10.2337/db06-1766
66. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi:10.1126/science.1170116
67. Offield MF, Jetton TL, Labosky PA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995.
68. Babu DA, Deering TG, Mirmira RG. A feat of metabolic proportions: pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Mol Genet Metab. 2007;92(1–2):43–55. doi:10.1016/j.ymgme.2007.06.008
69. Khoo C, Yang J, Weinrott SA, et al. Research resource: the pdx1 cistrome of pancreatic islets. Mol Endocrinol. 2012;26(3):521–533. doi:10.1210/me.2011-1231
70. Weir GC, Sharma A, Zangen DH, Bonner-Weir S. Transcription factor abnormalities as a cause of beta cell dysfunction in diabetes: a hypothesis. Acta Diabetol. 1997;34(3):177–184. doi:10.1007/s005920050071
71. Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC. Genetic regulation of metabolic pathways in beta-cells disrupted by hyperglycemia. J Biol Chem. 2002;277(13):10912–10921. doi:10.1074/jbc.M111751200
72. Leibowitz G, Ferber S, Apelqvist A, et al. IPF1/PDX1 deficiency and beta-cell dysfunction in Psammomys obesus, an animal With type 2 diabetes. Diabetes. 2001;50(8):1799–1806. doi:10.2337/diabetes.50.8.1799
73. Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl 3):S16–21. doi:10.2337/diabetes.53.suppl_3.S16
74. Fajans SS, Bell GI, Paz VP, et al. Obesity and hyperinsulinemia in a family with pancreatic agenesis and MODY caused by the IPF1 mutation Pro63fsX60. Transl Res. 2010;156(1):7–14. doi:10.1016/j.trsl.2010.03.003
75. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17(2):138–139. doi:10.1038/ng1097-138
76. Cockburn BN, Bermano G, Boodram LL, et al. Gene symbol: IPF1. Disease: MODY 4. Hum Genet. 2005;116(6):538.
77. Johnson JD, Bernal-Mizrachi E, Alejandro EU, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci U S A. 2006;103(51):19575–19580. doi:10.1073/pnas.0604208103
78. Fujimoto K, Hanson PT, Tran H, et al. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem. 2009;284(40):27664–27673. doi:10.1074/jbc.M109.041616
79. Fujimoto K, Chen Y, Polonsky KS, Dorn GW. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci U S A. 2010;107(22):10214–10219. doi:10.1073/pnas.0914209107
80. Fujimoto K, Ford EL, Tran H, et al. Loss of Nix in Pdx1-deficient mice prevents apoptotic and necrotic β cell death and diabetes. J Clin Invest. 2010;120(11):4031–4039.
81. Sun J, Mao L, Yang H, Ren D. Critical role for the Tsc1-mTORC1 pathway in β-cell mass in Pdx1-deficient mice. J Endocrinol. 2018;238(2):151–163. doi:10.1530/JOE-18-0015
82. Gannon M, Ables ET, Crawford L, et al. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol. 2008;314(2):406–417. doi:10.1016/j.ydbio.2007.10.038
83. Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–134. doi:10.1038/ng959
84. Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM. Predicting aberrant CpG island methylation. Proc Natl Acad Sci U S A. 2003;100(21):12253–12258. doi:10.1073/pnas.2037852100
85. Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG. Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes. 2009;58(1):185–193. doi:10.2337/db08-1150
86. Maganti AV, Maier B, Tersey SA, et al. Transcriptional activity of the islet β cell factor Pdx1 is augmented by lysine methylation catalyzed by the methyltransferase Set7/9. J Biol Chem. 2015;290(15):9812–9822. doi:10.1074/jbc.M114.616219
87. Demirtas L, Guclu A, Erdur FM, et al. Apoptosis, autophagy & endoplasmic reticulum stress in diabetes mellitus. Indian J Med Res. 2016;144(4):515–524. doi:10.4103/0971-5916.200887
88. Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. 2018;19(3):192–206. doi:10.1038/nrm.2017.94
89. Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116(2):259–272. doi:10.1016/S0092-8674(04)00044-3
90. Kitamura T, Nakae J, Kitamura Y, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110(12):1839–1847. doi:10.1172/JCI200216857
91. Rohatgi N, Aly H, Marshall CA, et al. Novel insulin sensitizer modulates nutrient sensing pathways and maintains β-cell phenotype in human islets. PLoS One. 2013;8(5):e62012. doi:10.1371/journal.pone.0062012
92. Zangen DH, Bonner-Weir S, Lee CH, et al. Reduced insulin, GLUT2, and IDX-1 in beta-cells after partial pancreatectomy. Diabetes. 1997;46(2):258–264. doi:10.2337/diab.46.2.258
93. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12(12):1763–1768. doi:10.1101/gad.12.12.1763
94. Pinzón-Cortés JA, Perna-Chaux A, Rojas-Villamizar NS, et al. Effect of diabetes status and hyperglycemia on global DNA methylation and hydroxymethylation. Endocr Connect. 2017;6(8):708–725. doi:10.1530/EC-17-0199
95. van Otterdijk SD, Binder AM, Szarc Vel Szic K, Schwald J, Michels KB. DNA methylation of candidate genes in peripheral blood from patients with type 2 diabetes or the metabolic syndrome. PLoS One. 2017;12(7):e0180955. doi:10.1371/journal.pone.0180955
96. Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4(6):1262–1275. doi:10.1016/j.celrep.2013.08.010
97. Kirchner H, Osler ME, Krook A, Zierath JR. Epigenetic flexibility in metabolic regulation: disease cause and prevention? Trends Cell Biol. 2013;23(5):203–209. doi:10.1016/j.tcb.2012.11.008
98. Dayeh T, Volkov P, Salö S, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160. doi:10.1371/journal.pgen.1004160
99. Volkmar M, Dedeurwaerder S, Cunha DA, et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012;31(6):1405–1426. doi:10.1038/emboj.2011.503
100. Volkov P, Bacos K, Ofori JK, et al. Whole-Genome Bisulfite Sequencing of Human Pancreatic Islets Reveals Novel Differentially Methylated Regions in Type 2 Diabetes Pathogenesis. Diabetes. 2017;66(4):1074–1085. doi:10.2337/db16-0996
101. Rahmani S, Vakhshiteh F, Hodjat M, et al. Gene-environmental interplay in bisphenol a subchronic animal exposure: new insights into the epigenetic regulation of pancreatic islets. Chem Res Toxicol. 2020;33(9):2338–2350. doi:10.1021/acs.chemrestox.0c00109
102. Bahrami G, Sajadimajd S, Mohammadi B, et al. Anti-diabetic effect of a novel oligosaccharide isolated from Rosa canina via modulation of DNA methylation in Streptozotocin-diabetic rats. Daru. 2020;28(2):581–590. doi:10.1007/s40199-020-00363-8
103. Khan F, Hodjat M, Rahimifard M, et al. Assessment of arsenic-induced modifications in the DNA methylation of insulin-related genes in rat pancreatic islets. Ecotoxicol Environ Saf. 2020;201:110802. doi:10.1016/j.ecoenv.2020.110802
104. Stresemann C, Bokelmann I, Mahlknecht U, Lyko F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol Cancer Ther. 2008;7(9):2998–3005. doi:10.1158/1535-7163.MCT-08-0411
105. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13. doi:10.1002/ijc.23607
106. Sahebi L, Niknafs N, Dalili H, et al. Iranian neonatal diabetes mellitus due to mutation in PDX1 gene: a case report. J Med Case Rep. 2019;13(1):258. doi:10.1186/s13256-019-2149-x
107. Boons G, Vandamme T, Ibrahim J, et al. PDX1 DNA methylation distinguishes two subtypes of pancreatic neuroendocrine neoplasms with a different prognosis. Cancers (Basel). 2020;12(6):1461. doi:10.3390/cancers12061461
108. Ferber S, Halkin A, Cohen H, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6(5):568–572. doi:10.1038/75050
109. Kojima H, Fujimiya M, Matsumura K, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9(5):596–603. doi:10.1038/nm867
110. Cao LZ, Tang DQ, Horb ME, Li SW, Yang LJ. High glucose is necessary for complete maturation of Pdx1-VP16-expressing hepatic cells into functional insulin-producing cells. Diabetes. 2004;53(12):3168–3178. doi:10.2337/diabetes.53.12.3168
111. Kaneto H, Nakatani Y, Miyatsuka T, et al. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes. 2005;54(4):1009–1022. doi:10.2337/diabetes.54.4.1009
112. Wang H, Iezzi M, Theander S, et al. Suppression of Pdx-1 perturbs proinsulin processing, insulin secretion and GLP-1 signalling in INS-1 cells. Diabetologia. 2005;48(4):720–731. doi:10.1007/s00125-005-1692-8
113. Koizumi M, Doi R, Fujimoto K, et al. Pancreatic epithelial cells can be converted into insulin-producing cells by GLP-1 in conjunction with virus-mediated gene transfer of pdx-1. Surgery. 2005;138(2):125–133. doi:10.1016/j.surg.2005.06.008
114. Gan Y, Dang N, Qu Z, et al. GLP-1-Exendin-4/IgG4 (Fc) fusion protein as a novel drug for diabetes treatment. Exp Clin Endocrinol Diabetes. 2015;123(6):371–375. doi:10.1055/s-0035-1548797
115. Hao T, Zhang H, Li S, Tian H. Glucagon-like peptide 1 receptor agonist ameliorates the insulin resistance function of islet β cells via the activation of PDX-1/JAK signaling transduction in C57/BL6 mice with high-fat diet-induced diabetes. Int J Mol Med. 2017;39(4):1029–1036. doi:10.3892/ijmm.2017.2910
116. Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia. 2011;54(10):2606–2614. doi:10.1007/s00125-011-2250-1
117. Kornelius E, Li HH, Peng CH, et al. Liraglutide protects against glucolipotoxicity-induced RIN-m5F β-cell apoptosis through restoration of PDX1 expression. J Cell Mol Med. 2019;23(1):619–629. doi:10.1111/jcmm.13967
118. Bernard H, Teijeiro A, Chaves-Pérez A, et al. Coxsackievirus B type 4 infection in β cells downregulates the chaperone prefoldin URI to induce a MODY4-like diabetes via Pdx1 silencing. Cell Rep Med. 2020;1(7):100125. doi:10.1016/j.xcrm.2020.100125
119. Zhou D, Chen L, Mou X. Acarbose ameliorates spontaneous type‑2 diabetes in db/db mice by inhibiting PDX‑1 methylation. Mol Med Rep. 2021;23(1):72. doi:10.3892/mmr.2020.11710
120. Gabr MM, Zakaria MM, Refaie AF, et al. Generation of insulin-producing cells from human bone marrow-derived mesenchymal stem cells: comparison of three differentiation protocols. Biomed Res Int. 2014;2014:832736. doi:10.1155/2014/832736
121. Xiao X, Guo P, Shiota C, et al. Endogenous reprogramming of alpha cells into beta cells, induced by viral gene therapy, reverses autoimmune diabetes. Cell Stem Cell. 2018;22(1):78–90.e74. doi:10.1016/j.stem.2017.11.020
122. Bramswig NC, Everett LJ, Schug J, et al. Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J Clin Invest. 2013;123(3):1275–1284. doi:10.1172/JCI66514
123. Wilcox CL, Terry NA, Walp ER, Lee RA, May CL. Pancreatic α-cell specific deletion of mouse Arx leads to α-cell identity loss. PLoS One. 2013;8(6):e66214. doi:10.1371/journal.pone.0066214
124. Courtney M, Gjernes E, Druelle N, et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013;9(10):e1003934. doi:10.1371/journal.pgen.1003934
125. Avrahami D, Li C, Zhang J, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015;22(4):619–632. doi:10.1016/j.cmet.2015.07.025
126. Akinci E, Banga A, Greder LV, Dutton JR, Slack JM. Reprogramming of pancreatic exocrine cells towards a beta (β) cell character using Pdx1, Ngn3 and MafA. Biochem J. 2012;442(3):539–550. doi:10.1042/BJ20111678
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.