")
Back to Journals » OncoTargets and Therapy » Volume 8
Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis and autophagy in oral cancer SSC-4 cells
Authors Irimie AI, Braicu C , Zanoaga O, Pileczki V, Gherman C, Berindan-Neagoe I, Campian RS
Received 29 November 2014
Accepted for publication 19 January 2015
Published 20 February 2015 Volume 2015:8 Pages 461—470
DOI https://doi.org/10.2147/OTT.S78358
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Alexandra Iulia Irimie,1 Cornelia Braicu,2 Oana Zanoaga,2 Valentina Pileczki,2,3 Claudia Gherman,2,4 Ioana Berindan-Neagoe,2,4–6 Radu Septimiu Campian7
1Department of Prosthodontics and Dental Materials, Faculty of Dental Medicine, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 2Research Center for Functional Genomics, Biomedicine and Translational Medicine, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 3Department of Analytical Chemistry, Faculty of Pharmacy, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 4Department of Functional Genomics and Experimental Pathology, The Oncology Institute “Prof Dr. Ion Chiricuta”, Cluj-Napoca, Romania; 5Department of Immunology, Faculty of Medicine, University of Medicine and Pharmacy, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 6Department of Experimental Therapeutics MD Anderson Cancer Center Houston, TX, USA; 7Department of Oral Rehabilitation, Faculty of Dental Medicine, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania
Abstract: Epigallocatechin-3-gallate (EGCG) is the major bioactive component of green tea. Our experimental data indicated that EGCG treatment suppresses cell proliferation of SSC-4 human oral squamous cell carcinoma (OSCC), the effect being dose- and time-dependent. In parallel was observed the activation of apoptosis and autophagy, in response to EGCG exposure in SSC-4 cells. Treatment with EGCG activates the expression of the BAD, BAK, FAS, IGF1R, WNT11, and ZEB1 genes and inhibits CASP8, MYC, and TP53. All of these results suggest that EGCG has an excellent potential to become a therapeutic compound for patients with OSCC, by inducing tumor cell death via apoptosis and autophagy.
Keywords: oral squamous carcinoma, time dependent cell proliferation, gene expression
Introduction
Oral cancer is a highly common malignant tumor of the oral cavity1,2 and the tenth most frequent common cancer in the world.3–5 The most frequent malignancy is oral squamous cell carcinoma (OSCC), which constitutes more than 90% of the oral malignancies6,7 and which has poor prognosis due to therapy-resistant locoregional recurrences and distant metastases.8 OSCC is defined as a neoplastic disorder in the oral cavity and is a complex malignancy, where environmental factors, viral infections, and genetic alterations most likely interact and thus give rise to the malignant condition.2 Development of oral cancer proceeds through epigenetic alteration and discrete molecular genetic changes that are acquired from the loss of genomic integrity after continued exposure to environmental or dietary risk factors.9,10
OSCC is characterized by serial epigenetic and genetic alteration. The accumulations of this alteration leads to uncontrolled cell proliferation of the mutated human oral squamous cells, and the accumulation of damaged genetic material leads to uncontrolled division of mutant oral keratinocytes cells.11 Multiple gene alterations result in oral carcinogenesis and cause aberrant expression and function of proteins in a number of cellular processes, including apoptosis and angiogenesis.12
Natural phytochemicals have received a significant interest for chemoprevention and treatment for a wide range of diseases, including OSCC.13 Phytochemicals, such as polyphenols, are known for their antioxidant capacity and free-radical scavenging properties. In the last years, phytochemicals have received significant attention and been proved to interfere in key cellular pathways.14 Epigallocatechin-3-gallate (EGCG) is the most abundant and most active phenolic constituent of green tea and has strong antioxidant properties, possessing chemotherapeutic and chemopreventive roles.15,16
Recently, the involvements of EGCG in apoptotic or autophagy-induced cell death have been increasingly appreciated.3,10,13,14 Consequently, our main interest was to understand the role of EGCG-mediated cell death, comprising both apoptotic and nonapoptotic cell death, but also, the impact on the gene expression pattern for the main genes involved in apoptosis, in SSC-4 cells, a relevant model for oral cancer. The studies were performed in a noncytotoxic dose in order to avoid prooxidant mechanisms of action that we observed previously (Irimie, unpublished data, 2014).
Materials and methods
Materials
EGCG was purchased from Sigma-Aldrich Corp (St Louis, MO, USA).
Cell culture
SCC-4 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/Nutrient Mixture F-12 Ham F12 (1:1), supplemented with 2 mM glutamine, 10% fetal bovine serum (FBS), and 0.5 μg/mL sodium hydrocortisone succinate (all procured from Sigma-Aldrich Corp). Cells were maintained at 37°C and 5% CO2, in a humidified incubator.
Evaluation of cell proliferation in real time, using the E-Plate16
Cell proliferation evaluation in the presence of different EGCG doses (0, 10, 20, 50, 100, and 200 μM) was evaluated using a real-time cell analyzer (RTCA) DP instrument (F Hoffman-La Roche Ltd, Basel, Switzerland), based on basic assay protocol. Background measurement was done using 100 μL of cell culture medium incubated for 30 minutes in CO2 incubator at 37°C. Once the E-Plate16 was equilibrated, it was placed in the RTCA DP station, and the background cell index values were measured. Afterwards, the E-Plate16 was removed from the RTCA DP station, and 15,000 cells/well were added in each well. Cell proliferation kinetics was registered by the RTCA DP instrument for over 49 hours.
Apoptosis evaluation
SSC-4 cells were seeded at a density of 0.5×106 cells in six-well plates in 2 mL culture media and treated for 24 and 48 hours with a single dose of 20 μM. The cells were harvested, then washed once with phosphate-buffered saline (PBS) and centrifuged at 500 RPM for 5 minutes. Then, the SSC-4 cells were resuspended and stained with Annexin-V-FITC solution (BD Biosciences, Franklin Lakes, NJ, USA) and incubated for 15 minutes at room temperature. Just before analysis, it was stained with propidium iodide (PI) in order to quantify the necrotic cells. Apoptotic and necrotic cells were evaluated on a BD FACSCanto™ II flow cytometer.
Autophagy test
The autophagy was evaluated using an Autophagy/Cytotoxicity Dual Staining Kit (Cayman Chemical Co, Ann Arbor, MI, USA), in accordance with the producer recommendation for inverted fluorescence microscopy, in a 96-well plate seeded with 20,000 cells/well. Microscopic evaluation of the autophagic vacuole staining intensity was confirmed by fluorescence evaluation, with an excitation wavelength of 335 nm and emission wavelength at 512 nm, using a BioTek Microplate Reader.
Gene expression evaluation
For gene expression evaluation, cells were plated in six-well plates at 0.5×106 cells/well and treated with a single dose of 20 μM EGCG. At 24 hours posttreatment cells were lysed using Tri Reagent® (Sigma-Aldrich Corp), and total RNA was extracted. RNA quantification was done using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA), and quality control was done using an Agilent Bioanalyzer 2100. For complementary DNA (cDNA) synthesis, we used 500 μg of total RNA and a Transcriptor First Strand cDNA Synthesis Kit (F Hoffman-La Roche Ltd), and for gene expression evaluation, we used the LightCycler® TaqMan® Master kit, on a LightCycler 480 instrument (F Hoffman-La Roche Ltd).
For gene expression normalization, we used β–actin (AGG AAT GGA AGC TTC CGG TA3′/AAT TTT CAT GGT GGA TGG TGC). Specific primers sequences for each gene of interest are presented below: AKT3: GGGAGGCCAAGGTAGATGA/TCA CAC CTA TAA TCC CAC ATG C; BAD: ACC AGC AGC AGC CAT CAT/GGT AGG AGC TGT GGC GAC T; BAK: CCT GCC CTC TGC TTC TGA/CTG CTG ATG GCG GTA AAA A; CASP8: TAG GGG ACT CGG AGA CTG C/TTT CTG CTG AAG TCC ATC TTT TT; FADD: CCG AGC TCA AGT TCC TAT GC/ AGG TCT AGG CCG CTC TGC; FAS: GGC CAA GTT GCT GAA TCA AT/GAG ACG AGC TCA CGA AAA GC; MYC:GCC GTA GGA AAT GAG CGA TA/AAC CTC GGT TGC TCT TCG T; NOD1: GAA TGC AAA GGC CTC ACG/GGC GAG ATA CTT CCC TCC TT; SMAD3: CAC CAC GCA GAA CGT CAA/GAT GGG ACA CCT GCA ACC; SMAD4: CCT GTT CAC AAT GAG CTT GC/GCA ATG GAA CAC CAA TAC TCA G; SMAD7: ACC CGA TGG ATT TTC TCA AA/AGG GGC CAG ATA ATT CGT TC; TNF-α: CAG CCT CTT CTC CTT CCT GAT/TGG GGA ACT CTT CCC TCT G; TP53: AGG CCT TGG AAC TCA AGG AT/CCC TTT TTG GAC TTC AGG TG; WNT11: AGC TCG CCC CCA ACT ATT/ATA CAC GAA GGC CGA CTC C; WNT3: CTC GCT GGC TAC CCA ATT T/ GAG CCC AGA GAT GTG TAC TGC; XIAP: GCA AGA GCT CAA GGA GAC CA/AAG GGT ATT AGG ATG GGA GTT CA; and ZEB1: TGA CTA TCA AAA GGA AGT CAA TGG/GTG CAG GAG GGA CCT CTT TA. For each target gene, we used the ΔΔCt method for quantification of the relative gene expression.17
Genes having a fold change ≤−1.25 or ≥1.25 and P-value <0.05 were considered to be genes of interest. The significant statistical genes were analyzed with Ingenuity® Pathway Analysis (IPA®) software (Ingenuity Systems, Redwood City, CA, USA).
Immunoblotting for protein validation
SSC-4 cells were treated with the same dose as for qRT-PCR evaluation, in order to validate gene expression data at the protein level. At 48 hours posttreatment, the cell culture medium was removed, and cells were washed twice with PBS and 150 μL cell lyse solution added, followed by a sonication step for 30 seconds; then cells were centrifuged at 16,000 g for clarification of the cellular lysate.
Cells were treated with 20 microM EGCG and seeded at a confluence of half a million/well in a six-well plate. After 48 hours from treatment, the medium was removed, and the cells were removed by scraping, in 200 μL of cell lyses solution (containing 2.5 mM tris(hydroxymethyl)aminomethane [Tris]-HCl [pH 6.8 at 25°C], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 50 mM dithiothreitol [DTT], and bromophenol blue) and sonicated for 30 seconds and then, centrifuged at 16,000 g for 5 minutes; then, the samples were transferred to a new tube. The protein determination was done using a BCA™ Protein Assay Kit (Thermo Fisher Scientific) in order to normalize the protein quantity. The protein separation was done using 7% acrylamide electrophoresis gel, based on a standard electrophoresis and nitrocellulose transfer. The immunoblotting was done for β-actin (1:1,000), p53 (1:1,000) (both antibodies from R&D Systems, Minneapolis, MN, USA) and FAS (1:500) from Cell Signaling Technology (Danvers, MA, USA). Membranes were washed and then incubated for 2 hours, with anti-mouse secondary antibody (1:1,000) (DakoCytomation, Glostrup, Denmark) for p53, and with anti-rabbit secondary antibody for β-actin. The staining was done by a colorimetric method using a Metal Enhanced DAB Substrate Kit (Thermo Fisher Scientific).
Results
The RTCA DP instrument is an innovative method for the monitoring of cell proliferation in real time, by using culture plates coated with gold microelectrodes at the base. These electrodes are used to measure the impedance from the electrical circuit, this being proportional with the cell proliferation index. Based on this device, we were able to register treatment response curves for the interval of time between 0–49 hours (Figure 1). As expected, we found a reduced proliferation rate in EGCG-treated SSC-4 cells.
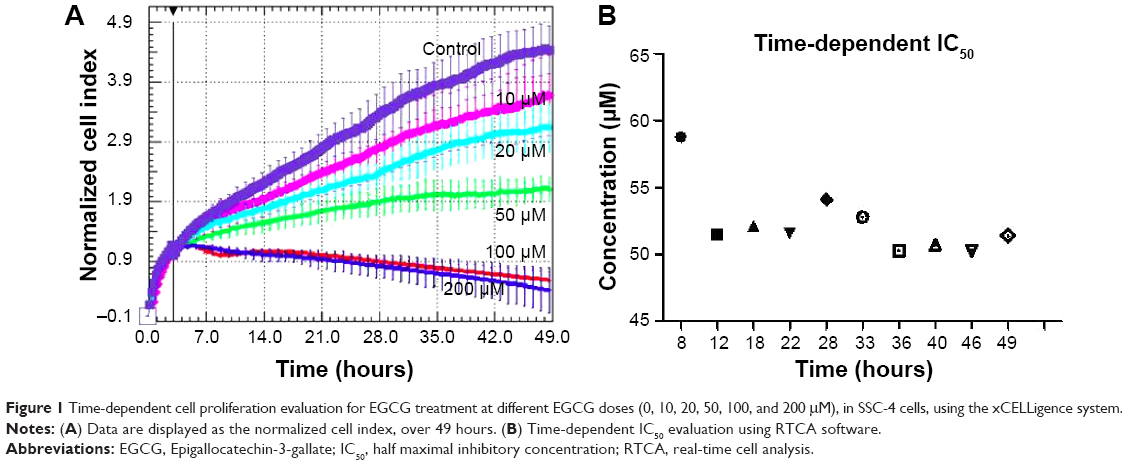 | Figure 1 Time-dependent cell proliferation evaluation for EGCG treatment at different EGCG doses (0, 10, 20, 50, 100, and 200 μM), in SSC-4 cells, using the xCELLigence system. |
The inhibitory effect was dose-dependent and accumulated over time, as displayed by the xCELLigence measurements in the 0–49 hours interval. Based on the cell proliferation data, the time-dependent half maximal inhibitory concentration (IC50) of EGCG was determined, and the average of was 52.3 μM in the case of SSC-4.
Apoptosis/necrosis evaluation in the SSC-4 cells treated with EGCG
The assessment of cellular apoptosis after 24 and 48 hours was done by comparing annexin V-fluorescein isothiocyanate (Annexin V-FITC) binding and PI staining of EGCG-treated cells (20 μM) versus that of a control (Figure 2). The results were expressed as % of apoptotic cells, and an increase of 28.6% of apoptotic cells was observed at 48 hours posttreatment. EGCG administration revealed that at 24 hours and 48 hours, 16.9% and 28.6% cells, respectively, underwent apoptosis. A relatively low percent of necrotic cells was observed for the same treatment (the values being 0.8% and 2.1%, respectively, at 24 hours and 48 hours posttreatment). This evaluation allowed differentiation among the different apoptosis stages and necrosis.
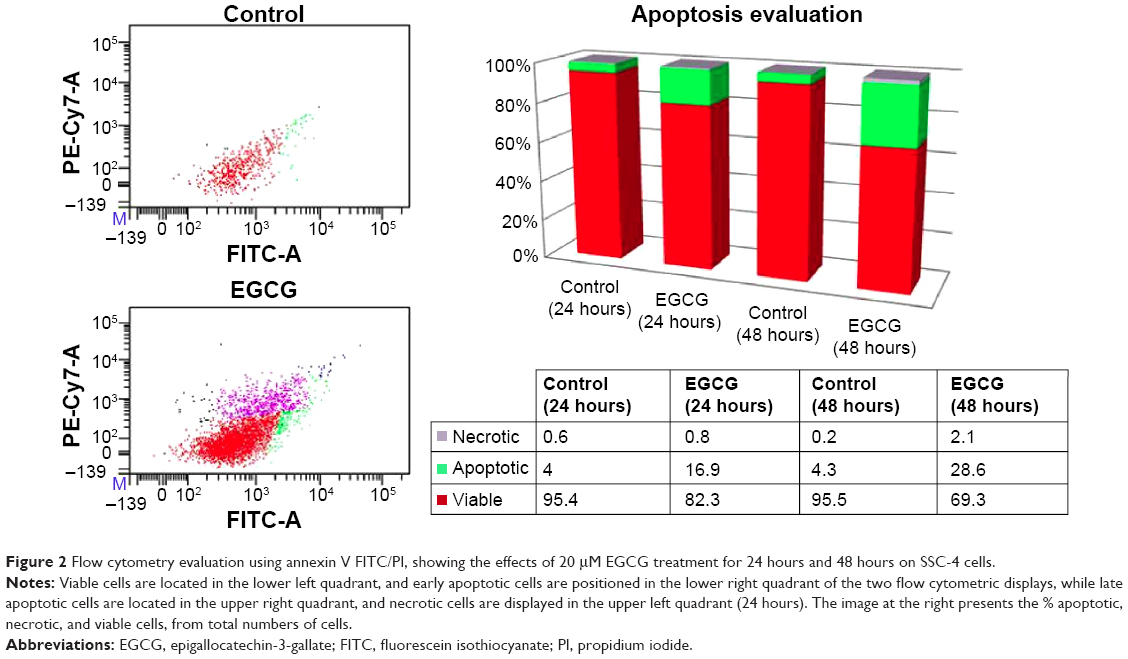 | Figure 2 Flow cytometry evaluation using annexin V FITC/PI, showing the effects of 20 μM EGCG treatment for 24 hours and 48 hours on SSC-4 cells. |
Autophagy evaluation in the SSC-4 cells treated with EGCG
Autophagy was proved to be related to the modulation of specific mechanisms of cell death. Using inverted fluorescence microscopy, we assessed whether EGCG treatment (20 μM) led to the modulation of the autophagy in SSC-4 human OSCC cells. Evaluation of the autophagy was done using a protocol based on monodansylcadaverine, a fluorescent product that is incorporated by the multilamellar bodies. Although SSC-4 is a cell line that manifests autophagy spontaneously, EGCG treatment intensified the level of autophagy (Figure 3B).
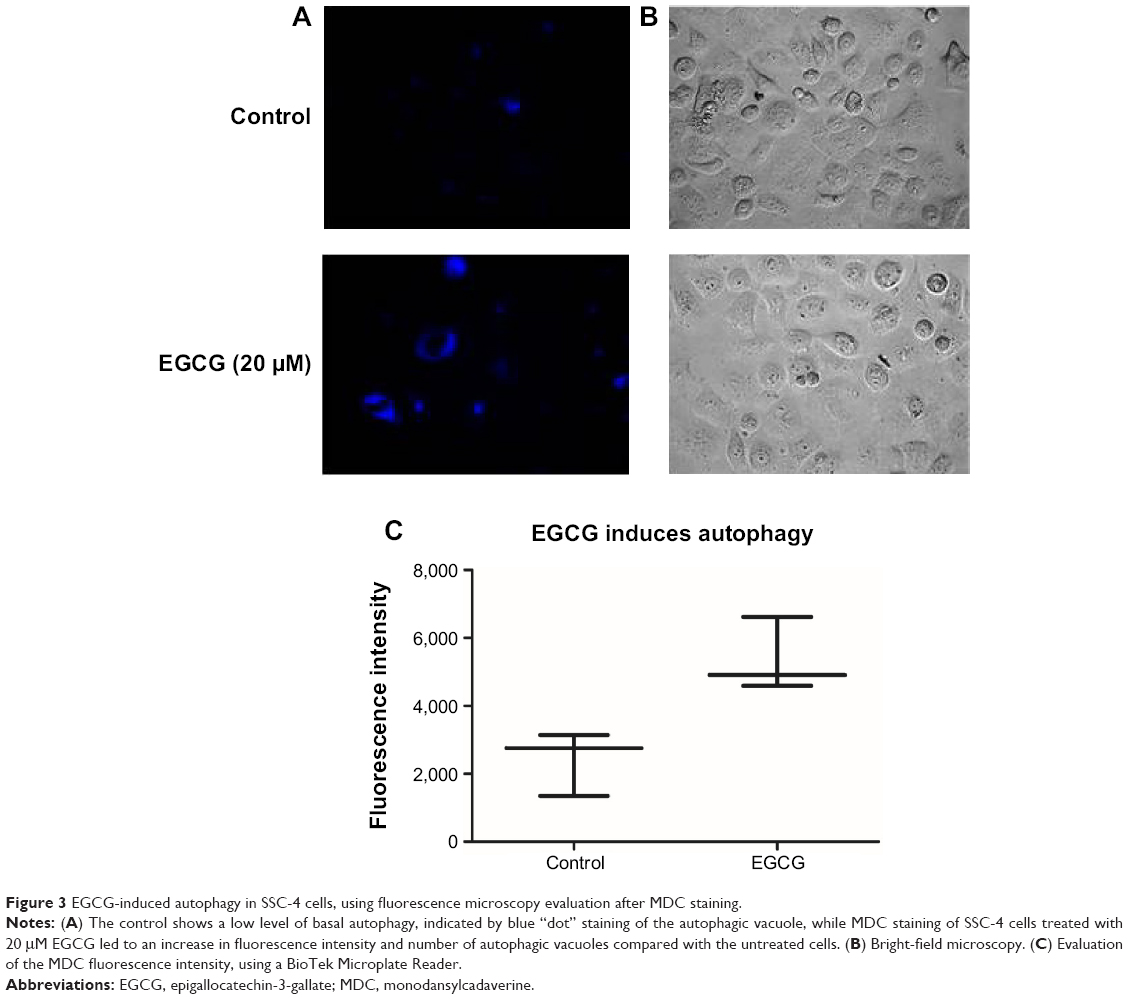 | Figure 3 EGCG-induced autophagy in SSC-4 cells, using fluorescence microscopy evaluation after MDC staining. |
EGCG treatment caused increase fluorescence intensity (Figure 3C), indicating that EGCG caused autophagy in SSC-4 cells.
Gene expression quantification as response to EGCG
To evaluate the relative gene expression in response to EGCG exposure, TaqMan technology was used for 20 genes. β-actin served as a housekeeping gene. Data were analyzed using the ΔΔCt method. Genes with a fold change of ≤−1.25 or ≥1.25 and P-value <0.05 were considered statistically significant. The fold change data are presented in Figure 4. Therefore, among the analyzed genes, we had six genes upregulated (BAD, BAK, FAS, IGF1R, WNT11, and ZEB1) and three genes downregulated (CASP8, MYC, and TP53).
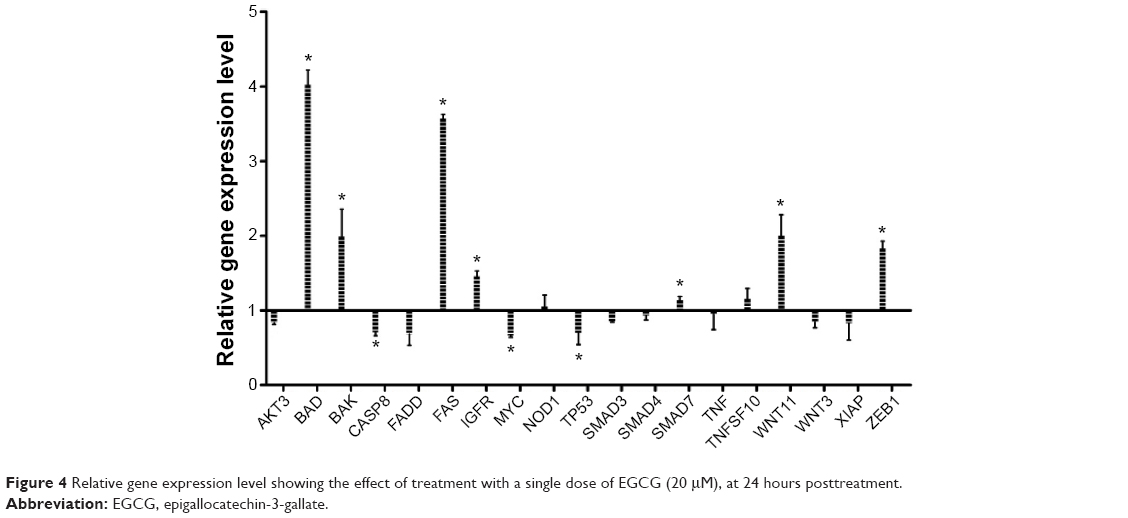 | Figure 4 Relative gene expression level showing the effect of treatment with a single dose of EGCG (20 μM), at 24 hours posttreatment. |
IPA analysis is a very useful program, integrating the altered genes as response to EGCG, in order to predict the altered biological processes and cellular functions (Figures 5 and 6). The color indicates altered genes (red for the upregulated and green downregulated genes) involved in disease progression or with prognostic value, all related to the alteration of cellular functions. EGCG treatment was activate proapoptotic genes (BAD, BAK or FAS) or cell cycle checkpoints (p53), all genes recognized as markers involved diagnostic or prognostic as displays IPA. It was observed downregulation of genes that decrease transmembrane potential of mitochondria (CASP8, MYC, TP53). The downregulation of genes that decrease the transmembrane potential of mitochondria (CASP8, MYC, and TP53) and that increase the damage by mitochondria (FAS) was observed, confirming the microscopic observation.
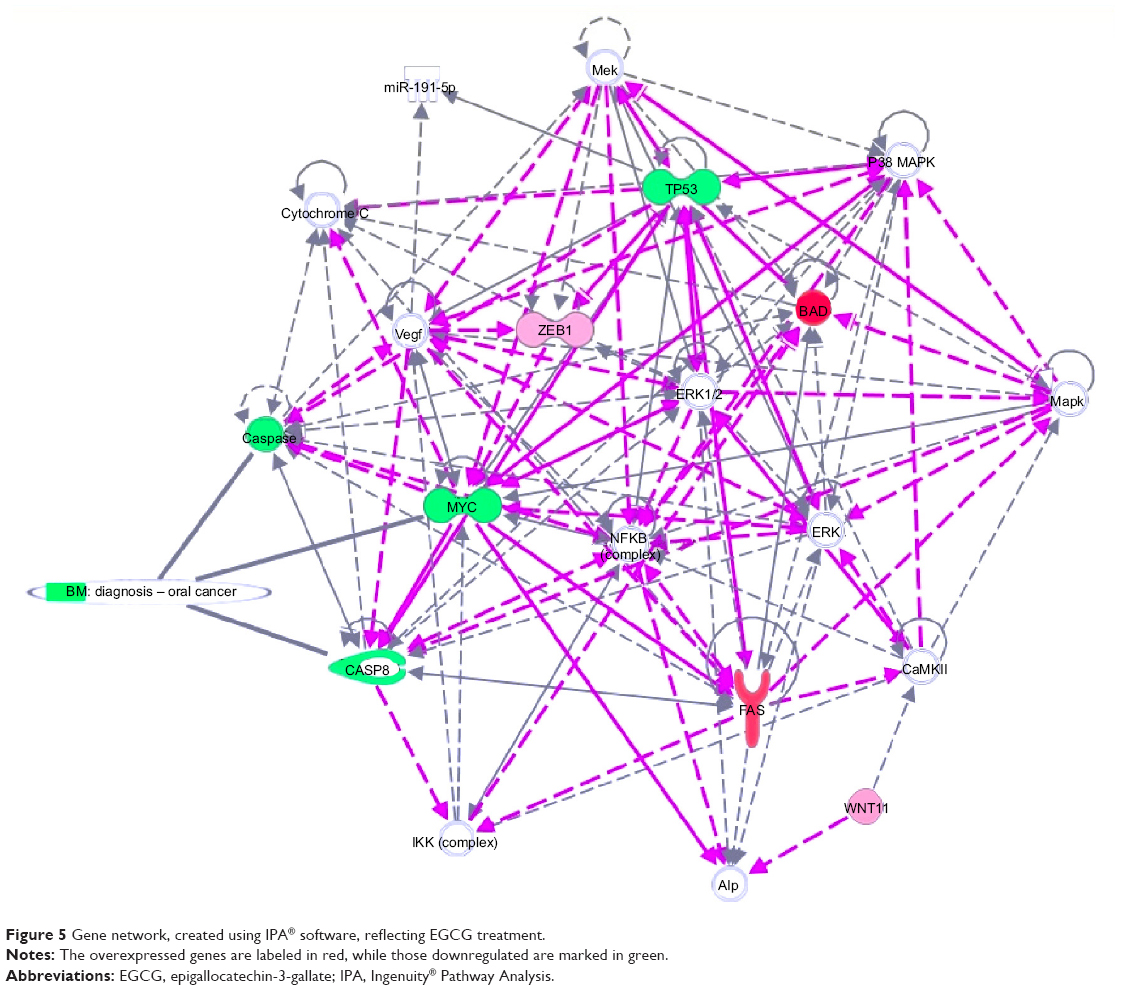 | Figure 5 Gene network, created using IPA® software, reflecting EGCG treatment. |
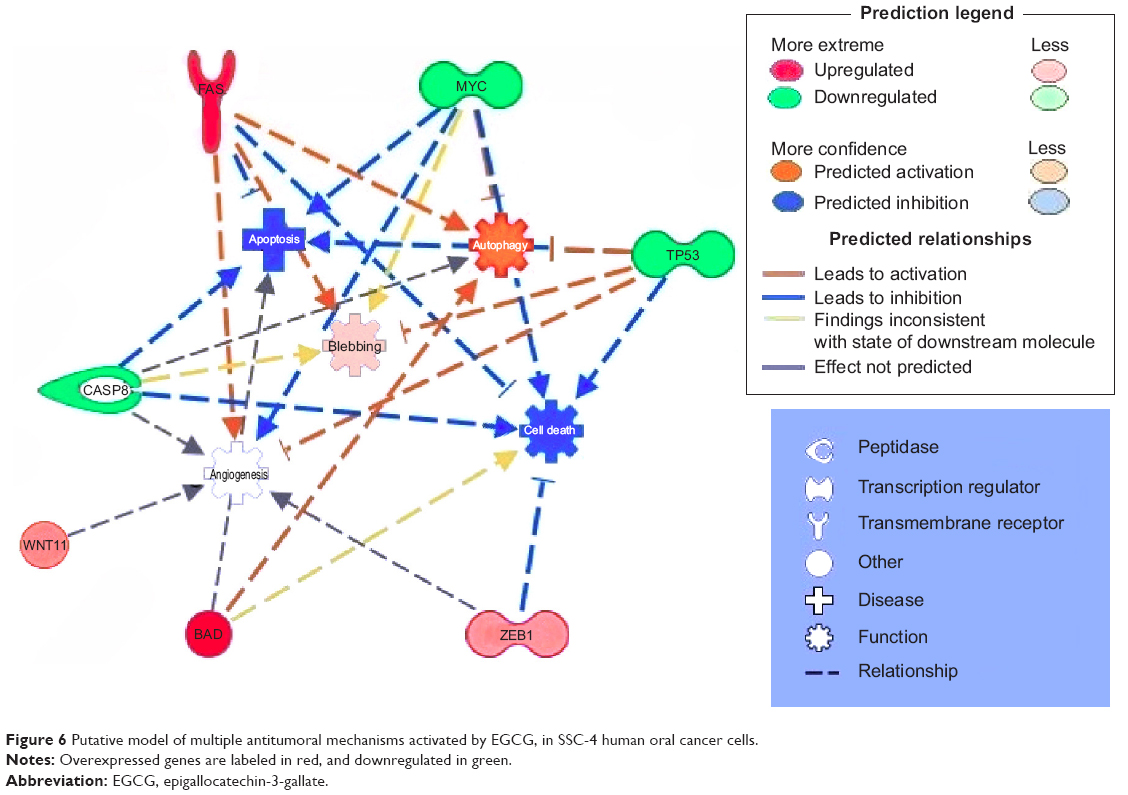 | Figure 6 Putative model of multiple antitumoral mechanisms activated by EGCG, in SSC-4 human oral cancer cells. |
Immunoblotting for gene expression validation of the data
ECGC treatment led to p53 downregulation and FAS overexpression, not only at the gene expression level but also, at protein level. The protein evaluation at 48 hours posttreatment is presented in Figure 7.
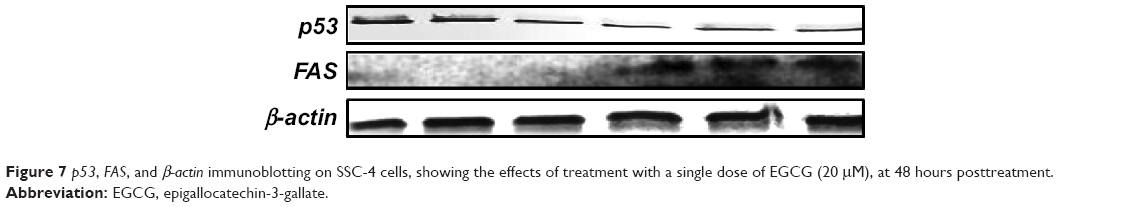 | Figure 7 p53, FAS, and β-actin immunoblotting on SSC-4 cells, showing the effects of treatment with a single dose of EGCG (20 μM), at 48 hours posttreatment. |
Discussion
EGCG reduces cell proliferation via apoptosis in a dose-dependent manner, as was confirmed in multiple cell lines. Autophagy is another important mechanism adding to apoptotic processes to reduce cell proliferation.2,18,19 In a previous study, the reduction of cell proliferation rate was shown to be related to the activation of the autophagy and induced reactive oxygen species (ROS).20,21 One of our previous studies demonstrated an essential function in ROS having the EGCG metabolic product.22,23 An important role in exerting biological active properties are the EGCG oxidation products, such as quinones and semiquinones.24 Consequently, our investigation presents the fact that the therapeutic actions of EGCG on the human squamous tumor cells involves, not only apoptosis but also, autophagy. This fact might have significant importance for chemoresistance mechanisms, based on the fact that EGCG is able to target multiple death pathways. It was proved that the autophagy enhances EGCG-induced cell death – this suggests the utility of autophagy inhibitors in enlarging the therapeutic response or in preventing activation of resistance to therapy.17
The Ingenuity software allowed identification of the biological processes and functions related to the affected up- and downregulated genes. As can be observed in Figures 5 and 6, EGCG treatment was able to target multiple genes involved in apoptosis, autophagy, and angiogenesis. This sustains the idea of developing novel therapeutic strategies based on EGCG. Gene expression data confirmed our previous findings concerning the activation of apoptosis and autophagy.22,25,26 The reduction of cell proliferation is related to these processes. We summarize the effect of EGCG treatment on different signaling genes and the potential effect on cell survival, apoptosis, and autophagy in SSC-4 cells, in Figure 6: Pharmacological activation of autophagy, by EGCG treatment, led to activation of apoptosis.
TP53, an essential regulator of tumor cell growth and proliferation, controlling cell cycle progression, cellular senescence, apoptosis, or autophagy of tumor cells, was downregulated by EGCG treatment.27 TP53 downregulation was associated with an increased response to chemotherapy.28
An interesting finding was the inhibition of CASP8, a key upstream mediator in death receptor–mediated apoptosis that participates in mitochondria-mediated apoptosis via cleavage of proapoptotic factors.29–32 In concert with the CASP8 activation by EGCG, an enhanced expression in the functional FAS/CD95 pathway has also been identified,33 which was confirmed in the present study on SSC-4 cells. The FAS gene plays important roles in oral cancer cell apoptotic mechanisms.34 Apoptosis in response to local FAS activation in vivo has also been demonstrated in epithelial cells.35,36 The MYC gene has been found to mediate apoptosis of oral squamous cell carcinoma in certain conditions, and this might be related to upregulation of FAS expression.37 MYC has two outputs in normal cells, the induction of apoptosis and proliferation.38–40
BAD and BAK are proapoptotic BCL-2 family member proteins that regulate the intrinsic apoptosis pathway.41 BAK is an essential effector of the intrinsic pathway of apoptosis,42 specifically activated by EGCG in SSC-4 cells. In OSCC, compared with oral epithelium, there is a decreased BCL-2 expression.43
ZEB1 overexpression, using lentiviral based delivery system for ZEB1 (lenti-siRNA/ZEB1) in lung adenocarcinoma cells, was previously found to be related to the inhibition of cell proliferation both in vitro and in vivo, and induces cell apoptosis.31 This has significance for the development of novel treatment strategies, particularly in context of the capacity for multitargeted therapy.
IGF1R expression increases with tumor progression,44 and downregulation of IGF1R expression causes apoptosis.45,46 The IGF1R gene also activates alternative pathways for protection from apoptosis and in some cases, is involved in cell proliferation and differentiation. The multiplicity of signaling pathways used by IGF1R may explain why its receptor has such a powerful and widespread antiapoptotic activity.47–49 Expression of IGF1R was shown to be significantly increased in the carcinogenesis of OSCC, with metabolically active regions of OSCC being strongly correlated to proliferating cancer cells without detection of apoptosis.50 IGF1R overexpression is related to familial breast cancer,51 and this factor might be related to resistance to therapy in the case of OSCC cells.52
The literature, to date, has presented WNT11 as overexpressed in OSCC.53–55 This might represent a therapeutic target for OSCC,53 based on the fact that WNT-pathway genes were related to the activation of tumorigenesis mechanisms,54 caused by epigenetic alteration.55
Our results showed that EGCG induces activation of cell death receptors, leading to activation of intrinsic apoptotic pathways. Therefore, data from this study have identified an important role of autophagy in the occurrence of a cell death mechanism induced by EGCG, in SSC-4 cells. All of these results suggest that EGCG has an excellent potential for treatment or as adjuvant therapy for patients with OSCC, by inducing cell death via apoptosis and autophagy.
Acknowledgments
Dr Gherman received a fellowship financed by the grant (number 159/1.5/s/138776) entitled “Model colaborativ institutional pentru translatarea cercetarii stiintifice biomedicale in practica clinica-TRANSCENT” [Institutional collaborative model of biomedical scientific research transposed in clinical practice – TRANSCENT].
Disclosure
The authors report no conflicts of interest in this work.
References
Parris TZ, Aziz L, Kovács A, et al. Clinical relevance of breast cancer-related genes as potential biomarkers for oral squamous cell carcinoma. BMC Cancer. 2014;14:324. | ||
Sand L, Jalouli J. Viruses and oral cancer. Is there a link? Microbes Infect. 2014;16(5):371–378. | ||
Bhavana SM, Lakshmi CR. Oral oncoprevention by phytochemicals – a systematic review disclosing the therapeutic dilemma. Adv Pharm Bull. 2014;4(Suppl 1):S413–S420. | ||
Lin LW, Lu YC, Wang CP, et al. Visfatin/pre-B cell colony-enhancing factor immunohistochemical overexpression in oral cancers. J Appl Biomed. 2014;12(4):255–226. | ||
González-Ramírez I, Soto-Reyes E, Sánchez-Pérez Y, Herrera LA, García-Cuellar C. Histones and long non-coding RNAs: the new insights of epigenetic deregulation involved in oral cancer. Oral Oncol. 2014;50(8):691–695. | ||
Islam MR, Jones SJ, Macluskey M, Ellis IR. Is there a pAkt between VEGF and oral cancer cell migration? Cell Signal. 2014;26(6):1294–1302. | ||
Güneri P, Epstein JB. Late stage diagnosis of oral cancer: Components and possible solutions. Oral Oncol. 2014;50(12):1131–1136. | ||
Liang X, Osman TA, Sapkota D, et al. Rapid adherence to collagen IV enriches for tumour initiating cells in oral cancer. Eur J Cancer. 2014;50(18):3262–3270. | ||
Papagerakis S, Pannone G, Zheng L, et al. Oral epithelial stem cells – implications in normal development and cancer metastasis. Exp Cell Res. 2014;325(2):111–129. | ||
Wang W, Yang Y, Zhang W, Wu W. Association of tea consumption and the risk of oral cancer: a meta-analysis. Oral Oncol. 2014;50(4):276–281. | ||
Chianeh YR, Prabhu K. Biochemical markers in saliva of patients with oral squamous cell carcinoma. Asian Pac J Trop Dis. 2014;4 Suppl 1: S33–S40. | ||
Supic G, Jovic N, Zeljic K, Kozomara R, Magic Z. Association of VEGF-A genetic polymorphisms with cancer risk and survival in advanced-stage oral squamous cell carcinoma patients. Oral Oncol. 2012;48(11):1171–1177. | ||
Mukhtar E, Adhami VM, Khan N, Mukhtar H. Apoptosis and autophagy induction as mechanism of cancer prevention by naturally occurring dietary agents. Curr Drug Targets. 2012;13(14):1831–1841. | ||
Bodhade AS, Dive AM. Chemoprevention of premalignant and malignant lesions of oral cavity: Recent trends. Eur J Dent. 2013;7(2):246–250. | ||
Lee UL, Choi SW. The chemopreventive properties and therapeutic modulation of green tea polyphenols in oral squamous cell carcinoma. ISRN Oncol. 2011;2011:403707. | ||
Braicu C, Ladomery MR, Chedea VS, Irimie A, Berindan-Neagoe I. The relationship between the structure and biological actions of green tea catechins. Food Chem. 2013;141(3):3282–3289. | ||
Hatok J, Babusikova E, Matakova T, Mistuna D, Dobrota D, Racay P. In vitro assays for the evaluation of drug resistance in tumor cells. Clin Exp Med. 2009;9(1):1–7. | ||
Li W, Zhu S, Li J, Assa A, et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol. 2011;81(9):1152–1163. | ||
Kim HS, Montana V, Jang HJ, Parpura V, Kim JA. Epigallocatechin gallate (EGCG) stimulates autophagy in vascular endothelial cells: a potential role for reducing lipid accumulation. J Biol Chem. 2013;288(31):22693–22705. | ||
Satoh M, Takemura Y, Hamada H, Sekido Y, Kubota S. EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy. Cancer Cell Int. 2013;13(1):19. | ||
Wang CT, Chang HH, Hsiao CH, et al. The effects of green tea (-)-epigallocatechin-3-gallate on reactive oxygen species in 3T3-L1 preadipocytes and adipocytes depend on the glutathione and 67 kDa laminin receptor pathways. Mol Nutr Food Res. 2009;53(3):349–360. | ||
Braicu C, Pilecki V, Balacescu O, Irimie A, Neagoe IB. The relationships between biological activities and structure of flavan-3-ols. Int J Mol Sci. 2011;12(12):9342–9353. | ||
Chedea VS, Braicu C, Chirilă F, et al. Antioxidant/Prooxidant and antibacterial/probacterial effects of a grape seed extract in complex with lipoxygenase. Biomed Res Int. 2014;2014:313684. | ||
Chedea VS, Braicu C, Socaciu C. Antioxidant/prooxidant activity of a polyphenolic grape seed extract. Food Chem. 2010;121(1):132–139. | ||
Braicu C, Gherman C. Epigallocatechin gallate induce cell death and apoptosis in triple negative breast cancer cells Hs578T. J Drug Target. 2013;21(3):250–256. | ||
Berindan-Neagoe I, Braicu C, Tudoran O, Balacescu O, Irimie A. Early apoptosis signals induced by a low dose of epigallocatechin 3-gallate interfere with apoptotic and cell death pathways. J Nanosci Nanotechnol. 2012;12(3):2113–2119. | ||
Hao Q, Cho WC. Battle against cancer: an everlasting saga of p53. Int J Mol Sci. 2014;15(12):22109–22127. | ||
Fiorini C, Cordani M, Padroni C, Blandino G, Di Agostino S, Donadelli M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim Biophys Acta. 2015;1853(1):89–100. | ||
Groebner AE, Schulke K, Unterseer S, et al. Enhanced proapoptotic gene expression of XAF1, CASP8 and TNFSF10 in the bovine endometrium during early pregnancy is not correlated with augmented apoptosis. Placenta. 2010;31(3):168–177. | ||
Lee SY, Kang HG, Yoo SS, et al. Polymorphisms in DNA repair and apoptosis-related genes and clinical outcomes of patients with non-small cell lung cancer treated with first-line paclitaxel-cisplatin chemotherapy. Lung Cancer. 2013;82(2):330–339. | ||
Liu J, Uematsu H, Tsuchida N, Ikeda MA. Essential role of caspase-8 in p53/p73-dependent apoptosis induced by etoposide in head and neck carcinoma cells. Mol Cancer. 2011;10:95. | ||
Chaopatchayakul P, Jearanaikoon P, Yuenyao P, Limpaiboon T. Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma. Am J Obstet Gynecol. 2010;202(3):281.e1–281.e9. | ||
Lin HY, Hou SC, Chen SC, et al. (-)-Epigallocatechin gallate induces Fas/CD95-mediated apoptosis through inhibiting constitutive and IL-6-induced JAK/STAT3 signaling in head and neck squamous cell carcinoma cells. J Agric Food Chem. 2012;60(10):2480–2489. | ||
de Carvalho-Neto PB, dos Santos M, de Carvalho MB, et al. FAS/FASL expression profile as a prognostic marker in squamous cell carcinoma of the oral cavity. PLoS One. 2013;8(7):e69024. | ||
Santiago B, Galindo M, Palao G, Pablos JL. Intracellular regulation of Fas-induced apoptosis in human fibroblasts by extracellular factors and cycloheximide. J Immunol. 2004;172(1):560–566. | ||
Oz HS, Ebersole JL. Green tea polyphenols mediated apoptosis in intestinal epithelial cells by a FADD-dependent pathway. J Cancer Ther. 2010;1(3):105–113. | ||
Supino R, Perego P, Gatti L, et al. A role for c-myc in DNA damage-induced apoptosis in a human TP53-mutant small-cell lung cancer cell line. Eur J Cancer. 2001;37(17):2247–2256. | ||
Watson AJ. An overview of apoptosis and the prevention of colorectal cancer. Crit Rev Oncol Hematol. 2006;57(2):107–121. | ||
Claassen G, Brin E, Crogan-Grundy C, et al. Selective activation of apoptosis by a novel set of 4-aryl-3-(3-aryl-1-oxo-2-propenyl)-2(1H)-quinolinones through a Myc-dependent pathway. Cancer Lett. 2009;274(2):243–249. | ||
Järvinen K, Hotti A, Santos L, Nummela P, Hölttä E. Caspase-8, c-FLIP, and caspase-9 in c-Myc-induced apoptosis of fibroblasts. Exp Cell Res. 2011;317(18):2602–2615. | ||
Gryko M, Pryczynicz A, Guzińska-Ustymowicz K, et al. Immunohistochemical assessment of apoptosis-associated proteins: p53, Bcl-xL, Bax and Bak in gastric cancer cells in correlation with clinical and pathomorphological factors. Adv Med Sci. 2012;57(1):77–83. | ||
Ma S, Hockings C, Anwari K, et al. Assembly of the Bak apoptotic pore: a critical role for the Bak protein α6 helix in the multimerization of homodimers during apoptosis. J Biol Chem. 2013;288(36):26027–26038. | ||
Jain M, Kasetty S, Udyavara Sridhara S, Jain N, Khan S, Desai A. Apoptosis and its significance in oral diseases: an update. J Oral Dis. 2013;2013:401049. | ||
Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res. 2001;61(19):7318–7324. | ||
Maloney EK, McLaughlin JL, Dagdigian NE, et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63(16):5073–5083. | ||
Hilmi C, Larribere L, Giuliano S, et al. IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin. J Invest Dermatol. 2008;128(6):1499–1505. | ||
Torres Aleman I. Role of insulin-like growth factors in neuronal plasticity and neuroprotection. Adv Exp Med Biol. 2005;567:243–258. | ||
Shelton JG, Steelman LS, White ER, McCubrey JA. Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF-1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle. 2004;3(3):372–379. | ||
Kuemmerle JF. IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G411–G422. | ||
Grimm M, Cetindis M, Lehmann M, et al. Association of cancer metabolism-related proteins with oral carcinogenesis – indications for chemoprevention and metabolic sensitizing of oral squamous cell carcinoma? J Transl Med. 2014;12:208. | ||
Jernström H, Sandberg T, Bågeman E, Borg A, Olsson H. Insulin-like growth factor-1 (IGF1) genotype predicts breast volume after pregnancy and hormonal contraception and is associated with circulating IGF-1 levels: implications for risk of early-onset breast cancer in young women from hereditary breast cancer families. Br J Cancer. 2005;92(5):857–866. | ||
Jiang L, Liu X, Chen Z, et al. MicroRNA-7 targets IGF1R (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem J. 2010;432(1):199–205. | ||
Vincent-Chong VK, Ismail SM, Rahman ZA, et al. Genome-wide analysis of oral squamous cell carcinomas revealed over expression of ISG15, Nestin and WNT11. Oral Dis. 2012;18(5):469–476. | ||
Andrade Filho PA, Letra A, Cramer A, et al. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J Dent Res. 2011;90(6):740–746. | ||
Pannone G, Bufo P, Santoro A, et al. WNT pathway in oral cancer: epigenetic inactivation of WNT-inhibitors. Oncol Rep. 2010;24(4):1035–1041. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.