")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 15
Epidemiology, Impact, and Management Strategies of Anti-Glomerular Basement Membrane Disease
Received 8 January 2022
Accepted for publication 15 March 2022
Published 7 April 2022 Volume 2022:15 Pages 129—138
DOI https://doi.org/10.2147/IJNRD.S326427
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Muhammad Asim,1 Mohammed Akhtar2
1Division of Nephrology, Department of Medicine, Hamad General Hospital, Hamad Medical Corporation, Doha, Qatar; 2Department of Laboratory Medicine and Pathology, Hamad General Hospital, Hamad Medical Corporation, Doha, Qatar
Correspondence: Muhammad Asim, Division of Nephrology, Department of Medicine, Hamad General Hospital, Hamad Medical Corporation, Doha, Qatar, Tel +97455838342, Email [email protected]
Abstract: Anti-glomerular basement membrane (anti-GBM) disease is a rare but serious autoimmune disease, which is characterized by the development of pathogenic antibodies to type IV collagen antigens in the glomerular and alveolar basement membranes. This results in rapidly progressive glomerulonephritis (GN), alveolar hemorrhage, or both. A variety of environmental factors can trigger the disease in genetically predisposed patients. Temporal associations with influenza, SARS-CoV-2 infection, and COVID-19 vaccination have been described although there is insufficient evidence to suggest causality. Anti-GBM disease accounts for approximately 20% of the cases of rapidly progressive GN cases secondary to crescentic GN, but is an uncommon cause of end-stage kidney disease. Early diagnosis by detection of circulating antibodies, increased awareness of atypical as well as complex clinical variants of the disease, and combined therapy with immunosuppression and plasma exchange has improved the prognosis of patients with this potentially fatal disease. Progress has been hampered by the rarity of anti-GBM disease, but new agents and therapeutic regimens are emerging.
Keywords: antibody, glomerulonephritis, rapidly progressive glomerulonephritis, plasma exchange, IdeS, rituximab
Anti-glomerular basement membrane (anti-GBM) disease is an autoimmune small- vessel vasculitis caused by immunoglobulin (Ig) G autoantibodies that are mainly directed against the non-collagenous domain of the α3 chain of type IV collagen in the glomerular basement membrane.1 These antibodies typically cause a rapidly progressive GN, often combined with pulmonary hemorrhage because the autoantibodies react with the same epitope in the alveolar basement membrane.2
The basement membranes in the glomerular and pulmonary alveolar capillaries are complex structures lined by endothelial and epithelial cells. The principal component of the basement membrane is type IV collagen (Figure 1), which includes three alpha subunits: alpha3, alpha4 and alpha5 chains (Figure 1A). Each chain comprises a long collagenous domain, a non-collagenous amino terminus (7S), and a non-collagenous domain (NC1) at the carboxyl terminus. NC1 domains of the alpha3, alpha4 and alpha5 chains join to form an extensively crosslinked helical molecule known as a protomer (Figure 1B).1,3 The protomers dimerize at NC1 domains to form hexamers, that further interconnect to form a collagenous meshwork. Deposition of additional proteins such as agrin, laminin, and nidogen completes the formation of the basement membrane.4 The anti-GBM antibodies are most strongly reactive to alpha3 (IV) NC1, although there may also be variable reactivity to alpha5 (IV) NC1 and a lesser extent, alpha4 (IV) NC1.1,5 The autoantibodies bind to two epitopes, designated EA and EB, located at the amino acid residues 17–31 and 127–141 respectively, in the NC1 domain of alpha3 (IV) (Figure1C).6 These epitopes are partly buried and hidden during the assembly of hexamers.
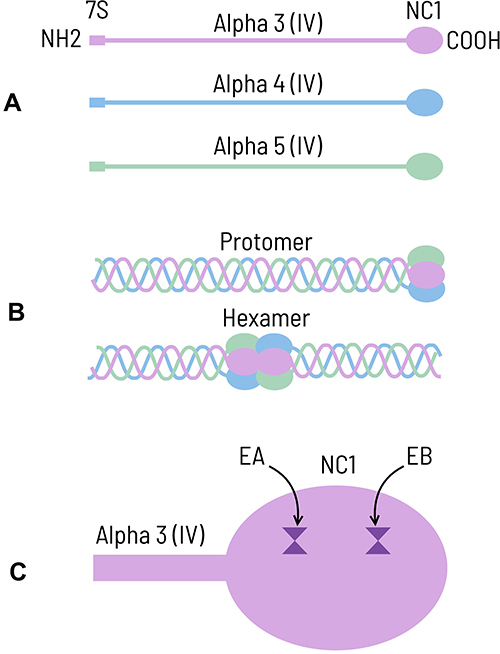 |
Figure 1 (A) Cartoon showing alpha3, alpha4 and alpha5 chains of type IV collagen. Each chain consists of collagenous component in the middle with non-collagenous domains (7S and NC1) at the ends. (B) NC1 domains of the alpha3, alpha4 and alpha5 chains join to form a triple helical extensively crosslinked molecule known as protomer. The protomers dimerize at NC1 domains to form alpha3.alpha4.alpha5 (IV)NC1 hexamers. (C) Cartoon depicting NC1 domain of the alpha3 chain of type IV collagen with two epitopes, namely EA (residues 17–31) and EB (residues 127–141). Copyright © 2022 Mohammed Akhtar, Corel Corporation and its licensors. All rights reserved. |
Immunofluorescent microscopy demonstrates characteristic intense and diffuse linear IgG deposition along the glomerular basement membrane (Figure 2). On light microscopy the glomeruli characteristically display segmental or global necrosis, and rupture of the capillary tufts, associated with prominent crescent formation in most of the cases (Figure 3).7,8 The glomerular changes are temporally homogeneous, with all glomeruli presenting the same stage of the disease process. Other findings usually include intratubular red cells, interstitial edema, and interstitial inflammatory infiltrate composed of lymphocytes, plasma cells, and macrophages. Periglomerular accentuation of the mononuclear infiltrate is commonly seen in association with variably sized ruptures of the Bowman capsules. Multinucleated giant cells or granulomas in and around the glomeruli may also be found.7 In the chronic phase, the glomeruli may show fibrous and fibro-cellular crescents, later leading to global glomerulosclerosis with interstitial fibrosis and tubular atrophy.
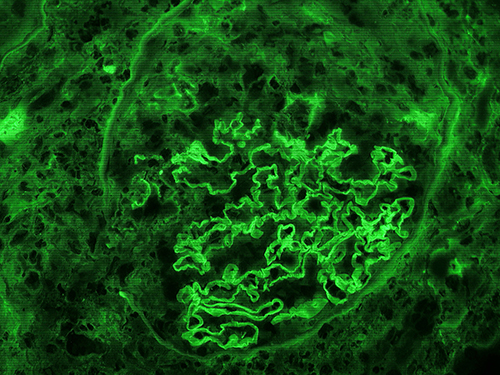 |
Figure 2 Immunofluorescent microscopic image showing intense linear deposition of IgG along the glomerular capillaries. The adjacent crescent is devoid of fluorescence (magnification 400 X). |
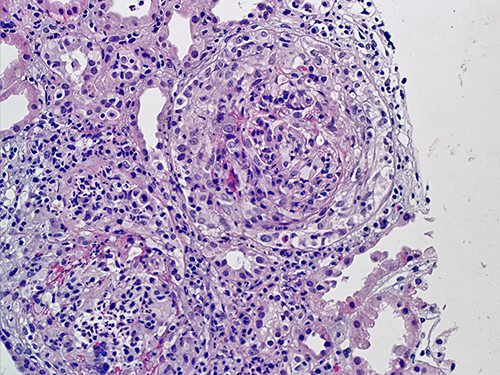 |
Figure 3 Light microscopy of the renal biopsy featuring cellular crescents in both the glomeruli, and a large number of inflammatory cells within. There is extensive destruction of the glomerular tufts (Hematoxylin and Eosin staining; magnification 300 X). |
Epidemiology
The annual incidence of anti-GBM disease is estimated at fewer than 2 cases per million population – about 10 times lower than that of ANCA-associated vasculitis.4 It has a predilection for European Caucasians, though it occurs across all racial groups. A bimodal distribution of age-specific incidence rates has been recognized that peaks in the third decade (characterized by male preponderance, frequent concomitant lung hemorrhage, high level of specific anti-GBM antibodies, but ANCA negativity) and seventh decade (demonstrating even gender distribution, infrequent lung hemorrhage, lower levels of anti-GBM antibodies with broader reactivity, but frequent ANCA positivity).9,10 DRB1*1501, DRB1*03, and DRB1*04 have been shown to be associated with increased susceptibility to the disease.11 In addition, environmental triggers have been proposed to incite the disease in susceptible individuals.12 Basement membrane injury from various etiological agents (including infection, smoking, organic solvents, nephrectomy/ extracorporeal shock wave lithotripsy) can either initiate the disease by exposing the cryptic epitopes in glomerular/alveolar basement membrane with consequent antibody production and immune-mediated glomerular and/or pulmonary damage; or unmask the covert disease by releasing autoantigens in patients who had pre-existing circulating anti-GBM antibodies. Consistent with these hypotheses is the fact that pulmonary hemorrhage occurs in nearly all current cigarette smokers but is uncommon in nonsmokers.13 Similarly, outbreaks of anti-GBM disease have been described during influenza epidemics.14–17 More recently, temporal clustering of anti-GBM disease during COVID-19 pandemic has been reported.18–20 The affected patients had prodromal viral illness 1–6 weeks before presentation with anti-GBM disease, and tested positive for viral RNA, or IgM and/or IgG antibodies to SARS-CoV-2 spike protein, raising the possibility of SARS-CoV-2 infection as a possible trigger for the disease.18–20 A case of recurrence of anti-GBM disease presenting with pulmonary hemorrhage and crescentic GN following COVID-19 has also been described.21 Several cases of anti-GBM disease related to SARS-CoV-2 vaccination have been published recently.22–24 Although a temporal relationship has been noted, there is insufficient evidence to postulate causality. Nevertheless, it is plausible that the immune response to the vaccine mimics the biological response to natural infection resulting in anti-GBM disease in susceptible patients. With the continuation of mass vaccination programs, the advent of more potent vaccines (ie mRNA), and recommendations of booster doses, long-term pharmacovigilance is required to determine the incidence of anti-GBM disease secondary to vaccination, and its clinical outcomes.
Impact
Anti-GBM disease is a rare disease and an uncommon cause of end-stage kidney disease [ESKD].25 It accounts for only up to 5% of all types of GN, and 20% of the cases of rapidly progressive GN cases secondary to crescentic GN.26,27 However, the disease has a major impact on those who are affected. Patient and renal survival depends largely on the severity of kidney dysfunction at presentation.28,29 Need for mechanical ventilation, oligo-anuria, >85% cellular crescents on renal biopsy, and dialysis dependency at presentation are among the strongest predictors of poor outcomes. One-year patient and renal survival rates following a diagnosis of anti-GBM disease have previously been reported to be 73% and 25%, respectively.30 However, timely diagnosis, increased cognizance of disease heterogeneity, and early institution of intensive therapy has changed the outlook of this disease, resulting in a two-fold higher kidney survival rate since 2007.28
Long-term patient and graft survival after kidney transplantation for anti-GBM patients are comparable to IgA nephropathy as a cause of ESKD.31 However, 5–10% of kidney transplant recipients with underlying Alport syndrome develop overt anti-GBM disease, which has a major impact on affected patients – the risk of irreversible graft failure approaches 90%, usually within a few weeks to months after diagnosis and re-transplantation is rarely successful.32
Management Strategies
Management of anti-GBM disease takes into consideration the following salient features of the disease:
1-Anti-GBM antibodies are considered pathogenic, and are detected in the majority of cases.33,34 A “seronegative” variant of anti-GBM disease is well recognized which is characterized by diffuse linear staining of GBM by IgG on immunofluorescence microscopy without detectable anti-GBM antibodies in the serum by enzyme-linked immunosorbent assay (ELISA), Western blot, or indirect immunofluorescence, and which has the potential of progressing to end-stage renal disease.35,36
2-Renal function diminishes rapidly in anti-GBM disease; early treatment is often the key to better clinical outcomes.4
3-It is typically a single-phase disease, unlike most other types of GN or vasculitis, with initially severe renal and pulmonary involvement, but infrequent subsequent relapses. Hence, maintenance immunosuppression therapy is usually not required.
4-There is substantial heterogeneity. At one end of the spectrum are patients with a milder clinical phenotype–“atypical anti-GBM disease”–who have subtle renal involvement (hematuria and/or proteinuria but relatively preserved renal function) without pulmonary hemorrhage.35 At the other end of the spectrum are the vast majority of patients with “classic” anti-GBM who present with a rapidly progressive crescentic GN.37 About 50% of the latter have a full blown pulmonary-renal syndrome with severe alveolar hemorrhage. Fewer than 10% present with pulmonary disease, although they often have renal disease histologically.38 Another interesting subset of anti-GBM patients, the “double-positive” patients, are those who exhibit co-existence of ANCA and anti-GBM antibodies. Acknowledging the entity of seronegative anti-GBM disease, “double positivity” has been more precisely defined as the association of ANCA and histological features of anti-GBM disease, with or without circulating anti-GBM antibodies.39 These patients demonstrate a hybrid phenotype, although the anti-GBM component is the dominant phenotype.4,39 Their initial clinical presentation is aggressive, similar to that of “single-positive” anti-GBM patients (RPGN and alveolar hemorrhage) but they display several features of ANCA associated vasculitis, such as older age, longer prodromal illness, systemic organ involvement, histological evidence of chronicity (interstitial fibrosis and glomerulosclerosis), and tendency to relapse.4,40
The primary therapeutic aim is to rapidly decrease the level of anti-GBM antibodies and reduce inflammation. This can be achieved by removing preformed circulating IgG by extracorporeal therapy, or by blocking the production of new antibodies using immunosuppressive therapy. An emerging approach is to hydrolyze and degrade the antibodies, using proteolytic enzymes. Supportive care, including dialytic and mechanical ventilatory support is provided when indicated.
Extracorporeal Therapies to Remove Preformed Anti-GBM Antibodies
Although never submitted to randomized controlled trials, plasma exchange (PLEX) has been considered standard of care since the 1970s, and there is convincing evidence of its clinical effectiveness.27,41–44 PLEX is recommended in all patients with renal limited disease except those who are dialysis-dependent at presentation, have 100% glomerular crescents or >50% globally sclerotic glomeruli.45 This grade 1B recommendation by KDIGO Glomerulonephritis Work Group is primarily based on overwhelming evidence from observational studies that showed better clinical outcomes compared with patients treated with immunosuppressive treatment alone. Similarly, the American Society for Apheresis advocates PLEX as the first line treatment for patients with anti-GBM disease who have diffuse alveolar hemorrhage or non-dialysis-dependent renal failure.46 PLEX might have an additional benefit of removing cytokines, complement components, or adhesion molecules from plasma, thereby attenuating inflammation and tissue injury.47 Prognosis of classical anti-GBM disease is strongly correlated with rapid reduction in anti-GBM antibody levels. Hence, it is vital that intensive PLEX is implemented early in the course of anti-GBM disease (60mL/kg; max 4 L of plasma processed per session) and continued on daily basis, if possible, till anti-GBM titer becomes negative/ markedly suppressed, and hemoptysis resolves. This usually occurs in two to three weeks in most patients.48,49 In patients with seronegative anti-GBM disease, who have no detectable circulating anti-GBM antibodies, PLEX should be continued until the resolution of evidence of ongoing pulmonary or glomerular injury.48
Immunoadsorption (IA) and double filtration plasmapheresis (DFPP) have been utilized at some centers to remove circulating antibodies in patients with crescentic GN, including those with anti-GBM disease. In DFPP, two types of filters are employed; the first filter separates the plasma, which then passes through a second “plasma component separator” before being returned to the patient. This makes DFPP a more selective modality for removing pathogenic antibodies compared to PLEX and lessens the need for blood product replacement. Clinical outcomes are comparable to those of PLEX.50 IA allows more profound antibody removal with efficiency exceeding that of PLEX. It might have a role in patients with very high anti-GBM antibody levels or those with persistence of anti-GBM antibodies despite PLEX therapy.51,52
Immunosuppressive Therapy to Block the Generation of New Antibodies
No randomized study has defined the optimal immunosuppressive regimen. The use of systemic glucocorticoids and cyclophosphamide, the mainstays of immunosuppressive treatment, has relied largely on historical comparisons and on data extrapolated from trials in other forms of GN and vasculitis. Oral prednisone is started at a dose of 1 mg/kg per day to a maximum of 60 mg/day, and oral cyclophosphamide at 2mg-3mg/kg/day (adjusted for GFR and age).49 Prednisone is slowly tapered down to zero by 6 months; cyclophosphamide is administered for 2–3 months with the dose titrated to maintain total leukocyte count to > 4 ×109/ L. Regimens of intravenous methylprednisolone, and intravenous cyclophosphamide derived from other forms of crescentic GN are used for fulminant anti-GBM disease at some centers though there is inadequate data to support this approach.4 Mycophenolate mofetil has been used successfully as an alternative to cyclophosphamide to prevent known toxicities of the latter.53
Rituximab is being increasingly used in the field of immune-mediated glomerular disease. It is a chimeric anti-CD20 monoclonal antibody that depletes CD-20 positive B cells via antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and apoptosis, thereby suppressing antibody production.54 Although it has demonstrated efficacy in ANCA-associated vasculitis and membranous nephropathy, its evidence-based use remains limited in anti-GBM disease due to a lack of controlled studies.
Proteolytic Cleavage of IgG: A Novel Strategy of Depleting Pathogenic Antibodies
The IgG-degrading enzyme of Streptococcus pyogenes (IdeS) is a novel therapeutic proteinase that cleaves human IgG preventing subsequent complement and neutrophil- induced injury.55 It works with dramatic efficacy and efficiency, decreasing the anti-GBM antibody titer to an undetectable/ non-toxic level within hours. An additional advantage of IdeS (over PLEX) is that it also degrades kidney-bound IgG. Initial reports in 3 patients with refractory anti-GBM nephritis revealed that IdeS led to rapid reduction in anti-GBM antibodies, but renal outcomes did not improve; none of the patients achieved dialysis independence.55 It is noteworthy that these patients had been on dialysis for an extended period of time before IdeS was administered, raising the possibility that accelerated breakdown of anti-GBM antibodies using IdeS earlier during the course of the illness might have led to favorable renal outcomes. This hypothesis was tested in an open-label multicenter Phase II study (GOOD-IDES). This study evaluated the efficacy and safety of a single dose of IdeS in 15 patients with severe anti-GBM disease (eGFR<15mL/min and high circulating anti-GBM antibodies) receiving corticosteroids, cyclophosphamide, and PLEX but absence of anuria for more than 48 hours, dialysis dependency of more than 5 days, or ongoing moderate-severe pulmonary hemorrhage.56,57 At inclusion, 10 patients were dialysis-dependent; the remaining 5 patients had eGFR of 7–14mL/min. Anti-GBM antibody titer normalized in all patients within 6 hours after IdeS administration, although a rebound of antibodies was observed in 10 patients, requiring PLEX. At six months, 10 patients were dialysis independent. The safety profile was good; no serious unexpected suspected adverse reactions were seen. Hence, IdeS might offer a significant clinical advantage over PLEX and immunoadsorption, in the management of patients with fulminant anti-GBM disease when swift clearance of pathogenic antibodies is desirable for abatement of inflammation.
Selecting Management Strategy According to the Clinical Status and Phenotype of the Disease
Renal function declines more rapidly in anti-GBM disease than in other forms of rapidly progressive GN because antibodies bind rapidly and tightly to the GBM.58 There is a direct correlation between anti-GBM antibody titer, serum creatinine at presentation, and the percentage of glomeruli with crescents.27 Prompt initiation of treatment is often the key to better clinical outcomes.4 Hence, if the clinical suspicion is high, it is preferable to initiate treatment with high-dose steroids (and PLEX in selected cases), rather than waiting for histological confirmation.
For most patients with anti-GBM associated RPGN, combination therapy of plasma exchange (PLEX), cyclophosphamide, and prednisone, introduced by the Hammersmith group remains the standard treatment.27 This includes patients with “double-positive” disease as the phenotype of their disease at presentation is comparable to that of classic anti-GBM disease.39,40 Renal outcomes are much better using a combination of PLEX and immunosuppressive therapies, compared to immunosuppressive treatment alone.59 Prophylaxis against peptic ulcer, pneumocystis and osteoporosis is employed in patients receiving this immunosuppressive regimen. Advanced renal failure with creatinine >600 μmol/l, oligo-anuria, or dialysis-dependency at presentation is usually associated with severe crescentic disease and renal scarring (>85% cellular crescents or >50% globally sclerotic glomeruli). These patients are unlikely to recover renal function, and, in the absence of pulmonary hemorrhage, they are not good candidates for intensive treatment.28,45,60 However, combined PLEX and immunosuppressive therapy should be considered if i) the renal biopsy reveals early non-fibrotic crescents and concomitant acute tubular necrosis,27 ii) the patient has a very short history of progressive renal dysfunction,61 iii) the patient has double-positive disease (more likely to recover independent kidney function even after initially requiring dialysis, as compared to single-positive anti-GBM patients),40 or iv) the patient has concomitant pulmonary hemorrhage, regardless of the degree of renal dysfunction.46
Patients who are refractory to standard management, or who have intolerable adverse effects from cyclophosphamide will be candidates for rituximab therapy.62 Rituximab has been shown to effectively induce complete remission of alveolar hemorrhage but, unfortunately, not improve renal outcomes in dialysis-dependent patients at presentation, even if antibody titer became negative.63 Data from the RAVE trial suggest the superiority of rituximab over cyclophosphamide in attenuating antibody production in ANCA-associated vasculitis. Hence, rituximab may have a special therapeutic role in double-positive patients.64 Varying doses and schedules of rituximab have been used in anti-GBM disease. Some used weekly doses of 375 mg/m2 for 2 to 6 weeks, while others administered a single or two doses of 1000 mg. It is noteworthy that rituximab is rapidly removed from circulation by PLEX, so the PLEX schedule needs to be devised carefully (allowing at least 48 hours after administration of rituximab prior to the next PLEX session).
Patients with atypical anti-GBM disease usually have mild renal disease and absence of diffuse crescentic GN. They do not require immunosuppressive therapy unless they develop signs of progressive GN.65
Anti-GBM disease usually follows a single-hit course, although relapses have been reported.66,67 Patients who respond to the initial immunosuppressive therapy, and recover do not need maintenance immunosuppression. However, double-positive patients carry the long-term risk of relapse similar to patients with ANCA-associated vasculitis, so maintenance therapy should be considered using either azathioprine (2mg/kg body weight), or rituximab (500–1000 mg each, every 6 months) for about 2 years.68,69 Rituximab might also have a role in patients who have a relapsing disease.
Anti-GBM patients with ESKD can be transplanted after 6 months of persistent negativity of anti-GBM antibodies. Clinically significant post-transplant disease recurrence is rare (approximately 2%) although histological recurrence in the form of linear IgG staining in the glomeruli without other glomerular lesions may be as high as 50%.4,70 Therapeutic strategies are similar to those used in the treatment of disease in the native kidneys. PLEX is instituted, immunosuppression is augmented (high dose steroids and a switch from antimetabolites to cyclophosphamide for 2–3 months), and higher CNI trough levels are maintained. Allograft failure from disease recurrence is exceedingly rare.
A de novo anti-GBM disease can develop in approximately 5% of patients with Alport syndrome who receive a kidney transplant, usually within the first year following transplantation. This occurs because the neo-antigens contained in the basement membrane in the kidney allograft (that were lacking in the native kidneys) trigger antibody production. The de novo alloantibodies are primarily directed against the alpha5 chains of type IV collagen in the donor kidney, as opposed to mainly alpha3 chains in anti-GBM disease involving native kidneys.71 Clinically, GN appears indistinguishable from anti-GBM disease of native kidneys; histology reveals prominent crescentic changes, necrosis, and inflammation. Unfortunately, these patients are less likely to respond to traditional treatment, so the risk of irreversible graft failure approaches 90% within a few weeks to months after diagnosis.32 Re-transplantation carries the same grim prognosis.
Concluding Remarks
Anti-GBM disease has emerged as a multidimensional entity; a spectrum of several phenotypes with distinct renal outcomes and response patterns to treatment modalities. Serological testing for both anti-GBM antibodies and ANCA, as well as renal histopathological evaluation is required for recognizing disease-variants, determining prognosis, and devising appropriate treatment plans. Progress toward new treatments has been hampered by the rarity of anti-GBM disease, but new therapies are emerging. Phase III clinical trials are required to confirm the therapeutic efficacy and safety of IdeS. Even if approved for clinical use in anti-GBM disease, IdeS is unlikely to completely obviate the need for any of the components of the established therapy. PLEX will still be required to curb the rebound of antibodies that often occurs after IdeS administration, and immunosuppression will remain obligatory to suppress antibody formation and inflammation. Orally administered complement component C5a receptor inhibitors have been found to be effective in replacing high-dose glucocorticoids in treating vasculitis.72 It will be interesting to see if such agents can also be used in anti-GBM disease to decrease glucocorticoid toxicity. A multipronged approach that utilizes all three modalities (immunosuppressive therapy, IdeS, and PLEX) might have the potential to effectively treat the disease and yet minimize the adverse effects of each individual component.
Abbreviations
ANCA, antineutrophil cytoplasmic antibodies; DFPP, double filtration plasmapheresis; ESKD, end-stage-kidney-disease; GBM, glomerular basement membrane; GFR, glomerular filtration rate; GN, glomerulonephritis; IA, immunoadsorption; IdeS, IgG-degrading enzyme of Streptococcus pyogenes; IgG, immunoglobulin G; KDIGO, Kidney Disease: Improving Global Outcomes; PLEX, plasma exchange; RPGN, rapidly progressive glomerulonephritis.
Acknowledgment
We thank Hamza Asim for his help with computer graphics.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the alpha 3 chain of type IV collagen. J Clin Invest. 1992;89(2):592–601. doi:10.1172/JCI115625
2. Yoshioka K, Iseki T, Okada M, Morimoto Y, Eryu N, Maki S. Identification of Goodpasture antigens in human alveolar basement membrane. Clin Exp Immunol. 1988;74(3):419–424.
3. Borza DB, Netzer KO, Leinonen A, et al. The goodpasture autoantigen. Identification of multiple cryptic epitopes on the NC1 domain of the alpha3(IV) collagen chain. J Biol Chem. 2000;275(8):6030–6037. doi:10.1074/jbc.275.8.6030
4. McAdoo SP, Pusey CD. Anti-glomerular basement membrane disease. Clin J Am Soc Nephrol. 2017;12(7):1162–1172. doi:10.2215/CJN.01380217
5. Saus J, Wieslander J, Langeveld JP, Quinones S, Hudson BG. Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem. 1988;263(26):13374–13380. doi:10.1016/S0021-9258(18)37714-7
6. Netzer KO, Leinonen A, Boutaud A, et al. The goodpasture autoantigen. mapping the major conformational epitope (s) of alpha3 (IV) collagen to residues 17–31 and 127–141 of the NC1 domain. J Biol Chem. 1999;274(16):11267–11274. doi:10.1074/jbc.274.16.11267
7. Fischer EG, Lager DJ. Anti-glomerular basement membrane glomerulonephritis: a morphologic study of 80 cases. Am J Clin Pathol. 2006;125(3):445–450. doi:10.1309/nptp-4ukv-7ju3-elmq
8. Akhtar M, Taha NM, Asim M. Anti-glomerular basement membrane disease: what have we learned? Adv Anat Pathol. 2021;28(1):59–65. doi:10.1097/PAP.0000000000000280
9. Shah MK, Hugghins SY. Characteristics and outcomes of patients with Goodpasture’s syndrome. South Med J. 2002;95(12):1411–1418. doi:10.1097/00007611-200295120-00012
10. Yang R, Hellmark T, Zhao J, et al. Antigen and epitope specificity of anti-glomerular basement membrane antibodies in patients with goodpasture disease with or without anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2007;18(4):1338–1343. doi:10.1681/ASN.2006111210
11. Fisher M, Pusey CD, Vaughan RW, Rees AJ. Susceptibility to anti-glomerular basement membrane disease is strongly associated with HLA-DRB1 genes. Kidney Int. 1997;51(1):222–229. doi:10.1038/ki.1997.27
12. Canney M, O’Hara PV, McEvoy CM, et al. Spatial and temporal clustering of anti-glomerular basement membrane disease. Clin J Am Soc Nephrol. 2016;11(8):1392–1399. doi:10.2215/CJN.13591215
13. Donaghy M, Rees AJ. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. Lancet. 1983;2(8364):1390–1393. doi:10.1016/s0140-6736(83)90923-6
14. Wilson CB, Smith RC. Goodpasture’s syndrome associated with influenza A2 virus infection. Ann Intern Med. 1972;76(1):91–94. doi:10.7326/0003-4819-76-1-91
15. Perez GO, Bjornsson S, Ross AH, Aamato J, Rothfield N. A mini-epidemic of Goodpasture’s syndrome clinical and immunological studies. Nephron. 1974;13(2):161–173. doi:10.1159/000180389
16. Savage CO, Pusey CD, Bowman C, Rees AJ, Lockwood CM. Antiglomerular basement membrane antibody mediated disease in the British Isles 1980–4. Br Med J. 1986;292(6516):301–304. doi:10.1136/bmj.292.6516.301
17. Williams PS, Davenport A, McDicken I, Ashby D, Goldsmith HJ, Bone JM. Increased incidence of anti-glomerular basement membrane antibody (anti-GBM) nephritis in the Mersey Region, september 1984-october 1985. Q J Med. 1988;68(257):727–733.
18. Prendecki M, Clarke C, Cairns T, et al. Anti-glomerular basement membrane disease during the COVID-19 pandemic. Kidney Int. 2020;98(3):780–781. doi:10.1016/j.kint.2020.06.009
19. Prema KSJ, Kurien A. Incidence of anti-glomerular basement membrane disease during the COVID-19 pandemic. Clin Kidney J. 2021;15(1):180–181. doi:10.1093/ckj/sfab204
20. Sebastian R, Arunachalam J, Rajendran M. Temporal clustering of antiglomerular basement membrane disease in COVID-19 pandemic: a case series. Int J Nephrol Renovasc Dis. 2021;14:393–398. doi:10.2147/IJNRD.S333894
21. Winkler A, Zitt E, Sprenger-Mähr H, Soleiman A, Cejna M, Lhotta K. SARS-CoV-2 infection and recurrence of anti-glomerular basement disease: a case report. BMC Nephrol. 2021;22(1):75. doi:10.1186/s12882-021-02275-4
22. Nagai K, Iwase M, Ueda A. A case of anti-GBM nephritis following centipede bites and COVID-19 vaccination [published online ahead of print, 2021 Sep 15]. CEN Case Rep. 2021;1–5. doi:10.1007/s13730-021-00646-2
23. Sacker A, Kung V, Andeen N. Anti-GBM nephritis with mesangial IgA deposits after SARS-CoV-2 mRNA vaccination. Kidney Int. 2021;100(2):471–472. doi:10.1016/j.kint.2021.06.006
24. Gupta RK, Ellis BK. Concurrent anti-GBM nephritis and ANCA-mediated glomerulonephritis after second dose of SARS-CoV-2 mRNA vaccination [published online ahead of print, 2021 Oct 30]. Kidney Int Rep. 2021;7(1):127–128. doi:10.1016/j.ekir.2021.10.020
25. Tang W, McDonald SP, Hawley CM, et al. Anti-glomerular basement membrane antibody disease is an uncommon cause of end-stage renal disease. Kidney Int. 2013;83(3):503–510. doi:10.1038/ki.2012.375
26. Kluth DC, Rees AJ. Anti-glomerular basement membrane disease. J Am Soc Nephrol. 1999;10(11):2446–2453. doi:10.1681/ASN.V10112446
27. Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001;134(11):1033–1042. doi:10.7326/0003-4819-134-11-200106050-00009
28. Van Daalen EE, Jennette JC, McAdoo SP, et al. Predicting outcome in patients with anti-GBM glomerulonephritis. Clin J Am Soc Nephrol. 2018;13(1):63–72. doi:10.2215/CJN.04290417
29. Cui Z, Zhao MH, Xin G, Wang HY. Characteristics and prognosis of Chinese patients with anti-glomerular basement membrane disease. Nephron Clin Pract. 2005;99(2):c49–c55. doi:10.1159/000083133
30. Chen M, Cui Z, Zhao MH. ANCA-associated vasculitis and anti-GBM disease: the experience in China. Nephrol Dial Transplant. 2010;25(7):2062–2065. doi:10.1093/ndt/gfq134
31. Singh T, Kharadjian TB, Astor BC, Panzer SE. Long-term outcomes in kidney transplant recipients with end-stage kidney disease due to anti-glomerular basement membrane disease. Clin Transplant. 2021;35(2):e14179. doi:10.1111/ctr.14179
32. Kashtan CE. Renal transplantation in patients with Alport syndrome. Pediatr Transplant. 2006;10(6):651–657. doi:10.1111/j.1399-3046.2006.00528.x
33. Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967;126(6):989–1004. doi:10.1084/jem.126.6.989
34. Almkuist RD, Buckalew VM
35. Nasr SH, Collins AB, Alexander MP, et al. The clinicopathologic characteristics and outcome of atypical anti-glomerular basement membrane nephritis. Kidney Int. 2016;89(4):897–908. doi:10.1016/j.kint.2016.02.001
36. Liang D, Liang S, Xu F, et al. Clinicopathological features and outcome of antibody-negative anti-glomerular basement membrane disease. J Clin Pathol. 2019;72(1):31–37. doi:10.1136/jclinpath-2018-205278
37. Ang C, Savige J, Dawborn J, et al. Anti-glomerular basement membrane (GBM)-antibody-mediated disease with normal renal function. Nephrol Dial Transplant. 1998;13(4):935–939. doi:10.1093/ndt/13.4.935
38. Serisier DJ, Wong RC, Armstrong JG. Alveolar haemorrhage in anti-glomerular basement membrane disease without detectable antibodies by conventional assays. Thorax. 2006;61(7):636–639. doi:10.1136/thx.2004.028985
39. Clerte M, Philip R, Levi C, et al. Renal and overall outcomes of double-positive (ANCA and anti-GBM antibodies) patients compared to ANCA-associated vasculitis patients with severe renal involvement: a multicenter retrospective study with systematic renal pathology analysis [published online ahead of print, 2021 Jun 25]. Scand J Rheumatol. 2021:1–9. doi:10.1080/03009742.2021.1920120
40. McAdoo SP, Tanna A, Hrušková Z, et al. Patients double-seropositive for ANCA and anti-GBM antibodies have varied renal survival, frequency of relapse, and outcomes compared to single-seropositive patients. Kidney Int. 2017;92(3):693–702. doi:10.1016/j.kint.2017.03.014
41. Lockwood CM, Rees AJ, Pearson TA, Evans DJ, Peters DK, Wilson CB. Immunosuppression and plasma-exchange in the treatment of Goodpasture’s syndrome. Lancet. 1976;1(7962):711–715. doi:10.1016/s0140-6736(76)93089-0
42. Couser WG. Rapidly progressive glomerulonephritis: classification, pathogenetic mechanisms, and therapy. Am J Kidney Dis. 1988;11(6):449–464. doi:10.1016/s0272-6386(88)80079-9
43. Madore F, Lazarus JM, Brady HR. Therapeutic plasma exchange in renal diseases. J Am Soc Nephrol. 1996;7(3):367–386. doi:10.1681/ASN.V73367
44. Cui Z, Zhao J, Jia XY, et al. Anti-glomerular basement membrane disease: outcomes of different therapeutic regimens in a large single-center Chinese cohort study. Medicine. 2011;90(5):303–311. doi:10.1097/MD.0b013e31822f6f68
45. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4S):S1–S276. doi:10.1016/j.kint.2021.05.021
46. Schwartz J, Padmanabhan A, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the writing committee of the American Society for apheresis: the seventh special issue. J Clin Apher. 2016;31(3):149–162. doi:10.1002/jca.21470
47. Reeves HM, Winters JL. The mechanisms of action of plasma exchange. Br J Haematol. 2014;164(3):342–351. doi:10.1111/bjh.12629
48. Padmanabhan A, Connelly-Smith L, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice - evidence-based approach from the writing Committee of the American Society for apheresis: the eighth special issue. J Clin Apher. 2019;34(3):171–354. doi:10.1002/jca.21705
49. Pusey CD. Anti-glomerular basement membrane disease. Kidney Int. 2003;64(4):1535–1550. doi:10.1046/j.1523-1755.2003.00241.x
50. Maxted AP, Connell R, Hussain F. Double filtration plasmapheresis - 10-year pediatric experience as an alternative to plasma exchange. Transfus Apher Sci. 2020;59(6):102928. doi:10.1016/j.transci.2020.102928
51. Biesenbach P, Kain R, Derfler K, et al. Long-term outcome of anti-glomerular basement membrane antibody disease treated with immunoadsorption. PLoS One. 2014;9(7):e103568. doi:10.1371/journal.pone.0103568
52. Zhang YY, Tang Z, Chen DM, Gong DH, Ji DX, Liu ZH. Comparison of double filtration plasmapheresis with immunoadsorption therapy in patients with anti-glomerular basement membrane nephritis. BMC Nephrol. 2014;15:128. doi:10.1186/1471-2369-15-128
53. Mori M, Nwaogwugwu U, Akers GR, McGill RL. Anti-glomerular basement membrane disease treated with mycophenolate mofetil, corticosteroids, and plasmapheresis. Clin Nephrol. 2013;80(1):67–71. doi:10.5414/cn107333
54. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22(47):7359–7368. doi:10.1038/sj.onc.1206939
55. Soveri I, Mölne J, Uhlin F, et al. The IgG-degrading enzyme of Streptococcus pyogenes causes rapid clearance of anti-glomerular basement membrane antibodies in patients with refractory anti-glomerular basement membrane disease. Kidney Int. 2019;96(5):1234–1238. doi:10.1016/j.kint.2019.06.019
56. An Open-Label Phase II Study to Evaluate the Efficacy and Safety of IdeS in Anti-GBM Disease (GOOD-IDES). ClinicalTrials.gov identifier: NCT03157037. Posted May 17, 2017. Accessed March 17, 2022. https://www.clinicaltrials.gov/ct2/show/NCT03157037
57. Segelmark M, Uhlin F, Sonesson E. The Immunoglobulin G Degrading Enzyme Imlifidase for the Treatment of Anti-GBM Disease: The GOOD-IDES 01 Trial. Late-Breaking Clinical Trials Poster presented at American Society of Nephrology Kidney week 2020; October 22, 2020; On Demand. Abstract PO2639. Accessed March 17, 2022. https://www.asn-online.org/education/kidneyweek/2020/program-abstract.aspx?controlId=3479229
58. Rutgers A, Meyers KE, Canziani G, Kalluri R, Lin J, Madaio MP. High affinity of anti-GBM antibodies from Goodpasture and transplanted Alport patients to alpha3 (IV) NC1 collagen. Kidney Int. 2000;58(1):115–122. doi:10.1046/j.1523-1755.2000.00146.x
59. Johnson JP, Jr MJ, Austin HA
60. Alchi B, Griffiths M, Sivalingam M, Jayne D, Farrington K. Predictors of renal and patient outcomes in anti-GBM disease: clinicopathologic analysis of a two-centre cohort. Nephrol Dial Transplant. 2015;30(5):814–821. doi:10.1093/ndt/gfu399
61. Maxwell AP, Nelson WE, Hill CM. Reversal of renal failure in nephritis associated with antibody to glomerular basement membrane. BMJ. 1988;297(6644):333–334. doi:10.1136/bmj.297.6644.333
62. Touzot M, Poisson J, Faguer S, et al. Rituximab in anti-GBM disease: a retrospective study of 8 patients. J Autoimmun. 2015;60:74–79. doi:10.1016/j.jaut.2015.04.003
63. Jain R, Dgheim H, Bomback AS. Rituximab for anti-glomerular basement membrane disease. Kidney Int Rep. 2018;4(4):614–618. doi:10.1016/j.ekir.2018.12.002
64. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–232. doi:10.1056/NEJMoa0909905
65. Kaplan AA, Appel GB, Pusey CE, et al. Anti-GBM (Goodpasture) disease: treatment and prognosis. Uptodate: evidence-based clinical decision support. Available from: www.uptodate.com.
66. Liu P, Waheed S, Boujelbane L, Maursetter LJ. Multiple recurrences of anti-glomerular basement membrane disease with variable antibody detection: can the laboratory be trusted? Clin Kidney J. 2016;9(5):657–660. doi:10.1093/ckj/sfw038
67. Gu B, Magil AB, Barbour SJ. Frequently relapsing anti-glomerular basement membrane antibody disease with changing clinical phenotype and antibody characteristics over time. Clin Kidney J. 2016;9(5):661–664. doi:10.1093/ckj/sfw048
68. Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771–1780. doi:10.1056/NEJMoa1404231
69. Smith RM, Jones RB, Guerry MJ, et al. Rituximab for remission maintenance in relapsing antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64(11):3760–3769. doi:10.1002/art.34583
70. Coche S, Sprangers B, Van Laecke S, et al. Recurrence and outcome of anti-glomerular basement membrane glomerulonephritis after kidney transplantation. Kidney Int Rep. 2021;6(7):1888–1894. doi:10.1016/j.ekir.2021.04.011
71. Brainwood D, Kashtan C, Gubler MC, Turner AN. Targets of alloantibodies in Alport anti-glomerular basement membrane disease after renal transplantation. Kidney Int. 1998;53(3):762–766. doi:10.1046/j.1523-1755.1998.00794.x
72. Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28(9):2756–2767. doi:10.1681/ASN.2016111179
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.