")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 8
Epidemiology, diagnosis, and optimal management of glioma in adolescents and young adults
Authors Diwanji TP, Engelman A, Snider JW, Mohindra P
Received 10 November 2015
Accepted for publication 28 March 2017
Published 22 September 2017 Volume 2017:8 Pages 99—113
DOI https://doi.org/10.2147/AHMT.S53391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Alastair Sutcliffe
Tejan P Diwanji, Alexander Engelman, James W Snider, Pranshu Mohindra
Department of Radiation Oncology, University of Maryland School of Medicine, Baltimore, MD, USA
Abstract: Neoplasms of the central nervous system (CNS) are the most frequently encountered solid tumors of childhood, but are less common in adolescents and young adults (AYA), aged 15–39 years. Gliomas account for 29%–35% of the CNS tumors in AYA, with approximately two-thirds being low-grade glioma (LGG) and the remaining being high-grade glioma (HGG). We review the epidemiology, work-up, and management of LGG and HGG, focusing on the particular issues faced by the AYA population relative to pediatric and adult populations. Visual pathway glioma and brainstem glioma, which represent unique clinical entities, are only briefly discussed. As a general management approach for both LGG and HGG, maximal safe resection should be attempted. AYA with LGG who undergo gross total resection (GTR) may be safely observed. As age increases and the risk factors for recurrence accumulate, adjuvant therapy should be more strongly considered with a strong consideration of advanced radiation techniques such as proton beam therapy to reduce long-term radiation-related toxicity. Recent results also suggest survival advantage for adult patients with the use of adjuvant chemotherapy when radiation is indicated. Whenever possible, AYA patients with HGG should be enrolled in a clinical trial for the benefit of centralized genetic and molecular prognostic review and best clinical care. Chemoradiation should be offered to all World Health Organization grade IV patients with concurrent and adjuvant chemotherapy after maximal safe resection. Younger adolescents with GTR of grade III lesions may consider radiotherapy alone or sequential radiotherapy and chemotherapy if unable to tolerate concurrent treatment. A more comprehensive classification of gliomas integrating pathology and molecular data is emerging, and this integrative strategy offers the potential to be more accurate and reproducible in guiding diagnostic, prognostic, and management decisions.
Keywords: glioma, adolescents, young adults, radiation, radiation therapy
Introduction
Neoplasms of the central nervous system (CNS) are the most frequently encountered solid tumors of childhood and among children younger than age 19 years; the average annual age-adjusted incidence is 5.4 per 100,000. For adolescents and young adults (AYA) aged 15–39, the annual incidence is reported at 3.41 per 100,000, with ~1.9% of all brain tumors occurring in adolescents 15–19 years old and 15% of all brain tumors occurring in AYA 15–39 years old. CNS tumors are less common in AYA (age 15–39) as compared to older adults. It is estimated that 3,690 adolescents and 51,118 AYA will be diagnosed with tumors of the CNS in the year 2015 and that the majority (75%) will be glial in origin.1
Among the CNS tumors in adolescents aged 15–19 years, the most common histologies include tumors of the pituitary (25.4%) followed by pilocytic astrocytoma (PA; 10.1%), a type of low-grade glioma (LGG). The most common histologies of CNS tumors in AYA (15–39) are tumors of the pituitary (29%), followed by meningioma (16%) and nerve sheath tumors (8.8%).2 As a group, gliomas account for 29%–35% of CNS tumors in this age group. Approximately two-thirds of the adolescent gliomas are low grade, with the remaining one-third being World Health Organization (WHO) grade III and IV tumors. Table 1 describes the top four most common histologies of brain tumors in various age groups from pediatrics to AYA as reported by the Central Brain Tumor Registry of the United States.
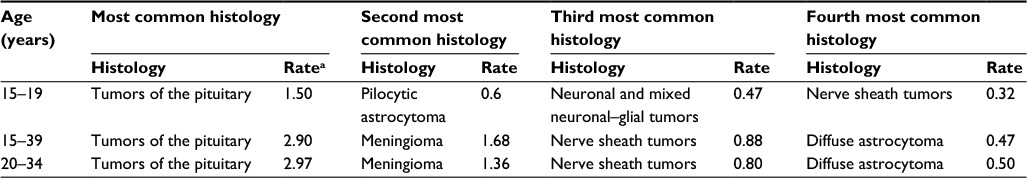 | Table 1 Most common primary brain and CNS tumors by age, CBTRUS statistical report: NPCR and SEER 2007–2011 Notes: aAge-adjusted per 100,000 individuals. Adapted from Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63, by permission of Oxford Univeristy Press.1 Abbreviations: CBTRUS, Central Brain Tumor Registry of the United States; CNS, central nervous system; NPCR, National Program of Cancer Registries; SEER, Surveillance, Epidemiology, and End Results. |
Gliomas are neuroepithelial tumors originating from the supporting glial cells of the CNS. Gliomas can range in behavior from benign LGGs, amenable to resection or observation, to aggressive histologies such as diffuse intrinsic pontine gliomas (DIPGs) and supratentorial glioblastomas (GBMs) with nearly uniform poor prognoses despite aggressive therapy. LGGs are the most frequently encountered CNS tumors of early childhood, while high-grade gliomas (HGGs) tend to occur at increasing rates with increasing age. LGGs (WHO grades I and II) include an array of astrocytomas: pilocytic, pilomyxoid, subependymal giant cell, pleomorphic xanthoastrocytoma and diffuse (fibrillary, gemistocytic, protoplasmic), as well as mixed gliomas, oligodendrogliomas, gangliogliomas, dysembroplastic neuroepithelial tumor, and desmoplastic infantile ganglioglioma.2 HGGs (WHO grades III and IV) include anaplastic astrocytoma (III), anaplastic oligodendroglioma (III), oligoastrocytoma (III), GBM (IV), and gliosarcoma (IV). The relative distribution of histology by age is given in Figure 1A (age 15–19 years) and Figure 1B (age 15–39 years). In adolescents aged 15–19 years, gliomas account for one-thirds of all tumors and nearly three-fourths of all malignant tumors. Among all AYA (15–39 years), gliomas account for a little less than a third of all tumors, but more than 80% of all malignant tumors.
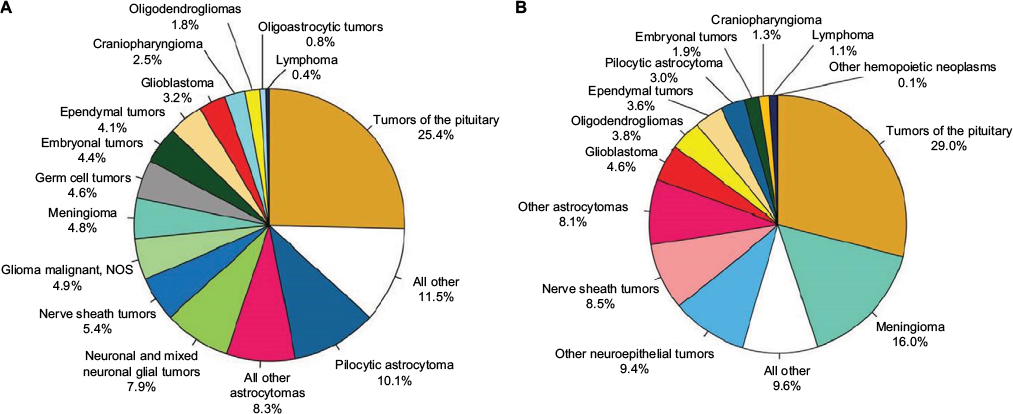 | Figure 1 Distribution in (A) adolescents (age 15–19 years) and (B) adolescents and young adults (age 15–39 years) of primary brain and CNS tumors by histology (n=6,491 and 51,118, respectively) CBTRUS statistical report: NPCR and SEER, 2007–2011. Notes: Adapted from Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63, by permission of Oxford Univeristy Press.1 Abbreviations: CBTRUS, Central Brain Tumor Registry of the United States; CNS, central nervous system; NOS, not otherwise specified; NPCR, National Program of Cancer Registries; SEER, Surveillance, Epidemiology, and End Results. |
Predisposing factors for increased glioma risk include genetic tumor predisposition syndromes such as neurofibromatosis (NF) 1 and 2, tuberous sclerosis, Li–Fraumeni syndrome, and Turcot’s syndrome. The only environmental exposure routinely linked to the development of glioma is exposure to ionizing radiation.3 AYA have higher rates of relative survival than adults over age 40 for all histologic types, but the survival rates are lower than those for children.1
We will review the epidemiology, investigation, and management of LGGs and HGGs in the AYA population. Visual pathway glioma and brainstem glioma, which represent unique clinical entities, are only briefly discussed. Clinical studies usually evaluate the pediatric (typically age <14 years) and adult (typically age >18 years) populations separately, with no consistent representation of the AYA population. Focusing on gliomas, the outcomes for the AYA can be expected to bridge those described for the perimeters of age with generally favorable outcomes in the pediatric population. The definition of AYA has varied, but typically includes ages 15–22 at a minimum. In the setting of limited information on epidemiologic information specifically for AYA, the comprehensive Central Brain Tumor Registry of the United States Statistical Report: primary Brain and Central Nervous System Tumors Diagnosed in the US was selected as the primary reference (tables and figures from the report are reproduced with permission), which defines AYA with age range of 15–39 years.1
Low-grade glioma
Pediatric and adult LGGs represent a heterogeneous group of tumors classified by the WHO according to their morphologic features into several groups that may or may not share similar clinical behavior. These include astrocytic, oligodendroglial, mixed oligoastrocytic, neuronal, or mixed glioneuronal morphology. Despite having a similar histologic appearance to adult LGG, pediatric LGGs have a distinct and frequently more favorable course, with notably lower incidence of malignant transformation.4 The majority of children diagnosed with an LGG are long-term survivors well into adulthood, and adult survivors of pediatric LGG have relatively high glioma-specific survival.5
Epidemiology
The annual incidence of pediatric LGGs is 1.3–2.1 per 100,000 in the US, accounting for an anticipated 1,000–1,600 new diagnoses in 2015. Adult LGG is more common and is estimated to occur at a rate of 9.1–12.5 cases per 100,000 individuals in the US; and accounted for ~5,000 new diagnoses in 2015. It occurs most commonly in the second through fourth decades of life. Table 2 depicts the incidence rates of several low-grade histologies specifically in AYA.
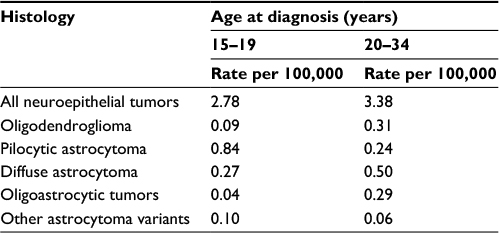 | Table 2 Average annual age-adjusted incidence rates for LGG in adolescents and young adults, brain and central nervous system tumors by major histology groupings, histology, and age at diagnosis, CBTRUS statistical report: NPCR and SEER, 2007–2011 Notes: Adapted from Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63, by permission of Oxford Univeristy Press.1 Abbreviations: CBTRUS, Central Brain Tumor Registry of the United States; LGG, low-grade glioma; NPCR, National Program of Cancer Registries; SEER, Surveillance, Epidemiology, and End Results. |
Presentation
The clinical presentation of LGG in children and adolescents depends on the age of the child as well as the location of the lesion and the histology. In AYA, localizing symptoms are common. Whereas low-grade lesions often present with an insidious onset over a period of months or with a long history of seizures which may be refractory to antiepileptic treatment, high-grade lesions generally present with more rapidly progressive symptoms. Seizures are the presenting symptom in ~30% of malignant gliomas, and more so with low-grade lesions.6 Signs of increased intracranial pressure such as headaches, gait/balance changes, mental status changes, and focal neurologic deficits such as hemiparesis, hemisensory deficits, or aphasia are often seen.
Diagnosis
Magnetic resonance imaging (MRI) is currently the mainstay of imaging and can suggest the grade of disease, though the final diagnosis is primarily histopathologic. LGGs are often homogeneous with low signal intensity on T1-weighted sequences and hyperintense on T2-weighted and fluid-attenuated inversion recovery sequences. Grade I gliomas are typically well circumscribed and demonstrate homogeneous enhancement following gadolinium administration. Grade II gliomas, especially diffuse astrocytomas, may be less circumscribed and are typically nonenhancing, although oligodendrogliomas may have some degree of contrast enhancement and may be associated with calcifications.5 LGGs are not usually associated with significant edema or restricted diffusion on diffusion-weighted sequences. MRI spectroscopy can help identify additional tumor extent beyond the T2 hyperintensity for surgical and radiotherapy (RT) planning.7 While lack of enhancement at baseline suggests low-grade histology, it does not rule out presence of high-grade elements in the final pathology. On the other hand, appearance of new enhancement in a previously nonenhancing lesion is highly suggestive of tumor progression.
The main goal of surgery is to obtain pathologic diagnosis and, when feasible, to achieve a gross total resection (GTR) which helps in reducing the mass effect from the lesion. Because of the potential heterogeneity of these tumors, stereotactic biopsy alone may not reflect the highest grade for diagnosis, with reported accuracy rates ranging from 51% to 83%.8 WHO grading is established based on histologic morphology. The common grade I tumors in pediatrics and young adults include PA and mixed neuroglial tumors. PAs are classically characterized by the presence of Rosenthal fibers, biphasic architecture (alternating microcystic and compact cellular areas), vascular proliferation, and eosinophilic granular bodies.9 The common grade II lesions include diffuse astrocytoma, oligodendroglioma, oligoastrocytoma, pilomyxoid astrocytoma, and pleomorphic xanthoastrocytoma. Diffuse astrocytomas are characterized by the presence of nuclear atypia, but maintain a low mitotic rate and absence of vascular proliferation or palisading necrosis. Oligodendroglial tumors contain monomorphic cells with uniform round nuclei, perinuclear halos (fried egg appearance), microcalcifications, and network of capillaries.9
A more comprehensive classification of gliomas integrating pathology and molecular data is emerging, and this integrative strategy offers the potential to be more accurate and reproducible in guiding diagnostic, prognostic, and management decisions. As it pertains to AYA, several genetic and epigenetic features commonly observed in adult gliomas are interestingly absent in their pediatric counterparts. Whether the presence or absence of these particular markers should influence the consideration of age-specific treatment recommendations remains to be seen. Further discussion of genetic and molecular signatures and their influence on prognosis follows.
Prognosis
Great advancement has been made in recent years augmenting the classic clinical prognostic factors with genetic and molecular details to further subdivide LGG into groups of tumors with distinct behaviors. In general, LGGs in children exhibit slow growth rates and have even been reported to spontaneously regress, especially in patients with NF type 1.10
Clinical prognostic factors
In the European Organization for Research and Treatment of Cancer (EORTC) clinical studies (22844 and 22845), favorable clinical prognostic factors identified include young age (<40), nonastrocytic tumor type, tumor size (<6 cm), tumor limited to one hemisphere (not crossing the midline), and preserved neurologic function.11 In general, prognosis also varies inversely with grade and proliferative index as well as declining performance status.12 There is growing consensus that the extent of surgical resection can also influence survival.13 Tumors able to be completely resected often require no further therapy. This results in biases when comparing outcomes between series, as superficial lesions in noneloquent areas are more likely to be completely resected than their counterparts, and therefore have more favorable outcomes.
Molecular classification
Genetic and epigenetic signatures are emerging as independent prognostic variables equally as important as clinical factors.
In genome-wide analyses of 293 histologically classified lower-grade gliomas from adults, Brat et al partitioned LGG into three distinct prognostic groups.14
Patients who had lower-grade gliomas with an IDH mutation and 1p/19q co-deletion had the most favorable clinical outcomes (median survival 8 years), while patients with LGGs with IDH mutations and no 1p/19q co-deletion frequently had mutations in TP53 and ATRX inactivation and had intermediate outcomes (median survival 6.3 years). LGGs without an IDH mutation had genomic aberrations and clinical behavior strikingly similar to those found in primary GBM (median survival 1.7 years). In a similar study,15 again in adults, the presence of three mutations (TERT promoter, mutations in IDH, and 1p/19q co-deletion) was used to define roughly five groups of clinical behavior: triple-positive as well as TERT and IDH mutant had the best outcomes, with IDH-only mutant and triple-negatives having intermediate survival, and TERT-only tumors having the worst survival. The mean age at diagnosis was lowest (37 years) among patients who had gliomas with only IDH mutations and was highest (59 years) among patients who had gliomas with only TERT mutations. However, in contrast to adult lesions, 1p/19q co-deletion is rare in children with oligodendrogliomas16 and may not confer similar responsiveness to therapy.17
In the pediatric population, the discovery of multiple previously unreported oncogenetic mechanisms and expansion of the known genetic profile for pediatric diffuse (WHO grade II) gliomas is driving new clinical trials. While the initial data were not tied to clinical outcomes per se, several actionable alterations were identified, including the potential therapeutic benefit of targeting upregulation of the MAPK/ERK and PI3K pathways in diffuse gliomas and BRAF fusion aberrations, that is, KIAA1549-BRAF18,19 fusions in Pas and BRAF:p.V600E20,21 mutations in pleomorphic xanthoastrocytomas.
Methylation of the MGMT promoter, which thereby silences the repair gene, is associated with prolonged progression-free survival (PFS) in adults with HGG treated with temozolomide (TMZ).22 Methylated MGMT is present in a substantial proportion of adults with high-grade lesions (30%–50%) and in series of selected patients, and has also been detected in up to 93% with low-grade lesions, conferring a favorable PFS.23 The frequency of MGMT promoter methylation in newly diagnosed pediatric gliomas is lower than the proportion seen in adult gliomas.24,25 Limited data suggest that the few pediatric patients with LGG and MGMT promoter methylation may benefit from TMZ, but the case series are small and not conclusive.25,26
Management
The management of LGG in AYA will represent a balance of sometimes complimentary, sometimes conflicting approaches for the pediatric and adult populations. The majority of pediatric patients with LGG have excellent overall survival (OS) and PFS, making the goal of therapy to achieve disease control while minimizing long-term tumor and treatment-related morbidity. Most patients require only surveillance after surgery, especially in the setting of gross or near-total resection, with chemotherapy reserved to delay definitive management in very young children with subtotal resection and RT upon progression/recurrence or in the case of unresectable disease. The use of radiation in the pediatric population is deferred as much as possible due to the risk of long-term side effects, including cognitive dysfunction, hormonal imbalance, and risk of second malignancy. In adult patients, there is evidence that routine adjuvant therapy after resection extends the time to progression and that sequential chemoradiotherapy extends survival in patients at high risk for recurrence/progression compared to RT alone.27 The management of specific subsets of LGG will be discussed individually below.
Pilocytic astrocytoma
Surgical resection is the primary treatment modality for this entity. Most peripheral, hemispheric, cerebellar, and even some deep-seated lesions, can be safely and completely resected due to major advances in microsurgical techniques. For patients achieving a GTR on postoperative imaging, outcomes are excellent and observation is appropriate.28 Long-term progression-free intervals may be seen even after partial resection, and tumors that recur or progress may be re-resected, followed by observation without further treatment if the risk of neurologic impairment from further growth is low and the tumor has undergone a significant interim period of dormancy. Long-term survival after partial resection is still favorable, in the range of 80%–95%.29 As PA is predominantly a tumor of young children, the management of young adults with this disease is not as well studied; however, similarly encouraging results are seen in the adult population. In a prospective study of 20 adults with supratentorial PA (mean age 32), where gross total or subtotal resection was followed by observation and biopsy alone followed by RT to 50.4 Gy, 10-year OS was 95%.30
Indications for RT include primary treatment for symptomatic disease in unresectable sites, tumor progression in unfavorable location following subtotal resection or chemotherapy, and postoperatively following several prior resections. Since many children will survive their disease, reduction of long-term RT-related morbidity with advanced RT techniques such as proton beam therapy should be given foremost consideration. As will be discussed further in the sections that follow, proton beam therapy allows for lower integral radiation dose to the uninvolved developing brain, and has been associated with improved health-related quality of life (QOL) compared to traditional photon-based irradiation.31 One series of proton RT for predominantly progressive, unresectable, or residual low-grade astrocytomas following subtotal resection included 14 patients with PA and resulted in acceptable 80% control rates at early follow-up without significant toxicity.32
Diffuse astrocytoma, oligodendroglioma, and other grade II LGGs
Treatment of newly diagnosed LGG in children and especially adults is one of the most controversial areas in neuro-oncology. The histologic heterogeneity, diverse natural history of these tumors, long life expectancy and concern regarding treatment morbidity, impact of novel molecular markers, and historical absence of prospective OS benefit from adjuvant treatment have resulted in disagreement regarding the timing and extent of surgery, timing and dose of RT, and role of chemotherapy.
Surgery is considered curative therapy for pediatric patients and adults under the age of 40, with LGGs amenable to complete resection. GTR is often feasible for patients with cerebellar and cerebral hemisphere lesions. In a series of 75 pediatric patients who attained a GTR, <15% had progressed at 4 years follow-up.33 Likewise, in an intergroup study (CCG-9891/POG-9130), LGG patients who received no adjuvant therapy maintained a >90% 5-year PFS if they underwent a GTR. The 5-year PFS decreased to ~55% for patients with any amount of radiographic residual tumor.34 These results not only suggest that adjuvant therapy may not be indicated for young patients with completely resected LGGs but also indicate that all patients with less than a GTR might benefit from additional therapy. Since the publication of the above studies, surgical advances including intraoperative MRI and functional pathway mapping, fluorescence-guided surgery, and neuronavigation have increased the range of potentially resectable patients.35 Despite these advances, a significant portion of individuals will present with incompletely resectable or unresectable tumors. The role of adjuvant therapy is the subject of much debate.
RT – young adults
The combination of generally favorable natural history and concern for treatment morbidity has led to the evaluation of observation or a “delayed RT” approach for favorable risk patients. The Radiation Therapy Oncology Group (RTOG) protocol 9802 prospectively observed patients deemed low risk for recurrence (age <40 and GTR). Less than ideal control rates were seen without detriment to survival. The 5-year PFS was 50% overall, while the survival was 94%.27 Similarly, the EORTC, in protocol 22845, essentially compared upfront RT to 54 Gy with “late” RT, reserved until the time of progression in patients with supratentorial low-grade astrocytoma, low-grade oligoastrocytoma, or low-grade oligodendroglioma. Patients who received immediate RT had significantly improved PFS (median 5.3 vs 3.4 years, hazard ratio [HR] 0.59, 95% confidence interval: 0.45–0.77; p<0.0001) without any impact on the OS (median 7.2–7.4 years).36 Although prospective QOL metrics were not performed, patients receiving RT suffered less seizures at 1 year than those in the observation arm (25% vs 41%). Ultimately, two-thirds of the patients in the observation arm underwent RT at progression. No differences were found in the incidence of malignant transformation in both the arms (~70%). For patients with asymptomatic supratentorial lesions, especially those under age 40 who undergo a GTR, observation is an acceptable approach. For those receiving RT, the dose and the timing of RT is a subject of debate. Two prospective randomized trials have evaluated dose escalation: one evaluating 50.4 with 64.8 Gy, and the other comparing 45 with 59.4 Gy. Neither study found improvement with dose escalation, but higher rates of toxicity were observed.36,37
RT – adolescents
The question of the impact of early radiation therapy examined prospectively in the adult population is also the subject of debate in the pediatric literature. As with adults, administration of early radiation did not appear to influence PFS or OS in a cohort of patients under 21 with LGG and without GTRs.38 The 5- and 10-year PFS for those not receiving early RT were 50% and 43%, respectively, as compared with 61% and 43%, respectively, for the early RT group. The 10-year OS rates were 92% and 74%, respectively. This lack of survival benefit and concern for impaired neurocognitive development resulted in a practice of deferring RT in young children. However, RT for incompletely resected LGG is associated with excellent disease control. In children aged 2.2–19.8 years with residual or recurrent LGG, conformal RT resulted in 10-year PFS rates as high as 74% with an OS of 96%.39
Proton RT
In an effort to reduce the cognitive risk of RT, the dosimetric advantages of proton RT have been used for children with LGG, and at a median follow-up of 3.3 years, it was not associated with any decline in performance status and no patient experienced a 10% or greater drop in QOL scores. In a retrospective analysis of pediatric patients receiving cranial irradiation at the MD Anderson Cancer Center,40 the use of proton RT was not significantly associated with long-term decline in IQ, a major concern with photon cranial irradiation in pediatric patients. As the concern for compromised neural development eases with increasing age, the AYA population may stand to gain more in disease control from early RT than the risk of detriment to cognitive function, especially as advanced radiation techniques are used.
Chemotherapy – young adults
Inspired by the activity seen in higher grade lesions, the addition of systemic therapy has been evaluated with recent long-term follow-up. For patients over the age of 40 or any age with less than a GTR, RTOG 9802 examined the impact of sequential RT followed by six cycles of three drugs chemotherapy combination: procarbazine (P), lomustine (CCNU - C), and vincristine (V). Patients in the RT+PCV arm had significantly longer median survival time compared to the RT alone arm (13.3 vs 7.8 years, p=0.03; HR=0.59), and longer median PFS (10.4 vs 4.0 years, p=0.002; HR=0.50). The 5- and 10-year OS rates for RT+PCV vs RT alone were 73% vs 64% and 62% vs 41%, respectively. The authors concluded that for grade 2 glioma patients with less than gross total tumor resection or >40 years of age, PCV+RT prolongs both OS and PFS compared with RT alone.27
The oral alkylating agent TMZ is largely replacing PCV, given its superior tolerability. EORTC 22033 randomized patients with progressive, symptomatic, or high-risk LGG to either primary chemotherapy with dose-dense TMZ (75 mg/m2 daily ×21 days, q28 days, max. 12 cycles) or to standard RT to 50.4 Gy.41 PFS was not significantly different between the groups, and median OS was not reached. 1p deletion and IDH mutation were positive prognostic factors irrespective of treatment. Patient with IDH-mutant/non-1p/19q co-deleted tumors achieved better PFS when treated with first-line RT, compared to treatment with TMZ.42
In concurrent combination, RT+TMZ seems to represent an improvement on historical controls for patients with higher-risk LGG. RTOG 0424, a Phase II trial of high-risk LGG patients with ≥3 risk factors as defined by Pignatti et al,11 delivered 54 Gy with concurrent and adjuvant TMZ. They found a 3-year OS rate of 73.1%, which was significantly improved on the expected control. The therapy was reasonably well tolerated (43% experienced grade 3 adverse events, 10% experienced a grade 4 adverse event), suggesting concurrent chemoradiotherapy may be a reasonable approach for patients at substantial risk for recurrence or symptomatic progression.43
Chemotherapy – adolescents/pediatrics
In the pediatric population, chemotherapy is often used to defer or replace RT in young children in whom treatment is felt to pose an unacceptably high risk of neurodevelopmental injury. In studies of young patients (typically <10 years of age) with residual or recurrent LGG, adjuvant systemic therapy (carboplatin and vincristine [CV] or thioguanine, procarbazine, lomustine, and vincristine [TPCV]) achieved 3–5-year event-free survival (EFS) of 45%–55% and 5-year OS rates of 80%–90%.44 As RT is considered more readily in the AYA population, the use of chemotherapy alone for these patients is not a routine. The addition of chemotherapy to RT, again as in the adult population, is a subject of much interest. A Children’s Cancer Group (CCG) study of combination chemotherapy and RT for HGG included patients diagnosed initially with HGG, subsequently treated according to protocol (CCG-945), and later shown to have had LGG after central review of pathology.45 The children in this study ranged in age from infants to 19 years, and 20% did not receive RT. At 5 years, the PFS rate for patients who received chemoradiotherapy was 68%, compared with 38% for patients who received chemotherapy only. As the majority of patients not receiving upfront RT received RT at progression, the 5-year OS rate was not significantly different between groups (79% and 77%, p=0.34).
Alternate regimens have been evaluated for patients unable to tolerate typical regimens, in the refractory setting, and in combination with promising agents used for adults. In a study alternating the administration of weekly carboplatin and vincristine with TMZ in the management of 66 children with progressive and/or symptomatic LGG, 79% were able to complete induction and four cycles of maintenance therapy without significant dose reduction, while 5-year EFS and OS were similar to the results obtained on CCG9952 (46% and 87%, respectively).46 Carboplatin hypersensitivity reactions can occur in up to 30% of patients. Weekly vinblastine has a good efficacy to toxicity ratio in the treatment of LGG and can be a valuable option for children who develop severe hypersensitivity reactions.47 Weekly vinblastine seems to be a reasonable alternative to radiation for pediatric patients with LGG who have experienced treatment failure with first-line chemotherapy.48
Summary
A summary of some of the larger prospective randomized data for the adjuvant treatment of LGG is shown in Table 3.
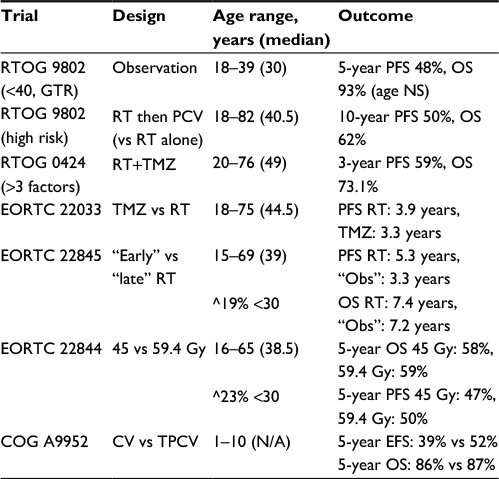 | Table 3 Results of large prospective trials of therapy for LGG and corresponding patient age inclusions Notes: ^indicates the proportion of the study population younger than 30 years old. The CV vs TPCV study can be referred to as Children’s Oncology Group Protocol A9952 (COG A9952) under Trial. The age range is <1–10. Abbreviations: CV, carboplatin and vincristine; COG, Children’s Oncology Group; EORTC, European Organization for Research and Treatment of Cancer; EFS, event-free survival; GTR, gross total resection; LGG, low-grade glioma; N/A, not applicable; NS, not significant; Obs, observation; OS, overall survival; PCV, procarbazine, lomustine, and vincristine; PFS, progression-free survival; RT, radiotherapy; RTOG, Radiation Therapy Oncology Group; TMZ, temozolomide; TPCV, thioguanine, procarbazine, lomustine, and vincristine. |
When considering all potential permutations of patient and disease status, some general rules of thumb emerge regarding the management of AYA with LGGs.
- Maximal safe resection should be attempted.
- AYA with LGG who undergo GTR may be safely observed.
- In younger adolescents and patients in whom the risk of cognitive detriment from RT is felt to be excessive, maximal safe surgical resection followed by close observation and delay in RT may be appropriate.
- As age increases and the risk factors for recurrence accumulate, adjuvant therapy should be more strongly considered. In this young age group, proton therapy or other modality capable of maximal neurocognitive structure sparing should be used. RT dose should be in the range of 45–54 Gy.
- If RT is to be delivered, strong consideration should be given to the role of adjuvant chemotherapy. Molecular marker data from the trials regarding the role of chemotherapy will help guide this treatment decision.
- The use of concurrent chemotherapy in young adult patients should be limited to clinical trials.
High-grade glioma
HGG in adults and pediatrics remains a therapeutic challenge with patients having a relatively poor prognosis. Despite progress in the understanding of the genetic and molecular diversity of these lesions and the resultant impact on outcomes, major advancements in therapeutic strategies have not emerged. Results with current therapies, including focal RT (often utilized in combination with chemotherapy and biologically targeted agents), remain disappointing with the vast majority of patients succumbing to their disease.
Epidemiology
GBM is the second most frequently reported primary intracranial tumor and the most common malignant tumor of the CNS when considering all ages. GBM accounts for 15.4% of all primary brain tumors and 45.6% of primary malignant brain tumors.1 This disease is less common in children, comprising ~2.9% of all brain and CNS tumors reported among 0–19 year olds and 3.2% of all brain and CNS tumors reported among 15–19 year olds.1
Presentation
AYA with HGG often present with similar symptoms as those with other CNS tumors. The sequelae of increased intracranial pressure, namely, persistent headaches, behavior changes, early morning nausea/emesis, diplopia, and papilledema, remain most common. Nevertheless, patients can also present with more specific localizing symptoms such as focal motor deficits, hemiplegia, pyramidal tract findings, dysmetria, and chorea, depending upon the tumor’s location.49 As compared to LGG, the duration of symptoms prior to presentation is often shorter with HGG, owing to the rapid rate of progression.
Diagnosis
Gadolinium-enhanced MRI remains the imaging modality of choice. Typically, HGG have less distinct and irregular borders on MRI as compared to other CNS tumors, sometimes with nodular enhancement. Classically, these lesions induce significant peritumoral edema, best visualized on fluid-attenuated inversion recovery sequences. Contrast enhancement is commonly seen, although the amount and degree of enhancement does not always correlate with tumor grade (see Diagnosis in LGG section). Perfusion-weighted MRI can aid in diagnosis as HGGs commonly demonstrate increased blood flow relative to cerebral blood pools. MR spectroscopy can be useful to distinguish the tumor from similar appearing lesions; as the tumor grade increases, there is an increase in the choline to N-acetyl aspartate ratio, reflecting increased metabolic activity.
Anaplastic astrocytomas are diffusely infiltrating tumors with increased cellularity, distinct nuclear atypia, and marked mitotic activity. GBMs have similar features to anaplastic astrocytomas, but with prominent microvascular proliferation and necrosis. They are classically described as having a “fried egg” appearance. Distinction of anaplastic variants of oligodendroglial tumors is based on the presence of mitotic activity, microvascularization, and necrosis.9 Malignant astrocytomas (non-brainstem) represent ~8%–10% of all pediatric CNS tumors. Due to the low incidence in children, studies on pediatric HGG have traditionally grouped grade III and IV tumors that are astrocytic or oligodendroglial in nature, including anaplastic astrocytoma (III), GBM (IV), anaplastic oligodendroglioma (III), oligoastrocytoma (III), and DIPGs. Although most DIPGs typically present as high-grade tumors and nearly without exception share a dismal prognosis, this term comprises tumors with varying histologic subtypes and degrees of differentiation. Anaplastic astrocytomas and GBMs often develop de novo in children, as opposed to progression from a lower-grade lesion, which can be observed in adults. High-grade oligodendrogliomas in children are rare and represent well-differentiated, diffusely infiltrating glial tumors.50
Prognosis
Histologic grade correlates with survival. Patients with a WHO grade III (anaplastic) tumor have improved survival, as compared to those with WHO grade IV tumors. In the aforementioned CCG-945 trial, patients with an anaplastic astrocytoma had statistically improved survival, with estimated 5-year PFS and OS of 28% and 29%, respectively, as compared to patients with GBM with the 5-year PFS and OS of 16% and 18%, respectively.51 Table 4 depicts the general survival rates by diagnosis and age for LGG and HGG.
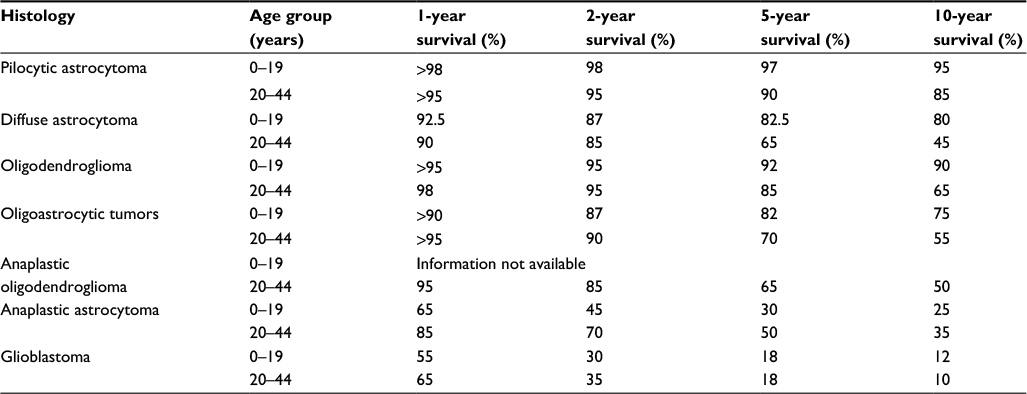 | Table 4 Average survival rates for brain and central nervous system tumors by major histology groupings and age at diagnosis, CBTRUS statistical report: NPCR and SEER, 2007–2011 (data approximated) Notes: Adapted from Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63, by permission of Oxford Univeristy Press.1 Abbreviations: CBTRUS, Central Brain Tumor Registry of the United States; NPCR, National Program of Cancer Registries; SEER, Surveillance, Epidemiology, and End Results. |
Emerging data has begun to elucidate distinct genetic profiles amidst HGGs in the pediatric population that seem to possess very different prognoses and clinical outcomes.52–54 Work from the German Cancer Research Center has delineated at least six separate subgroups based on age predilection, gene expression pattern, median survival, oncogenic drivers, and location within the brain.54 Median survival can vary widely between these groups, ranging from 6 months to multiple years. Mutations in histones H3.1, at position K27, and H3.3, at positions K27 and G34, have more recently been focused upon for their oncogenic tendencies and prognostic value.55 The K27, RTK-I, and PXA-like subtypes are characteristically encountered more often in the AYA and pediatric populations.
MGMT silencing through methylation is associated with improved outcomes in adults with HGG treated with TMZ.22 Likewise, MGMT overexpression was associated with adverse outcome independent of grade, age, amount of residual tumor, and tumor location in pediatric patients with HGG treated with an alkylating agent in the CCG study CCG-945.23,56 MGMT promoter methylation has been correlated with a survival benefit and sensitivity to TMZ in pediatric GBM.57 It is anticipated that across the spectrum of AYA ages, MGMT methylation remains a positive prognosticator.56
Also, in the CCG-945 study of pediatric HGG, patients with overexpression of p53 and/or a mutation in the TP53 gene had significantly lower PFS, as compared to children who had neither of these findings.49 TP53 mutations are thought to be one of several driving aberrations in the majority of adult HGG.
Data from whole-genome sequencing of HGG reveal that platelet-derived growth factor receptor A amplification is by far the most common genomic event identified in pediatric HGG and that this amplification seems to occur most often in older children and may have some prognostic significance.58
Epidermal growth factor receptor (EGFR) is found to be expressed in 30%–50% of adult GBM, mainly through amplification of the EGFR gene. Several EGFR gene mutations have been reported in GBMs, with the most common being the truncated EGFRvIII that can be found in 45% of EGFR-amplified GBM and in 8.5% patients without amplification.59 A pathologic review of adult patients with HGG showed on multivariate analysis that EGFR amplification was an independent and significant unfavorable predictor for OS, with EGFRvIII overexpression being the strongest indicator of a poor prognosis.59 EGFR gene amplification and mutation appear to occur less frequently in childhood gliomas.60
In adult anaplastic gliomas, loss of heterozygosity of 1p and 19q remains a key genetic marker that is highly prognostic of outcomes and response to therapy. As discussed above, in contrast to adult lesions, 1p/19q deletion is rare in children with anaplastic oligodendrogliomas and may not confer the same responsiveness to therapy.17
A similar story holds for IDH1 and IDH2 mutations, as they are associated with prolonged survival and enhanced sensitivity to TMZ in adults,61 but are less common in pediatric HGG. In younger adolescents, the presence of these mutations may reflect the biology of the adult form of the disease and not the more favorable infantile course.
Management
As the prognosis for AYA with HGGs is substantially different than that for low-grade lesions, the management tends to focus on maximal therapy to attempt cure, with less focus on delaying or omitting adjuvant therapy. As such, adolescents tend to be treated in paradigms similar to those developed for adults.
Maximal safe surgical resection remains the mainstay of definitive management, although complete removal remains challenging. Independent from tumor location and specific histology, the amount of surgical resection is one of the most important clinical prognostic factors identified to date in children with supratentorial HGG. The previously mentioned CCG-945 study showed that those children with HGG who underwent a surgical resection of 90% or greater had a PFS of 35%±7%, as compared to a 5-year PFS of 17%±4% in patients who did not.51
Radiotherapy
Five of six early randomized trials have demonstrated a survival advantage with the addition of postoperative RT over supportive care alone or single/multiagent chemotherapy alone in malignant gliomas.62 Historically, anaplastic gliomas have been included in clinical trials addressing HGGs. Thus, the treatment paradigm remains largely similar, including maximal safe surgical excision, followed by radiation with or without chemotherapy.
In adult HGG, the standard dose of postoperative radiation utilized in HGG is 60 Gy in 30 fractions. In pediatric and AYA patients, fractionation is often reduced to 1.8 Gy size delivering 59.4 Gy in 33 fractions to reduce the late effects of RT. Hypofractionated approaches such as 40 Gy in 15 fractions or 25 Gy in 5 fractions are considered in elderly or poor prognostic GBM patients treated with radiation alone.63,64 Such fractionations are rarely used in pediatric or AYA patients. Dose escalation for HGG beyond 60 Gy has not been shown to improve disease control and was associated with increased toxicity. An effort from the University of Michigan that was aimed to escalate the total fractionated dose as high as 90 Gy proved negative for its target outcome of local control. Strikingly, 95% of the recurrence volume after this approach was within the 90 Gy isodose line in over three-quarters of the treated patient setting.65
Chemotherapy
In 2005, Stupp et al published data showing that the addition of concurrent and adjuvant TMZ to RT for newly diagnosed GBM in adult patients resulted in a clinically meaningful and statistically significant survival benefit with minimal additional toxicity, as compared to radiation alone.66 TMZ increased median survival from 12.1 to 14.6 months, while increasing the 2-year survival from 10.4% to 26.5%. These results prompted the development of a pediatric trial, ACNS-0126, a Phase II trial of concurrent and adjuvant TMZ in children with newly diagnosed HGG.67 In this study, TMZ showed no survival benefit as compared to historic controls; however, the comparator was not to radiation alone, but to a prior evaluation of chemoradiotherapy.
While the Stupp trial was definitive in its findings for GBM, in patients with anaplastic oligodendroglioma or mixed cellularity tumors, the question of the addition of concurrent chemotherapy to radiation has not been formally addressed. There is evidence suggesting a benefit from the addition of systemic therapy sequentially with adjuvant RT. In 1994, the Phase III RTOG 9402 trial was undertaken by Cairncross et al. They randomized a total of 289 patients to RT alone, or four cycles of PCV (procarbazine, lomustine, and vincristine) followed by RT.68 The most recent update of their results, with a median follow-up of 11.3 years, showed no OS benefit for the whole cohort. This lack of benefit persisted in the subgroup of patients with 1p/19q intact tumors. However, unplanned subset analysis in the group of patients with 1p/19q co-deletions revealed a significant improvement in median survival from the addition of PCV (14.7 vs 7.3 years).68
Within 2 years of RTOG 9402 initiation, there was a separate European trial undertaken along a similar path. The EORTC 26951 trial examined the use of PCV following radiation therapy in the setting of grade III anaplastic oligodendroglioma or oligoastrocytoma (defined as having >25% oligodendroglial components on histology). At 5 years, van den Bent et al reported an improvement in PFS, but no significant difference in OS.65,69 More recently, in 2012, the EORTC updated their results with a median follow-up of 11.6 years and showed an improvement in median survival from 30.6 to 42.3 months with the addition of six cycles of PCV chemotherapy after completion of adjuvant RT to a dose of 59.4 Gy. Furthermore, similar to the RTOG study, they also reported that the benefit from adjuvant PCV was even greater in a subset of 80 patients harboring the 1p/19q co-deletion.70
The VEGF inhibitor bevacizumab is frequently used in the recurrent setting for adults with HGG. Studies of its value in the upfront setting have not shown a benefit in survival.55 Bevacizumab is being evaluated in children with newly diagnosed HGG in the currently open COG ACNS-0822 trial. There are ongoing trials in pediatric HGG exploring the use of PARP inhibitors in children with both newly diagnosed and recurrent HGG and DIPG as well as studies of immune therapies, including vaccine therapies in both adult and children with newly diagnosed and recurrent HGG.71,72
Tumor treating fields (TTF)
TTF are low-intensity, intermediate-frequency electric fields delivered by noninvasive transducer arrays. They are an antimitotic, physical treatment modality thought to act in metaphase, anaphase, and telophase of the cell cycle. In an international multicenter, prospective, randomized Phase III trial of TTF vs physician’s choice chemotherapy in recurrent GBM, TTF were shown to be essentially equivalent to systemic therapy in efficacy, with toxicity and QOL favoring TTF, though it did not demonstrate an improvement in the primary endpoint of OS.73 Due to improved QOL, the device is Food and Drug Administration approved for individuals over the age of 22 with recurrent/progressive supratentorial GBM after surgical and radiation options have been exhausted. Its use in younger AYA should be considered investigational. There are no currently enrolling trials in the pediatric population. The results of another Phase III trial were recently presented, this time in the newly diagnosed setting, randomized patients after completion of RT with concurrent TMZ, patients were randomized (2:1) to adjuvant TMZ with NovoTTF or adjuvant TMZ alone.74 The trial was closed to accrual after interim analysis showed improvement in median PFS (7.1 vs 4.0 months), which was the primary endpoint, and also OS (19.6 vs 16.6 months), and the 24-month survival rate (43% vs 29%) favoring the addition of TTF to TMZ in the adjuvant setting. The authors concluded that adjuvant TMZ chemotherapy and NovoTTF provided a clinically and statistically significant improvement in PFS and OS, and should become the new standard of care against GBM. The two Phase III trials allowed enrollment of individuals as young as 18; however, the median age in the newly diagnosed trial was 57 years.
Summary
Whenever possible, young patients with HGGs should be enrolled in clinical trial for the benefit of centralized genetic and molecular prognostic review and best clinical care. As with LGGs, several general recommendations can be established.
- Young adults and adolescents with WHO Grade IV Gliomas, deemed capable of tolerating chemoradiation, should be offered RT to 60 Gy with concurrent and adjuvant chemotherapy after maximal safe resection.
- Younger adolescents with GTR of grade III lesions may consider RT alone or sequential RT and chemotherapy if unable to tolerate concurrent treatment, especially if 1p/19q co-deleted mutation status is demonstrated.
Visual pathway gliomas
Tumors of the optic pathway can present with symptoms of neurologic, ophthalmic, neuropsychologic, and endocrine deficiencies, depending on their location and pattern of growth. These tumors are frequently (25%–40%) associated with NF1. In older children and young adults, visual complaints are the most common presenting symptom and coexisting hypopituitarism is often encountered. Histologically, >90% are LGGs and they occur predominately in young children, with 50% presenting before age 5.
Optic pathway gliomas are typically hypointense lesions on T1, contrast-enhancing, and hyperintense on T2. Typical chiasmatic lesions that involve components of the visual pathways beyond the optic chiasm and hypothalamic region may be managed without biopsy confirmation.
The management of gliomas of the visual pathways is controversial. Patients who are asymptomatic at presentation can be observed until clinical or radiographic disease progression. Surgical resection can be possible for small prechiasmatic lesions and is indicated in exophytic tumors with significant mass effect and in the case of single nerve involvement causing progressive disfiguring proptosis, blindness, or both. In most cases, the role of operative management is limited. Chemotherapy is frequently used as a first-line treatment and may delay RT in younger children. Radiation is used in adolescents with good effect: 10-year PFS exceeds 80% in the literature. Typical doses range from 50.4 to 54 Gy in 1.8 Gy per fraction.75 Visual improvement has been reported in 25%–35% of children after RT; however, visual deterioration is reported in 10%–20%, often related to cystic degeneration and consequent increased mass effect at the chiasm.76
The young age at diagnosis, central location, and often extensive optic involvement create a challenge when trying to deliver adequate RT while trying to preserve neurocognitive function. Proton RT may spare the contralateral temporal lobe and hippocampus, even if it is often difficult to exclude these ipsilateral structures from treatment volumes. Although a rare phenomenon, occlusive vasculopathy at the circle of Willis (Moyamoya syndrome) is encountered when treating younger children, especially those with NF1. Patients with NF1 can also be at substantial relative risk of second malignancy.77 This risk of second malignancy should be discussed in detail with the patient/family, with the use of RT preferred only when other options are exhausted including strong consideration for chemotherapy and/or clinical trial participation or for patients with bilateral involvement where the risk of negative impact of bilateral vision loss on QOL needs to be weighed against the risks with radiation. Modern radiation techniques such as proton beam therapy could also help mitigate some of this risk.
DIPG
DIPGs are typically tumors of childhood with extremely poor prognosis, and 77% of patients are under 20 years of age. Whereas low-grade lesions may present with focal deficits or symptoms of obstructive hydrocephalus, malignant lesions typically manifest with multiple rapidly progressive lower cranial nerve deficits and corticospinal tract impairments such as hemiparesis. Diffuse lesions can usually be diagnosed on MRI without the need for biopsy; however, the routine lack of tissue diagnosis in some ways hampers the investigation of the molecular underpinnings of these tumors. Recent efforts have demonstrated the safety of routine stereotactic biopsy in high-volume neurosurgical centers.78 Similar to HGGs, as discussed above, DIPGs also represent a diverse set of histologic subtypes which can correlate with outcomes and could potentially be targeted with tailored therapies in the future.79 Hence, a surgical attempt to obtain tissue diagnosis for biobanking or molecular analysis is strongly encouraged to further the knowledge of this terrible disease with poor outcomes.
Most frequently, though, the diagnosis is made based on imaging and clinical presentation without biopsy. Diffuse gliomas are treated with RT, typically in the range of 54 Gy with conventional fractionation. Despite efforts to explore dose escalation and hyperfractionation, outcomes remain dismal with median survival measured around 9–10 months.80,81 Unfortunately, the addition of TMZ has also failed to improve outcomes with median time to progression and death of 6 and 9 months in the COG-conducted ACNS-0126 study.82 Participation in clinical trials is strongly encouraged as future attempts explore combination with other systemic therapies and analysis of tumor tissue for better understanding of the disease.
Conclusion
Gliomas are a diverse family of tumors with varying prognoses depending on grade and age. In most cases, maximal safe resection should be undertaken. AYA who achieve a GTR for a low-grade lesion can, in general, be safely observed. Patients with low-grade lesions with residual or unresectable disease should be treated with external beam RT in the range of 45–54 Gy. Consideration should be given to adjuvant chemotherapy for patients with low-grade lesions who receive RT. Concurrent chemoradiation should only be performed for low-grade lesions in a clinical trial. HGGs in AYA should also receive maximal safe resection and should universally receive adjuvant or definitive RT depending on resectability. The decision to add concurrent chemotherapy to RT for grade III lesions should depend on histology, co-deletion of molecular markers such as 1p/19q, and the patient’s ability to tolerate therapy. For GBM, concurrent chemoradiation followed by adjuvant chemotherapy is the standard of care. Future trials will focus on the use of molecular signatures of individual patients’ glioma in guiding treatment approaches with the use of mutation-targeted therapies.
Acknowledgment
Both TPD and AE are co-first authors for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63. | ||
National Cancer Institute at the National Institutes of Health. Adolescents and young adults with cancer. Available from: http://www.cancer.gov/cancertopics/aya. Accessed February 26, 2016. | ||
Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006; 2(9):494–503 | ||
Unal E, Koksal Y, Cimen O, Paksoy Y, Tavli L. Malignant glioblastomatous transformation of a low-grade glioma in a child. Childs Nerv Syst. 2008;24(12):1385–1389. | ||
Strong JA, Hatten HP Jr, Brown MT, et al. Pilocytic astrocytoma: correlation between the initial imaging features and clinical aggressiveness. AJR Am J Roentgenol. 1993;161(2):369–372. | ||
Kerkhof M, Vecht CJ. Seizure characteristics and prognostic factors of gliomas. Epilepsia. 2013;54(Suppl 9):12–17. | ||
Pirzkall A, Nelson SJ, McKnight TR, et al. Metabolic imaging of low-grade gliomas with three-dimensional magnetic resonance spectroscopy. Int J Radiat Oncol Biol Phys. 2002;53(5):1254–1264. | ||
Pouratian N, Asthagiri A, Jagannathan J, Shaffrey ME, Schiff D. Surgery insight: the role of surgery in the management of low-grade gliomas. Nat Clin Pract Neurol. 2007;3(11):628–639. | ||
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | ||
Schmandt SM, Packer RJ, Vezina LG, Jane J. Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg. 2000;32(3):132–136. | ||
Pignatti F, van den Bent M, Curran D, et al; European Organization for Research and Treatment of Cancer Brain Tumor Cooperative Group; European Organization for Research and Treatment of Cancer Radiotherapy Cooperative Group. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002; 20(8):2076–2084. | ||
Forst DA, Nahed BV, Loeffler JS, Batchelor TT. Low-grade gliomas. Oncologist. 2014;19(4):403–413. | ||
Gunnarsson T, Olafsson E, Sighvatsson V, Hannesson B. Surgical treatment of patients with low-grade astrocytomas and medically intractable seizures. Acta Neurol Scand. 2002;105(4):289–292. | ||
Cancer Genome Atlas Research Network; Brat DJ, Verhaak RG, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. | ||
Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. | ||
Suri V, Jha P, Agarwal S, et al. Molecular profile of oligodendrogliomas in young patients. Neuro Oncol. 2011;13(10):1099–1106. | ||
Kreiger PA, Okada Y, Simon S, Rorke LB, Louis DN, Golden JA. Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol. 2005;109(4):387–392. | ||
Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67(9):878–887. | ||
Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. | ||
MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4(11):e7887. | ||
Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6(3):e17948. | ||
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. | ||
Everhard S, Kaloshi G, Criniere E, et al. MGMT methylation: a marker of response to temozolomide in low-grade gliomas. Ann Neurol. 2006; 60(6):740–743. | ||
Nakasu S, Fukami T, Jito J, Matsuda M. Prognostic significance of loss of O6-methylguanine-DNA methyltransferase expression in supratentorial diffuse low-grade astrocytoma. Surg Neurol. 2007;68(6):603–8; discussion 608–609. | ||
Adachi J, Suzuki T, Yanagisawa T, et al. Pl-02. mgmt promoter methylation status in newly diagnosed pediatric gliomas. Neuro Oncol. 2011;13(Suppl 3):102–106. | ||
Sardi I, Cetica V, Massimino M, et al. Promoter methylation and expression analysis of MGMT in advanced pediatric brain tumors. Oncol Rep. 2009;22(4):773–779. | ||
Buckner J, Pugh S, Shaw E, Gilbert M. Phase III study of radiation therapy (RT) with or without procarbazine, CCNU, and vincristine (PCV) in low-grade glioma: RTOG 9802 with alliance, ECOG, and SWOG. J Clin Oncol. 2014;32(5s):2000. | ||
Forsyth PA, Shaw EG, Scheithauer BW, O’Fallon JR, Layton DD Jr, Katzmann JA. Supratentorial pilocytic astrocytomas. A clinicopathologic, prognostic, and flow cytometric study of 51 patients. Cancer. 1993;72(4):1335–1342. | ||
Fernandez C, Figarella-Branger D, Girard N, et al. Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases. Neurosurgery. 2003;53(3):544–53; discussion 554–555. | ||
Brown PD, Anderson SK, Carrero XW, et al. Adult patients with supratentorial pilocytic astrocytoma: long-term follow-up of prospective multicenter clinical trial NCCTG-867251 (alliance). Neurooncol Pract. 2015;2(4):199–204. | ||
Yock TI, Bhat S, Szymonifka J, et al. Quality of life outcomes in proton and photon treated pediatric brain tumor survivors. Radiother Oncol. 2014;113(1):89–94. | ||
Hug EB, Muenter MW, Archambeau JO, et al. Conformal proton radiation therapy for pediatric low-grade astrocytomas. Strahlenther Onkol. 2002;178(1):10–17. | ||
Gajjar A, Sanford RA, Heideman R, et al. Low-grade astrocytoma: a decade of experience at st. jude children’s research hospital. J Clin Oncol. 1997;15(8):2792–2799. | ||
Wisoff JH, Sanford RA, Heier LA, et al. Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the children’s oncology group. Neurosurgery. 2011;68(6):1548–1554; discussion 1554–1555. | ||
Hollon T, Hervey-Jumper SL, Sagher O, Orringer DA. Advances in the surgical management of low-grade glioma. Semin Radiat Oncol. 2015;25(3):181–188. | ||
van den Bent MJ, Afra D, de Witte O, et al; EORTC Radiotherapy and Brain Tumor Groups and the UK Medical Research Council. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366(9490):985–990. | ||
Kiebert GM, Curran D, Aaronson NK, et al. Quality of life after radiation therapy of cerebral low-grade gliomas of the adult: results of a randomised phase III trial on dose response (EORTC trial 22844). EORTC radiotherapy co-operative group. Eur J Cancer. 1998; 34(12):1902–1909. | ||
Mishra KK, Puri DR, Missett BT, et al. The role of up-front radiation therapy for incompletely resected pediatric WHO grade II low-grade gliomas. Neuro Oncol. 2006;8(2):166–174. | ||
Merchant TE, Kun LE, Wu S, Xiong X, Sanford RA, Boop FA. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27(22):3598–3604. | ||
Kahalley LS, Ris MD, Grosshans DR, et al. Comparing intelligence quotient change after treatment with proton versus photon radiation therapy for pediatric brain tumors. J Clin Oncol. 2016;34(10):1043–1049. | ||
Baumert B, Mason W, Ryan G. Temozolomide chemotherapy versus radiotherapy in molecularly characterized (1p loss) low-grade glioma: a randomized phase III intergroup study by the EORTC/NCIC-CTG/TROG/MRC-CTU (EORTC 22033–26033). J Clin Oncol. 2013;31s:2007. | ||
Baumert B, Hegi M, Mason W. Radiotherapy in relation to temozolomide: subgroup analysis of molecular markers of the randomized phase III study by the EORTC/NCIC-CTG/TROG/MRC-CTU (EORTC 22033–26033) in patients with a high risk low-grade glioma. J Clin Oncol. 2015;33s:2006. | ||
Fisher BJ, Hu C, Macdonald DR, et al. Phase 2 study of temozolomide-based chemoradiation therapy for high-risk low-grade gliomas: preliminary results of radiation therapy oncology group 0424. Int J Radiat Oncol Biol Phys. 2015;91(3):497–504. | ||
Ater JL, Zhou T, Holmes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the children’s oncology group. J Clin Oncol. 2012; 30(21):2641–2647. | ||
Fouladi M, Hunt DL, Pollack IF, et al. Outcome of children with centrally reviewed low-grade gliomas treated with chemotherapy with or without radiotherapy on children’s cancer group high-grade glioma study CCG-945. Cancer. 2003;98(6):1243–1252. | ||
Chintagumpala M, Eckel SP, Krailo M, et al. A pilot study using carboplatin, vincristine, and temozolomide in children with progressive/symptomatic low-grade glioma: a Children’s Oncology Group study†. Neuro Oncol. 2015;17(8):1132–1138. | ||
Lafay-Cousin L, Holm S, Qaddoumi I, et al. Weekly vinblastine in pediatric low-grade glioma patients with carboplatin allergic reaction. Cancer. 2005;103(12):2636–2642. | ||
Bouffet E, Jakacki R, Goldman S, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol. 2012;30(12):1358–1363. | ||
Fangusaro J. Pediatric high grade glioma: a review and update on tumor clinical characteristics and biology. Front Oncol. 2012;2:105. | ||
Hargrave D. Paediatric high and low grade glioma: the impact of tumour biology on current and future therapy. Br J Neurosurg. 2009; 23(4):351–363. | ||
Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995;13(1):112–123. | ||
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. | ||
Diaz AK, Baker SJ. The genetic signatures of pediatric high-grade glioma: no longer a one-act play. Semin Radiat Oncol. 2014;24(4):240–247. | ||
Sturm D, Bender S, Jones DT, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014; 14(2):92–107. | ||
Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. | ||
Pollack IF, Hamilton RL, Sobol RW, et al. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 cohort. J Clin Oncol. 2006;24(21):3431–3437. | ||
Donson AM, Addo-Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer. 2007;48(4):403–407. | ||
Bax DA, Mackay A, Little SE, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2010;16(13):3368–3377. | ||
Shinojima N, Tada K, Shiraishi S, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63(20):6962–6970. | ||
Bax DA, Gaspar N, Little SE, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009;15(18):5753–5761. | ||
Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–1566. | ||
Laperriere N, Zuraw L, Cairncross G; Cancer Care Ontario Practice Guidelines Initiative Neuro-Oncology Disease Site Group. Radiotherapy for newly diagnosed malignant glioma in adults: a systematic review. Radiother Oncol. 2002;64(3):259–273. | ||
Roa W, Brasher PM, Bauman G, et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. J Clin Oncol. 2004;22(9):1583–1588. | ||
Roa W, Kepka L, Kumar N, et al. International atomic energy agency randomized phase III study of radiation therapy in elderly and/or frail patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2015; 33(35):4145–4150. | ||
Chan JL, Lee SW, Fraass BA, et al. Survival and failure patterns of high-grade gliomas after three-dimensional conformal radiotherapy. J Clin Oncol. 2002;20(6):1635–1642. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the children’s oncology group. Neuro Oncol. 2011;13(3):317–323. | ||
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. | ||
van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized european organisation for research and treatment of cancer phase III trial. J Clin Oncol. 2006; 24(18):2715–2722. | ||
van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. | ||
National Cancer Institute (NCI). A phase I/II study of ABT-888, an oral poly(ADP-ribose) polymerase inhibitor, and concurrent radiation therapy, followed by ABT-888 and temozolomide, in children with newly diagnosed diffuse pontine gliomas (DIPG) in: ClinicalTrials.gov [internet]. Bethesda, MD: National library of medicine (US). 2000NLM identifier: NCT01514201. Available from: https://clinicaltrials.gov/ct2/show/NCT01514201. Accessed February 26, 2016. | ||
University of Pittsburgh. A pilot study to evaluate the effects of vaccinations with HLA-A2-restricted glioma antigen-peptides with poly-ICLC for children with newly diagnosed malignant brain stem gliomas, non-brainstem high-grade gliomas, recurrent low-grade gliomas or recurrent high grade gliomas in: ClinicalTrials.gov [internet]. Bethesda, MD: National Library of Medicine (US); 2000 [cited February 27, 2016]. Available from: http://Clinicaltrials.gov/show/NCT01130077 NLM identifier: NCT01130077. Accessed February 27, 2016. | ||
Stupp R, Wong ET, Kanner AA, et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer. 2012;48(14):2192–2202. | ||
Stupp R, Wong E, Scott C, Tallibert S. NT-40 interim analysis of the EF-14 trial: a prospective, multi-center trial of NovoTTF-100A together with temozolomide compared to temozolomide alone in patients with newly diagnosed GBM. Neuro Oncol. 2014;16:167. | ||
Alshail E, Rutka JT, Becker LE, Hoffman HJ. Optic chiasmatic-hypothalamic glioma. Brain Pathol. 1997;7(2):799–806. | ||
Dalla Via P, Opocher E, Pinello ML, et al. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol. 2007;9(4):430–437. | ||
Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24(16):2570–2575. | ||
Puget S, Beccaria K, Blauwblomme T, et al. Biopsy in a series of 130 pediatric diffuse intrinsic pontine gliomas. Childs Nerv Syst. 2015; 31(10):1773–1780. | ||
Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. 2014;128(4):573–581. | ||
Marcus KJ, Dutton SC, Barnes P, et al. A phase I trial of etanidazole and hyperfractionated radiotherapy in children with diffuse brainstem glioma. Int J Radiat Oncol Biol Phys. 2003;55(5):1182–1185. | ||
Mandell LR, Kadota R, Freeman C, et al. There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: results of a pediatric oncology group phase III trial comparing conventional versus. hyperfractionated radiotherapy. Int J Radiat Oncol Biol Phys. 1999;43(5):959–964. | ||
Cohen KJ, Heideman RL, Zhou T, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the children’s oncology group. Neuro Oncol. 2011;13(4):410–416. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.