")
Back to Journals » Journal of Inflammation Research » Volume 15
Environmental Enrichment Protects Against Sepsis-Associated Encephalopathy-Induced Learning and Memory Deficits by Enhancing the Synthesis and Release of Vasopressin in the Supraoptic Nucleus
Authors Jiang S, Wang YQ, Tang Y, Lu X, Guo D
Received 3 November 2021
Accepted for publication 7 January 2022
Published 16 January 2022 Volume 2022:15 Pages 363—379
DOI https://doi.org/10.2147/JIR.S345108
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Shan Jiang,1,* Yong-Qiang Wang,2,* Yifei Tang,1 Xi Lu,1 Dan Guo1
1Department of Rehabilitation Medicine, the China-Japan Friendship Hospital, Beijing, 100029, People’s Republic of China; 2Department of Ophthalmology, the Sunshine Union Hospital, Weifang, Shandong, 261071, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shan Jiang, Department of Rehabilitation Medicine, the China-Japan Friendship Hospital, No. 2 Ying Hua Yuan East Street, Beijing, 100029, People’s Republic of China, Tel +86 10 84205288, Fax +86 10 64217749, Email [email protected]
Background: As a severe complication of sepsis, sepsis-associated encephalopathy (SAE) usually manifests as impaired learning and memory ability in survivors. Previous studies have reported that environmental enrichment (EE) can increase the learning and memory ability in different brain injury models. However, there has been no research on the possible positive effect of EE on SAE.
Aim: The present study aimed to test the effect of EE on SAE-induced impairment of learning and memory and its related mechanisms.
Methods: A Morris water maze test (MWM) was used to evaluate the learning and memory ability of SAE rats that received EE housing or not. The expression of vasopressin (VP) was assessed using immunofluorescence microscopy and enzyme-linked immunosorbent assays (ELISAs). The synthesis of VP in the supraoptic nucleus (SON) was determined using quantitative real-time reverse transcription-PCR analysis. Moreover, inflammatory markers and brain-derived neurotrophic factor (BDNF) were detected using ELISA.
Results: The results showed that SAE induced a decreased learning and memory ability, while EE reversed this impairment. EE also enhanced the synthesis and secretion of VP in the SON. Blocking the action of VP in the hippocampus interrupted the EE-induced amelioration of learning and memory impairment. Moreover, EE induced changes to the levels of BDNF and cytokines in the hippocampus and these effects were mediated by VP binding to the VP receptor 1a.
Conclusion: Our findings demonstrated that the enhanced synthesis and secretion of VP in the SON are a key determinant responsible for EE-induced alleviation of learning and memory deficits caused by SAE.
Keywords: sepsis associated encephalopathy, environmental enrichment, vasopressin, learning and memory, vasopressin receptor 1a
Introduction
Sepsis is induced by systemic responses to infection and has a high and growing prevalence worldwide.1–3 More than half of patients with sepsis have symptoms of sepsis-associated encephalopathy (SAE), which might be the first feature of sepsis before admission to the intensive care unit (ICU).4 Cognitive impairment is one of the major manifestations of SAE and can represent a serious social problem for survivors.5 SAE might pose substantial risks of long-term cognitive impairment, including deficits in learning and memory ability.6–10 These can lead to poor quality of life and an increased social burden. Therefore, how to improve the learning and memory deficits after SAE is a focus of current research.
Environmental enrichment (EE), a housing condition permitting exercise items, novelty, and social interaction, has been proven to ameliorate or attenuate functional deficits associated with various brain injuries.11,12 For example, sensory and memory impairment after traumatic brain injury (TBI) can be relieved by EE, possibly via cortical excitability and reorganization.13–17 EE was also shown to prevent stroke-induced learning and memory disorder.18–20 Moreover, in models of ethanol binge-drinking or perinatal asphyxia, accumulating evidence points to the positive effects of EE on learning and memory deficits.21–25 However, there has no research exploring the effect of EE on SAE.
Vasopressin (VP) is a neuroendocrine hormone that is produced mainly by magnocellular neurons (MONs) in the supraoptic nucleus (SON).26 Upon entering the systemic blood supply, VP exerts its endocrine actions, including the maintenance of stable osmolarity.27 In addition to its endocrine actions, VP is also involved in learning and memory ability.28 The hippocampus is generally regarded as the primary target structure for the enhanced effect of VP on learning and memory ability, and this enhanced effect is achieved via VP receptor 1a (V1aR).29 The hippocampus contains abundant VP-expressing fibers and V1aR can be detected in the whole hippocampus, particularly in the Cornu Ammonis 2 (CA2) region.30 However, septic cerebral dysfunction commonly causes impairment of the synthesis and release of VP in response to hypovolemia,31,32 which might be one of the explanations for SAE-induced learning and memory impairment. Recent research showed that EE increases the level of VP in pregnant SD rats, which suggested that EE might enhance the level of VP under SAE conditions.33
Accordingly, our study was designed to test the efficacy of EE on learning and memory impairment following SAE and its related mechanisms. To this end, SAE rats were placed in an enriched environment and changes in their learning and memory ability were evaluated. If it has a protective effect on learning and memory ability, EE could be an effective means to protect against the cognitive deficits after SAE and could be applied in clinical practice. In the present study, we also investigated the possible mechanism underlying the effect of EE. Considering that previous studies demonstrated the role of VP in learning and memory ability and SAE, we assessed whether VP mediates the effect of EE on learning and memory under SAE conditions.
Materials and Methods
Animals
Male Sprague-Dawley (SD) rats (60 days old, 200–250 g) were obtained from the Animal Center of the China-Japan Friendship Hospital. The experimental procedures were performed in compliance with the Guidelines for the use of animals in neuroscience research (published in the Membership Directory of the Society, pp 27–28, 1992), and with permission from the committee of Animal Use for Research and Education in China-Japan Friendship Hospital (Approval number: zryhyy61210324). All animals were housed in groups of four animals in polycarbonate cages and maintained under a 12-h light/dark cycle in a temperature-controlled room at 24 ± 1 °C with food and water available ad libitum.
SAE Induction via a Cecal Ligation and Perforation (CLP) Model
The cecal ligation and perforation (CLP) model was carried out to induce SAE, as previously described.34,35 Briefly, animals were anesthetized using sodium pentobarbital (50 mg/kg), administered intraperitoneally. The cecum and adjoining intestine were uncovered under aseptic conditions. The cecum was ligated using a 3.0 silk suture at its base, below the ileocecal valve and was perforated twice using a 14-gauge needle. The operator gently squeezed the cecum to extrude a small amount of feces through the perforation site into the peritoneal cavity. Finally, the cecum was then returned to the peritoneal cavity, and the laparotomy was closed using 4.0 silk sutures. Animals were resuscitated using regular saline (50 mL/kg) subcutaneously (s.c.) immediately after and at 12 h after CLP. All rats received antibiotics (ceftriaxone at 30 mg/kg and clindamycin 25 mg/kg) every 6 h s.c. for a maximum of 3 days. The animals were allowed to recover with free access to water and food for 24 h post-surgery until use. For the sham group, the laparotomy and the cecum exposure were performed, but without ligation and perforation. To exclude variability, the same investigator performed the experiments. The mortality of the rats in the model was about 53% at 10 days after surgery, and was compatible with severe sepsis.
Housing Conditions and Grouping
Two housing conditions were used in this study: standard environment conditions (SE) and enriched environment conditions (EE).
Standard environment (SE): Rats were housed in standard-sized polycarbonate cages (25 cm × 40 cm × 20 cm), with two rats per cage, located in a quiet room. The cages allowed for moderate activity and exploration.
Enriched environment (EE): Under these conditions, rats were reared in a large cage (40 cm × 54 cm × 30 cm) with six rats per cage. As described previously, the cages contained multiple objects, including wooden blocks, plastic bone-shaped toys, a running wheel, and a plastic tunnel.36,37 Moreover, the rats could complete climbing from one level to another through suspended ropes. All Objects were replaced twice a week. During the enrichment period, water and food were available ad libitum. After 30 days of EE housing, the objects were removed from the cages and no additional enrichment was given.
After 10 days of recovery, all animals were randomly divided into the following groups (six rats in each group): The sham group (Sham) (Rats received sham injury and were housed under SE conditions); the SAE group (Rats received CLP and were maintained under SE conditions); SAE-VP group (Rats received CLP, were given VP, and were housed under SE conditions); SAE-MIX group (Rats received CLP, were given a mixture of VP and SR49059 (the most potent nonpeptide competitive V1aR antagonist), and were housed under SE conditions); SAE-EE group (Rats received CLP and were maintained under EE conditions), SAE-EE-shRNA V1aR group (Rats received CLP, were given a short hairpin RNA (shRNA) targeting V1ar, and were housed under EE conditions); SAE-EE-anti-VP serum group (Rats received CLP, were given anti-VP serum, and were housed under EE conditions); SAE-EE-SR49059 group (Rats received CLP, were given SR49059, and were housed under EE conditions). The Morris water maze (MWM) is used widely to detect spatial learning and reference/working memory.38,39 Figure 1 shows the experimental process.
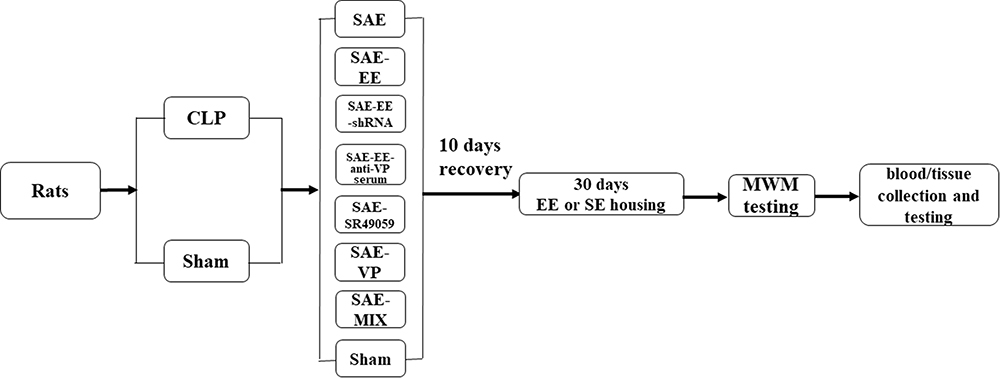 |
Figure 1 Flow chart of the experimental process. |
Learning and Memory Testing
This MWM was a dark circular tank (178 cm in diameter) that was separated into four quadrants (A, B, C, and D). The tank contained water to a depth of about 37 cm. A plexiglass platform (10.2 cm in diameter) was submerged to a depth of 2 cm below waterline (ie, invisible to the rat) and about 28 cm from the pool wall of quadrant C. Spatial learning was detected using a 5-day block comprising four tests each day. For each test, the rats were placed in the maze facing the wall in random quadrants. If the rats did not climb onto the platform within 120 s, they were physically guided to it. Once they reached the platform, the rats were kept on it for 30s and then given a 5-minute break before the next test. As a control, the platform was also elevated above the surface of the water to be visible to the rats on day 6 to identify the contributions of non-spatial factors. On the same day, memory retention was also measured through a single probe trial. We removed the platform and gave the rats 30s exploration time. The percent time spent in the target quadrant (quadrant C) and the number of crossings of the platform location were recorded.
Drugs Used and Their Administration
To examine the role of VP in EE protection against SAE-induced learning and memory impairment, we inserted two sterile polyethylene tubes into the bilateral hippocampus. With the help of a brain stereotaxic apparatus, the injection coordinates were determined as follows: dorsoventral, 2.7 mm; mediolateral, 1.5 mm; anteroposterior, 2.8 mm.
In this study, we used an shRNA for long term knockdown of V1ar. The shRNA sequences were inserted into adeno-associated virus (AAV) vectors, as reported previously.40 In this study, the AAVrh10 serotype was adopted because it possesses a highly neurotropic character and shows strong gene transfer into neurons. AAV-shRNA or the scrambled AAV control were injected bilaterally into the hippocampus of rats (300 nL/side). According to a previous study,40 the sequence of the shRNA was: sense sequence GCACGCCTCAGTACTTCATCT, hairpin loop CGAA, antisense sequence AGATGAAGTACTGAGGCGTGC; the sequence of the scrambled shRNA was: sense sequence GCTCCGTGACTTCACTACTCA, hairpin loop CGAA, antisense sequence TGAGTAGTGAAGTCACGGAGC. The vectors were injected at a titer of 1.0×1013 genomic particles/mL. The injection lasted over 10 min and the viral expression could persist for 3–4 months. Gel foam was used to fill the openings in the cranium, and the incision site was closed with absorbable antibiotic sutures.
Depending on the experimental design, VP (Sigma-Aldrich, St. Louis, MO, USA), anti-VP serum (Yu Xin Hui Ye Biotechnology Co., Ltd, Beijing, China), or SR49059 (Sigma-Aldrich) were also injected bilaterally into the hippocampus at daily doses of 0.3 μL (25 pg), 0.5 μL (1:10 dilution), and 0.3 μL (0.32 nmol),41 respectively, during EE or SE housing. The injection also lasted over 10 min.
Immunofluorescent Microscopy
Rats were killed and perfused transcardially with 100 mL of phosphate buffer (PB; 0.1 M, PH 7.4) followed by 500 mL of PB containing 4% paraformaldehyde. After being removed immediately, the brains were placed in 0.1 M PB containing 20% sucrose overnight at 4 °C. Forebrain slices through the SON were prepared for immunofluorescent staining, as described previously.42 The sections were incubated at 4 °C overnight with a mixture of rabbit anti-VP polyclonal antiserum (1:1000; Chemicon at Merck, Temecula, CA, USA; AB1565) in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA) and 0.3% Triton-X100. After washing for 1 h in 0.1 M PBS Triton X-100, the sections were then incubated with secondary antibodies (FITC-conjugated goat anti-rabbit IgG; 1:500; Sigma; AP132F) for 24 h at 48 °C. After further washing, the sections were mounted in Fluoromount (Sigma; F4680) and examined under a confocal laser microscope (LSM 510, Zeiss, Oberkochen, Germany).
Real-Time Quantitative Reverse Transcription-PCR Analysis
Real-time quantitative reverse transcription-PCR (qRT-PCR) analysis was carried out to detect the expression of Vp in the SON and the effect of AAV-shRNA on V1ar in the hippocampus. The rat SON or hippocampus was collected and total RNA was extracted using the Trizol reagent (Invitrogen, Waltham, MA, USA) and reverse transcribed into cDNA. The qPCR step was performed using the ABI 7901HT sequence detection system (Applied Biosystems, Foster City, CA, USA) with a Power SYBR green PCR Master Mix kit (Takara, Dalian, China) and the cDNA as the template. The sequences of the primers were as follows: Vp, 5′ primer (5′-CGGCAAAGGGCGCTGCTTCG-3′) and 3′ primer (5′-CCGGGGCTTGGCAGAATCCA-3′); V1ar, 5′ primer (5′-TACGTGKCTGGATGACCAG-3′) and 3′ primer (5′-AGCAACGCCGTGATTGTGAT-3′); Gapdh (encoding glyceraldehyde-3-phosphate dehydrogenase), 5′ primer (5′-ACAGCAACAGGGTGGTGGA-3′) and 3′ primer (5′-TTTGAGGGTGCAGCGAACT-3′). All data were normalized to the expression of Gapdh expression and further normalized to the control.
Enzyme-Linked Immunosorbent Assay
Inflammatory mediators and brain-derived neurotrophic factor (BDNF) levels in the hippocampus and VP levels in the SON and hippocampus were tested using enzyme-linked immunosorbent assays (ELISAs). Rats were killed and decapitated. Hippocampus or SON tissues were collected for ELISA as reported previously.43 In brief, the brains were microdissected and the hippocampus or SON was isolated. The tissue was homogenized in normal saline and centrifuged at 2000 rpm at 4 °C for 10 min. The supernatants were used to measure interleukin (IL)- 1β, IL-6, IL-10, tumor necrosis factor alpha (TNF-α), BDNF, or VP levels using ELISA kits (Nanjing Jiancheng, Nanjing, China) following the manufacturer’s instructions.
Inflammatory mediators or VP were also detected in serum using ELISA. Blood was collected from the right ventricle. After standing for 2 h at room temperature, the blood was centrifuged at 2000 rpm at 4 °C for 20 min and serum was collected. Serum IL-6, IL-1β, IL-10, TNF-α, and VP levels were tested using ELISA.
Statistical Analysis
All data are expressed as the mean ± the standard error of the mean (SEM). The Kolmogorov–Smirnov (K-S) test was used to test normality. The data were analyzed using analysis of variance (ANOVA). When a statistically significant difference was detected, Tukey’s post hoc analysis was carried out. P < 0.05 was considered statistically significant. The statistical analysis was performed using GraphPad Prism 9.0 software (GraphPad Inc. La Jolla, CA, USA).
Results
EE Ameliorated SAE-Induced Learning and Memory Impairment
In this section, the effect of EE on learning and memory was determined using the MWM, a hippocampus-dependent learning and memory task.
During the acquisition phase (learning testing), escape latency was recorded for 5 consecutive days. The findings revealed that there was a significant difference among sham group, SAE group, and SAE-EE group (F (2, 15) = 19.50, p < 0.0001; Figure 2A). Further post-hoc analysis indicated that both the sham rats and SAE-EE rats showed better latency to reach the hidden platform than the SAE rats at days 4 and 5, suggesting that SAE-impaired learning was ameliorated by EE housing (SAE-EE vs SAE group; day 4: p < 0.0001, day 5: p < 0.0001; Figure 2A). In addition, latency to the visible platform was also measured on day 6. The results exhibited no obvious differences among the groups (F (2, 15) = 1.764, p = 0.2051; Figure 2B), confirming that vision was not responsible for the differences observed in escape latencies. These results suggested that EE ameliorated the SAE-induced learning deficit.
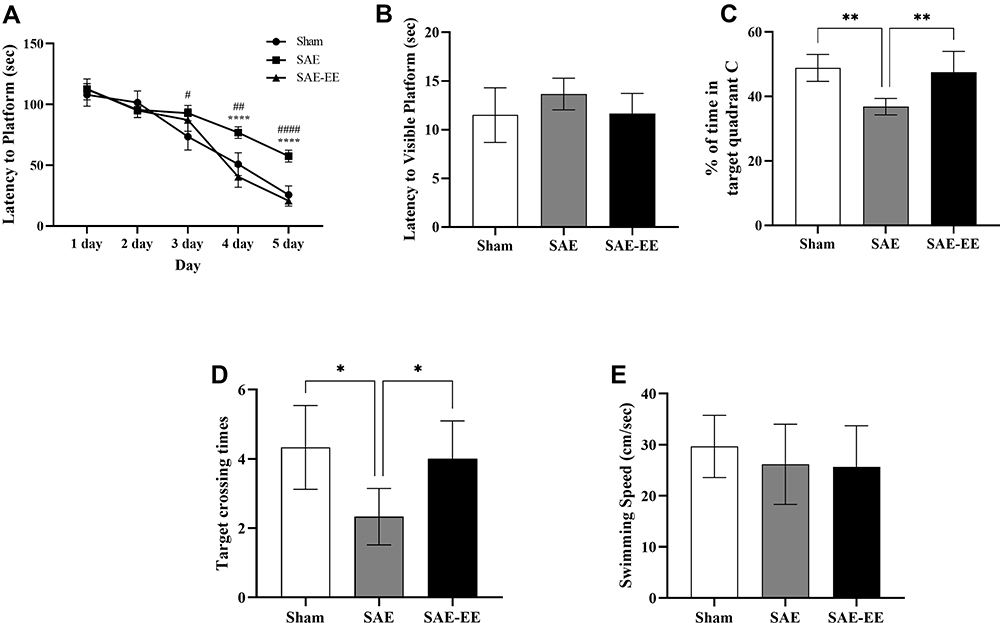 |
Figure 2 (A) From day 3 on, the rats with SAE had a longer latency time to the platform than the sham rats. From day 4 on, the SAE-EE rats reached the submerged platform quicker than the SAE rats. For A, Sham vs SAE: #p < 0.05, ##P < 0.01, ####P < 0.0001; SAE vs SAE-EE: ****p < 0.0001. (B) No differences were found in latency to the visible platform. (C) The SAE rats spent less time in target quadrant C compared with sham or SAE-EE rats. For C, **p < 0.01. (D) The SAE rats had fewer target crossing times when compared with the sham or SAE-EE rats. For D, *p < 0.05. (E) No differences were observed for swimming speed among the groups. The assignment of order was counterbalanced across the rats in this test. Data represent the means ± standard error of the mean. Abbreviations: SAE, sepsis-associated encephalopathy; EE, environmental enrichment. |
On day 6, the platform was removed and time spent in quadrant C and crossing times of the platform zone were recorded and analyzed for reference and/or working memory retention. The results showed that the rats in the Sham and SAE-EE group displayed better memory retention, as revealed by a higher percentage of allotted time spent in quadrant C, compared with that in the SAE rats (F (2, 15) = 11.83, p = 0.0008; Sham vs SAE group: p = 0.0013; SAE vs SAE-EE group: p = 0.0035; Figure 2C), which suggested that EE relieved the SAE-induced memory impairment. Analysis of crossing times of the platform zone showed similar results. The rats in the sham and SAE-EE group crossed the platform zone more often than those in the SAE group (F (2, 15) = 6.20, p = 0.0109; Sham vs SAE group: p = 0.0130; SAE vs SAE-EE group: p = 0.0382; Figure 2D).
Lastly, for swimming speed, no obvious differences was observed among the groups (F (2, 15) = 0.5217, p = 0.6039; Figure 2E), indicating that no motor deficits contributed to the above differences.
EE Attenuated the SAE-Induced Decrease in VP Synthesis and Release in the SON
As mentioned previously, sepsis commonly induces a persistent deficiency of VP.31 Moreover, VP is implicated in the learning and memory process.44 Thus, to verify the mechanism underlying the learning and memory enhancement caused by EE, we detected the effect of EE on VP synthesis and release after SAE.
As shown in Figure 3A, VP immunoreactivity was present on cells in the SON, as demonstrated previously.45 In the sham group, the mean fluorescence intensity (MFI) of VP was 352 ± 20.98 AU (Figure 3A and B). However, in the SAE group, the MFI decreased significantly to 162.7 ± 13.76 AU (Figure 3A and B), whereas this reduction was inhibited in the SAE-EE rats (398 ± 25.85 AU, Figure 3A and B). As expected, ELISA testing showed similar results (Figure 3C). Moreover, the qRT-PCR results also showed decreased Vp mRNA expression after SAE, while EE housing inhibited this decrease in Vp mRNA (Figure 3D). These results suggested that EE effectively relieves the impairment of VP synthesis resulting from SAE.
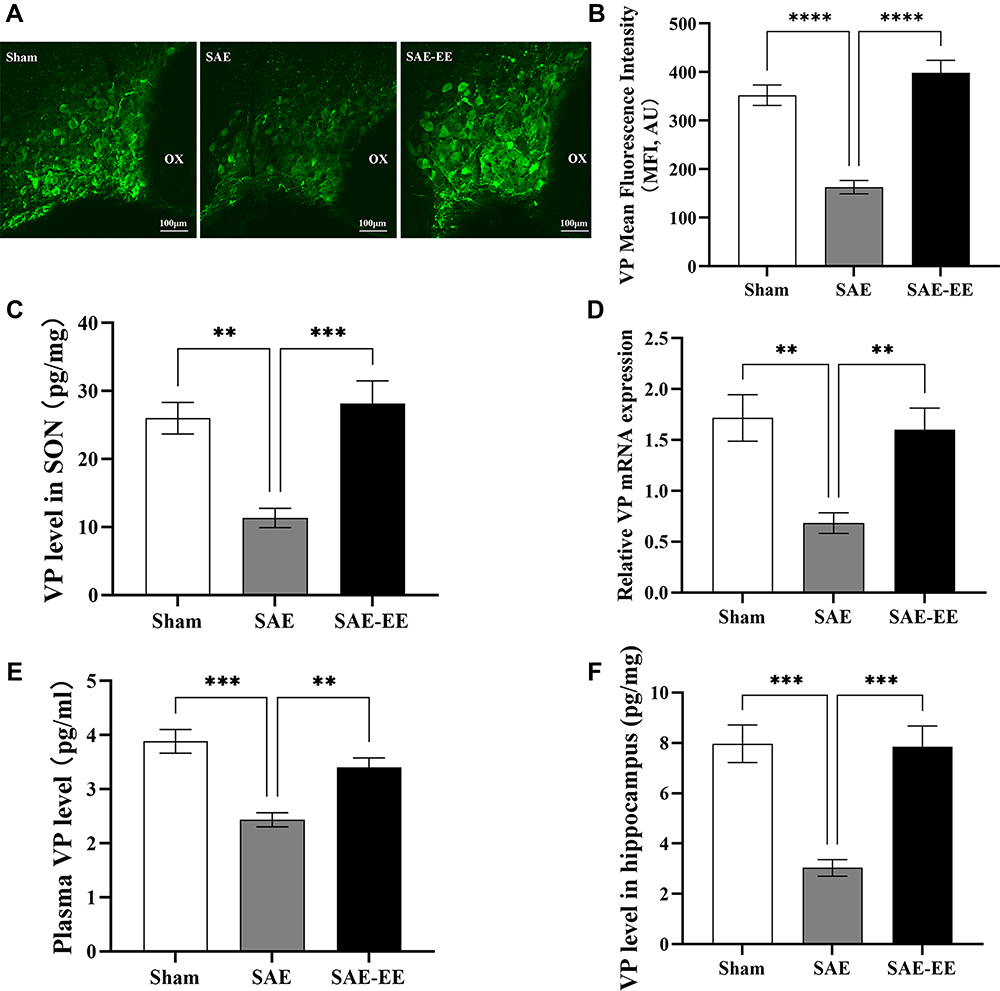 |
Figure 3 (A) VP staining in the SON. (B) The VP mean fluorescence intensity in the SAE rats was significantly lower than that in the sham or SAE-EE rats. (C) The amount of VP in the SON in the SAE rats was lower than that in the sham or SAE-EE rats. (D) The level of Vp mRNA in the SON was lower in the SAE rats compared with that in the sham or SAE-EE rats. (E) The plasma VP concentration in the SAE rats was lower than that in the sham or SAE-EE rats. (F) The amount of VP in the hippocampus of SAE rats was lower than that of the sham or SAE-EE rats. **p < 0.01, ***p < 0.001, ****p < 0.0001. Data represent the means ± standard error of the mean. Abbreviations: VP, vasopressin; SON, supraoptic nucleus; SAE, sepsis-associated encephalopathy; EE, environmental enrichment; Magnification, 40×. |
We also examined the changes in VP levels in the blood, which are considered an index of VP release. After SAE, the VP concentration decreased dramatically to 2.43 ± 0.13 pg/mL, which was markedly lower than that in the sham group (3.88 ± 0.22 pg/mL; Sham vs SAE: p = 0.0001; Figure 3E). As expected, the VP concentration in the SAE-EE group recovered to 3.40 ± 0.18 pg/mL (SAE vs SAE-EE group: p = 0.0045; Figure 3E). These findings implied that EE housing also ameliorated the SAE-induced impairment of VP release.
VP Accounts for EE-Induced Improvement of Learning and Memory After SAE
To gain an insight into how EE affects learning and memory after SAE, we examined the role of VP, which has been demonstrated to increase learning and memory ability by binding to V1aR in the hippocampus.
The hippocampus of rats contains VP-expressing fibers that arise from the SON.46 For the rats in the SAE-EE group, the ELISA results revealed that the content of VP in the hippocampus was markedly higher relative to that in the rats in the SAE group (Figure 3F), indicating that EE enhanced the delivery of VP to the hippocampus.
The shRNA for V1aR was capable of blocking the production of V1aR over the long term.47 As shown in the Supplemental Data (Supplemental Figure A), microinjection of the AAV-shRNA for V1ar led to a 60% decrease in the V1ar mRNA level in the hippocampus compared with that in rats transfected with the scrambled controls. Treatment with AAV-shRNA for V1ar markedly blocked the EE-induced amelioration of the learning and memory impairment caused by SAE (Figure 4A–C). In agreement with this, SR49059, a highly specific V1aR receptor antagonist, also abrogated the EE-induced enhancement of learning and memory after SAE (Figure 4A–C).
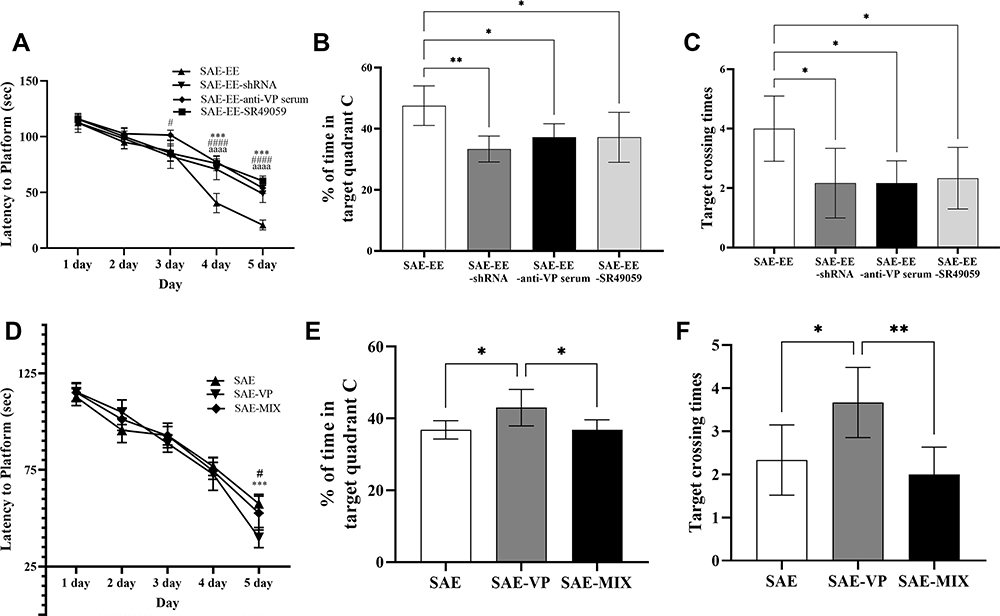 |
Figure 4 (A) The rats in the SAE-EE-anti-VP serum, SAE-EE-shRNA, or SAE-EE-SR49059 groups had a longer latency to platform than the rats in the SAE-EE group (from day 3 to day 5). SAE-EE vs SAE-EE-anti-VP serum: #p < 0.05, ####p < 0.0001; SAE-EE vs SAE-EE-shRNA:***p<0.001; SAE-EE vs SAE-EE-SR49059: aaaap < 0.0001. (B) The rats in the SAE-EE group spent more time in target quadrant C compared with the rats in the SAE-EE-anti-VP serum, SAE-EE-shRNA or SAE-EE-SR49059 groups. *p < 0.05,**p < 0.01. (C) The rats in the SAE-EE group had more target crossing times when compared with the rats in the SAE-EE-anti-VP serum, SAE-EE-shRNA or SAE-EE-SR49059 groups. *p < 0.05. (D) At day 5, the rats in the SAE and SAE-MIX group had a longer latency to the platform than the rats in the SAE-VP group. SAE vs SAE-VP: ***p < 0.001; SAE-VP vs SAE-MIX: #p < 0.05. (E) The rats in the SAE-VP group spent more time in target quadrant C compared with the rats in the SAE or SAE-MIX group. *p < 0.05. (F) The rats in the SAE-VP group had more target crossing times when compared with the rats in the SAE or SAE-MIX group. *p < 0.05,**p < 0.01. Data represent the means ± standard error of the mean. Abbreviations: EE, environmental enrichment; VP, vasopressin; SAE, sepsis-associated encephalopathy; MIX, a mixture of VP and SR49059; shRNA, short hairpin RNA. |
Additionally, for the SAE rats, microinjection of VP into the hippocampus mimicked the positive effect of EE on learning and memory (Figure 4D–F). By contrast, anti-VP serum blunted the EE-induced enhancement of learning and memory (Figure 4A–C). We also applied a mixture (MIX) of both VP and SR49059 to SAE rats. The results showed no obvious amelioration of the learning and memory deficit (Figure 4D–F). To rule out the possibility that the effect of AAV-shRNA, SR49059, or anti-VP serum was achieved through changing the expression of VP, we also detected the level of VP signaling in the hippocampus after treatment with shRNA, VP serum, and SR49059. We found no change of VP signaling (vs the SAE-EE group. See Supplemental Figure B).
Collectively, these findings suggested that EE increases the delivery of VP to the hippocampus and this increase in VP, via binding to V1aR, is a key factor responsible for the EE-induced improvement in learning and memory in SAE rats.
VP Mediates the EE-Induced Alleviation of Neuroinflammation in the Hippocampus
We have shown that VP contributes to the EE-induced improvement of learning and memory after SAE. In this section, we examined the underlying mechanism.
In the hippocampus, accumulating evidence revealed that inflammatory mediators play significant roles in the impairment of learning and memory.48–51 Accordingly, we examined the levels of inflammatory mediators in the hippocampus. For the rats in the SAE group, the levels of IL-1β, IL-6, and IL-10 increased in the hippocampus (Sham vs SAE group: IL-1β: p < 0.0001, IL-6: p < 0.0001, IL-10: p < 0.0001; Figure 5A–C). By contrast, in the SAE-EE group, there was no change in IL-1β or IL-6 levels; however, the IL-10 level was further increased compared with that in the SAE group (SAE vs SAE-EE group: IL-1β: p < 0.0001, IL-6: p < 0.0001; IL-10: p = 0.0317; Figure 5A–C), which indicated that EE counteracted the SAE-induced pro-inflammatory profile towards an anti-inflammatory profile in the hippocampus. Treatment with the shRNA for V1ar, SR49059, or anti-VP serum partly blocked the effects of EE on the IL-1β, IL-6, and IL-10 levels (SAE-EE vs SAE-EE-shRNA group: IL-1β: p = 0.021, IL-6: p = 0.0218; IL-10: p = 0.0390; SAE-EE vs SAE-EE-anti-VP serum group: IL-1β: p = 0.0004, IL-6: p = 0.0011; IL-10: p = 0.0478; SAE-EE vs SAE-EE-SR49059 group: IL-1β: p=0.0019, IL-6: p=0.0010; IL-10: p = 0.0390; Figure 5A–C). Additionally, microinjection of VP into the hippocampus of SAE rats ameliorated the SAE-induced changes in IL-1β, IL-6, and IL-10 levels (SAE vs SAE-VP group: IL-1β: p = 0.0008, IL-6: p = 0.0001; IL-10: p = 0.0256; Figure 5A–C). As expected, a mixture of VP and SR49059 had no effect on the IL-1β, IL-6, and IL-10 levels in the hippocampus of SAE rats (SAE vs SAE-MIX group: IL-1β: p = 0.9067, IL-6: p = 0.9949; IL-10: p = 1.000; Figure 5A–C).
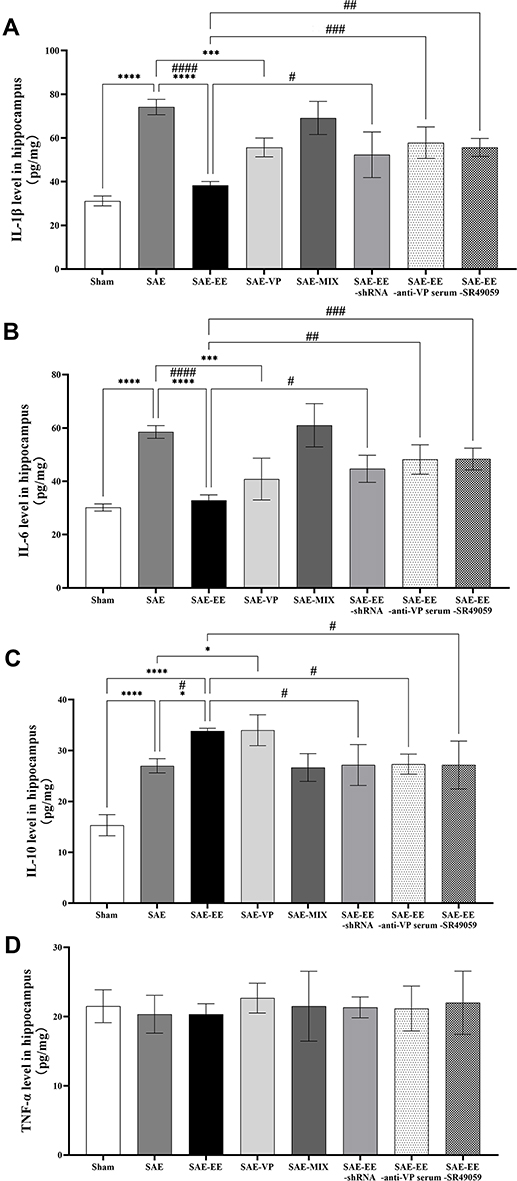 |
Figure 5 (A) The hippocampal IL-1β level in the SAE group was higher than that in the sham, SAE-EE group, or SAE-VP groups. No difference existed between the SAE group and SAE-MIX group. The hippocampal IL-1β level in the SAE-EE group was lower than that in the SAE-EE-anti-VP serum, SAE-EE-shRNA, or SAE-EE-SR49059 groups. (B) The hippocampal IL-6 level in the SAE group was higher than that in the Sham, SAE-EE, or SAE-VP groups. No difference existed between the SAE group and SAE-MIX group. The hippocampal IL-6 level in the SAE-EE group was lower than that in the SAE-EE-anti-VP serum, SAE-EE-shRNA, or SAE-EE-SR49059 groups. (C) The hippocampal IL-10 level in the SAE group was higher than that in the sham group, but lower than that in the SAE-EE group. No difference existed between the SAE group and SAE-MIX group. The hippocampal IL-10 level in the SAE-EE group was higher than that in the SAE-EE-anti-VP serum, SAE-EE-shRNA, or SAE-EE-SR49059 groups. (D) There was no difference of hippocampal TNF-α levels among these groups. vs SAE: *p < 0.05, ***p < 0.001,****p < 0.0001; vs SAE-EE: #p < 0.05, ##p < 0.01, ###p < 0.001,####p < 0.0001. Data represent the means ± standard error of the mean. Abbreviations: EE, environmental enrichment; VP, vasopressin; SAE, sepsis-associated encephalopathy; MIX, a mixture of VP and SR49059; shRNA, short hairpin RNA. |
However, we also found no difference in the TNF-α levels among these groups (F(7, 40) = 0.1888, p = 0.9862; Figure 5D), which suggested that EE or SAE had no influence on TNF-α levels after 30 days of housing.
In addition, we detected the levels of inflammatory mediators in serum. Unlike in the hippocampus, no differences were found in the levels of inflammatory cytokines among the three groups (IL-1β: F(7, 40) = 0.3393, p = 0.9310; IL-6: F(7, 40) = 1.450, p = 0.2131; IL-10: F(7, 40) =0.3322, p = 0.9346; TNF-α: F(7, 40) = 0.5661, p = 0.7788; Figure 6A–D).
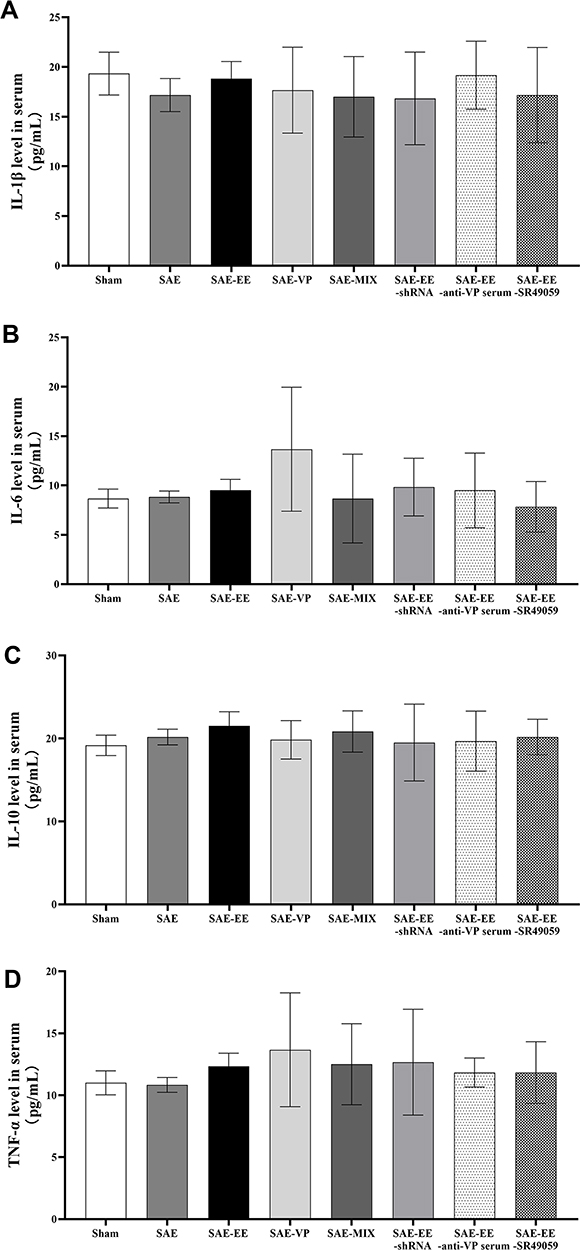 |
Figure 6 (A) The IL-1β level showed no differences in serum samples among the groups. (B) The IL-6 level showed no differences in serum samples among the groups. (C) The IL-10 level showed no differences in serum samples among the groups. (D) The TNF-α level showed no differences in serum samples among the groups. Data represent the means ± standard error of the mean. Abbreviations: EE, environmental enrichment; VP, vasopressin; SAE, sepsis-associated encephalopathy; MIX, a mixture of VP and SR49059; shRNA, short hairpin RNA. |
Taken together, these results indicated that VP induced by EE mediates the decrease in pro-inflammatory cytokines and an increase of anti-inflammatory cytokines by binding to V1aR.
VP Mediates the EE-Induced Increase in BDNF in the Hippocampus
EE is reported to change the expression of BDNF and BDNF can influence cognitive performance.52,53 Accordingly, we detected changes of BDNF levels in the hippocampus under SAE conditions.
The results revealed that the hippocampal BDNF level in the SAE rats was markedly lower than that in the Sham rats (Sham vs SAE group: p = 0.0138; Figure 7). However, in the SAE-EE group, the BDNF level was elevated to 262.7 ± 9.625 pg/mg, which was higher than the 194.5 ± 8.366 pg/mg of the SAE group (SAE vs SAE-EE group: p = 0.0311; Figure 7). In the SAE-EE-shRNA group, SAE-EE-SR49059 group, and SAE-EE-anti-VP serum group, the increase in the BDNF level was inhibited (SAE-EE vs SAE-EE-shRNA group: p = 0.0291; SAE-EE vs SAE-EE-anti-VP serum group: p = 0.0009; SAE-EE vs SAE-EE-SR49059 group: p = 0.0006; Figure 7). Moreover, microinjection of VP into the hippocampus of SAE rats increased the BDNF level in the hippocampus, while microinjection of a mixture of VP and SR49059 did not have this effect (SAE vs SAE-VP group: p = 0.0198; SAE vs SAE-MIX group: p = 1.000; Figure 7). These results strongly suggested that VP participates in the EE-induced increase in BDNF by binding to V1aR, which might be one of the mechanisms responsible for the EE-induced improvement in learning and memory under SAE conditions.
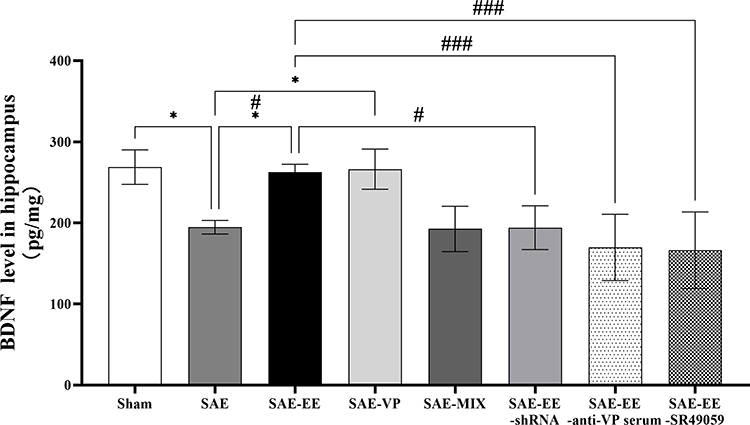 |
Figure 7 The hippocampal BDNF level in the SAE group was lower than that in the sham or SAE-EE group. The hippocampal BDNF level in the SAE-VP group was higher than that in the SAE group. No difference existed between the SAE group and SAE-MIX group. The hippocampal BDNF level in the SAE-EE group was higher than that in the SAE-EE-anti-VP serum, SAE-EE-shRNA, or SAE-EE-SR49059 groups. vs SAE: *p < 0.05, vs SAE: #p < 0.05, ###p <0.001. Data represent the means ± standard error of the mean. Abbreviations: EE, environmental enrichment; BDNF, brain-derived neurotrophic factor; VP, vasopressin; SAE, sepsis-associated encephalopathy; MIX, a mixture of VP and SR49059; shRNA, short hairpin RNA. |
Discussion
As a complication of sepsis, SAE is regarded as the result of neuroinflammation, destruction of blood brain barrier (BBB), and disordered neurotransmission.54,55 For the survivors of SAE, long–term cognitive dysfunction is very common, especially impairment of learning and memory.6 As a result, SAE is always associated with increased health care needs and costs, and increased mortality.
A previous study demonstrated that sepsis could induce the impairment of learning and memory ability.6 In agreement with these results, we showed that after CLP, the rats exhibited impaired learning and memory. EE intervention is a new, simple, and effective treatment that is widely used in rehabilitation for cognitive impairment.56,57 Our results showed that EE was effective to protect against the learning and memory impairment caused by SAE, as evidenced by the performance in the MWM task. This finding was consistent with similar research.70 MWM is used widely to evaluate the learning and memory ability, especially in Alzheimer’s disease research.58 In the MWM test, animals find the platform solely dependent on spatial features and working memory. Therefore, learning and memory ability can be reflected by the latency to the invisible platform. In the present study, we also ruled out the contribution of possible motor deficits to latency by demonstrating no difference in swimming speeds among the groups.
VP is an important neuropeptide that regulates fluid homeostasis and cardiovascular control.59 Recent research found that VP participates in the formation of cognitive functions, such as learning and memory ability, anxiety, addiction, nociception, feeding behavior, and processing of social information.60–63 Past studies have shown that the hippocampus receives projections from the MONs of the SON and VP-expressing fibers can be detected in all subfields of the hippocampus, suggesting the role of VP in learning and memory.63 In this study, we showed that EE increased the VP level in the SON and hippocampus, suggesting that EE enhances the delivery of VP from the SON to the hippocampus. Moreover, the results showed that both blocking V1aR and neutralizing VP abolished the effect of EE. Accordingly, we propose that VP mediates the positive effect of EE on SAE-induced learning and memory deficits by binding to V1aR. In fact, in other animal models, it has been proven that EE enhances hippocampus-dependent learning and memory via multiple mechanisms, such as increased neurogenesis,64,65 changes to dopaminergic and cholinergic systems,66,67 and activation of the vascular endothelial growth factor (VEGF)/kinase insert domain receptor (KDR) system,68 which might also be related to the EE-induced improvement of learning and memory ability under SAE conditions.
The results of the present study showed that: 1. SAE elevated the contents of pro-inflammatory mediators (IL-1β and IL-6) and an anti-inflammatory mediator (IL-10) in the hippocampus; 2. EE could decrease the pro-inflammatory mediator level and further increase the anti-inflammatory mediator level in the hippocampus. Previous studies reported the same results.69,70 However, other research revealed that SAE or EE did not change the IL-10 or IL-1β levels in the hippocampus. This contradiction might have been caused by the different SAE model and different experimental animals used. It has been proven that neuroinflammation is involved in psychological disorders.71,72 The receptor of IL-1β (IL-1R) is predominantly localized in the post-synaptic density on post-synaptic neurons and is tightly bound to the N-methyl D-aspartate (NMDA) receptor subtype 2B (NR2B) subunit of the NMDA receptor (NMDAR).73 Increased IL-1β levels result in the impairment of long-term potentiation (LTP), which is the electrophysiological marker of learning and memory.74 IL-6 is also widely reported to participate in the impairment of learning and memory. Blocking IL-6 led to a prolonged LTP and improved memory in a Y-maze task.75 A previous study further revealed that decreased extracellular regulated kinase 1/2 (ERK1/2) activation correlates with IL-6-induced impairment of LTP.76 IL-10 is the most important anti-inflammatory cytokine, which counteracts the damage caused by excessive inflammation. By acting on the IL-10 receptor in neurons, IL-10 facilitates the LTP via regulation of GABAB synaptic transmission.77 Therefore, our results indicated that EE might attenuate the impairment of learning and memory by changing the levels of inflammatory mediators in the hippocampus.
We also observed that treatment with a V1aR antagonist or anti-VP serum inhibited the EE-induced changes to inflammatory mediators in the hippocampus. These results suggested that VP binding to V1aR mediates the effect of EE on inflammatory mediators in the hippocampus. In the hippocampus, V1aR is mainly expressed in the dentate gyrus.63 Previous studies have shown that enhanced autophagy can be induced when VP binds to V1aR.78 Autophagy effectively regulates the production and release of inflammatory cytokines, such as IL-1β, IL-10, and IL-6,79 which might explain why the V1aR antagonist or anti-VP serum is capable of inhibiting the EE-induced changes to inflammatory mediators in hippocampus.
In this study, we did not observe changes in the content of TNF-α in the hippocampus between the three groups after 30 days of housing. We propose that the TNF-α level has returned to the normal level at this point and is not affected by housing conditions, such as EE. This result is similar to those of certain previous studies, but differs from the response of IL-1β.69,70 This difference might be due to the general responses of the rats to sepsis, such as fever, avoiding food, weight loss, and a second hit after initial recovery. Moreover, the role of TNF-α in learning and memory under inflammatory conditions is likely to be age-dependent.51 We speculated that the age of the rats used in present study might also be responsible for this difference. Thus, the mechanism needs further research. A previous study revealed that 10 days after sepsis induction, no sepsis signals could be observed in the plasma and the contents of inflammatory mediators in the plasma were similar to those of the control group.80 Our results further confirmed these findings by showing that the serum contents of inflammatory mediators showed no differences among the three groups.
BDNF is a widespread neurotrophin in the brain, which is believed to be important for synaptic consolidation and memory consolidation.81,82 The present study found that EE elevated the BDNF content in the hippocampus, which was similar to the results obtained using another animal model.52,53,83 This increased BDNF is responsible for the positive effect of EE on learning and memory. In this study, we found that blocking V1aR or applying anti-VP serum attenuated the EE-induced increase in BDNF. These results indicated that VP binding to V1aR also mediates the EE-induced increase of BDNF under SAE conditions. BDNF expression is regulated by numerous transcription factors, among which the cAMP-response element-binding (CREB) is the most important.84 A previous study demonstrated that the activation of V1aR can lead to CREB activation.85 Accordingly, we speculated that CREB is activated after VP binding to V1aR, which produces more BDNF in the hippocampus. Interestingly, studies also revealed that BDNF can contribute to the increased synthesis of VP.86,87 Combined with the results of the present study, that VP mediates the EE-induced increase in BDNF in the hippocampus under SAE conditions, we speculated that a mutual promotion mechanism might exist between BDNF and VP. This requires further investigation.
This study had some limitations. 1. Although we assessed inflammatory mediators, we did not detect oxidative stress biomarkers, which were also reported to be involved in SAE-induced learning and memory impairment.88,89 2. In this study, we assessed the learning and memory ability, VP level, cytokine levels, and BDNF levels at a single time point (ie, after 30 days of housing). Moreover, we lacked baseline data of the rats’ learning and memory abilities, which would decrease the comparability of the rats and increase the possibility of error. In a future study, we will measure the above indices at several time points to determine the temporal effect of EE on SAE rats. 3. In the present study, a sham-EE group should have been included. Nonetheless, the primary aim of our study was to identify the role of VP in the EE-induced learning and memory improvement under SAE conditions. We believe the evidence gained using the present experimental design is sufficient to determine the role of VP in this process. In fact, previous research showed that there were no differences in the outcomes of MWM or elevated plus maze (EPM) between the sham-EE group and sham group.13,69,70 Moreover, studies also revealed no differences in the level of BDNF and inflammatory factors between the sham-EE group and sham group.69,70,90 Therefore, we speculated that a sham-EE group might show the same results as the Sham group when compared with the other experimental groups. 4. In the present study, young (60 days old) rats were used. However, most SAE patients are least middle-aged and are typically much older. Although EE has a positive effect on rats of all ages, we believe the EE might have more benefits for young rats. In addition, VP is known to mediate social cognition and behavior in a sex-dependent manner.91 Therefore, the fact that we only used male rats in the present study might also have had some influence on the outcome. 5. In this study, we did not detect the level of endotoxins to reflect the level of infection. Endotoxins can stimulate the hypothalamic–pituitary–adrenal (HPA) axis and the ensuing hippocampal circuit is triggered.92 Therefore, we cannot rule out the influence of infection itself.
Sepsis-induced brain injury is another endpoint of sepsis-induced brain effects. The effect of EE or VP on sepsis-induced brain injury requires further study. Single Molecular Array (SIMOA) can be used to measure certain biomarkers of sepsis-induced brain injury (eg neurofilament proteins) to reveal the underlying mechanisms.
We believe that the present study reinforces the importance of EE intervention for the recovery of SAE. However, EE as a rehabilitation method is rarely applied in the cognitive rehabilitation of patients with SAE in clinical practice. We hypothesized that the lack of large, randomized controlled, prospective studies is the main reason. Moreover, considering that the present study demonstrated the role of VP in EE-induced alleviation of learning and memory deficits caused by SAE, it is possible that VP might be the next therapeutic drug to treat the sequela of SAE. Enhancing the synthesis of endogenous VP or supplementing with exogenous VP might be an alternative approach.
Conclusions
The present study showed that EE housing effectively ameliorated the learning and memory impairment caused by SAE, and this process is mediated by VP binding to V1aR. Moreover, VP binding to V1aR causes a decrease in pro-inflammatory cytokines, and an increase in anti-inflammatory cytokines and BDNF, ultimately resulting in learning and memory improvement. Therefore, these results support the hypothesis that VP plays a key role in the EE-mediated alleviation of learning and memory deficits caused by SAE.
Institutional Review Board Statement
The study was approved by the committee of Animal Use for Research and Education in China-Japan Friendship Hospital in Beijing (protocol code: zyhhyy61210324, date of approval: 8 January 2021).
Acknowledgments
We thank the staffs at the Clinical Research Institute of China-Japan Friendship Hospital and Trauma Research Center of Fourth Medical Center of the Chinese PLA General Hospital for providing experimental space, facilities, and technical services.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Natural Science Foundation of Beijing Municipality (CN) (Grant No.7212102), National Natural Science Foundation of China (CN) (Grant No.81201514 and Grant No.81472168) and Elite Medical Professionals project of China-Japan Friendship Hospital (Grant No.ZRJY2021-BJ04).
Disclosure
All five authors declare no conflict of interest.
References
1. Rello J, Valenzuela-Sánchez F, Ruiz-Rodriguez M, Moyano S. Sepsis: a review of advances in management. Adv Ther 2017;34:2393–2411. doi:10.1007/s12325-017-0622-8
2. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi:10.1097/00003246-200107000-00002
3. McBride MA, Patil TK, Bohannon JK, Hernandez A, Sherwood ER, Patil NK. Immune checkpoints: novel therapeutic targets to attenuate sepsis-induced immunosuppression. Front Immunol. 2021;3(11):624272. doi:10.3389/fimmu.2020.624272
4. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol 2012;8:557–566. doi:10.1038/nrneurol.2012.183
5. Imamura Y, Wang H, Matsumoto N, et al. Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience. 2011;187:63–69. doi:10.1016/j.neuroscience.2011.04.063
6. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–1794. doi:10.1001/jama.2010.1553
7. Semmler A, Widmann CN, Okulla T, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84:62–69. doi:10.1136/jnnp-2012-302883
8. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA. 2014;311:1308–1316. doi:10.1001/jama.2014.2637
9. Iwashyna TJ, Speelmon EC. Advancing a third revolution in critical care. Am J Respir Crit Care Med. 2016;194:782–783. doi:10.1164/rccm.201603-0619ED
10. Prescott HC. Variation in postsepsis readmission patterns: a cohort study of veterans affairs beneficiaries. Ann Am Thorac Soc. 2017;14:230–237. doi:10.1513/AnnalsATS.201605-398OC
11. Van, Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1:191–198. doi:10.1038/35044558
12. Rojas-Carvajal M, Sequeira-Cordero A, Brenes JC. The environmental enrichment model revisited: a translatable paradigm to study the stress of our modern lifestyle. Eur J Neurosci. 2021. doi:10.1111/ejn.15160
13. Alwis DS, Yan EB, Johnstone V, et al. Environmental enrichment attenuates traumatic brain injury: induced neuronal hyperexcitability in supragranular layers of sensory cortex. J Neurotrauma. 2016;33:1084–1101. doi:10.1089/neu.2014.3774
14. Redell JB, Maynard ME, Underwood EL, Vita SM, Dash PK, Kobori N. Traumatic brain injury and hippocampal neurogenesis: functional implications. Exp Neurol. 2020;331:113372. doi:10.1016/j.expneurol.2020.113372
15. Tang BL. Axon regeneration induced by environmental enrichment- epigenetic mechanisms. Neural Regen Res. 2020;15:10–15. doi:10.4103/1673-5374.264440
16. Gaulke LJ, Horner PJ, Fink AJ, McNamara CL, Hicks RR. Environmental enrichment increases progenitor cell survival in the dentate gyrus following lateral fluid percussion injury. Brain Res Mol Brain Res. 2005;141:138–150. doi:10.1016/j.molbrainres.2005.08.011
17. Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963–970. doi:10.1038/35039559
18. Gonçalves LV, Herlinger AL, Ferreira TAA, Coitinho JB, Pires RGW, Martins-Silva C. Environmental enrichment cognitive neuroprotection in an experimental model of cerebral ischemia: biochemical and molecular aspects. Behav Brain Res. 2018;348:171–183. doi:10.1016/j.bbr.2018.04.023
19. Zhang X, Yuan M, Yang S, et al. Enriched environment improves post-stroke cognitive impairment and inhibits neuroinflammation and oxidative stress by activating Nrf2-ARE pathway. Int J Neurosci. 2021;131:641–649. doi:10.1080/00207454.2020.1797722
20. Mestriner RG, Saur L, Bagatini PB, et al. Astrocyte morphology after ischemic and hemorrhagic experimental stroke has no influence on the different recovery patterns. Behav Brain Res. 2015;278:257–261. doi:10.1016/j.bbr.2014.10.005
21. Rodríguez-Ortega E, de la Fuente L, de Amo E, Cubero I. Environmental enrichment during adolescence acts as a protective and therapeutic tool for ethanol binge-drinking, anxiety-like, novelty seeking and compulsive-like behaviors in C57BL/6J mice during adulthood. Front Behav Neurosci. 2018;12:177. doi:10.3389/fnbeh.2018.00177
22. Rico-Barrio I, Peñasco S, Puente N, et al. Cognitive and neurobehavioral benefits of an enriched environment on young adult mice after chronic ethanol consumption during adolescence. Addict Biol. 2019;24:969–980. doi:10.1111/adb.12667
23. Diaz R, Miguel PM, Deniz BF, et al. Environmental enrichment attenuates the blood brain barrier dysfunction induced by the neonatal hypoxia-ischemia. Int J Dev Neurosci. 2016;53:35–45. doi:10.1016/j.ijdevneu.2016.06.006
24. Schuch CP, Diaz R, Deckmann I, Rojas JJ, Deniz BF, Pereira LO. Early environmental enrichment affects neurobehavioral development and prevents brain damage in rats submitted to neonatal hypoxia-ischemia. Neurosci Lett. 2016;617:101–107. doi:10.1016/j.neulet.2016.02.015
25. Galeano P, Blanco E, Logica Tornatore TM, et al. Life-long environmental enrichment counteracts spatial learning, reference and working memory deficits in middle-aged rats subjected to perinatal asphyxia. Front Behav Neurosci. 2015;8:406. doi:10.3389/fnbeh.2014.00406
26. Yuan H, Gao B, Duan L, et al. Acute hyperosmotic stimulus-induced Fos expression in neurons depends on activation of astrocytes in the supraoptic nucleus of rats. J Neurosci Res. 2010;88:1364–1373. doi:10.1002/jnr.22297
27. Brownstein MJ, Russell JT, Gainer H. Synthesis, transport, and release of posterior pituitary hormones. Science. 1980;207:373–378. doi:10.1126/science.6153132
28. Van Dessel P, Gawronski B, De Houwer J. Does explaining social behavior require multiple memory systems?. Trends Cogn Sci. 2019;23:368–369.
29. Alescio-Lautier B, Paban V, Soumireu-Mourat B. Neuromodulation of memory in the hippocampus by vasopressin. Eur J Pharmacol. 2000;405:63–72. doi:10.1016/S0014-2999(00)00542-2
30. Zhang L, Hernández VS. Synaptic innervation to rat hippocampus by vasopressin-immuno-positive fibres from the hypothalamic supraoptic and paraventricular nuclei. Neuroscience. 2013;228:139–162. doi:10.1016/j.neuroscience.2012.10.010
31. Mazeraud A, Pascal Q, Verdonk F, Heming N, Chrétien F, Sharshar T. Neuroanatomy and physiology of brain dysfunction in sepsis. Clin Chest Med. 2016;37:333–345. doi:10.1016/j.ccm.2016.01.013
32. Siami S, Bailly-Salin J, Polito A, et al. Osmoregulation of vasopressin secretion is altered in the postacute phase of septic shock. Crit Care Med. 2010;38:1962–1969. doi:10.1097/CCM.0b013e3181eb9acf
33. Sayiner FD, Öztürk DM, Ulupinar E, Velipasaoglu M, Corumlu EP. Stress caused by environmental effects on the birth process and some of the labor hormones at rats: ideal birth environment and hormones. J Matern Fetal Neonatal Med. 2021;34:2600–2608. doi:10.1080/14767058.2019.1670162
34. Sun X, Zhou R, Lei Y, Hu J, Li X. The ligand-gated ion channel P2X7 receptor mediates NLRP3/caspase-1-mediated pyroptosis in cerebral cortical neurons of juvenile rats with sepsis. Brain Res. 2020;1748:147109. doi:10.1016/j.brainres.2020.147109
35. Rocha M, Vieira A, Michels M, et al. Effects of S100B neutralization on the long-term cognitive impairment and neuroinflammatory response in an animal model of sepsis. Neurochem Int. 2021;142:104906. doi:10.1016/j.neuint.2020.104906
36. Briones TL, Klintsova AY, Greenough WT. Stability of synaptic plasticity in the adult rat visual cortex induced by complex environment exposure. Brain Res. 2004;1018:130–135. doi:10.1016/j.brainres.2004.06.001
37. Lippert-Gruener M, Maegele M, Garbe J, Angelov DN. Late effects of enriched environment (EE) plus multimodal early onset stimulation (MEOS) after traumatic brain injury in rats: ongoing improvement of neuromotor function despite sustained volume of the CNS lesion. Exp Neurol. 2007;203:82–94. doi:10.1016/j.expneurol.2006.07.025
38. Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi:10.1016/0165-0270(84)90007-4
39. Tucker LB, Velosky AG, McCabe JT. Applications of the Morris water maze in translational traumatic brain injury research. Neurosci Biobehav Rev. 2018;88:187–200. doi:10.1016/j.neubiorev.2018.03.010
40. Ross HE, Freeman SM, Spiegel LL, Ren X, Terwilliger EF, Young LJ. Variation in oxytocin receptor density in the nucleus accumbens has differential effects on affiliative behaviors in monogamous and polygamous voles. J Neurosci. 2009;29:1312–1318. doi:10.1523/JNEUROSCI.5039-08.2009
41. Serradeil-le Gal C, Wagnon J, Garcia C, et al. Biochemical and pharmacological properties of SR 49059, a new, potent, nonpeptide antagonist of rat and human vasopressin V1a receptors. J Clin Invest. 1993;92:224–231. doi:10.1172/JCI116554
42. Jiang S, Wang YQ, Xu CF, Li YN, Guo R, Li L. Involvement of connexin43 in the infrasonic noise-induced glutamate release by cultured astrocytes. Neurochem Res. 2014;39:833–842. doi:10.1007/s11064-014-1277-3
43. Porterfield VM, Gabella KM, Simmons MA, Johnson JD. Repeated stressor exposure regionally enhances beta-adrenergic receptor-mediated brain IL-1β production. Brain Behav Immun. 2012;26:1249–1255. doi:10.1016/j.bbi.2012.08.001
44. Alescio-Lautier B, Soumireu-Mourat B. Role of vasopressin in learning and memory in the hippocampus. Prog Brain Res. 1998;119:501–521.
45. Xiong Y, Liu R, Xu Y, et al. Effects of vagotomy, splanchnic nerve lesion, and fluorocitrate on the transmission of acute hyperosmotic stress signals to the supraoptic nucleus. J Neurosci Res. 2011;89:256–266. doi:10.1002/jnr.22548
46. Hernández VS, Hernández OR, Perez de la Mora M, et al. Hypothalamic vasopressinergic projections innervate central amygdala GABAergic neurons: implications for anxiety and stress coping. Front Neural Circuits. 2016;10:92. doi:10.3389/fncir.2016.00092
47. Barrett CE, Keebaugh AC, Ahern TH, Bass CE, Terwilliger EF, Young LJ. Variation in vasopressin receptor (Avpr1a) expression creates diversity in behaviors related to monogamy in prairie voles. Horm Behav. 2013;63:518–526. doi:10.1016/j.yhbeh.2013.01.005
48. Kartalou GI, Salgueiro-Pereira AR, Endres T, et al. Anti-inflammatory treatment with FTY720 starting after onset of symptoms reverses synaptic deficits in an AD mouse model. Int J Mol Sci. 2020;21:8957. doi:10.3390/ijms21238957
49. Li J, Cheng XY, Yang H, et al. Matrine ameliorates cognitive deficits via inhibition of microglia mediated neuroinflammation in an Alzheimer’s disease mouse model. Pharmazie. 2020;75:344–347. doi:10.1691/ph.2020.0395
50. Zhou Z, Hou J, Mo Y, et al. Geniposidic acid ameliorates spatial learning and memory deficits and alleviates neuroinflammation via inhibiting HMGB-1 and downregulating TLR4/2 signaling pathway in APP/PS1 mice. Eur J Pharmacol. 2020;869:172857. doi:10.1016/j.ejphar.2019.172857
51. Bourgognon JM, Cavanagh J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci Adv. 2020;4:2398212820979802. doi:10.1177/2398212820979802
52. Novkovic T, Mittmann T, Manahan-Vaughan D. BDNF contributes to the facilitation of hippocampal synaptic plasticity and learning enabled by environmental enrichment. Hippocampus. 2015;25:1–15.
53. Bekinschtein P, Oomen CA, Saksida LM, Bussey TJ. Effects of environmental enrichment and voluntary exercise on neurogenesis, learning and memory, and pattern separation: BDNF as a critical variable?. Semin Cell Dev Biol. 2011;22:536–542.
54. Comim CM, Cassol OJ
55. Adam N, Kandelman S, Mantz J, Chrétien F, Sharshar T. Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther. 2013;11:211–221. doi:10.1586/eri.12.159
56. Yuan M, Guo YS, Han Y, Gao ZK, Shen XY, Bi X. Effectiveness and mechanisms of enriched environment in post-stroke cognitive impairment. Behav Brain Res. 2021;410:113357.
57. Canning CG, Allen NE, Nackaerts E, Paul SS, Nieuwboer A, Gilat M. Virtual reality in research and rehabilitation of gait and balance in Parkinson disease. Nat Rev Neurol. 2020;16:409–425. doi:10.1038/s41582-020-0370-2
58. Terry AV
59. Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch. 2008;456:1005–1024. doi:10.1007/s00424-008-0498-1
60. Caldwell HK, Lee HJ, Macbeth AH, Young WS
61. de Wied D, Diamant M, Fodor M. Central nervous system effects of the neurohypophyseal hormones and related peptides. Front Neuroendocrinol. 1993;14:251–302. doi:10.1006/frne.1993.1009
62. Neumann ID, Landgraf R. Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012;35:649–659. doi:10.1016/j.tins.2012.08.004
63. Cilz NI, Cymerblit-Sabba A, Young WS. Oxytocin and vasopressin in the rodent hippocampus. Genes Brain Behav. 2019;18:e12535. doi:10.1111/gbb.12535
64. Olson AK, Eadie BD, Ernst C, Christie BR. Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus. 2006;16:250–260. doi:10.1002/hipo.20157
65. Segovia G, Yagüe AG, García-Verdugo JM, Mora F. Environmental enrichment promotes neurogenesis and changes the extracellular concentrations of glutamate and GABA in the hippocampus of aged rats. Brain Res Bull. 2006;70:8–14. doi:10.1016/j.brainresbull.2005.11.005
66. Segovia G, Del, Arco A, Mora F. Environmental enrichment, prefrontal cortex, stress, and aging of the brain. J Neural Transm. 2009;116:1007–1016. doi:10.1007/s00702-009-0214-0
67. Hilario WF, Herlinger AL, Areal LB, et al. Cholinergic and dopaminergic alterations in nigrostriatal neurons are involved in environmental enrichment motor protection in a mouse model of parkinson’s disease. J Mol Neurosci. 2016;60:453–464. doi:10.1007/s12031-016-0831-7
68. Cao L, Jiao X, Zuzga DS, et al. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36:827–835. doi:10.1038/ng1395
69. Gong X, Chen Y, Chang J, Huang Y, Cai M, Zhang M. Environmental enrichment reduces adolescent anxiety- and depression-like behaviors of rats subjected to infant nerve injury. J Neuroinflammation. 2018;5:262. doi:10.1186/s12974-018-1301-7
70. Keymoradzadeh A, Hedayati Ch M, Abedinzade M, Gazor R, Rostampour M, Taleghani BK. Enriched environment effect on lipopolysaccharide-induced spatial learning, memory impairment and hippocampal inflammatory cytokine levels in male rats. Behav Brain Res. 2020;394:112814. doi:10.1016/j.bbr.2020.112814
71. Osso LA, Chan JR. Astrocytes underlie neuroinflammatory memory impairment. Cell. 2015;163:1574–1576. doi:10.1016/j.cell.2015.12.001
72. Ji MH, Qiu LL, Tang H, et al. Sepsis-induced selective parvalbumin interneuron phenotype loss and cognitive impairments may be mediated by NADPH oxidase 2 activation in mice. J Neuroinflammation. 2015;12:182. doi:10.1186/s12974-015-0401-x
73. Gardoni F, Boraso M, Zianni E, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J Neuroinflammation. 2011;8:14. doi:10.1186/1742-2094-8-14
74. Hoshino K, Uchinami Y, Uchida Y, Saito H, Morimoto Y. Interleukin-1β modulates synaptic transmission and synaptic plasticity during the acute phase of sepsis in the senescence-accelerated mouse hippocampus. Front Aging Neurosci. 2021;13:637703. doi:10.3389/fnagi.2021.637703
75. Balschun D, Wetzel W, Del Rey A, et al. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi:10.1096/fj.04-1625fje
76. Tancredi V, D’Antuono M, Cafè C, et al. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi:10.1046/j.1471-4159.2000.0750634.x
77. Nenov MN, Malkov AE, Konakov MV, Levin SG. Interleukin-10 and transforming growth factor-β1 facilitate long-term potentiation in CA1 region of hippocampus. Biochem Biophys Res Commun. 2019;518:486–491. doi:10.1016/j.bbrc.2019.08.072
78. Yang C, Zhang X, Gao J, Wang M, Yang Z. Arginine vasopressin ameliorates spatial learning impairments in chronic cerebral hypoperfusion via V1a receptor and autophagy signaling partially. Transl Psychiatry. 2017;7:e1174. doi:10.1038/tp.2017.121
79. Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Mol Immunol. 2017;86:10–15. doi:10.1016/j.molimm.2017.02.013
80. Santos-Junior NN, Catalão CH, Costa LH, et al. Alterations in hypothalamic synaptophysin and death markers may be associated with vasopressin impairment in sepsis survivor rats. J Neuroendocrinol. 2018;e12604. doi:10.1111/jne.12604
81. Panja D, Kenney JW, D’Andrea L, et al. Two-stage translational control of dentate gyrus LTP consolidation is mediated by sustained BDNF-TrkB signaling to MNK. Cell Rep. 2014;9:1430–1445. doi:10.1016/j.celrep.2014.10.016
82. Bambah-Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM. A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J Neurosci. 2014;34:12547–12559. doi:10.1523/JNEUROSCI.0324-14.2014
83. Johansson BB, Zhao L, Mattsson B. Environmental influence on gene expression and recovery from cerebral ischemia. Acta Neurochir Suppl. 1999;73:51–55.
84. Esvald EE, Tuvikene J, Sirp A, Patil S, Bramham CR, Timmusk T. CREB family transcription factors are major mediators of BDNF transcriptional autoregulation in cortical neurons. J Neurosci. 2020;40:1405–1426. doi:10.1523/JNEUROSCI.0367-19.2019
85. Zhao L, Brinton RD. Vasopressin-induced cytoplasmic and nuclear calcium signaling in embryonic cortical astrocytes: dynamics of calcium and calcium-dependent kinase translocation. J Neurosci. 2003;23:4228–4239. doi:10.1523/JNEUROSCI.23-10-04228.2003
86. Balapattabi K, Little JT, Farmer GE, Cunningham JT. High salt loading increases brain derived neurotrophic factor in supraoptic vasopressin neurones. J Neuroendocrinol. 2018;30:e12639. doi:10.1111/jne.12639
87. Choe KY, Han SY, Gaub P, et al. High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron. 2015;23:549–560. doi:10.1016/j.neuron.2014.12.048
88. Giustina AD, de Souza Goldim MP, Danielski LG, et al. Lipoic acid and fish oil combination potentiates neuroinflammation and oxidative stress regulation and prevents cognitive decline of rats after sepsis. Mol Neurobiol. 2020;57:4451–4466. doi:10.1007/s12035-020-02032-y
89. Tian J, Tai Y, Shi M, et al. Atorvastatin relieves cognitive disorder after sepsis through reverting inflammatory cytokines, oxidative stress, and neuronal apoptosis in hippocampus. Cell Mol Neurobiol. 2020;40:521–530. doi:10.1007/s10571-019-00750-z
90. Ji MH, Tang H, Luo D, et al. Environmental conditions differentially affect neurobehavioral outcomes in a mouse model of sepsis-associated encephalopathy. Oncotarget. 2017;8:82376–82389. doi:10.18632/oncotarget.19595
91. Lu Q, Hu S. Sex differences of oxytocin and vasopressin in social behaviors. Handb Clin Neurol. 2021;180:65–88.
92. Salehzadeh M, Hamden JE, Li MX, Bajaj H, Wu RS, Soma KK. Glucocorticoid production in lymphoid organs: acute effects of lipopolysaccharide in neonatal and adult mice. Endocrinology. 2021;bqab244. doi:10.1210/endocr/bqab244
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.