")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Entrectinib and other ALK/TRK inhibitors for the treatment of neuroblastoma
Authors Pacenta HL, Macy ME
Received 22 June 2018
Accepted for publication 21 August 2018
Published 23 October 2018 Volume 2018:12 Pages 3549—3561
DOI https://doi.org/10.2147/DDDT.S147384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Holly L Pacenta, Margaret E Macy
Department of Pediatrics, University of Colorado Anschutz Medical Campus and Children’s Hospital Colorado, Aurora, CO, USA
Abstract: RTK plays important roles in many cellular signaling processes involved in cancer growth and development. ALK, TRKA, TRKB, TRKC, and ROS1 are RTKs involved in several canonical pathways related to oncogenesis. These proteins can be genetically altered in malignancies, leading to receptor activation and constitutive signaling through their respective downstream pathways. Neuroblastoma (NB) is the most common extracranial solid tumor in childhood, and despite intensive therapy, there is a high mortality rate in cases with a high-risk disease. Alterations of ALK and differential expression of TRK proteins are reported in a proportion of NB. Several inhibitors of ALK or TRKA/B/C have been evaluated both preclinically and clinically in the treatment of NB. These agents have had variable success and are not routinely used in the treatment of NB. Entrectinib (RXDX-101) is a pan-ALK, TRKA, TRKB, TRKC, and ROS1 inhibitor with activity against tumors with ALK, NTRK1, NTRK2, NTRK3, and ROS1 alterations in Phase I clinical trials in adults. Entrectinib’s activity against both ALK and TRK proteins suggests a possible role in NB treatment, and it is currently under investigation in both pediatric and adult oncology patients.
Keywords: neuroblastoma, entrectinib, ALK, TRK, ROS1
Introduction
The human genome contains 58 known RTK genes that activate common intracellular signaling cascades and are frequently altered in cancer.1 Different oncologic alterations in RTKs include activating mutations, gain of function mutations, fusion rearrangements, and overexpression. ALK, TRKA, TRKB, TRKC, and ROS1 are RTKs that may be altered or overexpressed in cancer, including neuroblastoma (NB).2–4
NB is the most common extracranial tumor of childhood that develops from progenitors arising from neural crest cells in the adrenal medulla or along the sympathetic chain.5 It accounts for 10% of all pediatric cancers and results in ~15% of pediatric cancer mortality.5,6 NB is known for its clinical heterogeneity, ranging from infants, where the disease spontaneously regresses or matures, to children with a highly aggressive metastatic disease.5 The treatment is risk stratified based on clinical and genetic features associated with outcome. Genetic alterations associated with outcomes include MYCN amplification, DNA ploidy, gain of chromosome 17q, and deletions of chromosome arms 1p or 11q.7–16 The current treatment for high-risk disease uses a multimodal approach incorporating chemotherapy, surgery, radiation therapy, autologous stem cell transplantation, and immunotherapy.5 Despite intensified regimens, ~50% of patients with a high-risk NB relapse or are treatment refractory, demonstrating a critical need for novel therapies to improve cure rates and decrease toxicities.17,18
The genetic landscape of NB has been widely studied, and several genetic aberrations have been identified. MYCN is a transcription factor located at 2p24 and is amplified in 20% of all patients at diagnosis.19,20 MYCN amplification is associated with metastatic disease and a poor prognosis; however, therapeutic inhibition of MYCN has been difficult due to the ubiquitous presence of this transcription factor and the lack of available drug-binding sites.19–21 Targetable genetic alterations such as ALK mutations/amplification are seen in 14% of NB cases.22 Less common alterations are mutations in ATRX, PTPN11, and NRAS genes; each is reported in fewer than 10% of NB cases.22–24 In addition to genetic alterations, there are genes that exhibit differential expression in NB, such as NTRK1/2/3.25 The overall low frequency of mutations combined with difficulty in targeting the more frequently altered genes has resulted in a paucity of molecularly targeted therapeutic options for NB to date.21,22,24,26 However, genes that are differentially expressed, such as NTRK1/2/3, and those that are genetically altered, such as ALK, are potential opportunities for molecularly driven therapy in NB.
ALK is an RTK in the insulin receptor superfamily and is located on chromosome 2. The ligands for ALK are pleiotrophin and midkine; their binding leads to receptor dimerization, autophosphorylation, adaptor protein recruitment, and downstream signal transduction through the RAS/MAPK, PI3K/AKT, and JAK/STAT pathways.27,28 ALK is expressed in both the murine and human nervous systems and not in other tissues.29–32 Studies in murine models identified high levels of ALK in the neonatal brain and low levels of ALK in adults, suggesting that this protein may be important in embryogenesis.29 Constitutive ALK activation through translocation or mutation occurs in multiple malignancies, supporting its role in oncogenesis.3 In fact, the ALK gene was initially discovered in the setting of anaplastic large cell lymphoma (ALCL) where most cases express a t(2;5) translocation, resulting in the fusion of ALK with NPM.33 ALK translocations are present in 50% of inflammatory myofibroblastic tumor (IMT) and in 3%–7% of non-small-cell lung cancer (NSCLC).34–37 ALK-activating mutations and amplification are also described in NB tumors and are more common in patients with a high-risk disease.38
TRK proteins, TRKA, TRKB, and TRKC, are another class of RTKs involved in oncogenesis. The proteins are encoded by NTRK1, NTRK2, and NTRK3, respectively. The ligand for TRKA is NGF; for TRKB is BDNF, NT3, and NT4/5; and for TRKC is NT3. Of note, some ligands like NT3 bind multiple TRK receptors.39–43 Ligand binding results in receptor homodimerization and activation, which lead to signaling through various canonical pathways including RAS/MAPK, AKT, PLCγ1, and PKC.44,45 TRK proteins are expressed in the human central and peripheral nervous system during embryogenesis.46 Studies in animal models have identified that the TRK proteins have different roles and functions, depending on the timing and location of their expression during development. For example, TRKB is expressed in early sensory neuron development, while TRKA in the later stages.47 Similarly, TRKC is expressed early in the development of sympathetic neurons of mouse embryos, while TRKA predominates later in development.48
Alterations in TRK proteins, including rearrangements and atypical expression, are described in a variety of cancers.49–55 Rearrangements of NTRK result in novel fusion proteins, which cause constitutive activation of the kinase. Such fusions are found in a majority of infantile fibrosarcomas but are also described in lung cancer, papillary thyroid carcinoma, glioblastoma, and colorectal carcinomas.49–53,55 Differential expression of TRK has also been reported in a variety of tumors including adrenal, pancreatic, ovarian, esophageal, bladder, pheochromocytoma, and NB.54 TRK expression levels have prognostic significance in some tumors; high levels of TRKB are associated with increased mortality in Wilms tumor, while TRKC expression is associated with a favorable outcome in medulloblastoma.56,57 Differential expression of TRK proteins in NB is also associated with disease severity and prognosis.58
ROS1 is a third RTK with an unknown ligand that thereby limits knowledge of its function.2 This protein is expressed primarily in epithelial cells and is found in a variety of tissues including the kidney, cerebellum, stomach, and intestine.2,59–61 ROS1 translocations leading to increased ROS1 activation have been reported in malignancies and were originally described in glioblastoma where an intrachromosomal deletion leads to the formation of a ROS1–FIG fusion protein.2,60–63 Other cancers where ROS1 translocations have been described include NSCLC, ovarian carcinoma, and cholangiocarcinoma.62,64–66 Of note, ROS1 translocations/alterations have not been reported in NB.67
To date, targeted inhibitors of ALK, TRKA/B/C, and/or ROS1 have shown effectiveness in the treatment of target-mutated malignancies in both preclinical and clinical settings.68–77 Entrectinib (RXDX-101, NMS-E628, NMS-01191372; Ignyta, San Diego, CA, USA) is a newly developed pan-TRK, ALK, and ROS1 inhibitor that has demonstrated preclinical efficacy in tumors with NTRK1/2/3, ALK, and ROS1 alterations, including NB (Figure 1). Entrectinib was well tolerated in Phase I adult clinical trials and demonstrated activity against tumors with NTRK1/2/3, ALK, and ROS1 translocations, providing the support for an ongoing Phase II study in adults.73,78
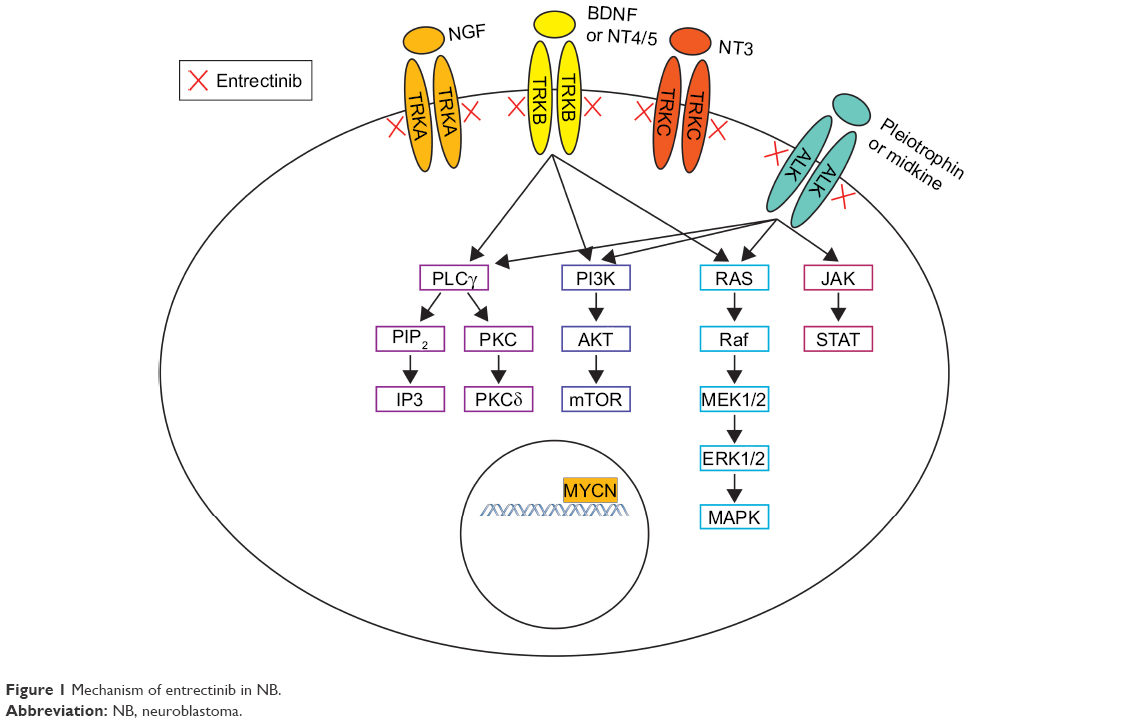 | Figure 1 Mechanism of entrectinib in NB. |
ALK expression and alterations in NB
ALK is recognized as an oncogenic driver of NB; and increased expression of ALK mRNA in NB is correlated with poor prognostic factors such as metastatic disease, MYCN amplification, and decreased survival.79,80 ALK alterations present in NB include copy number gain, amplification, and mutations. ALK copy number gain is seen in 15%–25% of NB, and amplification is seen in 4% of high-risk NB; both are associated with advanced-stage disease and decreased survival.81–85
ALK mutations have been identified in both familial and sporadic NB. ALK germline mutations are reported in 50% of cases of hereditary NB.85,86 These mutations are typically missense mutations within the kinase domain of ALK and lead to ALK hyperphosphorylation and constitutive activation of the kinase.82,84–86 Three different germline mutations have been identified: R1192P, G1128A, and the most frequent R1275Q.85,86 ALK mutations also occur in a small proportion (6%–10%) of somatic NB (Table 1).81–83,85–88 In all, 12 somatic ALK mutations have been identified in NB, the majority of which are missense mutations within the kinase domain.82,83,87–89 The three most common mutations are F1174L, R1275Q, and F1245C.81–83,86–88 Cells transduced with either the F1174L or R1275Q ALK mutation lead to cytokine-independent growth of IL-3-dependent Ba/F3 cells, supporting the role of ALK mutations as oncogenic drivers in NB.88 Of note, the F1174 mutation demonstrates increased oncogenic potential with faster transformation of Ba/F3 cells and stronger auto-phosphorylation compared to R1275Q.83 Similarly, while all ALK mutations in NB are correlated with lower survival rates, those associated with F1174 mutations lead to even worse outcome than those with the R1275Q mutation.81,83,87,90
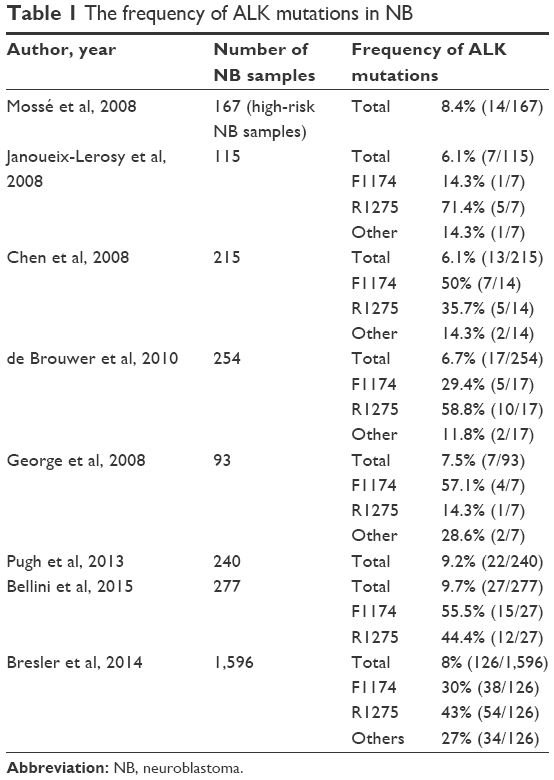 | Table 1 The frequency of ALK mutations in NB |
ALK alterations (amplification or mutations) are more common in cases with MYCN amplification. There is a strong correlation between the F1174L mutation and MYCN amplification in HR NB.81,83–85,89 The association between ALK alterations and MYCN amplification is felt to be due in part to close localization of ALK and MYCN on chromosome 2. MYCN regulates ALK expression, and ALK is a transcriptional target of MYCN.79 Additionally, ALK stimulates transcription of MYCN in NB cell lines, suggesting that the combination of MYCN amplification and ALK alterations may lead to increased oncogenic activity in NB.91
The presence of ALK alterations in NB and the association between both ALK overexpression and ALK alterations with decreased survival and more aggressive disease confirm that ALK is an important driver of NB and a potential therapeutic target.
TRK expression in NB
TRK proteins are differentially expressed in NB and have distinct roles in the pathogenesis of NB.58 TRKA expression is associated with favorable prognostic factors such as localized disease (stages 1, 2, MS), younger age, absence of MYCN amplification, and improved survival.92–94 Additionally, TRKA expression levels are decreased in patients with advanced disease and are inversely associated with MYCN amplification.92–94 When NGF is applied to low-risk NB cells in vitro, which typically have high levels of TRKA, they undergo terminal differentiation, suggesting that TRKA may have a role in the regression or maturation of low-risk NB.95 Similarly, high levels of TRKC in NB are associated with a low-risk disease and favorable prognosis and have a negative correlation with MYCN amplification.96,97 Low-risk NBs are more likely to express the full-length TRKC receptor, and high-risk cases more likely to have truncated TRKC or no TRKC expression at all.96,97 Furthermore, tumors with TRKC also tend to express high levels of TRKA.97
In contrast, TRKB expression is associated with a poor prognosis in NB, present in >50% of high-risk cases and correlates with MYCN amplification.45 TRKB activation leads to enhanced oncogenic potential in NB cells. When BDNF, the TRKB ligand, is applied to MYCN-amplified NB cells, there is improved cell survival and neurite growth.45 TRKB is also associated with the angiogenic factors, VEGF and bFGF, suggesting that it may promote angiogenesis and metastatic ability.25,98,99 Furthermore, TRKB expressing cell lines are less sensitive to doxorubicin, etoposide, and cisplatin, suggesting that TRKB may abrogate response to chemotherapy.100 The association between TRKB/BDNF and cell survival, angiogenesis, metastasis, and drug resistance suggests that TRKB in NB may be a useful therapeutic target.76,101
ALK agents in oncologic development
Preclinical studies
ALK inhibition has been evaluated as a therapeutic option in NB with ALK amplification, mutations, and wild-type ALK. Knockdown of ALK expression in NB cells lines resulted in growth inhibition but was more effective in cells with ALK alterations than in those with wild-type ALK.82,85,86 Although abrogation of ALK was less effective in the wild-type cells, several ALK inhibitors have been tested in models of NB with both wild-type and mutated ALK.
Crizotinib (Xalkori®; Pfizer, Inc., New York City, NY, USA) is a first-generation ALK inhibitor that competitively inhibits the binding of ATP to the active kinase site of ALK, MET, and ROS1.102 Crizotinib was evaluated in both ALK-altered and wild-type NB cell lines and xenografts.69 There was increased growth inhibition in vitro and decreased tumor growth in vivo in the cells with ALK alterations compared to wild-type ALK in response to crizotinib.69 However, there was differential sensitivity to crizotinib based on the type of mutation. NBs with ALK R1275Q mutations were more sensitive to crizotinib, whereas those with ALK F1174L and F1245C exhibited a relative resistance.69,103 The mechanism of relative resistance was due to the different ATP-binding affinities of the ALK mutations. For example, the F1174L ALK mutation demonstrates increased ATP binding compared to the R1275, thereby decreasing the ability of crizotinib to bind leading to the resistant phenotype.81 Crizotinib was also evaluated in combination with chemotherapy. In NB xenograft models with either ALK mutations (R1275Q, F1174L, F1245C) or amplification, crizotinib and cyclophosphamide/topotecan (C/T) in combination resulted in synergistic cytotoxicity and increased apoptosis compared to either agent alone.104 This suggests that crizotinib in conjunction with chemotherapy may be more effective in ALK-altered NB including those with crizotinib-resistant ALK mutations.
Second- and third-generation ALK inhibitors have also been tested in NB. Alectinib (Alecensa®; Chugai Pharmaceutical Co., Tokyo, Japan) is a second-generation ALK/RET inhibitor that has improved affinity for the ATP-binding site and thereby increased potency against the ALK kinase compared to crizotinib.105,106 Alectinib was evaluated in NB cell lines with both wild-type ALK and ALK F1174L mutations.107 Alectinib treatment resulted in growth inhibition in all cell lines, including cells with the F1174L mutations. Additionally, the combination of alectinib and doxorubicin led to enhanced cell death compared to alectinib alone in both ALK wild-type and mutant cell lines.107 While both crizotinib and alectinib demonstrate activity against ALK wild-type and mutant cell lines as a single agent and in combination with chemotherapy, alectinib has improved efficacy compared to crizotinib in the inhibition of the F1174L mutation.
Lorlatinib (PF-6463922; Pfizer, Inc.) is a third-generation ALK/ROS1 inhibitor designed to have improved inhibition of ALK compared to the previous agents.108 Treatment with lorlatinib resulted in decreased growth of ALK-amplified NB cell lines and NB cell lines with the R1275Q, F1174L, and F1245C ALK mutations.103 In xenograft models, complete and sustained tumor regression was seen in all animals with ALK mutations in response to lorlatinib therapy, whereas animals treated with crizotinib exhibited a more limited and transient delay in tumor growth.103 These results suggest that lorlatinib is not only effective against ALK amplifications and mutations in NB in vitro and in vivo but is also more effective than crizotinib.
Preclinical studies suggest that ALK inhibition is effective in NB with ALK alterations and has some activity in NBs with wild-type ALK. Furthermore, there is differential sensitivity of ALK mutations in response to ALK inhibitors with relative resistance of the F1174L and F1245C mutations to crizotinib. The later generation ALK inhibitors are able to overcome this resistance, suggesting that these agents may be effective in individuals with tumors that contain crizotinib-resistant mutations.
Clinical trials
In clinical trials, ALK inhibitors have been widely studied in the treatment of NSCLC and several have been approved for clinical use. Crizotinib was the first of these agents to be approved for the treatment of ALK- and ROS1-rearranged NSCLC.102 However, the clinical utility of crizotinib has been limited by the development of resistance and disease progression.109 Patients with ALK-rearranged tumors who are treated with crizotinib can acquire ALK mutations, such as those of the F1174L, which leads to the development of drug resistance.110 Furthermore, crizotinib does not cross the blood–brain barrier and brain metastases are a common location of disease progression in patients treated with crizotinib.111
The second- and third-generation ALK inhibitors (alectinib, ceritinib [Zykadia®; Novartis International AG, Basel, Switzerland], and lorlatinib) are able to overcome crizotinib resistance in clinical trials and have improved central nervous system (CNS) penetrance.109 While lorlatinib is under investigation in Phase III clinical trial for patients with NSCLC, both ceritinib and alectinib are approved for the treatment of ALK-positive NSCLC.112–114 However, the clinical utility of these agents is also limited by the eventual development of resistance. The mechanisms of acquired resistance include the addition of new mutations within the ALK or ROS1 kinase domain, which prevent the drug from binding to the active site, amplification of ALK itself, and activation of bypass signaling pathways.59,115–121 While the development of resistance remains a limitation of all ALK inhibitors, these agents are commonly used in the treatment of ALK-positive NSCLC and have been studied in pediatric malignancies.
Crizotinib was evaluated in a pediatric Phase I clinical trial that enrolled 79 patients with relapsed/refractory solid tumors, CNS tumors, or ALCL.102 The main side effects were nausea and vomiting, seen in 65% and 57% of patients, respectively, and mild visual disturbances in 37% of patients. Nine patients had a complete response (CR) and five had a partial response (PR). Responses were more common in patients with known ALK aberrations, with eight of nine patients with ALK-translocated ALCL demonstrating an objective response (CR or PR). In all, 34 NB patients were enrolled in this Phase I single-agent trial, 11 with known ALK mutations; of them, one patient had a CR and three patients demonstrated stable disease (SD). The individual with a CR had a germline R1275Q mutation, and the three patients with SD had a germline R1275Q mutation, a somatic mutation at R1275L, and a somatic mutation at F1174L. The other seven patients with ALK mutations had progressive disease, including three patients with F1174L mutations. Among the 23 patients with NB who had an unknown ALK mutation status, one patient had a CR and five had prolonged SD ranging from five to 39 cycles.102 These results suggest activity of crizotinib in NB in a subset of individuals with ALK mutations or unknown ALK status. Additionally, despite prior preclinical and clinical evidence that the F1174 mutation is crizotinib-resistant, there was activity in a patient with a F1174L mutation, suggesting that the resistance is not absolute.
Although there was some efficacy in the Phase I pediatric study, the preclinical evidence suggests that crizotinib may be more effective in combination with chemotherapy. A pediatric Phase I trial evaluated crizotinib in combination with conventional chemotherapy for relapsed or refractory solid tumors or ALCL.122 In this trial, crizotinib was combined with either C/T or vincristine/doxorubicin. Dose-limiting toxicities (DLTs) occurred in both groups and included nausea, diarrhea, dehydration, and prolonged QT in a total of four patients.123 It was suspected that these gastrointestinal (GI) adverse effects may have been related to the poor palatability of the oral solution of crizotinib, and subsequent patients received a capsule formulation.123 Neither the efficacy results nor the individual results of those receiving the capsule formulation are available. Crizotinib in combination with chemotherapy is also being evaluated in the current children’s oncology group high-risk NB study, which includes a cohort for patients with ALK-mutated or -amplified tumors who will receive crizotinib in combination with standard high-risk multiagent chemotherapy.124
The second- and third-generation ALK inhibitors may be more effective than crizotinib and are being studied in pediatrics. There is an open Phase I pediatric trial evaluating the second-generation ALK inhibitor ceritinib in patients with tumors who have ALK alterations.125 An interim report noted that 22 patients enrolled including seven patients with NB with ALK alterations. The adverse events were primarily GI related and included nausea, vomiting, diarrhea, transaminitis, abdominal pain, pyrexia, and fatigue.126 There were two DLTs, which were grade 3 elevation in ALT and grade 2 persistent abdominal pain. Reported responses included two patients with ALCL and four patients with IMT. The results were less favorable in the seven patients with NB, where one patient with an F1174L ALK mutation experienced a mixed response with decrease in the size of a retroperitoneal mass but progression of intracranial disease.126 It is difficult to draw conclusions about the efficacy of ceritinib in NB given the small number of patients. The third-generation ALK inhibitor lorlatinib may be more effective in NB due to increased potency against the ALK kinase and improved CNS penetrance. There is an ongoing Phase I pediatric trial studying lorlatinib in NB, but preliminary results are not yet available.127
TRK agents in oncologic development
Preclinical studies
There are several inhibitors of TRKA/B/C in development for the treatment of TRK-altered malignancies. Inhibition of TRKB in NB is particularly intriguing due to the association between TRKB expression and high-risk disease. Several preclinical studies have evaluated the efficacy of TRK inhibitors in NB models that express TRKB. CEP-751 (KT-6587; Cephalon, Inc., Frazer, PA, USA) is an inhibitor of TRKA/B/C and has activity against PDGFR, EGFR, and PKC.128 CEP-751 was evaluated in NB cells with varying levels of TRKB, both in vitro and in vivo, and was most effective in cells with high levels of TRKB.74,75 AZ64 (AstraZeneca plc, London, UK) and GNF-4256 (Genomics Institute of the Novartis Research Foundation, San Diego, CA, USA) are selective and potent inhibitors of TRKA/B/C; and lestaurtinib (CEP-701; Teva Pharmaceutical, Peta Tikva, Israel) is a potent inhibitor of Flt3 that also has activity against JAK2 and TRKA/B/C.71,76,129–131 Each of these agents was tested in a NB cell line that was transfected with TRKB. In these studies, drug treatment resulted in growth inhibition and decreased phosphorylation of TRKB, suggesting that the effect of the drugs was related to the inhibition of TRKB.71,76,130 When these agents were tested in xenograft models, there was decreased tumor growth and improved survival, suggesting that TRKB inhibition in NB may be beneficial.71,76,130
While multiple TRK inhibitors have demonstrated effectiveness as single agents in preclinical NB, they were also tested in combination with chemotherapy. AZ64 and GNF-4256 were evaluated in combination with irinotecan/temozolomide (I/T) in xenograft models that express TRKB.71,76 In both studies, the TRK inhibitor in combination with I/T led to decreased tumor growth and prolonged survival rates compared to single-agent TRK inhibition.71,76 Similarly, lestaurtinib led to improved antitumor efficacy in xenografts when administered in combination with C/T or with I/T.130 These studies suggest that inhibition of TRKB may be an important adjunct to conventional chemotherapy.
Clinical trials
While there are multiple preclinical reports of effective TRK inhibitors in NB, there are few agents in clinical trials. Neither GNF-4256 nor AZ64 was pursued for further development.67 Lestaurtinib was evaluated in a Phase I NB-specific clinical trial and enrolled 47 patients with refractory high-risk NB.77 The most notable adverse effects included grade 1/2 transaminitis in 63% of patients with dose-limiting transaminitis in four patients and reversible pancreatitis in three patients with prolonged drug exposure. In all, 26 patients were treated at a dose level of at least 70 mg/m2, two of whom experienced a PR and nine had an SD lasting a median of six cycles.77 This study demonstrated a clinical benefit for some of the patients treated at a biologically effective dose. The lack of significant toxicities and the preclinical evidence that lestaurtinib is more effective with chemotherapy provided evidence for trials evaluating lestaurtinib in combination with chemotherapy. Unfortunately, further development of lestaurtinib has been halted.
Larotrectinib (LOXO-101, ARRY-470; Loxo Oncology, Stamford, CT, USA) is a highly selective inhibitor of TRKA/B/C, which was studied preclinically in tumors with NTRK fusions and remains in clinical development.132 Larotrectinib was recently shown to be effective in early phase trials of patients (including children) with solid tumors who had NTRK fusions.72,133 A total of 55 patients with NTRK fusions were enrolled in the combined adult and pediatric trial. The overall response rate was 75%, and seven patients had a CR and 34 had a PR, with 71% of the responses persisting beyond 1 year.72 The pediatric specific Phase I study enrolled patients with and without NTRK fusions. There were 15 evaluable patients with infantile fibrosarcoma, papillary thyroid carcinoma, or other soft tissue sarcomas, which had documented NTRK fusions. Among the NTRK fusion-positive patients, the objective response rate (ORR) was 93% (four CR and 10 PR). The study enrolled seven patients without documented NTRK fusions including one patient with NB, and all developed progressive disease.133 Although there were no responses in the patients without NTRK fusions, the number of fusion-negative patients on the trial was small. The most common drug-related adverse effects were transaminitis, hematologic toxicities, and vomiting.72,133 In the pediatric-specific study, the only DLT was one grade 3 elevation in ALT.133 Additionally, as is seen with other TKIs, 10 patients in the combined adult/pediatric study and one patient in the pediatric study developed acquired resistance through the development of new mutations within the NTRK kinase domain.72,133 Larotrectinib was effective in treating tumors with NTRK fusions, which provided evidence for Phase II clinical trials studying larotrectinib in these patients.134,135 However, its role in tumors without NTRK fusions, such as NB, is unclear due to the small number of patients enrolled.
Entrectinib: a TRKA/B/C, ALK, and ROS1 inhibitor in oncologic development
Preclinical studies
Entrectinib is an ATP-competitive TKI with activity against TRKA, TRKB, TRKC, ALK, and ROS1.68 Entrectinib’s activity is specific to the TRK, ALK, and ROS1 targets. In vitro proliferation profiling of >200 tumor cell lines revealed that entrectinib’s antiproliferative effect was limited to cell lines dependent on entrectinib-specific RTK targets only.68 Entrectinib has 10 to 100 times more potency in inhibiting ROS1 and is seven to eight times more potent against ALK compared to crizotinib, and it is also more potent than lestaurtinib.67,68,136 Entrectinib’s ability to inhibit both ALK and TRKA/B/C may provide a therapeutic advantage over previous agents with specificity to either ALK or TRKA/B/C.
Entrectinib has also been studied in preclinical models of NB. To evaluate its ability to inhibit ALK-driven NB, ALK wild-type, -amplified, or -mutated cell lines were treated with entrectinib.137 Decreased cell proliferation and induction of apoptosis occurred in response to entrectinib as measured by Ki-67 and activation of caspase-3, respectively. The treated cell lines demonstrated decreases in ERK1/2 and STAT3 phosphorylation, supporting that entrectinib’s antiproliferative effects are mediated through downstream inhibition of the ALK signaling pathway. ALK-amplified cells were the most sensitive to entrectinib. Cells with ALK mutations, especially F1174L, were less sensitive to entrectinib due to the induction of autophagy, as measured by microtubule-associated protein 1 LC3. This relative resistance was abrogated when entrectinib was studied in combination with chloroquine, an inhibitor of autophagy. NB cells with F1174L mutations treated with entrectinib and chloroquine had greater growth inhibition compared to either agent alone, suggesting that this combination may lead to improved efficacy of the drug in ALK-mutated tumors.137
NB cell lines with the ALK F1174L mutation, and thereby resistant to entrectinib, were transduced to express TRKB.67 When entrectinib in increasing doses was applied to these transduced cells, there was decreased TRKB phosphorylation and cell viability suggesting that the effect was related to TRKB inhibition. Similarly, in xenograft models with the same ALK F1174 TRKB-transfected cell line, entrectinib therapy led to decreased tumor growth and improved survival compared to animals that were not treated. In the same in vitro model, combination treatment with chemotherapy resulted in increased growth inhibition compared to entrectinib or I/T alone. Furthermore, entrectinib in combination with I/T resulted in decreased tumor growth and improved xenograft survival compared to either regimen alone.67 These studies suggest that TRKB inhibition by entrectinib may provide a therapeutic benefit in NB, particularly when used in combination with chemotherapy.
Clinical trials
Entrectinib has been evaluated in two adult Phase I clinical trials, STARTRK-1 and ALKA-372-001, for patients with refractory or metastatic solid tumors and molecular alterations or rearrangements involving NTRK1, NTRK2, NTRK3, ROS1, or ALK. A total of 119 adult patients with advanced solid tumors were enrolled in these studies: 54 patients in ALKA-372-001 and 65 patients in STARTRK-1. The majority of patients had NSCLC (60%), and 15% had cancers of the GI tract. The drug was well tolerated, and the most common side effects were fatigue, dysgeusia, paresthesias, nausea, and myalgias. The majority of adverse events were grade 1 or 2, and all were reversible with dose modification. At the dose of 800 mg daily, there were three patients with DLTs: grade 3 cognitive disturbances, grade 3 fatigue, and grade 4 eosinophilic myocarditis. All the grade 3 and higher AEs resolved when the drug was held. The maximum tolerated dose was determined to be 600 mg daily and was the recommended Phase II dose (RP2D).73
A sub-analysis was performed on the patients with NTRK1/2/3, ROS1, or ALK fusions who were inhibitor naïve and received the RP2D. Of the 119 patients treated in the Phase I trials, 25 patients met these criteria and were evaluable for disease response. The ORR, those with PR or CR, was 79%. Responses were observed in patients with NSCLC, colorectal cancer, mammary analog secretory carcinoma, melanoma, and renal cell carcinoma.73 When responses were analyzed by the type of fusion, the response rate in each group remained high. Three patients had NTRK1/2/3 rearrangements. This cohort’s ORR was 100%. The 14 patients with ROS1 rearrangements had an ORR of 86% and included two patients with a CR. Patients with ALK-rearranged tumors had an ORR of 57% (n=7). The longest duration of response was 32 months in a patient with ROS1-rearranged lung cancer who remained on therapy at the time of study completion. Entrectinib also demonstrated antitumor activity within the CNS with five of eight (63%) patients with CNS disease having an objective response. This included one patient with a NTRK1 rearrangement who had a CR within the brain and an ongoing response at 15 months.73 These results demonstrate that entrectinib is well tolerated and may be beneficial in inhibitor-naïve patients with ALK, TRKA/B/C, and ROS1 fusions and patients with CNS involvement. However, there are two reports of acquired resistance to entrectinib.138,139 The first was a patient with mammary analog secretory carcinoma who had a NTRK3 fusion and developed a secondary NTRK3 mutation at G623R after treatment with entrectinib.138 The second was a patient with colorectal cancer who had a NTRK1 rearrangement and developed 2 NTRK1 point mutations at G595R and G667C following progression on entrectinib.139
While the responses were favorable in individuals with NTRK1/2/3, ROS1, and ALK translocations who had not previously received therapies targeting those molecular alterations, there were no responses in 25 patients with ROS1 or ALK translocations who had been previously treated with ROS1 or ALK inhibitors.73 This suggests that entrectinib was unable to overcome acquired resistance to other inhibitors and that this agent may be most effective when used upfront. Additionally, there was only one response in the other 59 patients who did not have a fusion. Notably, this response was in a patient with NB with the ALK F1245V mutation. This individual had a PR that lasted 8.3 months and was continued on the drug for 3.5 years due to a clinical benefit. Although only one patient without a fusion protein who had a response, it suggests that entrectinib may have some clinical efficacy against certain ALK mutations in NB.73
As a result of the favorable responses in the Phase I studies, entrectinib was granted US Food and Drug Administration (FDA) orphan drug designation for the treatment of TRKA/B/C, ALK, or ROS1 positive colorectal cancer, NSCLC, and NB.140 The Phase I adult trial also provided evidence for expanding further clinical studies evaluating entrectinib. STARTRK-2 is a global, multicenter, Phase II basket study for patients with NTRK1/2/3-, ROS1-, and ALK- rearranged cancers and is currently enrolling patients.78 There is also an open pediatric Phase I (RXDX-101-03) trial for children with refractory solid tumors and CNS tumors.141 The pediatric study includes both patients with and without NTRK1/2/3, ROS1, and ALK fusions or alterations. In an interim report from this study, there were 16 patients enrolled and 15 were evaluable, including 10 patients with NB, two with IMT, one with salivary gland adenocarcinoma, one with synovial sarcoma, and one with infantile fibrosarcoma.142 There were three DLTs reported. One was a grade 2 increase in creatinine for >7 days in one of the six patients at the 550 mg/m2 dose level. Two of the three patients at the 750 mg/m2 dose level had DLTs, one had grade 2 dysgeusia/fatigue >7 days and the other had grade 3 pulmonary edema. As a result, the RP2D was defined as 550 mg/m2. There were three fusion-positive patients (two IMT and one infantile fibrosarcoma) enrolled to date, and all demonstrated an objective response and continue on protocol therapy. The genetic alterations in the two IMT patients were ALK (DCTN1–ALK) or ROS1 (TFG–ROS1) translocations, and the patient with infantile fibrosarcoma had a TRKC (ETV6–NTRK3) translocation. The responses of the other patients were not reported, although there is one patient with NB who continues on protocol therapy.142 Although the outcome of the other nine NB patients has not been reported, the NB patient with an ongoing response suggests that there may be some efficacy of entrectinib in NB.
Conclusion
Both ALK and TRK play important roles in the pathogenesis of NB and are associated with aggressive disease and decreased survival. Inhibition of either ALK or TRK has been evaluated as a potential treatment for NB, and several agents demonstrated preclinical efficacy. However, relatively few NB patients have been treated with these agents in clinical trials and there has been limited efficacy. Furthermore, the ALK inhibitors tested in NB (crizotinib, ceritinib, alectinib, lorlatinib) do not inhibit TRKA/B/C, and similarly, the TRK inhibitors (CEP-751, AZ64, GNF-4256, lestaurtinib, larotrectinib) do not inhibit ALK (Table 2). Entrectinib is the first TKI that is highly selective for both ALK and TRK A/B/C and has increased potency compared to other ALK and TRK inhibitors and has demonstrated some preclinical efficacy in NB models.
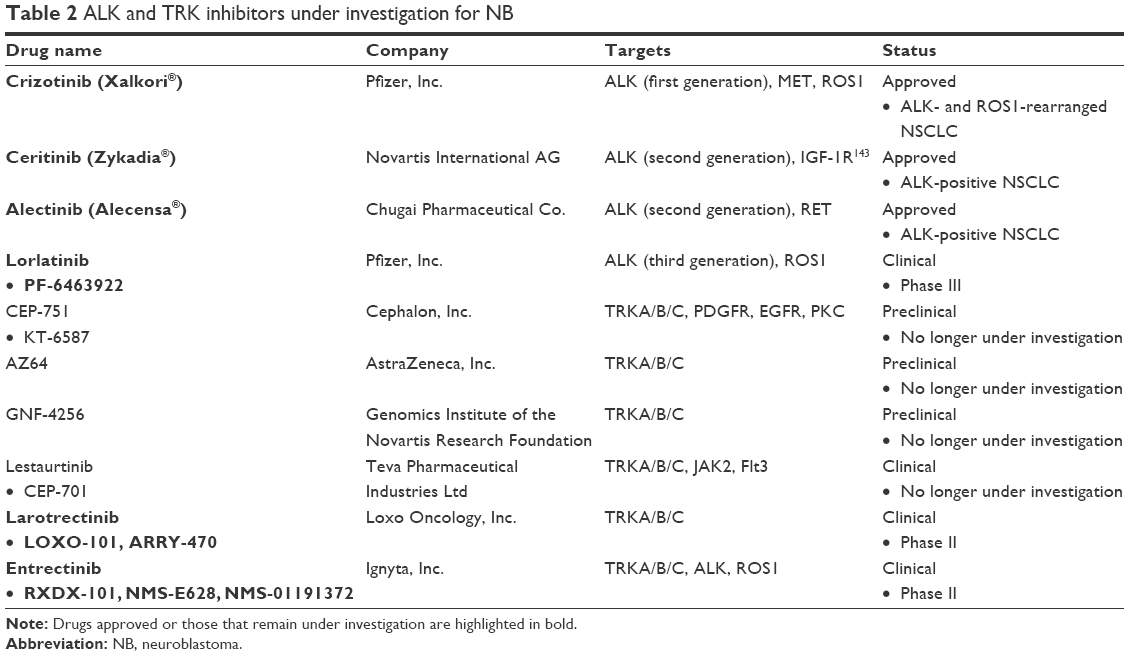 | Table 2 ALK and TRK inhibitors under investigation for NB |
This ability to potently inhibit dual pathways that may be activated in NB suggests entrectinib may have improved efficacy compared to other targeted inhibitors previously evaluated in NB. Treatment with entrectinib in ALK wild-type and ALK-amplified NB cells in vitro resulted in growth inhibition although ALK-mutated cells were generally less sensitive and the F1174L-mutated cells were resistant. The ability of entrectinib to inhibit TRKB in NB was also evaluated. Interestingly, entrectinib was effective in a NB model with the F1174L ALK mutation that also expresses TRKB. This suggests that entrectinib’s ability to inhibit TRKB may be sufficient to overcome resistance due to the F1174L ALK mutation. However, this requires further validation in preclinical studies. TRKB and TRKC expression are also important in the pathogenesis of NB and are seen in individuals with a low-risk disease, but entrectinib has not been studied in this setting.
Early preclinical data suggest that entrectinib may be most effective in combination with other therapies that may incorporate well into the current paradigm of multimodal therapy for high-risk NB. Further clinical trials evaluating entrectinib in combination with either chloroquine or with more standard cytotoxic chemotherapy are needed to confirm the utility of this regimen. If combination therapy proves effective, this could be used to improve outcomes for the 50% of patients who currently do not respond or relapse. Moreover, if the combination of chloroquine and entrectinib is effective, this could be particularly appealing, as it might be able to decrease or limit the use of cytotoxic chemotherapy, which current therapy relies on heavily.
While there is intriguing preclinical evidence for the use of entrectinib in the treatment of NB, particularly in patients with TRK, ALK and ROS1 alterations, the clinical efficacy in NB remains under investigation. As ALK expression/mutations and TRKB expression are associated with a high-risk disease and poor outcomes in NB, this agent is particularly exciting to consider as a potential treatment option. In the published Phase I clinical trials, there were relatively few patients with NB, and there is only one report of an individual with NB who had a PR to entrectinib. The ongoing pediatric phase I trial will provide necessary additional information regarding the efficacy as a single agent in this population. Additional clinical studies are needed, both as a single agent and in combination, to determine whether this is a beneficial and tolerable therapy for NB and which subset of patients is most likely to benefit.
Acknowledgment
Children’s Hospital Colorado (MEM) receives funding from Igntya for clinical trial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. | ||
Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 1795;2009:37–52. | ||
della Corte CM, Viscardi G, di Liello R, et al. Role and targeting of anaplastic lymphoma kinase in cancer. Mol Cancer. 2018;17(1):30. | ||
Khotskaya YB, Holla VR, Farago AF, Mills Shaw KR, Meric-Bernstam F, Hong DS. Targeting TRK family proteins in cancer. Pharmacol Ther. 2017;173:58–66. | ||
Pizzo PA, Poplack DG. Principles and Practice of Pediatric Oncology. 6 ed. Philadelphia: Lippincott Williams & Wilkins; 2016. | ||
Howlader NNA, Krapcho M, Miller D, et al. SEER cancer statistics review, 1975–2014 based on November 2016 SEER data submission, posted to the SEER web site, April 2017. | ||
Attiyeh EF, London WB, Mossé YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353(21):2243–2253. | ||
Bourhis J, Benard J, Devathaire F, et al. Combined analysis of DNA ploidy index and N-myc genomic content in neuroblastoma. Prog Clin Biol Res. 1991;366:107–113. | ||
Bown N, Cotterill S, Łastowska M, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. 1999;340(25):1954–1961. | ||
Bown N, Lastowska M, Cotterill S, et al. 17q gain in neuroblastoma predicts adverse clinical outcome. U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. Med Pediatr Oncol. 2001;36:14–19. | ||
Caron H, van Sluis P, van Hoeve M, et al. Allelic loss of chromosome 1p36 in neuroblastoma is of preferential maternal origin and correlates with N–myc amplification. Nat Genet. 1993;4(2):187–190. | ||
Cohn SL, Pearson ADJ, London WB, et al. The international neuroblastoma risk group (INRG) classification system: an INRG task force report. Journal of Clinical Oncology. 2009;27(2):289–297. | ||
George RE, London WB, Cohn SL, et al. Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: a pediatric oncology group study. Journal of Clinical Oncology. 2005;23(27):6466–6473. | ||
Look AT, Hayes FA, Shuster JJ, et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: a Pediatric Oncology Group study. Journal of Clinical Oncology. 1991;9(4):581–591. | ||
Maris JM, Weiss MJ, Guo C, et al. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a children’s cancer group study. Journal of Clinical Oncology. 2000;18(9):1888–1899. | ||
Schleiermacher G, Mosseri V, London WB, et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer. 2012;107(8):1418–1422. | ||
Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. Journal of Clinical Oncology. 2009;27(7):1007–1013. | ||
Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in risk classification and treatment strategies for neuroblastoma. Journal of Clinical Oncology. 2015;33(27):3008–3017. | ||
Brodeur G, Seeger R, Schwab M, Varmus H, Bishop J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):1121–1124. | ||
Seeger RC, Brodeur GM, Sather H, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313(18):1111–1116. | ||
Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016;122(1):20–33. | ||
Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–284. | ||
Cheung N-KV, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307(10):1062–1071. | ||
Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589–593. | ||
Brodeur GM, Minturn JE, Ho R, et al. Trk receptor expression and inhibition in neuroblastomas. Clinical Cancer Research. 2009;15(10):3244–3250. | ||
Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45(1):12–17. | ||
Stoica GE, Kuo A, Aigner A, et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. Journal of Biological Chemistry. 2001;276(20):16772–16779. | ||
Stoica GE, Kuo A, Powers C, et al. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. Journal of Biological Chemistry. 2002;277(39):35990–35998. | ||
Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):439–449. | ||
Morris SW, Naeve C, Mathew P, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997;14(18):2175–2188. | ||
Pulford K, Lamant L, Morris SW, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. 1997;89:1394–1404. | ||
Vernersson E, Khoo NKS, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expression Patterns. 2006;6(5):448–461. | ||
Morris S, Kirstein M, Valentine M, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. | ||
Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KSJ, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Modern Pathology. 2001;14(6):569–576. | ||
Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59:2776–2780. | ||
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | ||
Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. Journal of Thoracic Oncology. 2009;4(12):1450–1454. | ||
Azarova AM, Gautam G, George RE. Emerging importance of ALK in neuroblastoma. Semin Cancer Biol. 2011;21(4):267–275. | ||
Soppet D, Escandon E, Maragos J, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65(5):895–903. | ||
Klein R, Lamballe F, Bryant S, Barbacid M. The trk B tyrosine protein kinase is a receptor for neurotrophin-4. Neuron. 1992;8(5):947–956. | ||
Lamballe F, Klein R, Barbacid M. trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell. 1991;66(5):967–979. | ||
Kaplan D, Hempstead B, Martin-Zanca D, Chao M, Parada L. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252(5005):554–558. | ||
Klein R, Jing S, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65(1):189–197. | ||
Cunningham ME, Stephens RM, Kaplan DR, Greene LA. Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. Journal of Biological Chemistry. 1997;272(16):10957–10967. | ||
Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14(1):759–767. | ||
Muragaki Y, Timothy N, Leight S, et al. Expression of trk receptors in the developing and adult human central and peripheral nervous system. J Comp Neurol. 1995;356(3):387–397. | ||
Pinon LG, Minichiello L, Klein R, Davies AM. Timing of neuronal death in trkA, trkB and trkC mutant embryos reveals developmental changes in sensory neuron dependence on Trk signalling. Development. 1996;122:3255–3261. | ||
Fagan AM, Zhang H, Landis S, Smeyne RJ, Silos-Santiago I, Barbacid M. TrkA, but not TrkC, receptors are essential for survival of sympathetic neurons in vivo. J Neurosci. 1996;16(19):6208–6218. | ||
Ardini E, Bosotti R, Borgia AL, et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol Oncol. 2014;8(8):1495–1507. | ||
Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149. | ||
Greco A, Pierotti MA, Bongarzone I, Pagliardini S, Lanzi C, della Porta G. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene. 1992;7:237–242. | ||
Kim J, Lee Y, Cho H-J, et al. NTRK1 Fusion in Glioblastoma Multiforme. PLoS One. 2014;9(3):e91940. | ||
Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319(6056):743–748. | ||
Narayanan R, Yepuru M, Coss CC, et al. Discovery and preclinical characterization of novel small molecule TRK and ROS1 tyrosine kinase inhibitors for the treatment of cancer and inflammation. PLoS One. 2013;8(12):e83380. | ||
Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–1472. | ||
Eggert A, Grotzer MA, Ikegaki N, et al. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in wilms’ tumor. Journal of Clinical Oncology. 2001;19(3):689–696. | ||
Rutkowski S, von Bueren A, von Hoff K, et al. Prognostic relevance of clinical and biological risk factors in childhood medulloblastoma: results of patients treated in the prospective multicenter trial HIT’91. Clinical Cancer Research. 2007;13(9):2651–2657. | ||
Brodeur GM, Iyer R, Croucher JL, Zhuang T, Higashi M, Kolla V. Therapeutic targets for neuroblastomas. Expert Opin Ther Targets. 2014;18(3):277–292. | ||
Davies KD, Mahale S, Astling DP, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One. 2013;8(12):e82236. | ||
El-Deeb IM, Yoo KH, Lee SH. ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Med Res Rev. 2011;31:794–818. | ||
Legare C, Sullivan R. Expression and localization of c-ros oncogene along the human excurrent duct. Mol Hum Reprod. 2004;10(9):697–703. | ||
Charest A, Lane K, Mcmahon K, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes, Chromosomes and Cancer. 2003;37(1):58–71. | ||
Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clinical Cancer Research. 2013;19(15):4040–4045. | ||
Bergethon K, Shaw AT, Ignatius Ou S-H, Sh O, et al. ROS1 rearrangements define a unique molecular class of lung cancers. Journal of Clinical Oncology. 2012;30(8):863–870. | ||
Birch AH, Arcand SL, Oros KK, et al. Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One. 2011;6(12):e28250. | ||
Tl G, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6:e15640. | ||
Iyer R, Wehrmann L, Golden RL, et al. Entrectinib is a potent inhibitor of Trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. 2016;372(2):179–186. | ||
Ardini E, Menichincheri M, Banfi P, et al. Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. 2016;15(4):628–639. | ||
Bresler SC, Wood AC, Haglund EA, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3(108):108ra14. | ||
Claeys S, Denecker G, Cannoodt R, et al. Early and late effects of pharmacological ALK inhibition on the neuroblastoma transcriptome. Oncotarget. 2017;8(63):106820–106832. | ||
Croucher JL, Iyer R, Li N, et al. TrkB inhibition by GNF-4256 slows growth and enhances chemotherapeutic efficacy in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2015;75(1):131–141. | ||
Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378(8):731–739. | ||
Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–409. | ||
Evans AE, Kisselbach KD, Liu X, et al. Effect of CEP-751 (KT-6587) on neuroblastoma xenografts expressing TrkB. Med Pediatr Oncol. 2001;36(1):181–184. | ||
Evans AE, Kisselbach KD, Yamashiro DJ, et al. Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clinical cancer research. 1999;5:3594–3602. | ||
Iyer R, Varela CR, Minturn JE, et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2012;70(3):477–486. | ||
Minturn JE, Evans AE, Villablanca JG, et al. Phase I trial of lestaurtinib for children with refractory neuroblastoma: a new approaches to neuroblastoma therapy consortium study. Cancer Chemother Pharmacol. 2011;68(4):1057–1065. | ||
ClinicalTrials.gov [homepage on the Internet]. Basket study of entrectinib (RXDX-101) for the treatment of patients with solid tumors harboring NTRK 1/2/3 (Trk A/B/C), ROS1, or ALK gene rearrangements (Fusions). Available from: https://ClinicalTrials.gov/show/NCT02568267. Accessed April 15, 2018. | ||
Hasan MK, Nafady A, Takatori A, et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci Rep. 2013;3(1):3450. | ||
Passoni L, Longo L, Collini P, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69(18):7338–7346. | ||
Bresler SC, Weiser DA, Huwe PJ, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26(5):682–694. | ||
Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–974. | ||
de Brouwer S, de Preter K, Kumps C, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clinical Cancer Research. 2010;16(17):4353–4362. | ||
Duijkers FAM, Gaal J, Meijerink JPP, et al. Anaplastic lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly correlated with ALK mutation status, ALK mRNA and protein levels. Cellular Oncology. 2011;34(5):409–417. | ||
Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–935. | ||
Janoueix-Lerosey I, Lequin D, Brugières L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455(7215):967–970. | ||
Bellini A, Bernard V, Leroy Q, et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clinical Cancer Research. 2015;21(21):4913–4921. | ||
George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–978. | ||
Chen L, Humphreys A, Turnbull L, et al. Identification of different ALK mutations in a pair of neuroblastoma cell lines established at diagnosis and relapse. Oncotarget. 2016;7(52):87301–87311. | ||
Uryu K, Nishimura R, Kataoka K, et al. Identification of the genetic and clinical characteristics of neuroblastomas using genome-wide analysis. Oncotarget. 2017;8(64):107513–107529. | ||
Schönherr C, Ruuth K, Kamaraj S, et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012;31(50):5193–5200. | ||
Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328(12):847–854. | ||
Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res. 1992;52:1364–1368. | ||
Kogner P, Barbany G, Dominici C, Castello MA, Raschella G, Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53:2044–2050. | ||
Nakagawara A, Brodeur GM. Role of neurotrophins and their receptors in human neuroblastomas: a primary culture study. Eur J Cancer. 1997;33(12):2050–2053. | ||
Rydén M, Sehgal R, Dominici C, Schilling FH, Ibáñez CF, Kogner P. Expression of mRNA for the neurotrophin receptor trkC in neuroblastomas with favourable tumour stage and good prognosis. Br J Cancer. 1996;74(5):773–779. | ||
Yamashiro DJ, Liu X-G, Lee CP, et al. Expression and function of Trk-C in favourable human neuroblastomas. Eur J Cancer. 1997;33(12):2054–2057. | ||
Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cellular and Molecular Life Sciences. 2006;63(7–8):755–759. | ||
Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM. Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Res. 2002;62:1802–1808. | ||
Ho R, Eggert A, Hishiki T, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 2002;62:6462–6466. | ||
Matsumoto K, Wada RK, Yamashiro JM, Kaplan DR, Thiele CJ. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995;55:1798–1806. | ||
Mossé YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–480. | ||
Infarinato NR, Park JH, Krytska K, et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016;6(1):96–107. | ||
Krytska K, Ryles HT, Sano R, et al. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin Cancer Res. 2016;22(4):948–960. | ||
Kodama T, Tsukaguchi T, Satoh Y, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther. 2014;13(12):2910–2918. | ||
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | ||
Lu J, Guan S, Zhao Y, et al. The second-generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH-MYCN transgenic neuroblastoma mouse model. Cancer Lett. 2017;400:61–68. | ||
Basit S, Ashraf Z, Lee K, Latif M. First macrocyclic 3rd-generation ALK inhibitor for treatment of ALK/ROS1 cancer: Clinical and designing strategy update of lorlatinib. Eur J Med Chem. 2017;134:348–356. | ||
Cameron L, Solomon B. Treatment of ALK-rearranged non-small cell lung cancer: recent progress and future directions. Drugs. 2015;75(10):1059–1070. | ||
Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70(24):10038–10043. | ||
Gainor JF, Ou SH, Logan J, Borges LF, Shaw AT. The central nervous system as a sanctuary site in ALK-positive non-small-cell lung cancer. J Thorac Oncol. 2013;8(12):1570–1573. | ||
ClinicalTrials.gov. A study of lorlatinib versus crizotinib in first line treatment of patients with ALK-positive NSCLC. Available from: https://clinicaltrials.gov/ct2/show/NCT03052608. Accessed September 26, 2018. | ||
Food and Drug Administration [homepage on the Internet]. FDA broadens ceritinib indication to previously untreated ALK-positive metastatic NSCLC; 2017. Available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560873.htm. Accessed September 26, 2018. | ||
Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res. 2016;22(21):5171–5176. | ||
Awad MM, Katayama R, Mctigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–2401. | ||
Cargnelutti M, Corso S, Pergolizzi M, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6(7):5182–5194. | ||
Choi YL, Soda M, et al ALK Lung Cancer Study Group, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | ||
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | ||
Drilon A, Somwar R, Wagner JP, et al. A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res. 2016;22(10):2351–2358. | ||
Dziadziuszko R, Le AT, Wrona A, et al. An activating KIT mutation induces crizotinib resistance in ROS1-positive lung cancer. J Thorac Oncol. 2016;11(8):1273–1281. | ||
Song A, Kim TM, Kim DW, et al. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res. 2015;21(10):2379–2387. | ||
National Cancer Institute (NCI) [homepage on the Internet]. Crizotinib and combination chemotherapy in treating younger patients with relapsed or refractory solid tumors or anaplastic large cell lymphoma. Available from: https://ClinicalTrials.gov/show/NCT01606878. Accessed April 13, 2018. | ||
Greengard EG, Mosse YP, Liu X, et al. Safety and tolerability of crizotinib in combination with chemotherapy for relapsed or refractory solid tumors or anaplastic large cell lymphoma: a Children’s Oncology Group phase I consortium study. J Clin Onco. 2015;33 (Suppl 15):10058. | ||
ClinicalTrials.gov [homepage on the Internet]. Testing the addition of 131I-MIBG or crizotinib to intensive therapy in people with high-risk neuroblastoma (NBL). Available from: https://ClinicalTrials.gov/show/NCT03126916. Accessed June 14, 2018. | ||
ClinicalTrials.gov [homepage on the Internet]. Phase I study of LDK378 in pediatric, malignancies with a genetic alteration in anaplastic lymphoma kinase (ALK). Available from: https://ClinicalTrials.gov/show/NCT01742286. Accessed April 29, 2018. | ||
Geoerger B, Schulte J, Zwaan CM, et al. Phase I study of ceritinib in pediatric patients (Pts) with malignancies harboring a genetic alteration in ALK (ALK+): Safety, pharmacokinetic (PK), and efficacy results. Journal of Clinical Oncology. 2015;33:10005. | ||
ClinicalTrials.gov [homepage on the Internet]. Lorlatinib Sof. (PF-06463922). Available from: https://ClinicalTrials.gov/show/NCT03107988. Accessed April 26, 2018. | ||
Camoratto AM, Jani JP, Angeles TS, et al. CEP-751 inhibits TRK receptor tyrosine kinase activity in vitro exhibits anti-tumor activity. Int J Cancer. 1997;72(4):673–679. | ||
Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111(12):5663–5671. | ||
Iyer R, Evans AE, Qi X, et al. Lestaurtinib enhances the antitumor efficacy of chemotherapy in murine xenograft models of neuroblastoma. Clin Cancer Res. 2010;16(5):1478–1485. | ||
Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99(11):3885–3891. | ||
Doebele RC, Davis LE, Vaishnavi A, et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015;5(10):1049–1057. | ||
Laetsch TW, Dubois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–714. | ||
ClinicalTrials.gov. Larotrectinib in Treating Patients With Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphoma, or Histiocytic Disorders With NTRK Fusions (A Pediatric MATCH Treatment Trial). Available from: https://clinicaltrials.gov/ct2/show/NCT03213704. Accessed September 26, 2018. | ||
ClinicalTrials.gov. Study of LOXO-101 (Larotrectinib) in Subjects With NTRK Fusion Positive Solid Tumors (NAVIGATE). Available from: https://clinicaltrials.gov/ct2/show/NCT02576431. Accessed September 26, 2018. | ||
Rolfo C, Ruiz R, Giovannetti E, et al. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert Opin Investig Drugs. 2015;24(11):1493–1500. | ||
Aveic S, Pantile M, Seydel A, et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2016;7(5):5646–5663. | ||
Drilon A, Li G, Dogan S, et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol. 2016;27(5):920–926. | ||
Russo M, Misale S, Wei G, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6(1):36–44. | ||
Ignyta [webpage on the Internet]. Ignyta Receives Orphan Drug Designation and Rare Pediatric Disease Designation from FDA for Entrectinib for the Treatment of Neuroblastoma; 2014. Available from: https://ignyta.com/investors/press-release-article/888962/. Accessed September 26, 2018. | ||
ClinicalTrials.gov [homepage on the Internet]. Study of RXDX-101 in children with recurrent or refractory solid tumors and primary CNS tumors, with or without TRK, ROS1, or ALK fusionsat. Available from: https://ClinicalTrials.gov/show/NCT02650401. Accessed April 12, 2018. | ||
Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. Journal of Clinical Oncology. 2018;36:10536. | ||
Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.