")
Back to Journals » International Journal of Nanomedicine » Volume 15
Enhancing Anti-Tumor Activity of Sorafenib Mesoporous Silica Nanomatrix in Metastatic Breast Tumor and Hepatocellular Carcinoma via the Co-Administration with Flufenamic Acid
Authors Li ZY, Yin YF, Guo Y, Li H, Xu MQ, Liu M, Wang JR, Feng ZH, Duan XC, Zhang S, Zhang SQ, Wang GX, Liao A, Wang SM, Zhang X
Received 28 November 2019
Accepted for publication 21 February 2020
Published 16 March 2020 Volume 2020:15 Pages 1809—1821
DOI https://doi.org/10.2147/IJN.S240436
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Zhuo-Yue Li,1,2 Yi-Fan Yin,1,2 Yang Guo,1,2 Hui Li,1,2 Mei-Qi Xu,1,2 Man Liu,1,2 Jing-Ru Wang,1,2 Zhen-Han Feng,1,2 Xiao-Chuan Duan,1,2 Shuang Zhang,1,2 Shuai-Qiang Zhang,1,2 Guang-Xue Wang,2 Ai Liao,2 Shu-Min Wang,3 Xuan Zhang1,2
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, School of Pharmaceutical Sciences, Peking University, Beijing 100191, People’s Republic of China; 2Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing 100191, People’s Republic of China; 3Department of Ultrasound, Peking University Third Hospital, Peking University, Beijing 100191, People’s Republic of China
Correspondence: Xuan Zhang
Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Xueyuan Road 38, Beijing 100191, People’s Republic of China
Tel/Fax +86-10-82805765
Email [email protected]
Shu-Min Wang
Department of Ultrasound, Peking University Third Hospital, Peking University, Xueyuan Road 38, Beijing 100191, People’s Republic of China
Email [email protected]
Introduction: Because tumor-associated inflammation is a hallmark of cancer treatment, in the present study, sorafenib mesoporous silica nanomatrix (MSNM@SFN) co-administrated with flufenamic acid (FFA, a non-steroidal anti–inflammatory drug (NSAID)) was investigated to enhance the anti-tumor activity of MSNM@SFN.
Methods: Metastatic breast tumor 4T1/luc cells and hepatocellular carcinoma HepG2 cells were selected as cell models. The effects of FFA in vitro on cell migration, PGE2 secretion, and AKR1C1 and AKR1C3 levels in 4T1/luc and HepG2 cells were investigated. The in vivo anti-tumor activity of MSNM@SFN co-administrating with FFA (MSNM@SFN+FFA) was evaluated in a 4T1/luc metastatic tumor model, HepG2 tumor-bearing nude mice model, and HepG2 orthotopic tumor-bearing nude mice model, respectively.
Results: The results indicated that FFA could markedly decrease cell migration, PGE2 secretion, and AKR1C1 and AKR1C3 levels in both 4T1/luc and HepG2 cells. The enhanced anti-tumor activity of MSNM@SFN+FFA compared with that of MSNM@SFN was confirmed in the 4T1/luc metastatic tumor model, HepG2 tumor-bearing nude mice model, and HepG2 orthotopic tumor-bearing nude mice model in vivo, respectively.
Discussion: MSNM@SFN co-administrating with FFA (MSNM@SFN+FFA) developed in this study is an alternative strategy for improving the therapeutic efficacy of MSNM@SFN via co-administration with NSAIDs.
Keywords: sorafenib mesoporous silica nanomatrix, MSNM@SFN, flufenamic acid, FFA, PGE2, AKR1C1, AKR1C3, 4T1/luc, HepG2
Introduction
Tumor-associated inflammation is recognized as an emerging hallmark of cancer treatment.1 Inflammation significantly contributes to tumor progression probably due to the stimulation of prostanoid production.2 Clinical findings have shown that cyclooxygenase-2 (COX-2), a key isoenzyme for the production of prostaglandins (PGs), is overexpressed in many tumors.3,4 COX-2 is also a critical enzyme for synthesizing prostaglandin E2 (PGE2) in cancer cells and promoting PGE2 release.5,6 The released PGE2 in tumor microenvironments can activate existing quiescent cancer stem cells (CSCs) to proliferate5,7 and drive the identity conversion of non-CSCs tumor cells to be CSC-like cells.8 PGE2 can also promote cancer cell dissemination to cell migration and invasion.9 Thus, the COX-2/PGE2 axis can be selected as an anti-tumor target.
Non-steroidal anti–inflammatory drugs (NSAIDs) can be used as potential chemo-preventive agents against tumor cells due to their role in inhibiting the activity of cyclooxygenases (COXs).10 Unfortunately, NSAIDs seem to exercise their anti-cancer adjuvant effects only through the inhibition of inducible COX2.11 Therefore, the combination of NSAIDs with chemotherapeutic drugs may enhance the anti-tumor effects.12,13 The combination effects of NSAIDs with chemotherapeutic drugs were confirmed in many studies.14
Flufenamic acid (FFA) is a non-steroidal anti–inflammatory drug (NSAID). The anti–inflammatory and analgesic effects of FFA were confirmed in the 1960s.15 The anti–inflammatory mechanism of FFA was recognized mainly through the reduction in prostaglandin synthesis from arachidonic acid by inhibiting COXs.16
Sorafenib (SFN), an orally administered multi-kinase inhibitor, was approved by the FDA for the clinical treatment of metastatic renal cell carcinoma and advanced hepatocellular carcinoma.17 The therapeutic effects of SFN in lung, breast, and other cancers were also investigated.18,19 SFN is a Biopharmaceutical Classification System (BCS) class II drug due to its poor solubility characteristics. Many strategies have been used to enhance the solubility of SFN, including SFN-loaded nanoparticles,20,21 solid dispersion,22 liposome,23 polymersomes,24 and inclusion compounds,25 among others. In our previous research, we prepared SFN mesoporous silica nanomatrix (MSNM@SFN) to improve the solubility, dissolution, and bioavailability of SFN.26 The enhanced anti-tumor activity of MSNM@SFN compared with SFN suspension was confirmed in MDA-MB-231 cells in vitro and in vivo.26
Because tumor-associated inflammation is a hallmark of cancer treatment, in the present study, sorafenib mesoporous silica nanomatrix (MSNM@SFN) co-administrated with flufenamic acid (FFA, a non-steroidal anti–inflammatory drug (NSAID)) was investigated to enhance the anti-tumor activity of MSNM@SFN. Metastatic breast tumor 4T1/luc cells and hepatocellular carcinoma HepG2 cells were selected as cell models. The effects of FFA in vitro on cell migration, PGE2 secretion, and Aldo-keto reductases (AKR1C1 and AKR1C3, which catalyze NADPH-dependent reductions and are involved in biosynthesis, intermediary metabolism, and detoxification) levels in 4T1/luc and HepG2 cells were investigated. The in vivo anti-tumor activity of MSNM@SFN co-administrating with FFA (MSNM@SFN+FFA) was evaluated in a 4T1/luc metastatic tumor model, HepG2 tumor-bearing nude mice model, and HepG2 orthotopic tumor-bearing nude mice model, respectively.
Materials and Methods
Materials
SFN was obtained from MERYER Chemical Technology Co. Ltd. (Shanghai, China). FFA was purchased from J&K Scientific Ltd. (Beijing, China). Mesoporous silica (Sylysia 350) was supplied by Fuji Silysia Chemicals (Kasugai, Japan). Hydroxypropyl methylcellulose (HPMC, 7500–14,000 mPa.s) was purchased from Alfa Aesar Chemical Co. Ltd. (Tianjin, China). Distearoylphosphatidylethanolaminepoly (ethylene glycol) 2000 (DSPE-PEG) was supplied by NOF Corporation (Tokyo, Japan). Hoechst 33342 was obtained from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). A One-Step TUNEL Apoptosis Assay Kit was purchased from Beyotime Biotechnology Co., Ltd. (Nanjing, China). Cell culture medium, RPMI 1640, and DMEM medium were obtained from Macgene Biotech Co. Ltd. (Beijing China). Fetal bovine serum (FBS) was supplied from GIBCO (Invitrogen Co., Carlsbad, CA, USA).
Cell Lines
Both the 4T1/luc cells (a murine metastatic breast tumor cell line) and HepG2 cells (a human hepatocellular carcinoma cell line) were obtained from the Cell Resource Center, Peking Union Medial College (Beijing, China). The cells were cultured according to conditions recommended by the American Type Culture Collection (ATCC).
Animals
Female BALB/c mice (18–20 g) and female BALB/C nude mice (18–20 g) were obtained from the Experimental Animal Center of Peking University Health Science Center. All care and handling of the animals were performed with the approval of the Experimental Animal Center of Peking University Health Science Center. Additionally, this study was performed following the National Institutes of Health guidelines for the use of experimental animals.
The Effect of FFA on 4T1/luc and HepG2 Cell Migration
The cell migration assay was performed via a wound healing assay according to our previous research.27,28 A straight line was drawn across the bottom of a 24-well plate. A volume of 1 mL cell suspension with a concentration of 4 × 105 cells/mL was plated into the well and incubated for 24 h before the cells were scraped with a sharp tip vertically by the marked lines to create a cell-free space. The floating cells were washed with PBS three times to remove them. The cells were then incubated with non-serum containing cell culture medium without or with FFA for another 24 h. The width of the cell-free space at 0 h and 24 h was imaged using an EVOS microorganism online observation instrument (EVOS xl, AMG, USA). The distance between the scratches was analyzed using ImageJ software. The migration rate was calculated via the following formula: migration rate (%) = (distance of scratch at 0 h - distance of scratch at 24 h) × 100%/distance of scratch at 0 h.
The Effect of FFA on PGE2 in 4T1/luc and HepG2 Cells
Cells were seeded onto 12-well plates at a density of 4 × 105 cells/well and routinely cultured in l mL of RPMI 1640 medium for 24 h. Then fresh medium containing 2% serum was added and the cells were incubated for an additional 6 h. Subsequently, the cells were pre-stimulated with TNF-α (20 ng/mL) for 2 h. Then FFA, MSNM@SFN, or MSNM@SFN+FFA was added, respectively. After 6 h, the media were collected and the PGE2 level was determined according to the manufacturer’s protocols.
The Effect of FFA on AKR1C1 and AKR1C3 in 4T1/luc and HepG2 Cells
4T1/luc or HepG2 cells (1 × 105 cells/well) were seeded in confocal dishes (covered glass-bottom dishes) and incubated at 37 °C for 24 h. The cells were then treated with FFA (20 μg/mL) at 37 °C for 24 h. The cells were then washed three times with cold phosphate-buffered saline (PBS, pH 7.4). These cells were incubated with primary antibody (rabbit polyclonal to AKR1C1 and ab192785 and rabbit polyclonal to AKR1C3 and ab84327) and secondary antibodies (goat anti-rabbit IgG/Alexa Fluor 488, bs-0295G-AF488) according to the manufacturers’ standard protocols. The nuclei were counterstained with Hoechst 33342. The cells were imaged with a confocal laser scanning microscope to observe the fluorescence signal distribution of AKR1C1 and AKR1C3 in the cells. For the control group, cells that were treated as previously described only without FFA were observed via confocal laser scanning microscopy.
The Anti-Tumor Activity of MSNM@SFN+FFA in a 4T1/luc Metastatic Tumor Model
The 4T1/luc cells (0.2 mL, 1 × 106 cells) were intravenously injected into the tail vein of female BALB/C mice. Four days after 4T1/luc cell injection, the 4T1/luc tumor-bearing mice were randomly divided into three groups (12 animals per group).
We prepared SFN mesoporous silica nanomatrix (MSNM@SFN) at the ratio that SFN:Sylysia:HPMC:DSPE-PEG was 1:3:3:3 (w/w/w/w) as our previous research.26 For preparation of MSNM@SFN+FFA, the MSNM@SFN was mixed with FFA at the ratio of MSNM@SFN:FFA 40:1 (w/w) or SFN:FFA 4:1(w/w). The dose of MSNM@SFN was 40 mg/kg of SFN similar with that in MDA-MB-231 tumor-bearing nude mice model.26 And we selected the dose of FFA at 10 mg/kg according to previously reported references.29,30
Each group of mice was orally administered physiological saline as a control, MSNM@SFN (at a dose of 40 mg/kg SFN), or MSNM@SFN+FFA (at a dose of 40 mg/kg SFN and 10mg/kg FFA) every day for 14 days. For bioluminescent imaging, on days 6, 8, 10, 14, 17, and 20 after the 4T1/luc cell injection, a filter-sterilized solution (0.2 μm) of d-luciferin in PBS (15 mg/mL) was intraperitoneally injected (10 μL/g) 15 min before measuring the luminescence. The mice were then anesthetized with an initial dose of 5% isoflurane and a maintenance dose of 1.5% isoflurane. The mice were then imaged using the IVIS Spectrum (Xenogen, USA), and the bioluminescence signals were quantified by Living Image software 4.3.1. The survival time was calculated from the day of 4T1/luc cell injection (0 day) to the day of death. Kaplan-Meier survival curves were plotted for each group.
The Anti-Tumor Activity of MSNM@SFN+FFA HepG2 Tumor-Bearing Nude Mice Model
Male BALB/C nude mice were subcutaneously inoculated in the right armpits with 0.2 mL of the HepG2 cell suspension (5 × 106 cells/animal). Once the volume of tumors reached approximately 100–150 mm3 on day 14, the mice were randomly assigned to four groups (five animals per group): group 1 was given physiological saline as a control, group 2 was given SFN suspension (40 mg/kg of SFN, oral gavage, once a day for 18 days), group 3 was given MSNM@SFN (40 mg/kg of SFN, oral gavage, once a day for 18 days), and group 4 was given MSNM@SFN+FFA (40 mg/kg of SFN, 10 mg/kg of FFA oral gavage, once a day for 18 days). Throughout the study, the mice were weighed regularly and their tumors were measured using calipers every other day. The tumor volumes were calculated using the following formula: V = length (cm) × width2 (cm2) × 0.5236. On day 34, all the mice were sacrificed, and the tumor tissues were removed. Tumor sections were used for TUNEL straining experiments. TUNEL staining was preformed according to the standard protocols provided by the manufacturers.
The Anti-Tumor Activity of MSNM@SFN+FFA on HepG2 Orthotopic Tumor-Bearing Nude Mice
Male BALB/C nude mice were surgically inoculated in the median liver lobe through a midline abdominal incision with 25 μL of the HepG2 cell suspension (5 × 106 cells/animal). On day 14, the mice were randomly assigned to three groups (3 animals per group): group 1 was given physiological saline as a control, group 2 was given MSNM@SFN (40 mg/kg of SFN, oral gavage, once a day for 18 days), and group 3 was given MSNM@SFN+FFA (40 mg/kg of SFN, 10 mg/kg of FFA oral gavage, once a day for 18 days). On day 34, all of the animals were sacrificed and the livers excised. The tumors were measured using calipers. The tumor volumes were calculated using the following formula: V = length (cm) × width2 (cm2) × 0.5236.
Statistical Analysis
All of the data are shown as the mean ± SD. The significance among groups was determined using a one-way analysis of variance (ANOVA), after which post hoc tests with the Bonferroni correction for comparisons between individual groups were performed. Statistical significance was p < 0.05.
Results
The Effect of FFA on 4T1/luc and HepG2 Cell Migration
The effect of FFA on 4T1/luc or HepG2 cell migration was investigated by a wound healing assay. As shown in Figure 1, the migration of the 4T1/luc cells was significantly inhibited by FFA depending on the concentration. Untreated 4T1/luc migrated into the denuded areas. The calculated migration rate was 60.6%. In the FFA treatment group, few migrated 4T1/luc cells were observed in the denuded areas (Figure 1A). The calculated migration rate was 18.0% at a concentration of 0.39 μg/mL (Table 1). At the highest concentration (3.13 μg/mL), the calculated migration rate was 5.7%. Compared with the untreated 4T1 group as a percentage of control (60.6%), the value of the inhibited ratio in the FFA 0.39 μg/mL and 3.13 μg/mL treatment groups was 70.3% and 90.6%, respectively.
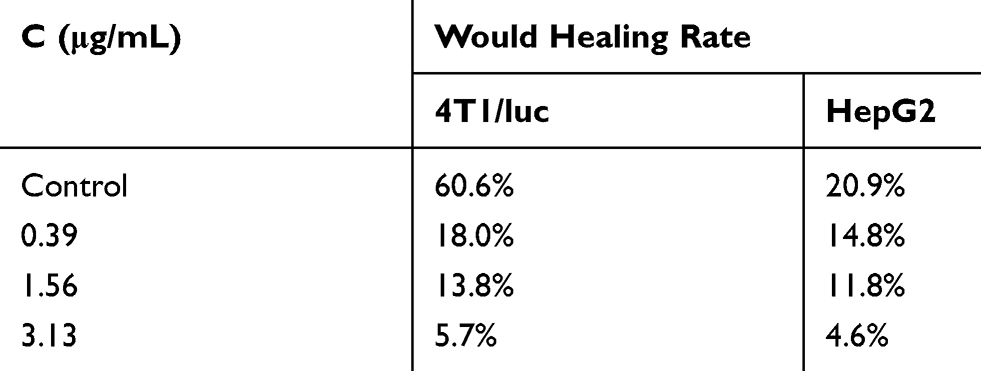 |
Table 1 The Wound Healing Rate in the 4T1/luc and HepG2 Cells After Treatment with FFA at 24 h |
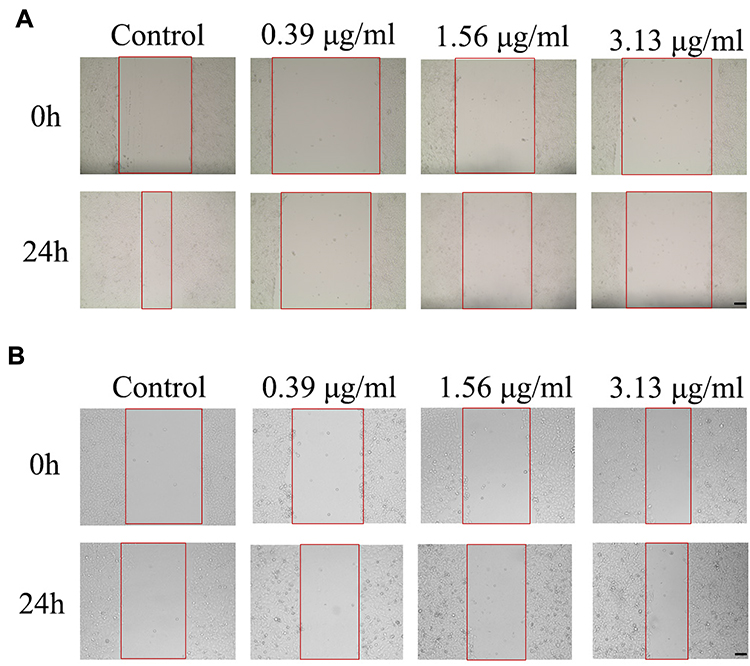 |
Figure 1 In vitro cell migration in the 4T1/luc cells (A) and HepG2 cells (B) after treatment with FFA for 24 h. The red lines were scratches which were scraped with a sharp tip vertically by the marked lines to create a cell-free space. The scale bar is 100 μm. |
Conversely, the amount of untreated HepG2 that migrated into the denuded areas was very low (Figure 1B). The calculated migration rate was only 20.9% (Table 1). In the FFA treatment group, the calculated migration rate was 14.8% at a concentration of 0.39 μg/mL and 4.6% at the highest concentration (3.13 μg/mL). Compared with the untreated HepG2 group as a percentage of control (20.9%), the value of the inhibited ratio in the FFA 0.39 μg/mL and 3.13 μg/mL treatment groups was 29.2% and 78.0%, respectively.
The Effect of FFA on the Secretion of PGE2 in the 4T1/luc and HepG2 Cells
The effect of FFA on the secretion of PGE2 in the 4T1/luc and HepG2 cells was evaluated. As indicated in Figure 2, in the 4T1/luc and HepG2 cells, FFA significantly decreased the level of PGE2 secretion compared to that of control groups (p < 0.01).
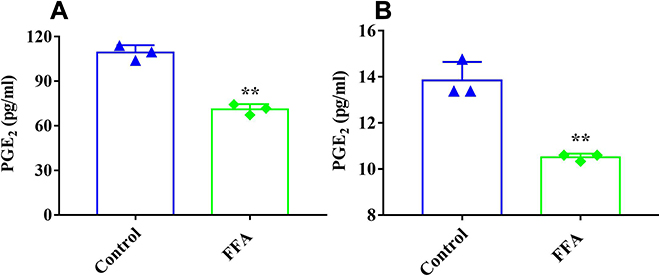 |
Figure 2 PGE2 secretion from 4T1/luc cells (A) and HepG2 cells (B) after treatment with FFA for 6 h. **p < 0.01 vs control treatment group. |
The Effect of FFA on AKR1C1 and AKR1C3 in the 4T1/luc and HepG2 Cells
The effect of FFA on AKR1C1 and AKR1C3 in the 4T1/luc and HepG2 cells was investigated. As shown in Figures 3A and 4A, the immunofluorescence analysis results indicated that the fluorescence of labeling AKR1C1 (green) was present in the 4T1/luc and HepG2 cells, showing that AKR1C1 was in the 4T1/luc and HepG2 cells. FFA significantly decreased the AKR1C1 levels in both the 4T1/luc and HepG2 cells compared with that of the control groups (p < 0.01) (Figures 3 and 4). Similarly, AKR1C3 was in the HepG2 cells (Figure 5A) and FFA also significantly decreased the levels of ARK1C3 in HepG2 cells compared with the control groups (p < 0.01) (Figure 5). We did not investigate the effect of FFA on AKR1C3 in the 4T1/luc because the murine AKR1C3 antibody was not been obtained.
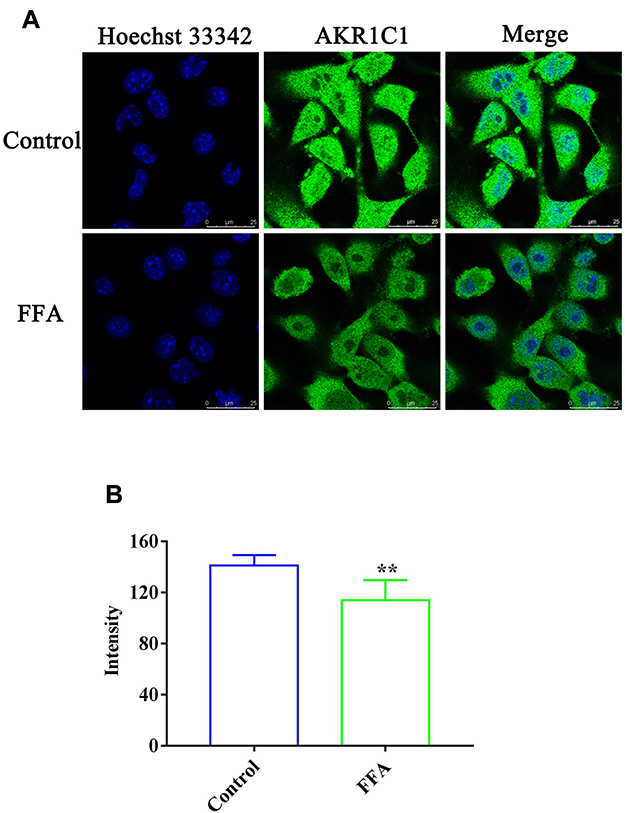 |
Figure 3 The expression of AKR1C1 in the 4T1/luc cells. (A) Confocal microscopy images of 4T1/luc cells incubated with or without FFA for 24 h. Green fluorescence denotes AKR1C1 and blue fluorescence denotes nuclei staining; (B) The fluorescence intensity in the 4T1/luc cells with or without FFA treatment. **p < 0.01 vs control treatment cells. |
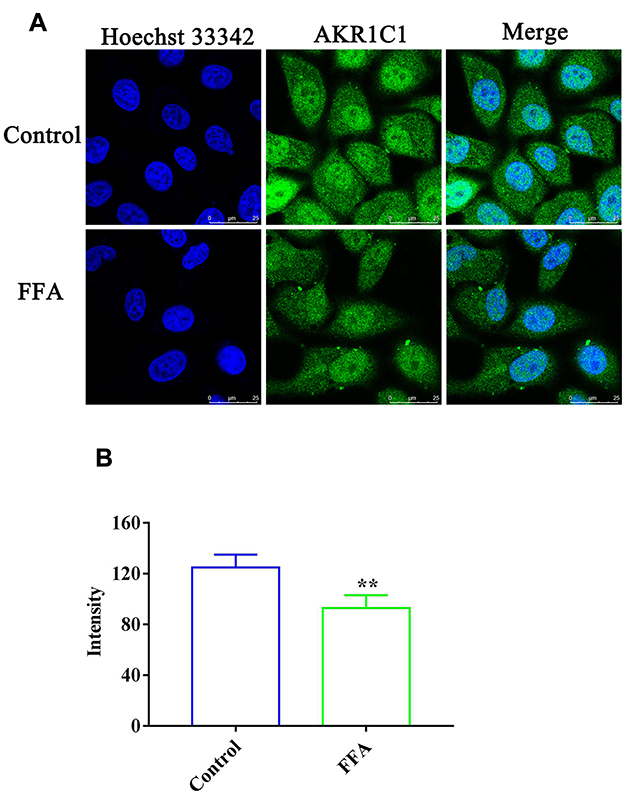 |
Figure 4 The expression of AKR1C1 in the HepG2 cells. (A) Confocal microscopy images of HepG2 cells incubated with or without FFA for 24 h. Green fluorescence denotes AKR1C1 and blue fluorescence denotes nuclei staining; (B) The fluorescence intensity in the HepG2 cells with or without FFA treatment. **p < 0.01 vs control treatment cells. |
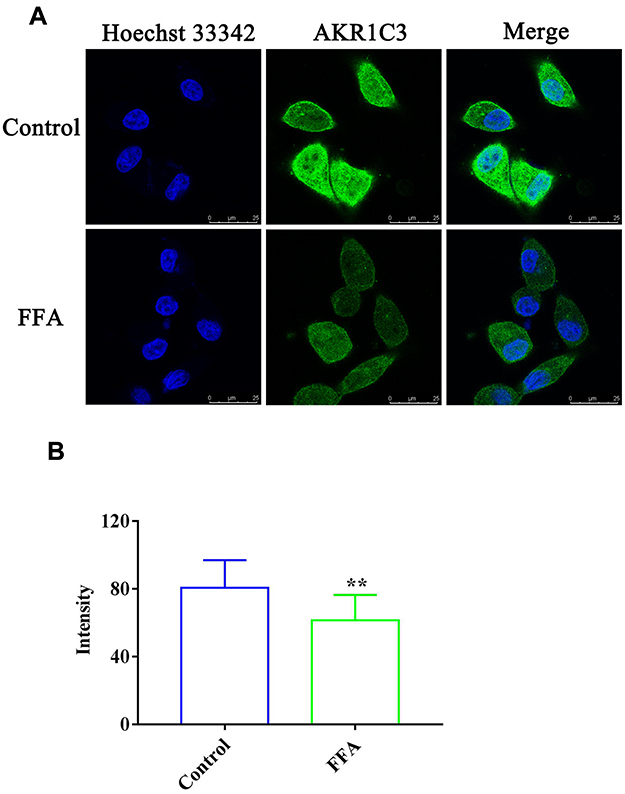 |
Figure 5 The expression of AKR1C3 in the HepG2 cells. (A) Confocal microscopy images of HepG2 cells incubated with or without FFA for 24 h. Green fluorescence denotes AKR1C3 and blue fluorescence denotes nuclei staining; (B) The fluorescence intensity in the HepG2 cells with or without FFA treatment. **p < 0.01 vs control treatment cells. |
The Anti-Tumor Activity of MSNM@SFN+FFA in a 4T1/luc Metastatic Tumor Model
As shown in Figure 6, the fluorescence signal was observed in the control group mice on day 14 after 4T1/luc cell injection and gradually increased with time, demonstrating that 4T1/luc cells were localized in the mice lungs. In the MSNM@SFN treatment group, the fluorescence signal was also observed in the mice on day 14 after 4T1/luc cell injection and gradually increased with time, demonstrating that 4T1/luc cells were also localized in the mice lungs after MSNM@SFN administration. In contrast, the fluorescence signal was not observed in the MSNM@SFN+FFA treatment mice on days 14 and 17 after 4T1/luc cell injection, and only a slight fluorescence signal was observed on day 20, indicating that after MSNM@SFN+FFA treatment, the metastasized 4T1/luc cells in the mice lungs were minor. As shown in Figure 7, the animals in the control group died on day 17 and all sacrificed on day 23 after 4T1/luc cell administration. The animals in the MSNM@SFN treatment group died on day 19 and 75% of the animals sacrificed on day 24. In the MSNM@SFN+FFA treatment group, the animals died on day 21 and only 33% of the animals sacrificed on day 24.
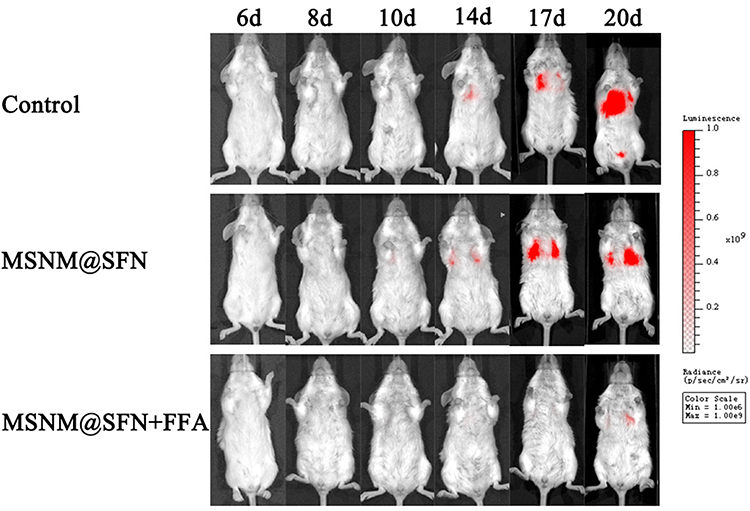 |
Figure 6 In vivo bioluminescence imaging in the 4T1/luc metastatic tumor mice. |
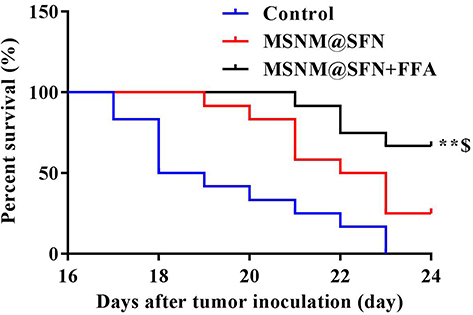 |
Figure 7 In vivo anti-tumor activity of MSNM@SFN+FFA in the 4T1/luc metastatic tumor mice. Kaplan-Meier survival curves of 4T1/luc metastatic tumor mice treated with physiological saline, MSNM@SFN (40 mg/kg of SFN, oral gavage, once a day for 14 days), and MSNM@SFN+FFA (40 mg/kg of SFN, 10 mg/kg of FFA, oral gavage, once a day for 14 days). **p < 0.01 vs control treatment group; $p < 0.05 vs MSNM@SFN treatment group. |
The Anti-Tumor Activity of MSNM@SFN+FFA HepG2 Tumor-Bearing Nude Mice Model
The in vivo anti-tumor activity of MSNM@SFN+FFA was also evaluated in HepG2 tumor-bearing nude mice. As shown in Figure 8, tumor growth was significantly inhibited in the SFN treatment groups compared with the physiological saline treatment group (p < 0.05 or p< 0.01), but the effect varied. Anti-tumor activity demonstrated no significant difference between the MSNM@SFN and SFN suspension treatment groups. However, MSNM@SFN+FFA significantly inhibited the growth of HepG2 tumors compared with that of the MSNM@SFN and SFN suspension treatment groups (p < 0.01). The mean tumor sizes on day 34 after implantation in the MSNM@SFN+FFA, MSNM@SFN, and SFN suspension treatment groups were 107 ± 33, 240 ± 59, and 397 ± 240 mm3, respectively, compared with 1014 ± 332 mm3 in the control group. The corresponding tumor growth inhibition rates in the SFN suspension, MSNM@SFN, and MSNM@SFN+FFA treatment groups were 60.8%, 76.4%, and 89.4% compared with the control group, respectively. We also evaluated the effect of tumor cell apoptosis by TUNEL analysis staining of tumor tissue sections. As shown in Figure 9A, tumors from the MSNM@SFN+FFA treatment group exhibited more advanced cell apoptosis compared with the groups treated with physiological saline, SFN suspension, and MSNM@SFN treatment groups, respectively. The calculated results are shown in Figure 9B.
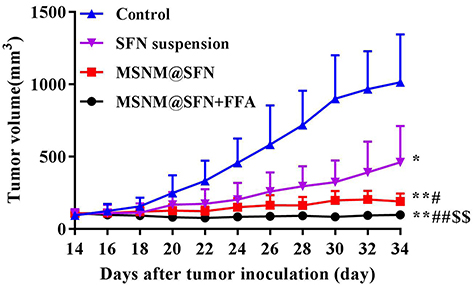 |
Figure 8 In vivo anti-tumor activity of MSNM@SFN+FFA in the HepG2 tumor-bearing nude mice. HepG2 xenograft tumor-bearing nude mice were treated with physiological saline, SFN suspension (40 mg/kg of SFN, oral gavage, once a day for 18 days), MSNM@SFN (40 mg/kg of SFN, oral gavage, once a day for 18 days), and MSNM@SFN+FFA (40 mg/kg of SFN, 10 mg/kg of FFA, oral gavage, once a day for 18 days). *p < 0.05 or **p < 0.01 vs control treatment group; #p < 0.05 or ##p < 0.01 vs SFN suspension treatment group; $$p < 0.01 vs MSNM@SFN treatment group. |
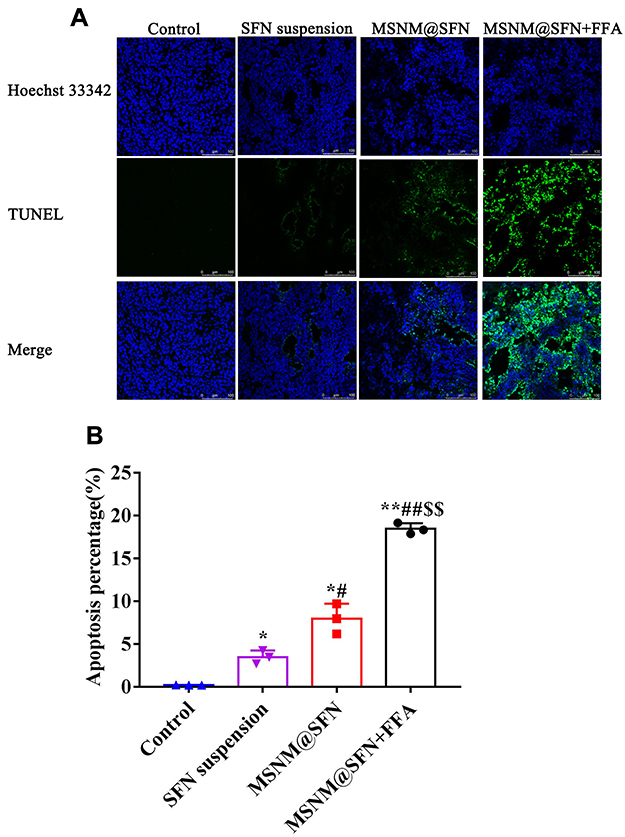 |
Figure 9 Effects of MSNM@SFN+FFA on apoptosis in the HepG2 tumors. (A) TUNEL staining of the paraffin-embedded tumors was performed according to the standard protocols provided by the manufacturers. Tumor apoptosis cells were detected by TUNEL. DNA strand breaks were labeled (green) and nuclei were stained with Hoechst 33342 (blue). Apoptotic cells exhibited a turquoise color as a result of color merging of these two labels. (B) The fluorescence area of each group was used for the statistical analysis of apoptosis activity. *p < 0.05 or **p < 0.01 vs control treatment group; #p < 0.05 or ##p < 0.01 vs SFN suspension treatment group; $$p < 0.01 vs MSNM@SFN treatment group. |
The Anti-Tumor Activity of MSNM@SFN+FFA on HepG2 Orthotopic Tumor-Bearing Nude Mice
The in vivo anti-tumor activity of MSNM@SFN+FFA was also investigated in HepG2 orthotopic tumor-bearing nude mice. As shown in Figure 10, the tumor sizes on day 34 after implantation in the MSNM@SFN+FFA and MSNM@SFN treatment groups were 34 and 46 ± 32 mm3, respectively, significantly lower than in the control group (289 ± 75 mm3) (p < 0.01). Furthermore, in the MSNM@SFN+FFA treatment group, the tumors completely disappeared in 2 of 3 HepG2 orthotopic tumor-bearing nude mice on day 34 after HepG2 cell implantation.
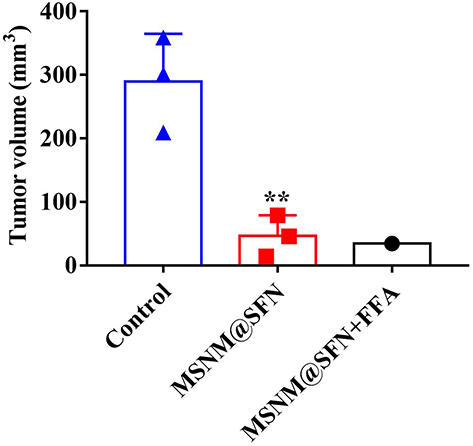 |
Figure 10 In vivo anti-tumor activity of MSNM@SFN+FFA in the HepG2 orthotopic tumor-bearing nude mice. HepG2 orthotopic tumor-bearing nude mice were treated with physiological saline, MSNM@SFN (40 mg/kg of SFN, oral gavage, once a day for 18 days), and MSNM@SFN+FFA (40 mg/kg of SFN, 10 mg/kg of FFA, oral gavage, once a day for 18 days). **p < 0.01 vs control treatment group. |
Discussion
Many anti-cancer drugs are co-administered with SFN to improve their anti-tumor activity, such as combretastatin A4,31 cisplatin,32 doxorubicin,33 paclitaxel,34 trichostatin A,35 cyclophosphamide,1 and gemcitabine.36 In addition, some natural products, such as usnic acid,37 oleanolic acid,38 betulinic acid,39 capsaicin,40 have also been reported for co-administration with SFN to enhance the anti-tumor effect.
Considering the tumor-associated inflammation microenvironment, in the present study, FFA, a COX2 specific inhibitor, was co-administered with SFN. Unlike the previously reported SFN co-administered drug delivery systems, our SFN was MSNM@SFN, which was a solubilized system. We previously investigated and confirmed that there were no effects of FFA on the solubility, dissolution, and pharmacokinetic behavior of SFN in MSNM@SFN.41 Considering our previous results of the anti-tumor activity of MSNM@SFN in MDA-MB-231 cells in vitro and in vivo,26 in the present study, the anti-tumor activity of MSNM@SFN co-administrating with FFA was investigated in 4T1/luc and HepG2 cells in vivo.
Evidence suggests that the expression of COXs might be involved in breast cancer pathogenesis.42 In breast cancer, COX-2 can be used as a biomarker to evaluate breast cancer risk in women with atypical hyperplasia.43 The overexpression of COXs and PGE2 might stimulate breast tumor cell migration and metastasis.44 Our results indicated that FFA could effectively inhibit 4T1/luc cell migration and decrease the level of PGE2 secretion in 4T1/luc cells, indicating the role of FFA in inhibiting COXs activity. Our in vivo anti-tumor activity results indicated the inhibiting effect and prolonging lifetime activity of MSNM@SFN+FFA in the 4T1/luc metastatic tumor model.
The COX/PGE2 axis acts as a regulator of HIF-2α expression and activity to promote hepatocellular carcinoma (HCC) development and reduce SFN sensitivity by constitutively activating the TGF-α/EGFR pathway. Therefore, the potential of COX2 specific inhibitors for HCC treatment would enhance the response to SFN treatment.45 It has been reported that the co-administration of SFN with celecoxib has synergistic anti-tumor activity in HCC.46 The present study confirmed that FFA can effectively inhibit HepG2 cell migration and decrease the level of PGE2 secretion in HepG2 cells. The in vivo anti-tumor activity of the MSNM@SFN+FFA experiments was evaluated in a HepG2 tumor-bearing nude mice model and a HepG2 orthotopic tumor-bearing nude mice model. The in vivo anti-tumor activity of MSNM@SFN+FFA was confirmed in both the HepG2 tumor-bearing nude mice model and HepG2 orthotopic tumor-bearing nude mice model.
Many metabolic enzymes participated in the initiation, progression, and prognosis of cancers.47 Aldo-keto reductases, comprised of AKR1C1, AKR1C2, AKR1C3, and AKR1C4, can catalyze NADPH-dependent reductions and are involved in biosynthesis, intermediary metabolism, and detoxification.48 The overexpression of AKR1C1 has been reported in many cancers, including breast cancer, non-small cell lung cancer, and bladder cancer.49–51 AKR1C3 is also reported to be abundantly expressed in breast cancer, prostate cancer, and T cell acute lymphoblastic leukemia (T-ALL).52–54 Controlling AKR1C activity using its inhibitors may lead to favorable therapeutic outcomes.55 FFA is known to potently inhibit AKR1C3 in a non-selective manner as COX off-target effects.56,57 The present study indicated that FFA could significantly decrease the level of ARK1C1 and ARK1C3 in 4T1/luc or HepG2 cells.
To determine the effects of FFA on 4T1/luc or HepG cell proliferation, 4T1/luc or HepG2 cells were treated with FFA for 48 h. The proliferation of 4T1/luc or HepG2 cells was determined via SRB. The results indicated that the proliferative ratio of 4T1/luc or HepG cells treated with FFA (50 μg/mL) was 60.99 ± 2.72% or 51.47 ± 2.93%, respectively (Fig S1).
FFA has slight to moderate toxicity (10–40% of cell death corresponding to 100 µM) in uterine cervical cancer cell lines.58 The present study demonstrated that FFA had moderate anti-tumor activity (30‑40% of cell death corresponding to 50 µg/mL) in 4T1/luc or HepG2 cells. FFA can also be used as a free radical scavenger and gap junction blocker in cancer cells. However, for normal cells in tumor surroundings, FFA has potential protective effect on normal cells in comparison to the role of FFA in cancerous cells.59
The in vitro anti-tumor activity of MSNM@SFN+FFA on 4T1/luc and HepG2 cells was investigated. As shown in Table S1, the IC50 values of MSNM@SFN+FFA in 4T1/luc and HepG2 cells were similar to that of MSNM@SFN and significantly lower than that of free SFN (p < 0.01), indicating that the anti-tumor activity of the MSNM@SFN+FFA in the 4T1/luc and HepG2 cells was significantly higher than that of the free SFN.
Conclusion
In the present study, we co-administrated sorafenib mesoporous silica nanomatrix (MSNM@SFN) with flufenamic acid (FFA) to improve the anti-tumor activity of SFN. Our results indicated that the FFA can significantly decrease the cell migration, PGE2 secretion, and AKR1C1 and AKR1C3 levels in both 4T1/luc and HepG2 cells. The in vivo anti-tumor activity of MSNM@SFN+FFA was confirmed in a 4T1/luc metastatic tumor model, HepG2 tumor-bearing nude mice model, and HepG2 orthotopic tumor-bearing nude mice model. MSNM@SFN co-administrating with FFA is an alternative strategy for improving the therapeutic efficacy of MSNM@SFN via co-administration with NSAIDs.
Acknowledgments
We gratefully acknowledge the financial support from the National Key Research and Development Program of China (2017YFA0205600), Major New Drug Innovation and Development Program of China (2018ZX09721003-004) and the Innovation Team of the Ministry of Education (No. BMU2017TD003).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zanotto-Filho A, Rajamanickam S, Loranc E, et al. Sorafenib improves alkylating therapy by blocking induced inflammation, invasion and angiogenesis in breast cancer cells. Cancer Lett. 2018;1(425):101–115.
2. Cerón-Carrasco JP, Pérez-Sánchez H, Zúñiga J, Requena A. Antibodies as carrier molecules: encapsulating anti-inflammatory drugs inside herceptine. J Phys Chem B. 2018;122(7):2064–2072. doi:10.1021/acs.jpcb.7b10749
3. Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277(21):18649–18657. doi:10.1074/jbc.M111415200
4. Farzaneh S, Zeinalzadeh E, Daraei B, Shahhosseini S, Zarghi A. New ferrocene compounds as selective Cyclooxygenase (COX-2) inhibitors: design, synthesis, cytotoxicity and enzyme-inhibitory activity. Anticancer Agents Med Chem. 2018;18(2):295–301. doi:10.2174/1871520617666171003145533
5. Kurtova AV, Xiao J, Mo Q, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517(7533):209–213. doi:10.1038/nature14034
6. Rong Y, Yuan CH, Qu Z, et al. Doxorubicin resistant cancer cells activate myeloid-derived suppressor cells by releasing PGE2. Sci Rep. 2016;6:23824. doi:10.1038/srep23824
7. Guo Z, Jiang J-H, Zhang J, et al. COX-2 promotes migration and invasion by the side population of cancer stem cell-like hepatocellular carcinoma cells. Medicine (Baltimore). 2015;94(44):e1806. doi:10.1097/MD.0000000000001806
8. Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology. 2015;149(7):1884–1895. doi:10.1053/j.gastro.2015.07.064
9. Kim MJ, Kim HS, Lee SH, Yang Y, Lee MS, Lim JS. NDRG2 controls COX-2/PGE2-mediated breast cancer cell migration and invasion. Mol Cells. 2014;37(10):759–765. doi:10.14348/molcells.2014.0232
10. Zhu W, Smith A, Young CY. A nonsteroidal anti-inflammatory drug, flufenamic acid, inhibits the expression of the androgen receptor in LNCaP cells. Endocrinology. 1999;140(11):5451–5454. doi:10.1210/endo.140.11.7246
11. Ravera M, Zanellato I, Gabano E, et al. Antiproliferative activity of Pt(IV) conjugates containing the Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) ketoprofen and naproxen. Int J Mol Sci. 2019;20(12):E3074. doi:10.3390/ijms20123074
12. Hiľovská L, Jendželovský R, Fedoročko P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol Clin Oncol. 2015;3(1):3–12. doi:10.3892/mco.2014.446
13. Sun W, Chen G. Impact and mechanism of non-steroidal anti-inflammatory drugs combined with chemotherapeutic drugs on human lung cancer-nude mouse transplanted tumors. Oncol Lett. 2016;11(6):4193–4199. doi:10.3892/ol.2016.4493
14. Duffy CP, Elliott CJ, O’Connor RA, et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDs). Eur J Cancer. 1998;34(8):1250–1259. doi:10.1016/S0959-8049(98)00045-8
15. WINDER CV, WAX J, SERRANO B, JONES EM, McPHEE ML. Anti-inflammatory and antipyretic properties of N-(alpha,alpha,alpha-trifluoro-m-tolyl) anthranilic acid (CI-440; flufenamic acid). Arthritis Rheum. 1963;6:36–47. doi:10.1002/art.1780060105
16. Flower R, Gryglewski R, Herbaczyńska-Cedro K, Vane JR. Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nat New Biol. 1972;238(82):104–106. doi:10.1038/newbio238104a0
17. Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther. 2010;9(2):319–326. doi:10.1158/1535-7163.MCT-09-0663
18. Takimoto CH, Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol. 2008;61(4):535–548. doi:10.1007/s00280-007-0639-9
19. Baselga J, Segalla JG, Roché H, et al. Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol. 2012;30(13):1484–1491. doi:10.1200/JCO.2011.36.7771
20. Wang H, Wang H, Yang W, Yu M, Sun S, Xie B. Improved oral bioavailability and liver targeting of sorafenib solid lipid nanoparticles in rats. AAPS PharmSciTech. 2018;19(2):761–768. doi:10.1208/s12249-017-0901-3
21. Park SY, Kang Z, Thapa P, et al. Development of sorafenib loaded nanoparticles to improve oral bioavailability using a quality by design approach. Int J Pharm. 2019;20(566):229–238. doi:10.1016/j.ijpharm.2019.05.064
22. Chen Y, Chen H, Wang S, Liu C, Qian F. A single hydrogen to fluorine substitution reverses the trend of surface composition enrichment of sorafenib amorphous solid dispersion upon moisture exposure. Pharm Res. 2019;36(7):105. doi:10.1007/s11095-019-2632-5
23. Zhao M, Lee SH, Song JG, Kim HY, Han HK. Enhanced oral absorption of sorafenib via the layer-by-layer deposition of a pH-sensitive polymer and glycol chitosan on the liposome. Int J Pharm. 2018;544(1):14–20. doi:10.1016/j.ijpharm.2018.04.020
24. Khan MA, Ali S, Venkatraman SS, Sohail MF, Ovais M, Raza A. Fabrication of poly (butadiene-block-ethylene oxide) based amphiphilic polymersomes: an approach for improved oral pharmacokinetics of Sorafenib. Int J Pharm. 2018;542(1–2):196–204. doi:10.1016/j.ijpharm.2018.03.023
25. Giglio V, Viale M, Bertone V, Maric I, Vaccarone R, Vecchio G. Cyclodextrin polymers as nanocarriers for sorafenib. Invest New Drugs. 2018;36(3):370–379. doi:10.1007/s10637-017-0538-9
26. Guo Y, Zhong T, Duan XC, et al. Improving anti-tumor activity of sorafenib tosylate by lipid- and polymer-coated nanomatrix. Drug Deliv. 2017;24(1):270–277. doi:10.1080/10717544.2016.1245371
27. Huang Y, Chen XM, Zhao BX, et al. Antiangiogenic activity of Sterically Stabilized Liposomes containing Paclitaxel (SSL-PTX): in vitro and in vivo. AAPS PharmSciTech. 2010;11(2):752–759. doi:10.1208/s12249-010-9430-z
28. Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. doi:10.1016/j.biomaterials.2012.10.029
29. Lee Y, Kim IH, Kim J, et al. Evaluation of dextran-flufenamic acid ester as a polymeric colon-specific prodrug of flufenamic acid, an anti-inflammatory drug, for chronotherapy. J Drug Target. 2011;19(5):336–343. doi:10.3109/1061186X.2010.499462
30. Mizuno H, Liang RF, Kawabata A. Effects of oral administration of various non-steroidal anti-inflammatory drugs on bone growth and bone wound healing in mice. Meikai Daigaku Shigaku Zasshi. 1990;19(2):234–250.
31. Wang Y, Yu H, Zhang D, et al. Co-administration of combretastatin A4 nanoparticles and sorafenib for systemic therapy of hepatocellular carcinoma. Acta Biomater. 2019;92:229–240. doi:10.1016/j.actbio.2019.05.028
32. Zhang W, Cai J, Wu B, Shen Z. pH-responsive hyaluronic acid nanoparticles coloaded with sorafenib and cisplatin for treatment of hepatocellular carcinoma. J Biomater Appl. 2019;34(2):219–228. doi:10.1177/0885328219849711
33. Ye J, Zhang R, Chai W, Du X. Low-density lipoprotein decorated silica nanoparticles co-delivering sorafenib and doxorubicin for effective treatment of hepatocellular carcinoma. Drug Deliv. 2018;25(1):2007–2014. doi:10.1080/10717544.2018.1531953
34. Lei M, Ma G, Sha S, et al. Dual-functionalized liposome by co-delivery of paclitaxel with sorafenib for synergistic antitumor efficacy and reversion of multidrug resistance. Drug Deliv. 2019;26(1):262–272. doi:10.1080/10717544.2019.1580797
35. Chen JC, Chuang HY, Liao YJ, et al. Enhanced cytotoxicity of human hepatocellular carcinoma cells following pretreatment with sorafenib combined with trichostatin A. Oncol Lett. 2019;17(1):638–645. doi:10.3892/ol.2018.9582
36. Zafrakas M, Papasozomenou P, Emmanouilides C. Sorafenib in breast cancer treatment: a systematic review and overview of clinical trials. World J Clin Oncol. 2016;7(4):331–336. doi:10.5306/wjco.v7.i4.331
37. Yurdacan B, Egeli U, Guney Eskiler G, Eryilmaz IE, Cecener G, Tunca B. Investigation of new treatment option for hepatocellular carcinoma: a combination of sorafenib with usnic acid. J Pharm Pharmacol. 2019;71(7):1119–1132. doi:10.1111/jphp.2019.71.issue-7
38. Liese J, Hinrichs TM, Lange M, Fulda S. Cotreatment with sorafenib and oleanolic acid induces reactive oxygen species-dependent and mitochondrial-mediated apoptotic cell death in hepatocellular carcinoma cells. Anticancer Drugs. 2019;30(3):209–217. doi:10.1097/CAD.0000000000000750
39. Kutkowska J, Strzadala L, Rapak A. Synergistic activity of sorafenib and betulinic acid against clonogenic activity of non-small cell lung cancer cells. Cancer Sci. 2017;108(11):2265–2272. doi:10.1111/cas.2017.108.issue-11
40. Bort A, Spínola E, Rodríguez-Henche N, Díaz-Laviada I. Capsaicin exerts synergistic antitumor effect with sorafenib in hepatocellular carcinoma cells through AMPK activation. Oncotarget. 2017;8(50):87684–87698. doi:10.18632/oncotarget.v8i50
41. Yin YF, Li ZY, Li H, Guo Y, Zhang Q, Zhang X. Effect of flufenamic acid on solubility, dissolution and pharmacokinetic behavior of sorafenib in MSNM@SFN. Acta Pharm Sinica. 2019;54(01):48–53.
42. Basu S, Combe K, Kwiatkowski F, et al. Cellular expression of cyclooxygenase, aromatase, adipokines, inflammation and cell proliferation markers in breast cancer specimen. PLoS One. 2015;10(10):e0138443. doi:10.1371/journal.pone.0138443
43. Visscher DW, Pankratz VS, Santisteban M, et al. Association between cyclooxygenase-2 expression in atypical hyperplasia and risk of breast cancer. J Natl Cancer Inst. 2008;100(6):421–427. doi:10.1093/jnci/djn036
44. Allison SE, Petrovic N, Mackenzie PI, Murray M. Pro-migratory actions of the prostacyclin receptor in human breast cancer cells that over-express cyclooxygenase-2. Biochem Pharmacol. 2015;96(4):306–314. doi:10.1016/j.bcp.2015.06.002
45. Dong XF, Liu TQ, Zhi XT, et al. COX-2/PGE2 axis regulates HIF2α activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res. 2018;24(13):3204–3216. doi:10.1158/1078-0432.CCR-17-2725
46. Morisaki T, Umebayashi M, Kiyota A, et al. Combining celecoxib with sorafenib synergistically inhibits hepatocellular carcinoma cells in vitro. Anticancer Res. 2013;33(4):1387–1395.
47. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi:10.1038/nrc2981
48. Zeng CM, Chang LL, Ying MD, et al. Aldo-Keto reductase AKR1C1-AKR1C4: functions, regulation, and intervention for anti-cancer therapy. Front Pharmacol. 2017;8:119. doi:10.3389/fphar.2017.00119
49. Ji Q, Aoyama C, Nien YD, et al. Selective loss of AKR1C1 and AKR1C2 in breast cancer and their potential effect on progesterone signaling. Cancer Res. 2004;64(20):7610–7617. doi:10.1158/0008-5472.CAN-04-1608
50. Zhu H, Chang LL, Yan FJ, et al. AKR1C1 activates STAT3 to promote the metastasis of non-small cell lung cancer. Theranostics. 2018;8(3):676–692. doi:10.7150/thno.21463
51. Matsumoto R, Tsuda M, Yoshida K, et al. Aldo-keto reductase 1C1 induced by interleukin-1β mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci Rep. 2016;6:34625. doi:10.1038/srep34625
52. Yamashita N, Kanno Y, Saito N, et al. Aryl hydrocarbon receptor counteracts pharmacological efficacy of doxorubicin via enhanced AKR1C3 expression in triple negative breast cancer cells. Biochem Biophys Res Commun. 2019;516(3):693–698. doi:10.1016/j.bbrc.2019.06.119
53. Dozmorov MG, Azzarello JT, Wren JD, et al. Elevated AKR1C3 expression promotes prostate cancer cell survival and prostate cell-mediated endothelial cell tube formation: implications for prostate cancer progression. BMC Cancer. 2010;10:672. doi:10.1186/1471-2407-10-672
54. Evans K, Duan J, Pritchard T, et al. OBI-3424, a novel AKR1C3-activated prodrug, exhibits potent efficacy against preclinical models of T-ALL. Clin Cancer Res. 2019;25(14):4493–4503. doi:10.1158/1078-0432.CCR-19-0551
55. Shiiba M, Yamagami H, Yamamoto A, et al. Mefenamic acid enhances anticancer drug sensitivity via inhibition of aldo-keto reductase 1C enzyme activity. Oncol Rep. 2017;37(4):2025–2032. doi:10.3892/or.2017.5480
56. Pippione AC, Giraudo A, Bonanni D, et al. Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur J Med Chem. 2017;139:936–946. doi:10.1016/j.ejmech.2017.08.046
57. Pippione AC, Carnovale IM, Bonanni D, et al. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur J Med Chem. 2018;150:930–945. doi:10.1016/j.ejmech.2018.03.040
58. Soriano-Hernandez AD, Madrigal-Pérez D, Galvan-Salazar HR, et al. Anti-inflammatory drugs and uterine cervical cancer cells: antineoplastic effect of meclofenamic acid. Oncol Lett. 2015;10(4):2574–2578. doi:10.3892/ol.2015.3580
59. Lara GG, Andrade GF, Cipreste MF, et al. Protection of normal cells from irradiation bystander effects by silica-flufenamic acid nanoparticles. J Mater Sci Mater Med. 2018;29(8):130. doi:10.1007/s10856-018-6134-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.