")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Enhanced therapeutic efficacy of budesonide in experimental colitis with enzyme/pH dual-sensitive polymeric nanoparticles
Authors Naeem M, Cao J, Choi M, Kim W, Moon HR, Lee BL, Kim M, Jung Y, Yoo J
Received 2 May 2015
Accepted for publication 13 June 2015
Published 16 July 2015 Volume 2015:10(1) Pages 4565—4580
DOI https://doi.org/10.2147/IJN.S87816
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Muhammad Naeem, Jiafu Cao, Moonjeong Choi, Woo Seong Kim, Hyung Ryong Moon, Bok Luel Lee, Min-Soo Kim, Yunjin Jung, Jin-Wook Yoo
College of Pharmacy, Pusan National University, Busan, South Korea
Abstract: Current colon-targeted drug-delivery approaches for colitis therapy often utilize single pH-triggered systems, which are less reliable due to the variation of gut pH in individuals and in disease conditions. Herein, we prepared budesonide-loaded dual-sensitive nanoparticles using enzyme-sensitive azo-polyurethane and pH-sensitive methacrylate copolymer for the treatment of colitis. The therapeutic potential of the enzyme/pH dual-sensitive nanoparticles was evaluated using a rat colitis model and compared to single pH-triggered nanoparticles. Clinical activity scores, colon/body weight ratios, myeloperoxidase activity, and proinflammatory cytokine levels were markedly decreased by dual-sensitive nanoparticles compared to single pH-triggered nanoparticles and budesonide solution. Moreover, dual-sensitive nanoparticles accumulated selectively in inflamed segments of the colon. In addition, dual-sensitive nanoparticle plasma concentrations were lower than single pH-triggered nanoparticles, and no noticeable in vitro or in vivo toxicity was observed. Our results demonstrate that enzyme/pH dual-sensitive nanoparticles are an effective and safe colon-targeted delivery system for colitis therapy.
Keywords: azo-polyurethane, methacrylate copolymer, budesonide, colon-targeted nanoparticles, colitis
Introduction
Ulcerative colitis (UC) and Crohn’s disease are the two main forms of inflammatory bowel disease (IBD), which is increasing in incidence worldwide. IBD is caused by unknown genetic, environmental, and bacterial factors.1 Symptoms of IBD range from bloody diarrhea and weight loss to ulceration and complete obstruction of the gastrointestinal (GI) tract, which can negatively affect daily life.2 Furthermore, IBD is a risk factor for the development of colon cancer.3 In many patients, IBD has been successfully treated with glucocorticoids, such as prednisolone, due to their potent anti-inflammatory activity.1 However, prolonged administration of glucocorticoids is often restricted to patients with severe disease, due to multiple serious side effects, including hypertension, diabetes mellitus, infection, osteoporosis, and glaucoma.4,5
Budesonide is a glucocorticoid with anti-inflammatory properties and few side effects; however, it has limited systemic bioavailability due to extensive first-pass hepatic metabolism by CYP3A enzymes.6 Therefore, many groups have sought to develop efficient colon-targeted delivery systems for budesonide to overcome this limitation.7 Various approaches have been used to develop colon-targeted delivery, including prodrugs, pH-dependent release systems, time-dependent release systems, and enzyme-dependent release systems. Among these, single pH-dependent systems are commercially available for budesonide (Budenofalk® and Entocort®).8 However, single pH-dependent products release budesonide in the ileum and proximal colon. Therefore, they are not ideal for UC treatment, which occurs in the distal colon and rectum.9 Recent studies showed that pH-dependent budesonide delivery did not effectively induce UC remission, likely due to unreliable colonic release in patients with altered intestinal pH caused by inter- and intraindividual gut pH variability.10,11 Therefore, an ideal colon-specific delivery system for UC therapeutics should prevent premature drug release before reaching the colon and deliver sufficient amounts of the drug to the inflamed area of the distal colon. Fine tuning of the formulation may be necessary to account for longer transit times in the gut, as well as slightly higher pH values (around 7.3) in the distal small intestine.12
In our previous study, we designed and developed an enzyme/pH dual-sensitive polymeric mixture nanoparticle (NP) system composed of an enzyme-sensitive azo-polyurethane (Azo-pu) and a pH-sensitive methacrylate copolymer (Eudragit S100 [ES]) for targeted drug delivery to the inflamed colon, thereby overcoming the limitations of single-triggered release systems.13 The prepared NPs remained intact in the stomach, and released a drug in a sustained manner in the ileum (pH >7.0), followed by enzyme-triggered release in the presence of the cecal content of rats with colitis. Higher fluorescence levels were also found in the colon compared to the other GI segments after oral administration of the NPs.
In this study, we employed novel enzyme/pH dual-sensitive NPs to enhance the therapeutic efficacy of budesonide for colitis treatment. Budesonide was loaded into Azo-pu ES NPs using quasiemulsion solvent diffusion. Single pH-sensitive NPs (ES NPs) and budesonide solution were also prepared for comparison. The size, shape, and drug-loading capability of the NPs were characterized. The NPs drug-release profiles were evaluated in different pH environments resembling those of the GI tract. After physicochemical characterization, the in vivo therapeutic efficacy of Azo-pu ES NPs was evaluated using a rat colitis model and compared to ES NPs and budesonide solution. Further, the accumulation of NPs in inflamed colon tissues was evaluated. Finally, in vivo and in vitro biocompatibility studies were performed to evaluate the safety of the novel NPs.
Materials and methods
Materials
Budesonide, coumarin 6 (C-6), polyvinyl alcohol (molecular weight 30,000–70,000), isophorone diisocyanate, 1,2-propanediol, polyethylene glycol (molecular weight 2,000), tin octanoate, and MTT were purchased from Sigma-Aldrich (St Louis, MO, USA). Methacrylate copolymer (Eudragit S100) was generously donated by Evonik Korea Ltd (Seoul, South Korea). m,m′-Di(hydroxymethyl)azobenzene (DAB) was prepared as described in the literature.14 The 2,4,6-Trinitrobenzenesulfonic acid (TNBS) was purchased from Wako Pure Chemicals (Osaka, Japan). Dulbecco’s Modified Eagle’s Medium, trypsin, fetal bovine serum, and penicillin–streptomycin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents and solvents were of the highest analytical grade commercially available.
Synthesis of azo-containing polyurethane
Azo-pu was synthesized as previously reported.13 Briefly, 5.85 g (24.2 mmol) DAB and 15.7 g (7.8 mmol) polyethylene glycol were placed in a 300 mL round-bottom flask equipped with a mechanical stirrer and dropping funnel. The flask was evacuated using a vacuum pump for several hours and flushed with dry nitrogen to dry the contents. The 1,2-Propanediol (6.7 g, 88 mmol) and 0.11 g tin octanoate were then added, and the flask was heated to 120°C with stirring under nitrogen flow. Isophorone diisocyanate (6.7 g, 88 mmol) was added dropwise over 4 hours using a dropping funnel. Stirring was maintained until the contents reached the required viscosity. Several grams of ethanol were added to stop the polymerization, and the product was dissolved in 80 mL of ethanol and poured into 1,000 mL of diethyl ether for precipitation. After filtration, the precipitate was dried in a vacuum oven.
Preparation of NPs
ES NPs and Azo-pu ES NPs were prepared by a quasiemulsion solvent diffusion with some modifications.15 Briefly, 100 mg of ES or Azo-pu ES (1:1 w/w) was dissolved with budesonide (10 mg) in 10 mL of acetone/ethanol solution (7:3 v/v). This solution was slowly injected using a syringe pump at a flow rate of 0.33 mL/minute into 40 mL citrate buffer (pH 5.0) containing 0.1% w/v polyvinyl alcohol solution with stirring. After evaporation of the residual solvent under a fume hood, the NPs were collected by centrifugation at 20,000 g for 30 minutes and washed with deionized water three times. The obtained NPs were immediately used for the following experiments. Budesonide was replaced by the hydrophobic fluorescent marker C-6 (2 mg) to facilitate the evaluation of enzyme-triggered release in rat cecal content, in vivo localization, and bioavailability studies. The same procedure was followed for C-6-loaded NP preparation.
Characterization of NPs
Scanning electron microscopy and particle-size analysis
External NP morphology was analyzed by scanning electron microscopy (SEM). NPs suspended in water were dropped on a carbon tape and air-dried at room temperature in a fume hood or desiccator. Samples were then coated with platinum for 2 minutes in a vacuum and viewed by field-emission SEM (S4800; Hitachi Ltd, Tokyo, Japan) at an acceleration voltage of 1–5 kV. Particle size and size distribution were determined using a qNano size analyzer (Izon Science, Christchurch, New Zealand) coupled with an air-based variable pressure module. All measurements were obtained under the same conditions of applied voltage, stretch, and pressure.
Loading efficiency and entrapment efficiency
The loading and entrapment efficiency of budesonide in NPs was determined by high-performance liquid chromatography (HPLC) according to an established method in the literature.16 The HPLC system used for the budesonide analysis was an LC-20AT (Shimadzu, Kyoto, Japan) equipped with an autosampler processor, an SPD-20A ultraviolet (UV) detector, and a Luna C18 column (5 μm, 150×4.6 mm; Phenomenex, Torrance, CA, USA). The UV-detector wavelength was set at 254 nm, and a combination of methanol and water (70:30) at a flow rate of 0.8 mL/min was used as the mobile phase. A calibration curve using standard budesonide solution was obtained, and was linear (R2=0.9997) over the range of 0.125–250 μg/mL. Specific amounts of ES NPs or Azo-pu ES NPs were dissolved in methanol, and the budesonide content was determined using the calibration curve. Samples were prepared in triplicate, and drug-loading efficiency (LE) and encapsulation efficiency (EE) were calculated using the following equations:
|
|

|
|
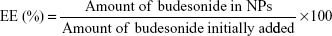
Differential scanning calorimetry study
The physical status of the NP-entrapped drugs was analyzed by differential scanning calorimetry (DSC; N-650; Scinco, Seoul, South Korea). Samples (6 mg) of budesonide, ES, Azo-pu, and the NPs were accurately weighed in aluminum pans and hermetically sealed with aluminum lids. Sample DSC thermograms were obtained by heating from 25°C to 300°C at a scanning rate of 10°C/min under dry nitrogen. Empty pans were used as a reference.
In vitro drug release in different pH
In vitro drug release from ES NPs and Azo-pu ES NPs was evaluated in buffers with gradually changing pH values of 1.2, 6.8, and 7.4, corresponding to the stomach, the upper small intestine, and the ileum and colon, respectively.17 NPs (10 mg) were added to 50 mL of the release medium and incubated in a shaking water bath (60 rpm, 37°C). Tween 80 (0.2% w/v) was added to the release medium to facilitate the solubilization of budesonide. At predetermined time intervals, 150 μL aliquots were sampled, and an equal volume of fresh buffer was added. The aliquots were centrifuged at 17,000 g for 30 minutes, and supernatants containing budesonide released from the NPs were analyzed using HPLC as described previously.
Animal studies
All animal experiments were approved by the Ethical Committee for Animal Care of the Health Science Sector of the Pusan National University, and performed in accordance with the regulations of Pusan National University and Korean legislation on animal studies. Male Sprague Dawley rats (250–290 g, 9 weeks old) were purchased from Samtako Bio Korea (Osan, South Korea) and housed in the university animal facility at 25°C±3°C under a 12-hour light/dark cycle.
TNBS-induced colitis
The TNBS colitis model was chosen as a well-recognized experimental model that allows induction of colitis at an exact location.18 Colitis was induced following a previously reported method.19 Briefly, rats were starved with free access to water for 24 hours before colitis induction. All colonic instillations were performed under isoflurane anesthesia. A rubber cannula (outer diameter 2 mm) was inserted rectally into the colon, such that the tip was 8 cm proximal to the anus, at the splenic flexure. TNBS dissolved in 50% (v/v) aqueous ethanol was instilled into the colon via the rubber cannula (15 mg/0.3 mL/rat). The healthy control group received only 50% aqueous ethanol. Rats were weighed and randomly divided into five groups: healthy control, colitis control, budesonide solution-treated, ES NP-treated, and Azo-pu ES NP-treated (n=5). The rats were monitored for 3 days without treatment to allow for the development of colitis. Each treated group received an equal dose of budesonide (0.168 mg/kg) either in the form of drug solution or suspended NPs orally by gavage. The healthy control and the colitis control groups received normal saline.
Clinical activity score
The degree of inflammation was evaluated by clinical score, assessing weight loss, stool consistency, and rectal bleeding, as previously described.20 Inflammation was scored on a scale of 0–4. No weight loss was 0 points, 1%–5% 1 point, 5%–10% 2 points, 10%–20% 3 points, and >20% 4 points. For stool consistency, 0 points were given for well-formed pellets, 2 points for pasty and semiformed stools that did not stick to the anus, and 4 points for liquid stools that stuck to the anus. Bleeding was scored as 0 points for no blood, 2 points for positive findings, and 4 points for gross bleeding. The mean of these scores formed the clinical score, ranging from 0 (healthy) to 4 (maximal colitis).
Colon macroscopic appearance, length, and colon/body weight ratio
All rats were killed 24 hours after the last treatment and their colons resected. Macroscopic changes, including colon length, ulceration, and severity of colitis, were evaluated. Distal colon tissue samples (10 cm) were resected, opened longitudinally, and rinsed with iced buffer to remove the luminal content. The ratio of the colon sample wet weight to body weight was calculated, and the colon/body weight ratio was determined as an index of colonic inflammation.21
Histological assessment of colitis
For the histological examinations, small segments of the colon were fixed in 10% buffered formalin phosphate and embedded in paraffin. Sections (5 μm) were cut with a microtome (Reichert, Munich, Germany) and stained with hematoxylin and eosin (H&E), followed by light microscopy analysis (Carl Zeiss Meditec AG, Jena, Germany). The degree of inflammation and epithelial injury on microscopic cross sections of the colon were graded semiquantitatively from 0 to 5.22 Grading was done in a blinded fashion on coded slides.
Myeloperoxidase activity
Myeloperoxidase (MPO) is an enzyme that is abundantly expressed in activated neutrophils in inflamed tissue, and its activity is measured as an index of inflammation. MPO activity was measured according to an established method.11 Briefly, distal colon specimens were minced with a sharp scissor in 1 mL of hexadecyltrimethylammonium bromide (HTAB) buffer (0.5% in 50 mM phosphate buffer, pH 6.0) on ice and homogenized. HTAB was added to adjust the pooled homogenate concentration to 100 mg of tissue per milliliter, followed by sonication for 10 seconds, three freeze–thaw cycles, and centrifugation at 14,000 rpm at 4°C for 3 minutes. Supernatants (0.1 mL) were added to 2.9 mL of 50 mM phosphate buffer solution (pH 6.0) containing 0.167 mg/mL O-dianisidine dihydrochloride and 0.0005% hydrogen peroxide, and the change in absorbance at 460 nm was measured by a UV spectrophotometer (Shimadzu) for 5 minutes at 25°C. One unit of MPO activity is defined as the amount of enzyme degrading 1 μmol of peroxide per minute at 25°C.
Determination of proinflammatory cytokines in the colon
The concentrations of the proinflammatory cytokines TNF-α and IL-6 in the colons of untreated and treated rats were determined by a sandwich-type enzyme-linked immunosorbent assay (ELISA) using kits (BioLegend, San Diego, CA, USA; R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Briefly, the frozen distal colon specimens were homogenized with potassium phosphate buffer (pH 6.0) and centrifuged at 2,500 g and 4°C for 5 minutes. Supernatants were centrifuged at 10,000 g and 4°C for 10 minutes, and were analyzed by ELISA for TNF-α and IL-6 quantification. Total protein concentration was determined by the Lowry protein assay using BCA kit (Thermo Fisher Scientific).
Accumulation of NPs in inflamed colon tissues
Colitis was induced in rats using TNBS as described previously. The hydrophobic fluorescent marker C-6 was loaded in NPs to facilitate in vivo detection and quantification in the colon after oral administration. After induction of colitis, rats were orally administered C-6 (50 μg/rat), either as a solution or suspended NPs. Colons were resected after 24 hours, and the inflamed and noninflamed parts were separated and rinsed in cold saline to remove the luminal content. For confocal microscopy, colon specimens were embedded in Tissue-Tek® optimum cutting temperature compound and cut into 5 μm sections using a cryostat (Reichert). Images were captured by confocal microscopy (FV10i FluoView; Olympus Corporation, Tokyo, Japan). For the quantitative study, the colon samples were freeze-dried and homogenized into powder. Subsequently, 50 mg powder was used to extract C-6 with 1 mL of chloroform/methanol (1:1, v/v) and quantified by a fluorescence plate reader (TriStar LB 941; Berthold Technologies, Bad Wildbad, Germany).
C-6 plasma-concentration study
Rats were kept in an environmentally controlled breeding room for 3 days before the experiment. The rats were starved for 12 hours prior to C-6 administration. Budesonide was replaced with C-6 formulation to facilitate plasma analysis.23 Rats were divided into three groups (n=3): the C-6 solution group, ES NP group, and Azo-pu ES NP group. The formulations were orally administered (50 μg/rat) by oral gavage under mild isoflurane anesthesia. Venous blood (300 μL) was obtained from the tail vein and collected in heparin-pretreated test tubes at 0, 0.5, 1, 2, 3, 4, 5, 6, 8, and 24 hours after administration. All blood samples were immediately centrifuged, and the obtained plasma was stored at −20°C before analysis. The C-6 content in the plasma was extracted with a chloroform/methanol (1:1 v/v) mixture and quantified by the fluorescence plate reader.21
Blood glucose level
The blood glucose level in the colitis control and budesonide-treated rats was measured using a blood glucose-monitoring system (CareSens II; ICENS Co Ltd, Wonju, South Korea) to compare the colon-specific drug-delivery potential of the developed novel Azo-pu ES NPs to ES NPs and the budesonide solution.
In vivo and in vitro biocompatibility study
The in vivo toxicity of budesonide-loaded Azo-pu ES NPs after 5 days’ treatment was evaluated in colitis rats. After treatment with Azo-pu ES NPs, liver and kidney sections were prepared, and H&E staining was performed for histological evaluations. Untreated healthy rats were used as a control group. The in vivo toxicity of blank Azo-pu ES NPs was also evaluated by administering NPs for 7 days. Body weight changes were recorded, and H&E-staining sections of the small intestine and colon were prepared for histological evaluation. In vitro biocompatibility of blank Azo-pu ES NPs was determined by MTT assay using the HT29 and HCT116 cell lines, obtained from the Korean Cell Line Bank (Seoul, South Korea). Cells were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and 100 U/mL penicillin–streptomycin. The cells were seeded in 96-well plates at a density of 5,000 cells/well. After 24 hours, the culture medium was replaced with varying concentrations of blank Azo-pu ES NP suspensions and incubated for 24 hours. Cell toxicity was determined by MTT assay.
Statistical analysis
Statistical analysis of the in vitro and in vivo data was performed using Student’s t-test in Sigma Plot 10.0 (Systat Software, Chicago, IL, USA). The data are presented as means ± standard deviation, and P-values less than 0.05 were accepted as a statistically significant difference.
Results
Preparation and physicochemical characterization of NPs
In the current study, budesonide was loaded in ES NPs and Azo-pu ES NPs using the oil-in-water emulsion/solvent-evaporation method.15 The prepared NPs were evaluated by SEM, which revealed a spherical morphology (Figure 1A) with uniform size and high yields and encapsulation efficiency, as shown in Table 1. The mean sizes of the ES NPs and Azo-pu ES NPs were 191 nm and 195 nm, respectively, with a narrow size distribution (Table 1 and Figure 1B). The physical status of the entrapped drugs in the NPs was evaluated by DSC, as shown in Figure 1C. The endothermic peak of budesonide in the DSC curves was observed at 262°C, whereas no budesonide peaks were found in the NPs, indicating that budesonide was present in the molecularly dispersed state and well encapsulated in NPs.24
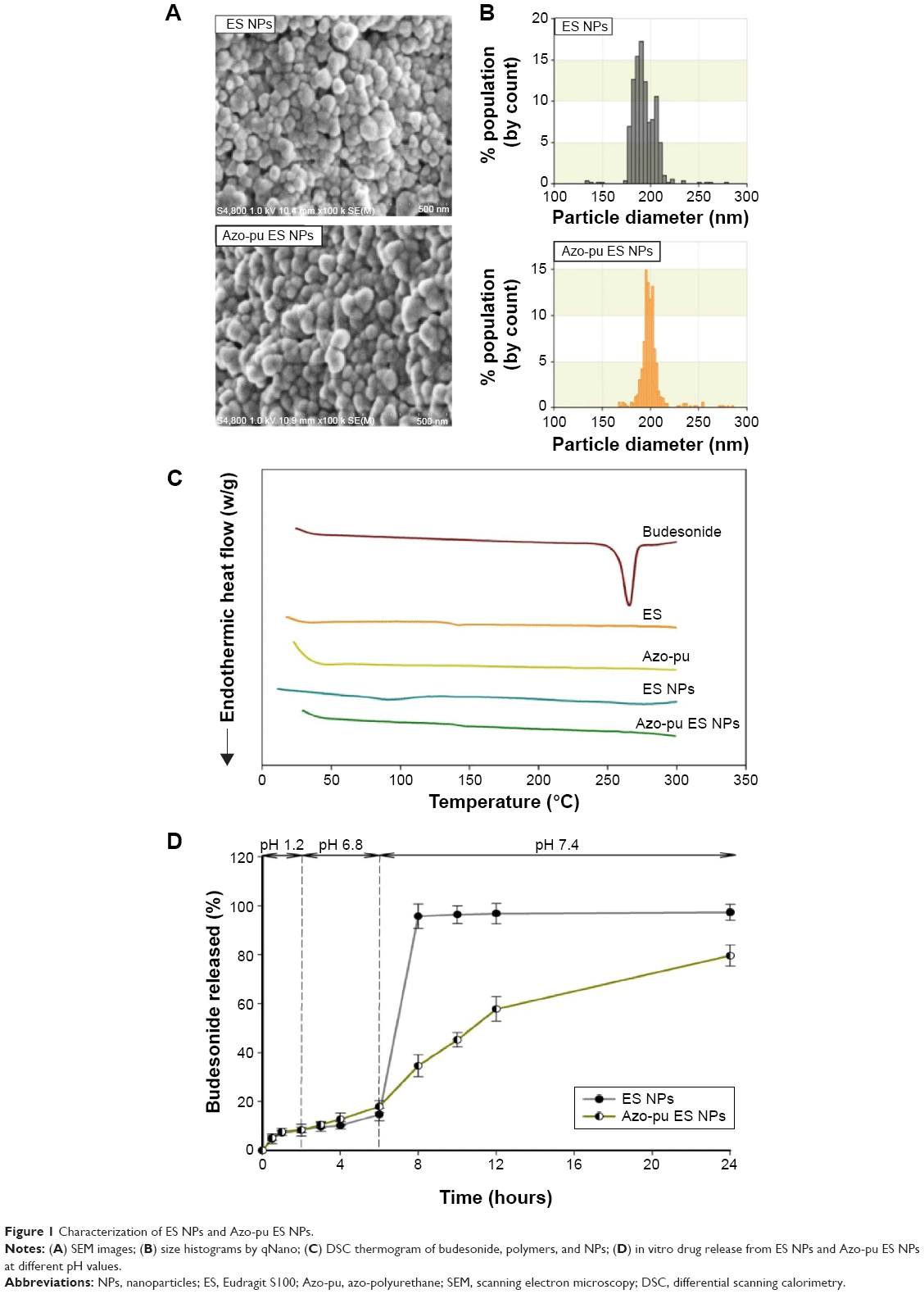 | Figure 1 Characterization of ES NPs and Azo-pu ES NPs. |
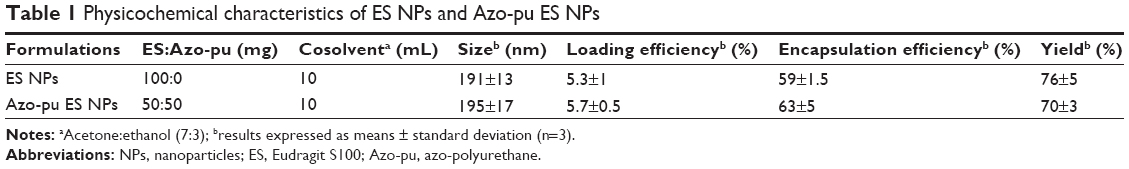 | Table 1 Physicochemical characteristics of ES NPs and Azo-pu ES NPs |
In vitro drug release
The profiles of budesonide release from ES NPs and Azo-pu ES NPs were determined in gradually pH-changing medium (pH 1.2, 6.8, and 7.4) for 24 hours, as shown in Figure 1D. At pH 1.2 and 6.8, which represent the pH of the stomach and the upper part of the small intestine, respectively, there was no significant difference in drug-release profile between the NPs, with less than 20% of the drug released during the first 6 hours. At the ileum pH (pH 7.4), ES NPs and Azo-pu ES NPs showed markedly different release profiles. ES NPs showed a sudden burst release (nearly 100%) of the drug, due to their complete dissolution at pH >7.0. In contrast, there was no drastic change in drug release from Azo-pu ES NPs at pH 7.4; however, the drug was released slightly faster upon exposure to pH 7.4 (Figure 1D).
In vivo therapeutic efficacy
Clinical activity score of colitis
Figure 2A shows the clinical activity indices, which incorporate weight loss, stool consistency, and GI bleeding. In the colitis control group, the clinical activity score increased within 24 hours in response to intestinal inflammation. All treated groups showed reduced clinical activity scores compared to the colitis control group. The Azo-pu ES NP-treated group showed the most prominent reduction in the clinical activity score, with twofold and fivefold reduction compared to ES NPs and budesonide solution, respectively.
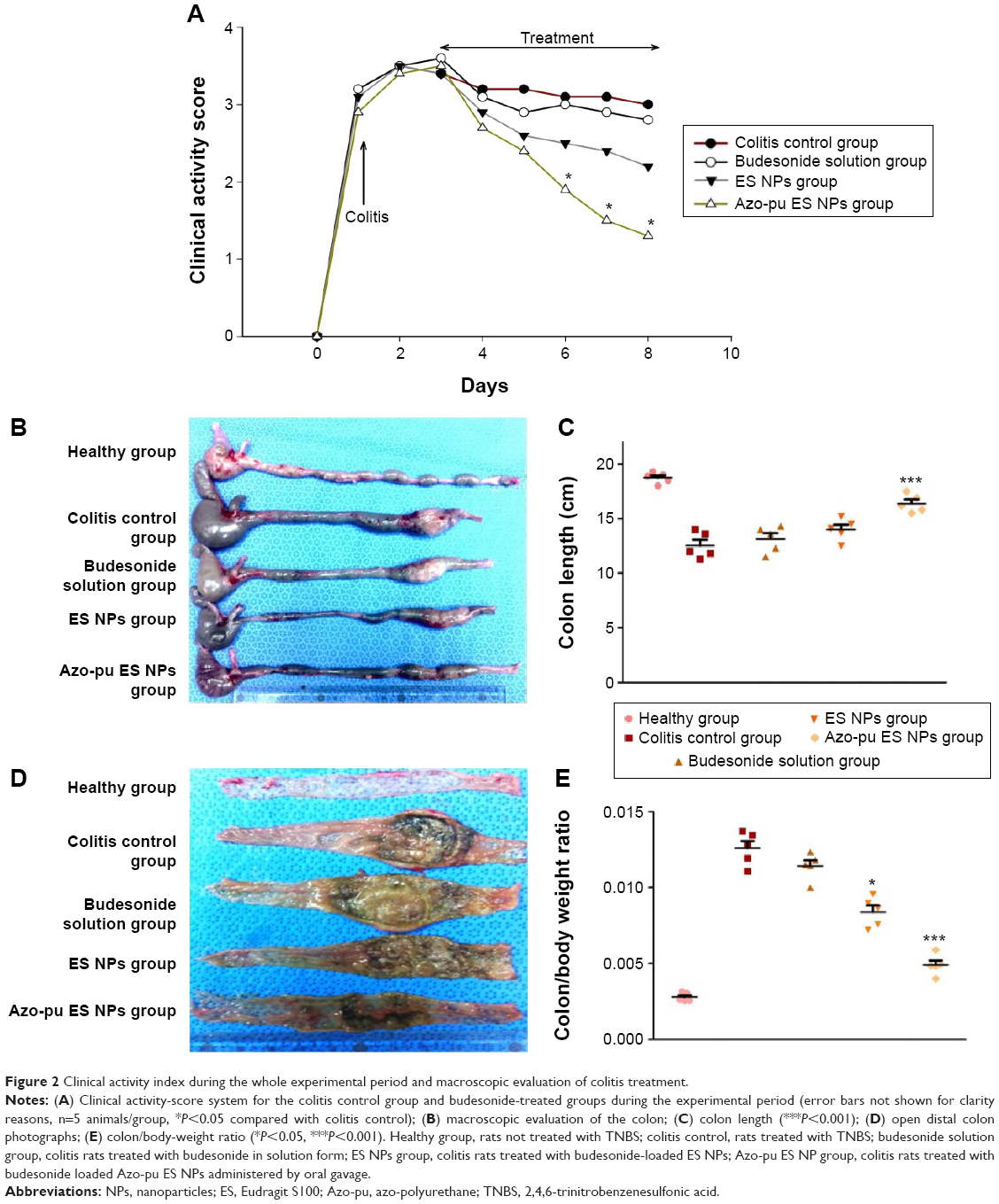 | Figure 2 Clinical activity index during the whole experimental period and macroscopic evaluation of colitis treatment. |
Macroscopic and microscopic assessment of colitis
All animals were killed after treatment. Inflammation was visible due to TNBS-induced colitis, as shown in Figure 2B. Rats treated with NPs showed reduced inflammation compared to the colitis control. As inflammation was alleviated, colon length also increased, as shown in Figure 2C. Similarly, colon/body weight ratio was significantly lower in the Azo-pu ES NP-treated group than in the colitis control group and other treated groups (Figure 2E). As shown in the distal colon photographs in Figure 2D, the Azo-pu ES NPs ameliorated colitis to a greater extent, indicating that the therapeutic effect of Azo-pu ES NPs was greater than ES NPs and budesonide solution. To confirm the drug-delivery potential of Azo-pu ES NPs for UC treatment, histological evaluation of treated and untreated colon tissue was also performed. As shown in Figure 3A, the mucosa from the healthy control group showed no signs of disrupted morphology. In contrast, tissue samples from the untreated colitis control exhibited necrosis, disruption, and irregular morphology. Tissue damage was also observed in the budesonide solution-treated rats. However, inflammation subsided substantially in the group treated with NPs. Histological findings from the Azo-pu ES NP-treated group demonstrated morphological tissue structures resembling those of healthy tissue, indicating epithelial restoration and reduced swelling of the bowel wall. Histological scoring of the tissue sections confirmed these findings, indicating that inflammation was most significantly alleviated (P<0.001) in the Azo-pu ES NP-treated group compared to the ES NP-treated group (P<0.05) (Figure 3B). Overall, these findings indicate that Azo-pu ES NPs deliver sufficient drug to the inflamed colon, thereby significantly alleviating inflammation.
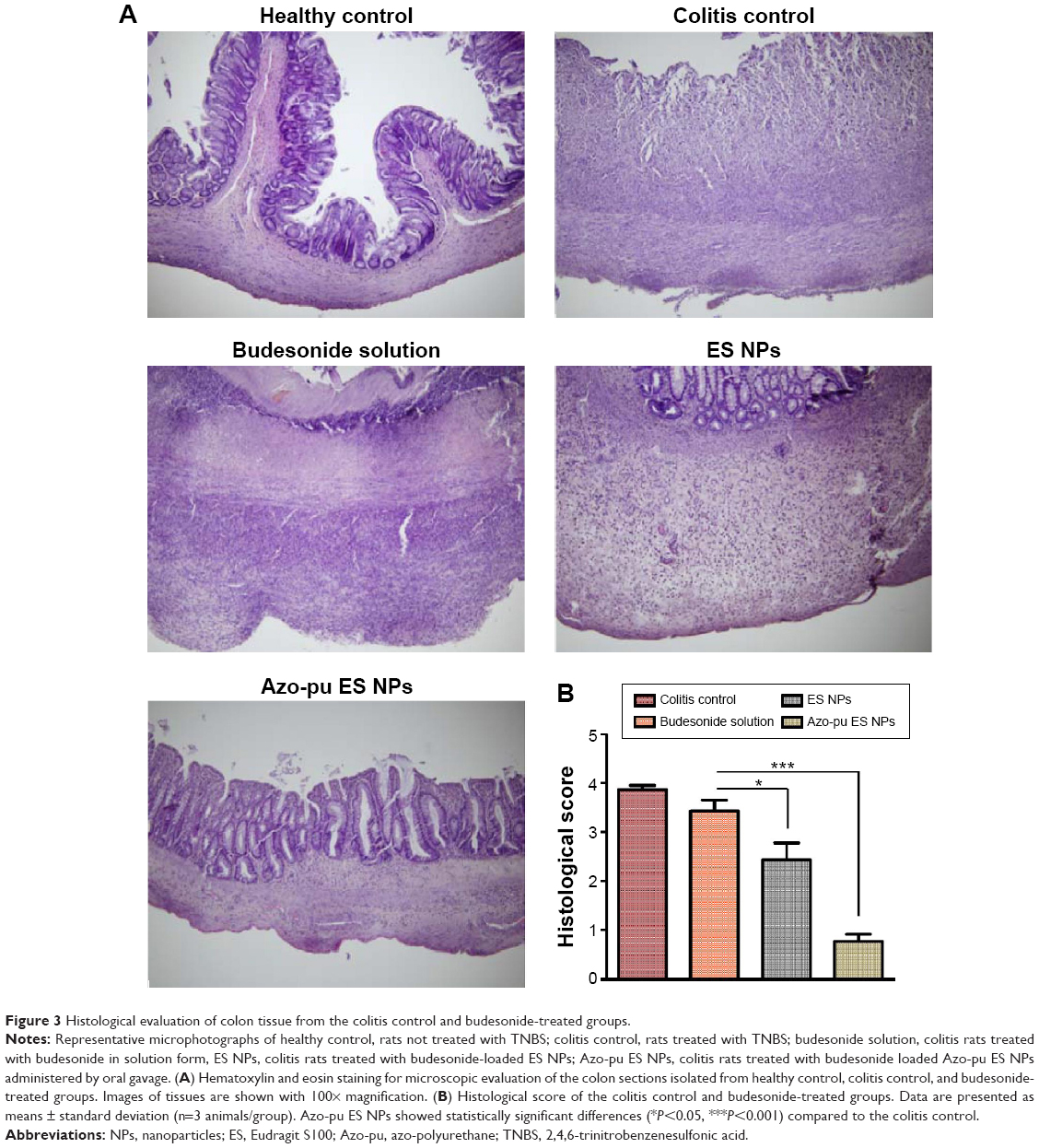 | Figure 3 Histological evaluation of colon tissue from the colitis control and budesonide-treated groups. |
MPO activity and proinflammatory cytokine levels
All rats were killed 24 hours after the last treatment (5 days), and their colons were resected for MPO and cytokine analysis. MPO activity in colonic tissues from healthy, untreated colitis control, and budesonide-treated rats is shown in Figure 4A. The expression profiles of the proinflammatory cytokines IL-6 and TNF-α in the treated and control groups are shown in Figure 4B and C, respectively. All animals suffering from colitis in this study exhibited elevated MPO activity and proinflammatory tissue-cytokine expression compared to healthy rats. However, MPO activity and proinflammatory cytokine expression was substantially decreased in the treated group compared to the untreated colitis group. It is noteworthy that MPO activity and cytokine concentrations were more significantly decreased in the Azo-pu ES NP-treated group (P<0.01) than the ES NP-treated (P<0.05) group or the drug solution-treated group.
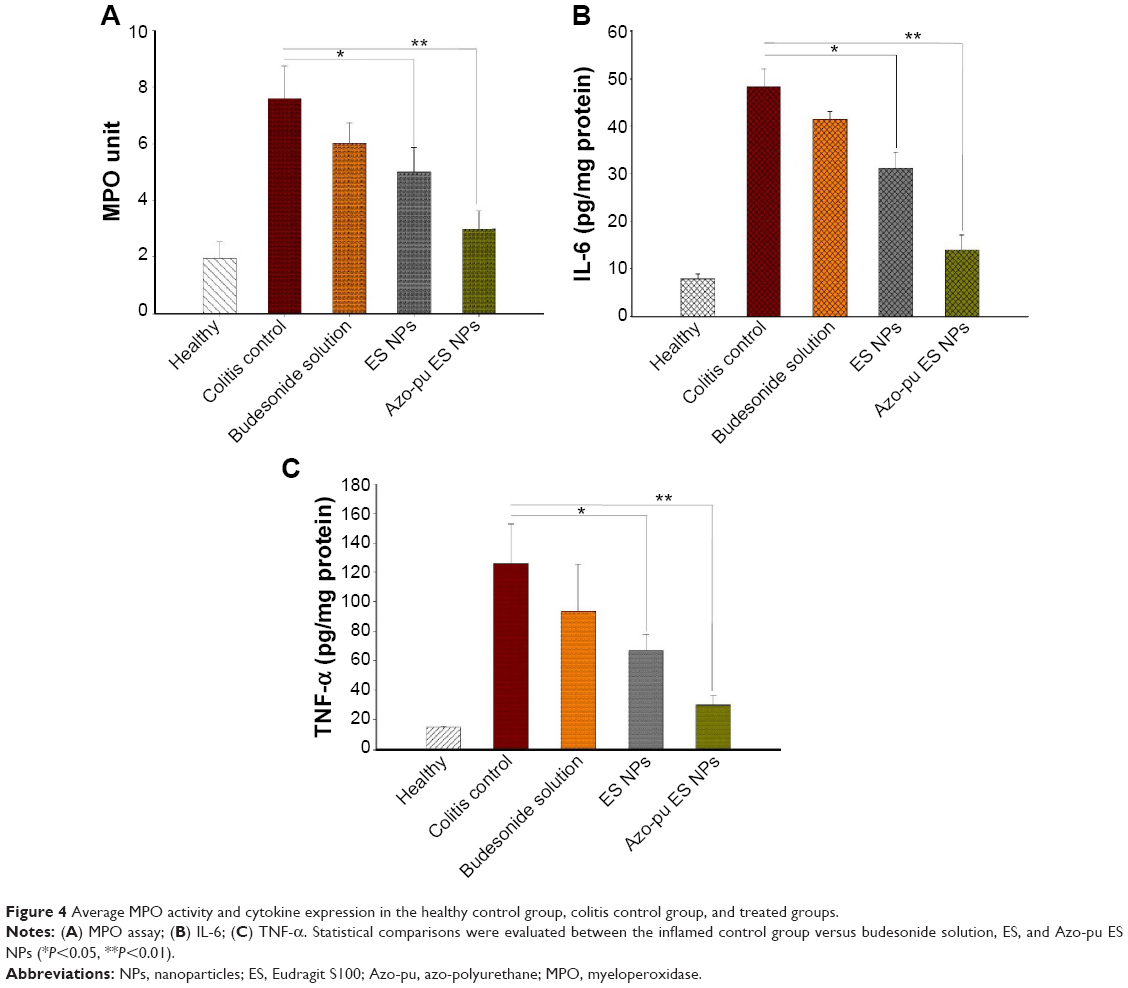 | Figure 4 Average MPO activity and cytokine expression in the healthy control group, colitis control group, and treated groups. |
NP accumulation in inflamed colon tissue
Nanoscale drug-delivery systems are believed to increase colonic residence time due to increased permeability of colitis tissue, leading to selective accumulation of NPs in inflamed tissue.25 The specific adhesion and accumulation of C-6-loaded NPs in healthy and inflamed colon tissue was evaluated after 24 hours, as shown in Figure 5. Confocal images revealed that the Azo-pu ES NPs were spread along the healthy colon tissue; however, the NPs were more abundant at the site of inflammation (Figure 5A). The ES NPs displayed negligible fluorescence intensity in both healthy and inflamed colon tissue. This observation further supported our hypothesis that enzyme/pH dual-sensitive NPs prevent burst drug release in the stomach and the small intestine, and could deliver sufficient amounts of the drug to the inflamed tissue compared to single-triggered pH-sensitive NPs. Quantitative fluorescence analysis revealed that the Azo-pu ES NPs achieved significantly higher C-6 than the ES NPs in healthy and inflamed colon tissues (Figure 5B). Moreover, the C-6 concentration in inflamed colon tissues was fourfold higher than concentrations in healthy colon tissue after administration of Azo-pu ES NPs.
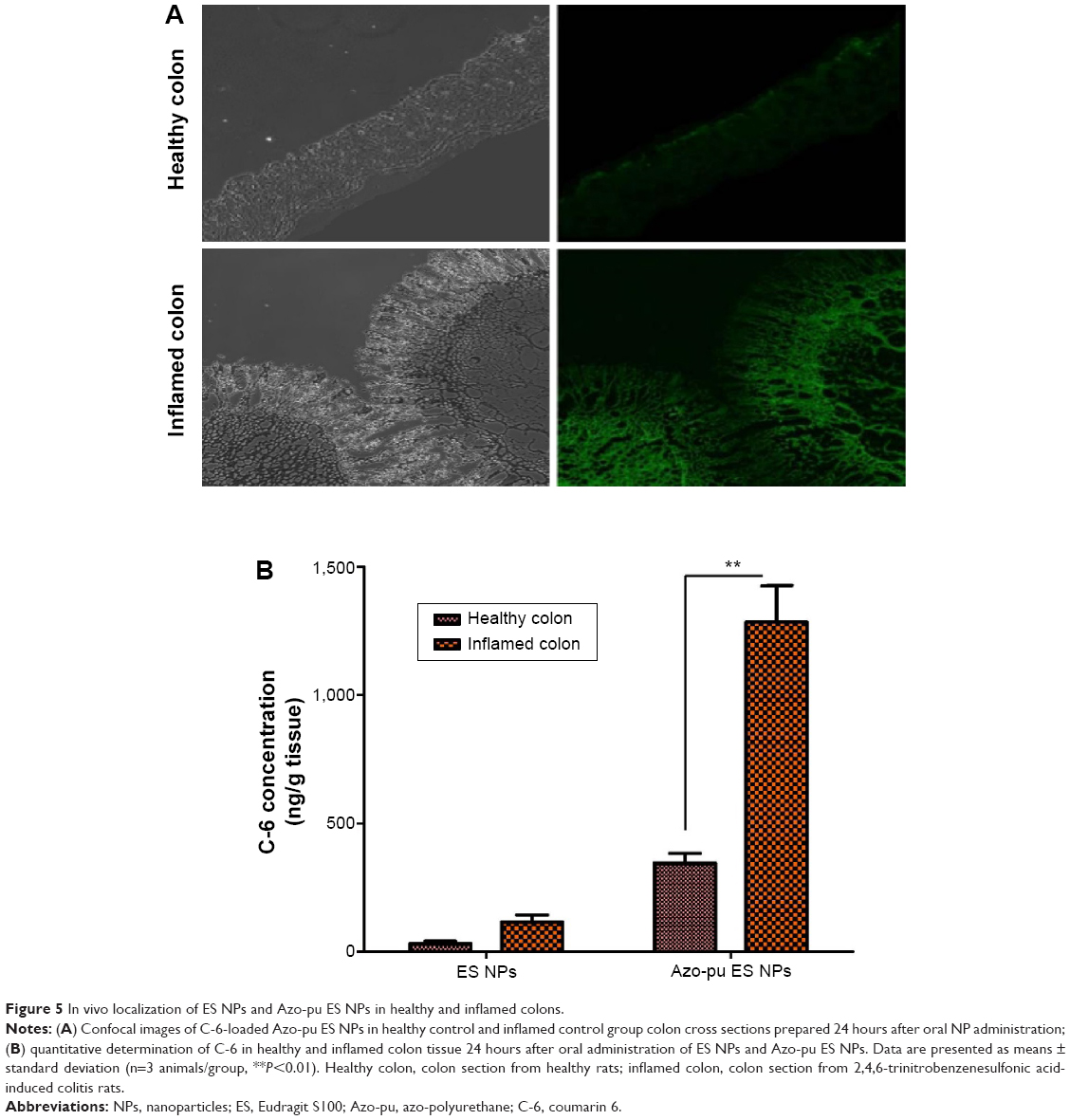 | Figure 5 In vivo localization of ES NPs and Azo-pu ES NPs in healthy and inflamed colons. |
Plasma concentration of C-6 after oral administration of NPs
C-6 solution or C-6-loaded NPs were administered, and systemic absorption was assessed by measuring the plasma C-6 concentration (Figure 6A). The highest plasma concentration of C-6 after oral administration was achieved after 1 hour for the solution group and 3–4 hours for the ES NP group. In contrast, the peak value was delayed until 5–6 hours after the oral administration of Azo-pu ES NPs, and the systemic availability was reduced by about 50%, implying that Azo-pu ES NPs avoid the complete release of budesonide in the small intestine. Therefore, a lower amount of drug is available for systemic absorption.
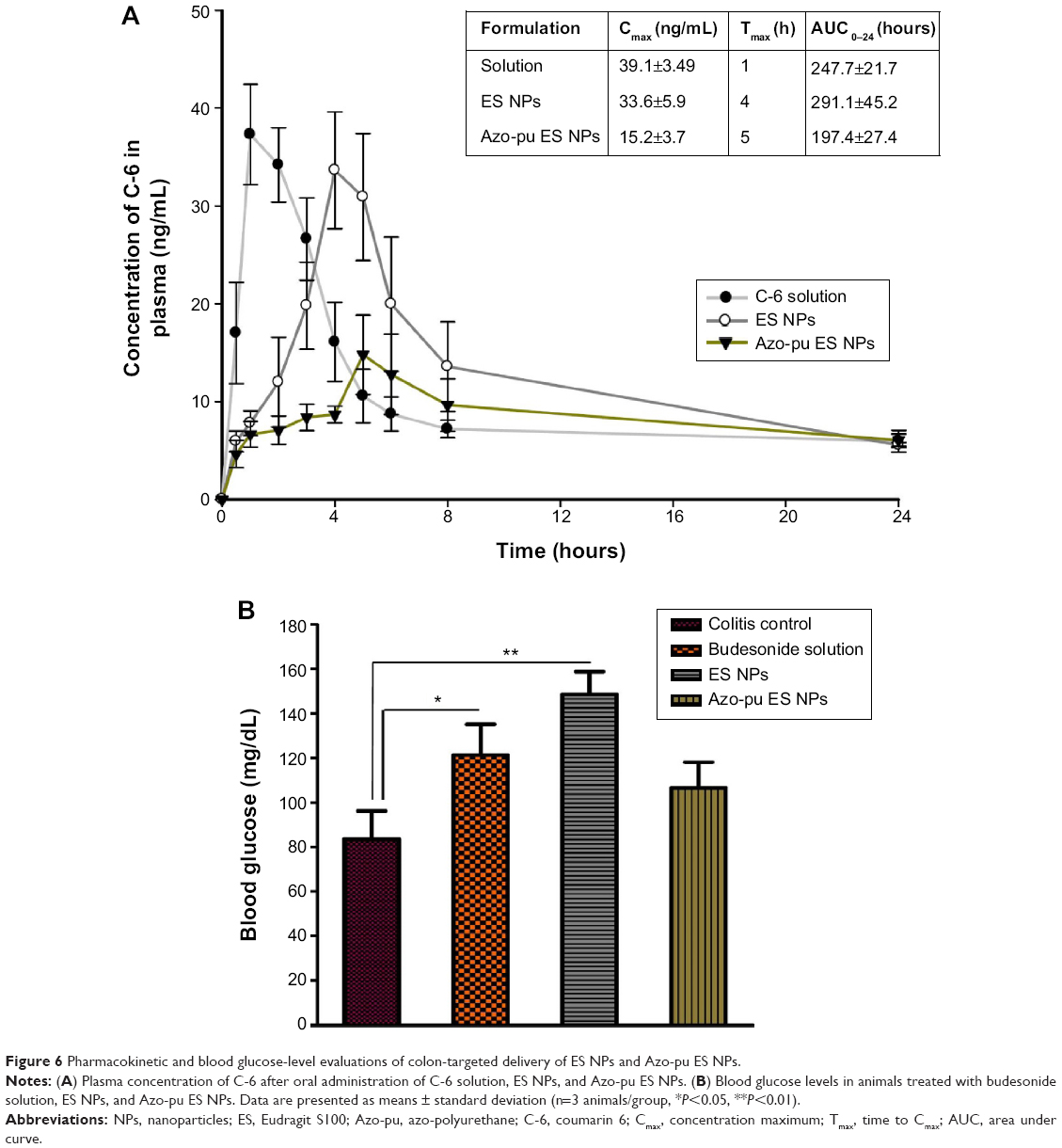 | Figure 6 Pharmacokinetic and blood glucose-level evaluations of colon-targeted delivery of ES NPs and Azo-pu ES NPs. |
Blood glucose levels
The intestinal sugar uptake and transport function of the intestines is known to be enhanced by locally acting glucocorticoids.26 As shown in Figure 6B, blood glucose levels increased after treatment with budesonide solution and ES NPs compared to the untreated colitis control group. However, the animals treated with Azo-pu ES NPs did not show a prominent increase in blood glucose level, indicating that budesonide was delivered efficiently to the colon in a sustained manner while avoiding complete release in the small intestine.
In vivo and in vitro biocompatibility
To determine whether oral administration of Azo-pu ES NPs would have any toxicity, in vivo biocompatibility was evaluated in colitis rats, which mimicked UC patient conditions. The liver and kidney H&E sections of colitis rats treated with Azo-pu ES NPs showed no pathological symptoms, and looked similar to healthy control sections (Figure 7A). Furthermore, macroscopic and microscopic assessment of the small intestine and colon segments revealed no pathological changes. Microphotographs of the small intestine and colon sections stained with H&E are shown in Figure 7B. To evaluate in vivo toxicity of blank Azo-pu ES NPs, animals were treated orally with blank Azo-pu ES NPs (10 mg/rat) for 7 days. There were no significant differences in clinical signs, such as diarrhea, fever, or other symptoms, between the treated and untreated groups. Furthermore, no mortality was observed. Additionally, there were no significant differences in body weight between the treated and untreated groups (Figure 7C). According to the in vitro cytotoxicity study (Figure 7D), varying concentrations of blank Azo-pu ES NPs exhibited no cell toxicity.
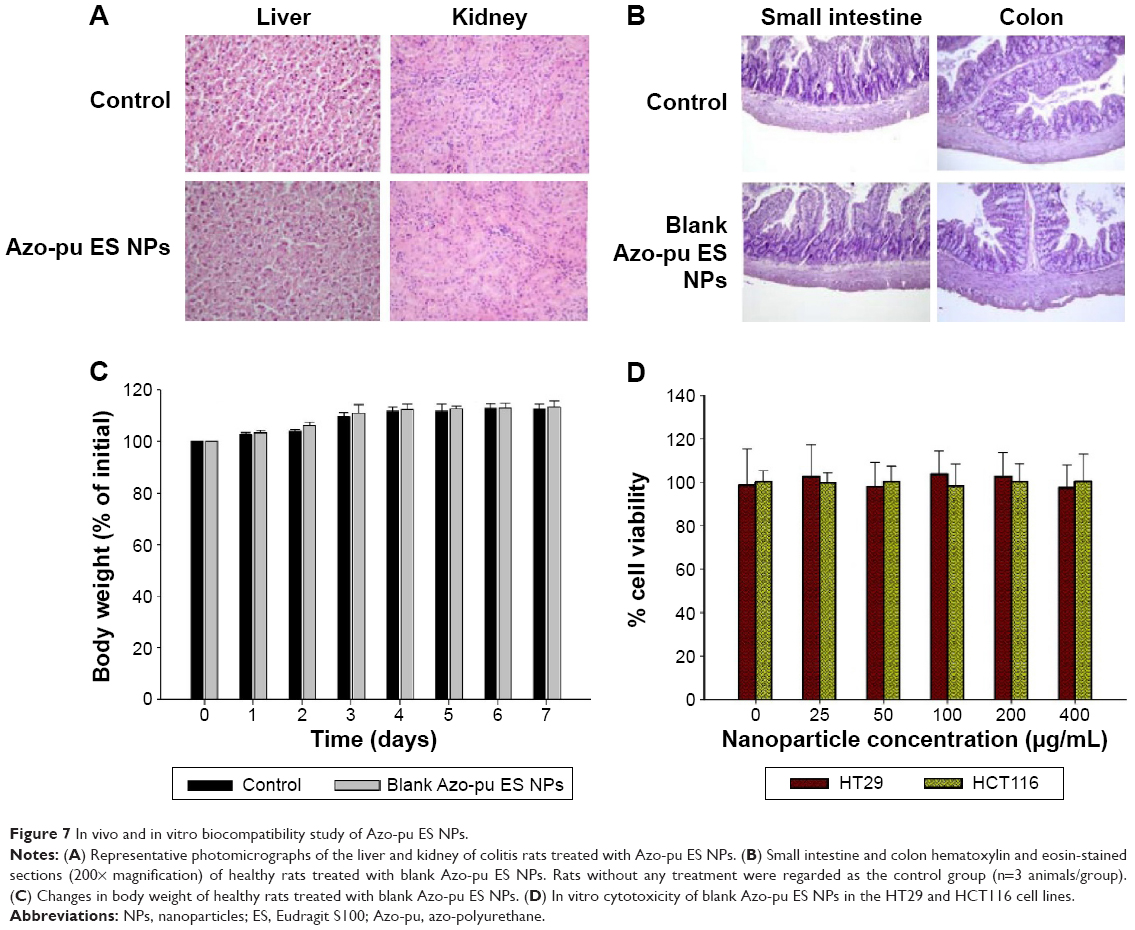 | Figure 7 In vivo and in vitro biocompatibility study of Azo-pu ES NPs. |
Discussion
In a previous study, improved colon-specific delivery of novel enzyme/pH dual-sensitive NPs (Azo-pu ES NPs) was observed compared to single-triggered NPs using a fluorescent marker as a model drug.13 In the current study, we evaluated the therapeutic applications of Azo-pu ES NPs for colitis therapy using a clinically relevant glucocorticoid – budesonide. Since current budesonide formulations using a single pH-dependent strategy are not satisfactory for treating UC, due to premature drug release in the ileum before reaching the inflamed area of the distal colon,9 we hypothesized that enzyme/pH dual-sensitive Azo-pu ES NPs loaded with budesonide could retain budesonide and deliver the drug specifically and sufficiently to the distal inflamed segments of the colon, thereby enhancing therapeutic efficacy of budesonide.
Results from the in vitro drug-release study in medium with a gradually changing pH that mimics the environment of the GI tract supported our hypothesis that Azo-pu ES NPs minimize burst release in ileum-like conditions (pH 7.4), and show sustained release thereafter (Figure 1D). The initial drug release (<20%) at acidic pH over the first 6 hours was likely due to unentrapped drug molecules present on the surface of the NPs or slow diffusion. These results demonstrated that unlike ES NPs, Azo-pu ES NPs can efficiently retain entrapped drugs from the stomach until reaching the colon to increase the drug availability in the inflamed colon, which is a key feature for colon-targeted delivery. Enzyme-triggered release from Azo-pu ES NPs in the presence of rat cecal content, which mimics the UC patient colonic environment, was shown in our previous study.13 C-6 was chosen as a model drug to facilitate analysis, due to rapid degradation of budesonide in the presence of cecal content.27
The therapeutic efficacy of budesonide by Azo-pu ES NPs was evaluated in a TNBS-induced rat colitis model compared to ES NPs and budesonide solution. The clinical activity score, colon/body weight ratio, and colon damage were markedly increased in the colitis control group compared to the treated group (Figure 2). However, NP treatment ameliorated inflammation better than budesonide solution treatment. The negligible efficacy of budesonide solution might have been due to absorption and extensive hepatic metabolism before reaching the inflamed colon. Furthermore, in all cases, the Azo-pu ES NPs exhibited significantly better therapeutic efficacy than ES NPs. The severity of inflammation and recovery was histologically evaluated in the untreated and treated groups (Figure 3A). TNBS-induced colitis changed the structure of the colon, with a loss in the protective epithelial layer and extensive swelling and thickening of the bowel wall. Colitis was alleviated with NPs or budesonide solution treatment; however, NPs had a greater effect. Interestingly, the general microscopic appearance of the group treated with Azo-pu ES NPs was quite close to the healthy control group, with epithelium recovery and no severe swelling of the bowel wall. The grade of tissue inflammation and recovery was also determined by MPO and proinflammatory cytokine assays (Figure 4). MPO, which is the most abundant protein in neutrophils, is widely used as a standard test in IBD animal models and in the feces of IBD patients.28 In the colitis control group, the MPO level was significantly higher, whereas MPO was downregulated in the colonic tissues of treated groups. However, the Azo-pu ES NP-treated group displayed significantly lower MPO levels than the other treatment groups. In the inflamed area, the activation of macrophages leads to proinflammatory cytokine secretion, including IL-6 and TNF-α.29 Therefore, quantification of IL-6 and TNF-α was essential in colon tissues, and was measured by ELISA, as shown in Figure 4B and C, respectively. The expression of both IL-6 and TNF-α was reduced in the colon tissues of treated rats when compared to untreated colitis rats. However, Azo-pu ES NPs suppressed IL-6 and TNF-α more prominently than the other treatments, indicating enhanced therapeutic efficacy of budesonide in colitis.
Unlike conventional colon-specific delivery systems that release the drug contents to the colonic epithelium independently of their healthy or inflamed state, NP-based drug carriers could represent a promising colon-targeted delivery system, due to their selective adhesion to inflamed segments.30,31 Since the dual-sensitive Azo-pu ES NPs are designed to reach the area of colitis, we assumed that Azo-pu ES NPs are selectively accumulated in the inflamed segment of the distal colon. Confocal imaging revealed a greater accumulation of fluorescent Azo-pu ES NPs in the inflamed colon tissue of colitis rats than in the healthy colon tissue (Figure 5). According to previous reports, there are several pathophysiological changes due to mucosal inflammation that are involved in preferential NP accumulation, including elevated mucus production, disrupted intestinal barriers, and infiltration of immune-related cells.32 A subsequent increase in residence time is postulated for NPs compared to existing drug-delivery systems, allowing for a dose reduction. Therefore, size is considered an important factor in the development of colon-specific drug-delivery strategies for the treatment of IBD, because carrier size impacts their accumulation in the inflamed colon.33
The therapeutic goal of colon-targeted delivery for colitis therapy is to maximize the drug availability in the inflamed site while minimizing systemic drug absorption and pharmacological activity in healthy areas of the GI tract. Our pharmacokinetic results showed that plasma C-6 levels in rats treated with Azo-pu ES NPs were significantly lower than in rats treated with ES NPs and C-6 solution (Figure 6A). These results can be attributed to the different release patterns between the single- and dual-sensitive NPs. ES NPs were expected to release all C-6 content in the early part of the GI tract or the ileum, leading to higher systemic absorption from the small intestine before reaching the colon. However, in the case of Azo-pu ES NPs, the slow release of the C-6 in the ileum resulted in lower plasma concentrations and lower systemic bioavailability, implying Azo-pu ES NPs are a safer treatment option than a single pH-dependent system in terms of systemic side effects. The low and delayed peak values of C-6 after oral administration of Azo-pu ES NPs may be attributed to the slow and incomplete absorption of drugs from the colon compared to the small intestine. To further elucidate colon-specific delivery, the blood glucose level was also measured after the oral administration of budesonide-loaded NPs and budesonide solution (Figure 6B). Orally administered budesonide can increase blood glucose levels by enhancing sugar uptake.26 We observed an increase in the blood glucose level after treatment with budesonide solution and ES NPs compared to the untreated control group. However, animals treated with Azo-pu ES NPs did not show a significant enhancement in blood glucose level from the other treatment groups, further supporting the bioavailability study. Taken together, this delivery system appears to avoid complete drug release in the early parts of the GI tract and unwanted systemic side effects.
Since local and systemic toxicity of Azo-pu, especially in a comixture with ES and budesonide, has not been previously reported, we performed in vivo and in vitro biocompatibility studies of Azo-pu ES NPs for colon-targeted applications. Since ES is a US Food and Drug Administration-approved polymer for oral applications, its biocompatibility was not a concern. In vivo toxicity studies in the liver and kidney were performed, because the accumulation of Azo-pu ES NPs in the inflamed colon, as shown in Figure 5A, implies that NPs may be arrested inside the inflamed tissue and absorbed into systemic circulation after degradation. No noticeable toxicity was observed in rats treated with Azo-pu ES NPs (Figure 7A). Budesonide is a safe locally acting steroid drug, due to extensive hepatic metabolism by CYP3A enzymes.6 Our main concern was to evaluate the toxicity of our carrier system (Azo-pu ES NPs), which has not been reported before. Therefore, we further evaluated blank particles in high concentrations in healthy rats and in cell biocompatibility studies. No inflammation was observed in healthy sections of the small intestine or colon (Figure 7B). No apparent body weight change in healthy rats was observed when treated with blank NPs, even with a fivefold-higher dose (10 mg/rat) than was used for the evaluation of therapeutic efficacy. Biocompatibility of Azo-pu ES NPs was further confirmed by in vitro cytotoxicity tests in the colon-originated cell lines HT29 and HCT116 (Figure 7D). Overall, neither the in vitro nor the in vivo biocompatibility study showed any signs of toxicity with Azo-pu ES NPs. However, further investigations are warranted to evaluate the potential toxicity with long-term use.
Conclusion
Based on our findings, we propose that prematurely released budesonide from a pH-dependent system in the ileum can be systemically absorbed, resulting in drug loss before reaching the colon and unwanted systemic side effects. However, Azo-pu ES NPs have the ability to avoid burst release in the ileum and deliver a sufficient amount of budesonide specifically to the inflamed colon, followed by a sustained release of budesonide by enzymatic reduction of the azo group in Azo-pu, resulting in enhanced therapeutic efficacy of budesonide compared to single-triggered ES NPs. The proposed drug-release mechanisms of the novel enzyme/pH-sensitive NPs in different GI-tract segments are summarized in Figure 8. These autonomous and complementary release mechanisms incorporated into the devised polymer system should overcome the limitations associated with the single-triggered release approach and improve site specificity. The enzyme/pH dual-sensitive NPs presented in this study appear to be a promising drug-delivery system for IBD therapy.
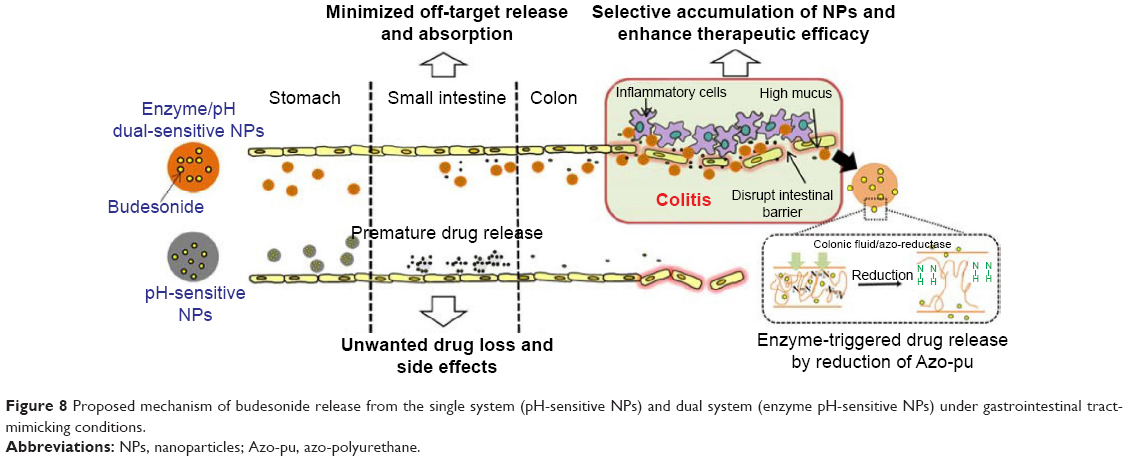 | Figure 8 Proposed mechanism of budesonide release from the single system (pH-sensitive NPs) and dual system (enzyme pH-sensitive NPs) under gastrointestinal tract-mimicking conditions. |
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the South Korean government (MSIP; number 2009-0083538).
Disclosure
The authors report no conflicts of interest in this work.
References
Travis SP, Danese S, Kupcinskas L, et al. Once-daily budesonide MMX in active, mild-to-moderate ulcerative colitis: results from the randomised CORE II study. Gut. 2014;63(3):433–441. | ||
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–13785. | ||
Knutson CG, Mangerich A, Zeng Y, et al. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc Natl Acad Sci U S A. 2013;110(26):E2332–E2341. | ||
Seow CH, Benchimol EI, Griffiths AM, Otley AR, Steinhart AH. Budesonide for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2008;(3):CD000296. | ||
Nakase H, Okazaki K, Tabata Y, et al. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J Pharmacol Exp Ther. 2000;292(1):15–21. | ||
Tromm A, Bunganič I, Tomsová E, et al. Budesonide 9 mg is at least as effective as mesalamine 4.5 g in patients with mildly to moderately active Crohn’s disease. Gastroenterology. 2011;140(2):425.e1–434.e1. | ||
Lautenschläger C, Schmidt C, Fischer D, Stallmach A. Drug delivery strategies in the therapy of inflammatory bowel disease. Adv Drug Deliv Rev. 2014;71:58–76. | ||
Friend DR. New oral delivery systems for treatment of inflammatory bowel disease. Adv Drug Deliv Rev. 2005;57(2):247–265. | ||
Sandborn WJ, Travis S, Moro L, et al. Once-daily budesonide MMX® extended-release tablets induce remission in patients with mild to moderate ulcerative colitis: results from the CORE I study. Gastroenterology. 2012;143(5):1218–1226.e1–e2. | ||
Gross V, Bunganic I, Belousova EA, et al. 3 g Mesalazine granules are superior to 9 mg budesonide for achieving remission in active ulcerative colitis: a double-blind, double-dummy, randomised trial. J Crohns Colitis. 2011;5(2):129–138. | ||
Hong S, Yum S, Yoo HJ, et al. Colon-targeted cell-permeable NFκB inhibitory peptide is orally active against experimental colitis. Mol Pharm. 2012;9(5):1310–1319. | ||
Zhu Q, Talton J, Zhang G, et al. Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat Med. 2012;18(8):1291–1296. | ||
Naeem M, Kim W, Cao J, Jung Y, Yoo JW. Enzyme/pH dual sensitive polymeric nanoparticles for targeted drug delivery to the inflamed colon. Colloids Surf B Biointerfaces. 2014;123:271–278. | ||
Bigelow H, Robinson D. Azobenzene. Organic Synth. 1942;22:28. | ||
Yoo JW, Giri N, Lee CH. pH-sensitive Eudragit nanoparticles for mucosal drug delivery. Int J Pharm. 2011;403(1–2):262–267. | ||
Krishnamachari Y, Madan P, Lin S. Development of pH-and time-dependent oral microparticles to optimize budesonide delivery to ileum and colon. Int J Pharm. 2007;338(1):238–247. | ||
Vandamme TF, Lenourry A, Charrueau C, Chaumeil JC. The use of polysaccharides to target drugs to the colon. Carbohydr Polym. 2002;48(3):219–231. | ||
Niebel W, Walkenbach K, Béduneau A, Pellequer Y, Lamprecht A. Nanoparticle-based clodronate delivery mitigates murine experimental colitis. J Control Release. 2012;160(3):659–665. | ||
Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96(3):795–803. | ||
Hartmann G, Bidlingmaier C, Siegmund B, et al. Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. J Pharmacol Exp Ther. 2000;292(1):22–30. | ||
Makhlof A, Tozuka Y, Takeuchi H. pH-sensitive nanospheres for colon-specific drug delivery in experimentally induced colitis rat model. Eur J Pharm Biopharm. 2009;72(1):1–8. | ||
Weigmann B, Lehr HA, Yancopoulos G, et al. The transcription factor NFATc2 controls IL-6-dependent T cell activation in experimental colitis. J Exp Med. 2008;205(9):2099–2110. | ||
Lamprecht A, Yamamoto H, Takeuchi H, Kawashima Y. A pH-sensitive microsphere system for the colon delivery of tacrolimus containing nanoparticles. J Control Release. 2005;104(2):337–346. | ||
Mezzena M, Scalia S, Young PM, Traini D. Solid lipid budesonide microparticles for controlled release inhalation therapy. AAPS J. 2009;11(4):771–778. | ||
Lamprecht A, Schäfer U, Lehr CM. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–793. | ||
Thiesen A, Wild G, Tappenden K, et al. The locally acting glucocorticosteroid budesonide enhances intestinal sugar uptake following intestinal resection in rats. Gut. 2003;52(2):252–259. | ||
Yadav V, Gaisford S, Merchant HA, Basit AW. Colonic bacterial metabolism of corticosteroids. Int J Pharm. 2013;457(1):268–274. | ||
Mendoza J, Abreu M. Biological markers in inflammatory bowel disease: practical consideration for clinicians. Gastroenterol Clin Biol. 2009;33 Suppl 3:S158–S173. | ||
Ali H, Weigmann B, Neurath M, Collnot E, Windbergs M, Lehr CM. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. Journal of Controlled Release. 2014;183:167–177. | ||
Ulbrich W, Lamprecht A. Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases. J R Soc Interface. 2010;7 Suppl 1:S55–S66. | ||
Lamprecht A. IBD: selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastroenterol Hepatol. 2010;7(6):311–312. | ||
Lamprecht A, Yamamoto H, Takeuchi H, Kawashima Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J Pharmacol Exp Ther. 2005;315(1):196–202. | ||
Schmidt C, Lautenschlaeger C, Collnot EM, et al. Nano- and microscaled particles for drug targeting to inflamed intestinal mucosa – first in vivo study in human patients. J Control Release. 2013;165(2):139–145. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.