")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Enhanced oral bioavailability of silymarin using liposomes containing a bile salt: preparation by supercritical fluid technology and evaluation in vitro and in vivo
Authors Yang G, Zhao Y, Zhang Y, Dang B, Liu Y, Feng N
Received 18 July 2015
Accepted for publication 20 September 2015
Published 22 October 2015 Volume 2015:10(1) Pages 6633—6644
DOI https://doi.org/10.2147/IJN.S92665
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Gang Yang,1 Yaping Zhao,2 Yongtai Zhang,1 Beilei Dang,1 Ying Liu,1 Nianping Feng1
1School of Pharmacy, Shanghai University of Traditional Chinese Medicine, 2School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
Abstract: The aim of this investigation was to develop a procedure to improve the dissolution and bioavailability of silymarin (SM) by using bile salt-containing liposomes that were prepared by supercritical fluid technology (ie, solution-enhanced dispersion by supercritical fluids [SEDS]). The process for the preparation of SM-loaded liposomes containing a bile salt (SM-Lip-SEDS) was optimized using a central composite design of response surface methodology with the ratio of SM to phospholipids (w/w), flow rate of solution (mL/min), and pressure (MPa) as independent variables. Particle size, entrapment efficiency (EE), and drug loading (DL) were dependent variables for optimization of the process and formulation variables. The particle size, zeta potential, EE, and DL of the optimized SM-Lip-SEDS were 160.5 nm, -62.3 mV, 91.4%, and 4.73%, respectively. Two other methods to produce SM liposomes were compared to the SEDS method. The liposomes obtained by the SEDS method exhibited the highest EE and DL, smallest particle size, and best stability compared to liposomes produced by the thin-film dispersion and reversed-phase evaporation methods. Compared to the SM powder, SM-Lip-SEDS showed increased in vitro drug release. The in vivo AUC0-t of SM-Lip-SEDS was 4.8-fold higher than that of the SM powder. These results illustrate that liposomes containing a bile salt can be used to enhance the oral bioavailability of SM and that supercritical fluid technology is suitable for the preparation of liposomes.
Keywords: silymarin, solution-enhanced dispersion by supercritical fluids, liposomes, bile salt, bioavailability
Introduction
Silymarin (SM), a hepatoprotective agent obtained from the herb Silybum marianum (L.), is widely used in the treatment of liver diseases such as cirrhosis, hepatitis, and fatty infiltration due to alcohol and toxins. A mixture of flavonolignan isomers, namely, silybin, isosilybin, silydianin, and silychristin, collectively constitute SM.1,2 Among these isomers, silybin is the major component of SM, representing approximately 60%–70%, and is responsible for its pharmacological activity.3,4 However, the therapeutic effects of SM are restricted owing to its poor enteral absorption (23%–47%), instability in gastric juices, and poor solubility.5,6 Some commercial SM products, such as Yiganling tablets, show poor into the blood. In recent years, different strategies have been investigated to improve the solubility and bioavailability of SM, such as self-microemulsifying drug delivery systems,7 solid dispersions,8 porous silica nanoparticles,9 phospholipid complexes,10 solid lipid nanoparticles,11 nanostructured lipid carriers,12 and liposomes.13
Liposomes, as a delivery system, can improve the therapeutic activity and safety of drugs. They consist of lipid bilayers with an inside water phase that can encapsulate both water-soluble and lipophilic drugs. In the past decade, some research efforts have demonstrated the effectiveness of including bile salts in improving the in vivo stability and performance of drugs in orally administered liposomes.14 The ability of bile salts to improve the passage of lipophilic drug molecules across biological membranes, and thus augment their oral bioavailability for many drug candidates, has been reported.15 Liposomes have also been employed successfully in drug delivery systems for improving the solubility and bioavailability of some poorly soluble drugs.16
Liposomes can be prepared using a wide range of methods, such as thin-film dispersion (TFD),17 reversed-phase evaporation (RPE),18 alcohol injection,19 and spray-freeze-drying.20 Although variable, almost all these methods require a large quantity of organic solvents. The use of organic solvents is problematic in the preparation of medicinal liposomes because of undesirable effects on the human body and environment that might be caused by the residual solvent.21 Moreover, some of the methods need complicated additional steps for proliposomal formulation, together with the use of a cryoprotectant and high process temperature, which limits their wide application.
Recently, supercritical fluid of carbon dioxide (SCF-CO2) technology has emerged as a major technique for the preparation of liposomes. The solvent properties of SCFs can be adjusted by altering the experimental conditions (temperature and pressure). In particular, CO2 offers considerable potential as an environmentally friendly alternative solvent that can replace organic solvents because it has a low critical temperature (Tc=31°C) and pressure (Pc=7.38 MPa) and is nontoxic, nonflammable, and inexpensive. The SCF-CO2 method also has a notable advantage in that it controls the physicochemical properties of liposomes, including their size, shape, and yield, all of which can be altered by the solvent properties of the SCF resulting in liposomes that are more stable to store.22,23 Solution-enhanced dispersion by supercritical fluids (SEDS) is one of the SCF-CO2 methods used widely for the preparation of liposomes. This method uses semi-continuous processes to atomize the solution into a supercritical atmosphere. If the drug is sparingly soluble in the SCF, which is highly soluble in the solvent, droplets with SCF in the solvent can produce an antisolvent effect. This process produces supersaturated solutions, facilitating precipitation of the solid in the form of small particles.24
According to the US Food and Drug Administration and the European Agency for the Evaluation of the Medical Products guidelines for liposomal drug products, the physicochemical properties of liposomes are critical to ensure the quality of the drug products. Our study was designed to prepare the SM-loaded liposomes containing a bile salt (sodium glycocholate [SGC]) by solution-enhanced dispersion using the SCF technique (SM-Lip-SEDS) and optimizing the preparation conditions by response surface methodology. Comparisons were made with liposomes prepared by conventional methods such as TFD and RPE. Parameters of particle size, zeta potential, morphology, encapsulation efficiency, drug loading (DL), and stability were compared between liposomes prepared by all methods. Moreover, both in vitro dissolution and in vivo pharmacokinetics were evaluated with the aim to enhance the oral bioavailability of SM.
Materials and methods
Materials
SM (purity >80%) was purchased from Dalian Meilun Biotech Co, Ltd (Dalian, People’s Republic of China). Soybean phosphatidylcholine (purity >90%) and hydrogenated soybean phosphatidylcholine (purity >95%) were obtained from Sigma-Aldrich (St Louis, MO, USA). SGC was supplied by Sinopharm Chemical Reagent Co Ltd (Shanghai, People’s Republic of China). Yiganling tablets were provided by Zhaohui Pharmaceutical Company (Shanghai, People’s Republic of China). CO2 with a purity of 99.99% was obtained from Shanghai Jiao Tong University (Shanghai, People’s Republic of China). All other chemicals were reagent grade and used as received.
Animals
Animal studies with male Wistar rats weighing 250±10 g were conducted with the approval of the Animal Ethical Committee, Shanghai University of Traditional Chinese Medicine. The animals were kept in a controlled environment with free access to a rodent diet and water and were acclimatized for at least 1 week before the start of the study.
High-performance liquid chromatography analysis
The content of SM (based on silybin) was determined using a LC-2010A HT HPLC system (Shimadzu, Kyoto, Japan) with an Agilent Eclipse XDB-C18 column (5 μm, 4.6×250 mm) (Agilent Technologies, Santa Clara, CA, USA). The mobile phase consisted of methanol and pure water (46:54, v/v) at a flow rate of 0.8 mL/min. The effluent was monitored at 288 nm.
Preparation of SM liposomes containing sodium deoxycholate
Preparation of SM-Lip-SEDS
The supercritical pilot plant at Nantong Huaxing Petroleum Devices Co, Ltd (Nantong, People’s Republic of China) (Figure 1A) was employed in this study. Briefly, this apparatus included three major components: a CO2 delivery system, an organic solution delivery system, and a precipitation system. The supercritical CO2 and organic solutions were separately pumped into the high-pressure vessel through different inlets of the coaxial nozzle (the inner and outer tubule diameters were 0.2 mm and 1 mm, respectively) and continuously discharged from the bottom. The inner structure of the nozzle is shown in Figure 1B. For the preparation of SM-Lip-SEDS, CO2 from the cylinder (Figure 1A) (1) was refrigerated (2) and compressed by the high-pressure pump (3) before the temperature controlling system (6) was activated to increase the temperature. The pressure of the precipitation system was increased by injection of CO2 to the set pressure. After the pressure and temperature of the view vessel (9) reached the required values, valve C was adjusted to maintain constant pressure in the vessel. Then, SM and the excipients with the required weight ratios were separately dissolved or dispersed in a mixture of ethanol and dichloromethane (12/13, v/v). The solution was aspirated by a high-pressure constant flow pump (11) (LC100; Nantong, People’s Republic of China) at a constant flow rate. SCF-CO2 and the organic solution mixed and diffused rapidly. Solutes originally dissolved in the organic solvent rapidly reached supersaturation, resulting in the precipitation of SM proliposomes in the vessel. Once the solution was exhausted, valve B was closed and SCF-CO2 was pumped continuously for ~40 minutes to remove the residual organic solvent from the SM proliposomes. Valve A was then closed, while valve C remained open. The pressure of the precipitation vessel was slowly reduced to 0 MPa, and the product in the vessel was collected. The precipitated proliposomes were hydrated with distilled water at 50°C to form liposomes with a concentration of 20 mg/mL.
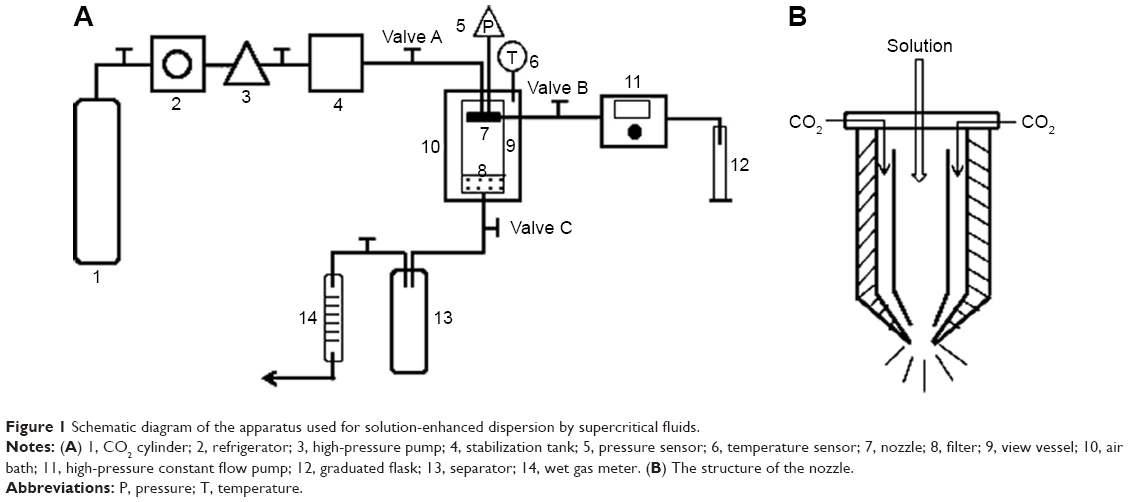 | Figure 1 Schematic diagram of the apparatus used for solution-enhanced dispersion by supercritical fluids. |
Drug EE and DL
The EE of SM liposomes was calculated by determining the amount of free drug using an ultrafiltration technique. SM liposomes were placed in the upper chamber of a centrifuge tube matched with an ultrafilter (10 kDa; Pall Corporation, Port Washington, NY, USA) and centrifuged for 10 minutes at 5,000 rpm. The total and separated drug contents were measured using high-performance liquid chromatography (HPLC), and the EE and DL were calculated by the following equations:
|
|
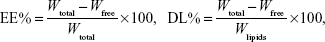
where Wfree is the analyzed weight of the untrapped drug, Wtotal is the weight of the drug in the liposome dispersion, and Wlipids is the weight of lipids added in the system.
Experimental design
On the basis of previous experiments to select influence factors, three critical process factors known to affect the results were adopted for a central composite design (CCD), which is a popular template for response surface methodology, using Design Expert software (Version 8.0.6). The three independent variables were the ratio of SM to phospholipids (w/w), flow rate of solution (mL/min), and pressure (MPa), which were labeled as X1, X2, and X3, respectively. Each variable was coded at the levels corresponding to the preliminary study results. The parameters and their levels are shown in Table 1. The dependent variables, Y1, Y2, and Y3, were the particle size, EE, and DL, respectively. The complete design consisted of 20 experimental points to establish the optimum conditions for the preparation of SM-Lip-SEDS. The nonlinear quadratic model generated by the design is
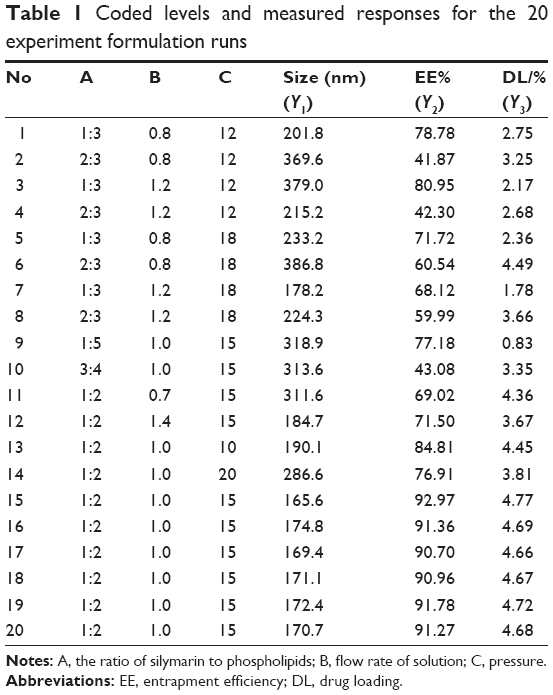 | Table 1 Coded levels and measured responses for the 20 experiment formulation runs |
|
|
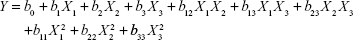
where Y is the measured response (dependent variable) associated with each factor-level combination, expressed in terms of the particle size, EE, and DL for the formulation variables; b0 is an intercept; and b1, b2, b3, b12, b13, b23, b11, b22, and b33 are the regression coefficients.
The P-values related to the regression coefficients indicated the significance of the factors on the response. Analysis of variance and the coefficient of determination (R2) were also applied to determine the suitability of the model.25
Preparation of SM-Lip-TFD
SM-Lip-TFD was prepared according to Bahia et al26 with modifications to optimize the conditions. Optimization was confirmed by the following step: SM, soybean phosphatidylcholine, hydrogenated soybean phosphatidylcholine, and SGC were first dissolved in a mixture of ethanol and dichloromethane (12/13, v/v). The solution was then transferred to a 50 mL round flask, and the organic solvent was removed by a rotary evaporator (R-205; Shanghai, People’s Republic of China) at 40°C for 1 hour under vacuum to form a thin lipid film on the inner wall of the flask. The thin film was hydrated with 10 mL of distilled water at 30°C for 1 hour to obtain a dispersion of liposomes. The liposome dispersion was stored at room temperature until analysis.
Preparation of SM-Lip-RPE
SM-Lip-RPE was prepared according to previous reports.27,28 Using the optimal condition, lipid materials and SGC were dissolved in 10 mL of a solvent mixture containing ethanol and dichloromethane (12/13, v/v) and sonicated to ensure homogeneous mixing. The water phase, which was the SM solution in a mixture of ethanol and distilled water (1:10, v/v), was added to the organic solvent phase, and the mixture was sonicated for 5 minutes. The organic solvent was removed under vacuum on a rotary evaporator at 40°C for ~1 hour to obtain SM liposomes.
Determination of particle size and zeta potential
The mean diameter and zeta potential of the SM-loaded liposomes were measured using a Nano ZS90 Zetasizer (Malvern Instruments, Malvern, UK). The zeta potential was calculated using the Smoluchowski equation. The samples were appropriately diluted with distilled water, and the particle size was evaluated using volume distribution.
Transmission electron microscopy
The general morphology of the SM-loaded liposomes was observed by transmission electron microscopy (TEM) (JEM-2100; JEOL, Tokyo, Japan). The samples were diluted initially with distilled water, followed by placement on a film-coated copper grid. The samples were then stained with a drop of 2% phosphotungstic acid and allowed to dry before examination by TEM.
X-ray diffraction
Samples were analyzed using an X-ray polycrystalline diffractometer (D8 ADVANCE; BrukerOptik GmbH, Ettlingen, Germany). Cu Kα radiation was used as the X-ray source. Samples were scanned over the range of 5°–60° 2è at a scanning speed of 6°/min and a step size of 0.02°. The voltage was set at 40 kV, and the current was 40 mA.
Differential scanning calorimetry
The thermal behaviors of the samples were analyzed using a differential scanning calorimeter (Q2000; New Castle, DE, USA). Samples (5–10 mg) were sealed in a specialized aluminum pan using an aluminum lid with a pinhole. Measurements were performed over 0°C–230°C under a dry nitrogen atmosphere at a heating rate of 10°C/min.
Stability study
Accelerated drug leakage studies were performed by sonication of liposomes in a pH 6.8 sodium phosphate buffer using an ultrasonic processor (Vibra Cell, Sonics and Materials Inc., Newton, CT, USA). Samples were first put into dialysis bags and closed with strings. Then, separate 2 mL samples were sonicated in 1-minute increments in conical flasks with 20 mL of pH 6.8 buffer solution for up to 8 minutes. Drug leakage was followed by determining the free SM concentration in the buffer solution by using HPLC. The drug leakage rate was calculated according to the following equation:
|
|
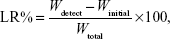
where Wdetect is the analyzed weight of the drug in the buffer solution at any time in minutes, Winitial is the analyzed weight of the drug in the buffer solution at 0 minute, and Wtotal is the analyzed weight of the drug in the liposome dispersion.
In vitro dissolution
In vitro release studies were performed using the dialysis bag method as reported previously.29 The dialysis bag (molecular weight cutoff 8,000–14,000) was soaked in distilled water for 12 hours before use. Then, a volume of suspended SM powder (dispersed in distilled water) and SM-loaded liposomes with the same amount of drug were placed in the dialysis bag. The receptor compartment was filled with 100 mL phosphate buffer solution (pH 6.8) containing 0.3% (w/v) sodium dodecyl sulfate and maintained at 37°C±0.5°C with stirring at 100 rpm. Samples of 1 mL were withdrawn after 10 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 24 hours, 36 hours, 48 hours, and 72 hours and replaced by the same volume of fresh medium at 37°C±0.5°C. The samples were filtered through a 0.45-μm membrane. The SM content was determined by HPLC, as described in Section 2.3.2. Each experiment was carried out in triplicate.
In vivo pharmacokinetics
Rats were assigned randomly to five groups (n=6/group). The animals were starved for 12 hours before the experiment but had free access to water. One group of rats was treated orally with an aqueous suspension of SM (prepared by adding SM to purified water). The second group was treated orally with a commercial SM product (Yiganling tablets) that was dispersed in distilled water. The other three groups of rats received SM-Lip-SEDS, SM-Lip-RPE, and SM-Lip-TFD orally. All rats were treated orally with 20 mg/kg SM equivalent. Blood samples (0.4 mL) were collected from the eye socket vein and placed in heparinized tubes at the indicated time points after dosing. Blood samples were centrifuged (Eppendorf, Hamburg, Germany) at 5,000 rpm for 10 minutes to isolate the plasma. One milliliter of ethyl acetate was added to the plasma, vortex mixed for 1 minute, and then centrifuged at 10,000 rpm for 10 minutes. The supernatant layer was dried under nitrogen and redissolved in 100 μL methanol prior to HPLC analyses for SM as described previously.
Statistical analysis
Data are presented as means ± standard deviation. Statistical data were analyzed by multivariate linear regression for uniform design experiments, and comparisons were made with one-way analysis of variance using SPSS software (v19.0; IBM Corporation, Armonk, NY, USA). A P-value of <5% was considered statistically significant.
Results and discussion
Results of the formulation design
Before beginning the CCD, we investigated the influencing factors including pressure, temperature, flow rate of the solution, the ratio of SM to phospholipids, the ratio of phospholipids to bile salt, and hydration time. Three factors, the ratio of SM to phospholipids (w/w), flow rate of the solution (mL/min), and pressure (MPa), which significantly influenced the results were selected to conduct the following experiments. For evaluating the influence of these parameters on the production of SM-Lip-SEDS and its final properties, 20 experiments were constructed by Design Expert 8.0.6 software and are listed in Table 1. The data were fitted to a quadratic polynomial model using the Design Expert software, and the equations are shown in terms of coded factors.
From the CCD runs, the following models were established:
|
|
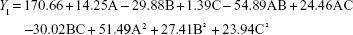
|
|
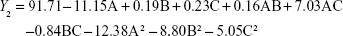
|
|
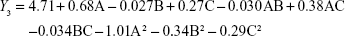
Items were ignored when the P-value was >0.05 because the smaller the P-value the more significant the corresponding coefficient. The model was further developed with only significant items.30 The determination coefficients R2 of Y1, Y2, and Y3 were 0.8590, 0.9632, and 0.9533, respectively. This indicated that 85.9%, 96.3%, and 95.3% of the variation was attributed to the independent variable. Comparisons between predicted and experimental values of the responses using diagnostic case statistic reports are listed in Table 2. These data show that the experimental values are very close to the predicted values with low percentage bias. This suggests that the optimized formulation is reliable and reasonable.
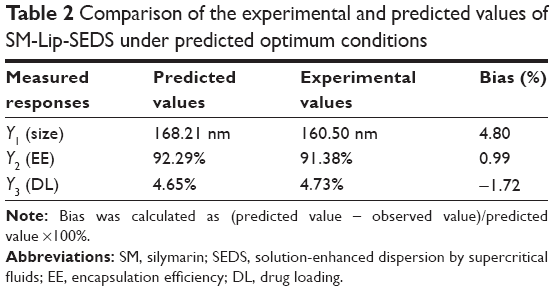 | Table 2 Comparison of the experimental and predicted values of SM-Lip-SEDS under predicted optimum conditions |
Contours and three-dimensional surface plots of the factors that most affect the responses are shown in Figure 2. As shown in Figure 2A and following the Y1 equation, the effect of the factors levels on particle size was significantly influenced by the ratio of SM to phospholipids (A) and the flow rate of solution (B), with large coefficients (14.25 and -29.88). This suggested that the particle size increases with increasing ratios of SM to phospholipids. In contrast, as the flow rate of the solution increases, the particle size decreases. This is due to solute concentration competing against volume expansion. When the solute concentration increased, the nucleation obtained at the lower volume expansion was diminished. Therefore, nucleation allowed to grow for a longer time formed larger particles. In contrast, increasing the flow rate reduced the resident time of a particle inside the nozzle and in the expansion chamber, decreasing particle growth time and leading to a decreased particle size.31,32
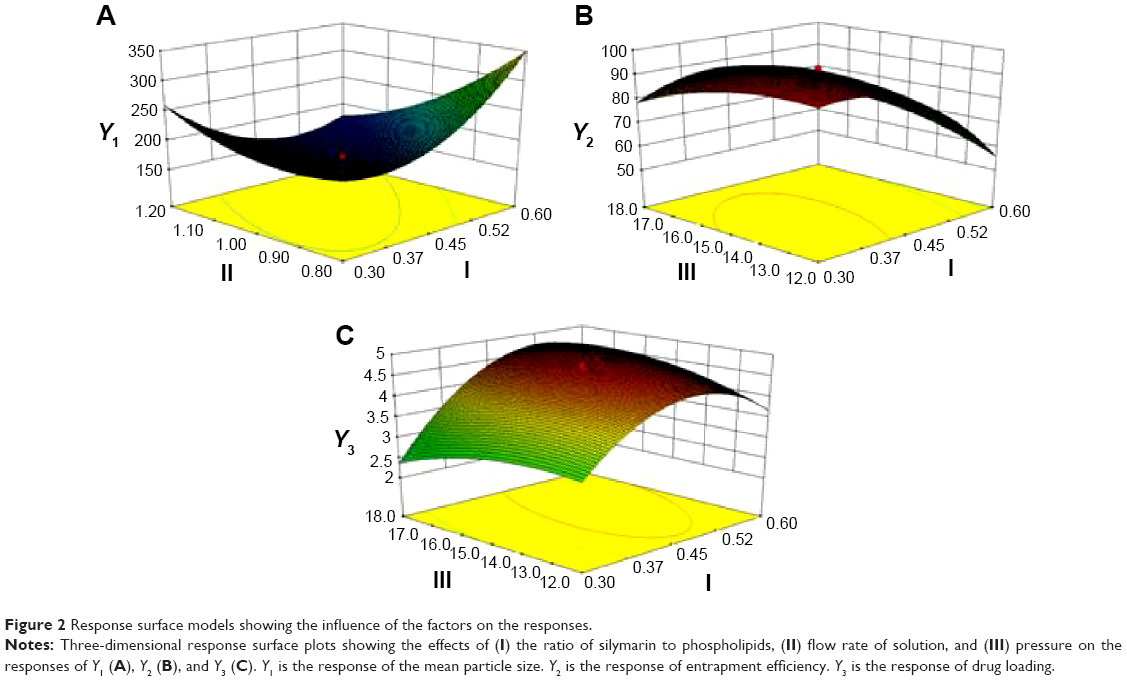 | Figure 2 Response surface models showing the influence of the factors on the responses. |
EE is one of the most important evaluation parameters for liposome carriers. The Y2 equation and Figure 2B indicated that the ratio of SM to phospholipids (A) had a primary influence on the EE for its large negative coefficient (-11.15). This suggested that increasing the amount of SM in the formulation would decrease the EE, which might be related to liposomes lacking sufficient spare space to accommodate excessive drug as the ratio of SM to phospholipids increased.33 In contrast, as the pressure increased, the EE increased gradually. This tendency could be attributed to the higher solubility of SM when the pressure increased.
DL is another important parameter for the evaluation of liposomes. The effect of formulation type and process variables on DL can be evaluated by observing the Y3 equation and Figure 2C. All the variables, except the solution flow rate, exhibited a statistically significant influence on the DL. As the ratio of SM to phospholipids increased, the DL increased gradually to its maximum point and then decreased, exhibiting an asymptotic curve. Pressure exerted a positive influence on DL (0.27). This might be interpreted that, at low ratios of SM to phospholipids, more drugs would be packaged with increasing drug concentrations. However, high levels of encapsulated drug might weaken the stability of liposomes and result in drug leakage.34 The influence of pressure on the DL implied that, by increasing pressure, the CO2–ethanol–dichloromethane ternary system shifts toward a single-phase equilibrium, leading to faster precipitation and more drugs in the liposomes.35
Comparison of liposomes prepared by different methods
The particle size and zeta potential of liposomes produced by the SEDS, TFD, and RPE methods are shown in Figure 3. The SEDS method resulted in the formation of the smallest particles with the greatest absolute zeta potential compared to those obtained by the TFD or RPE methods. TEM images of liposomes obtained by all three methods are shown in Figure 3. These results confirm that SM-Lip-SEDS has the smallest size. The EE and DL of liposomes produced by the different processes are shown in Figure 4. EE and DL were 35.1% and 0.84% and 34.9% and 1.13% for the TFD and RPE methods, respectively. The results were improved by the SEDS method with EE and DL values of 91.4% and 4.7%, respectively. Therefore, the SEDS method was more effective for liposome formation than the conventional methods.
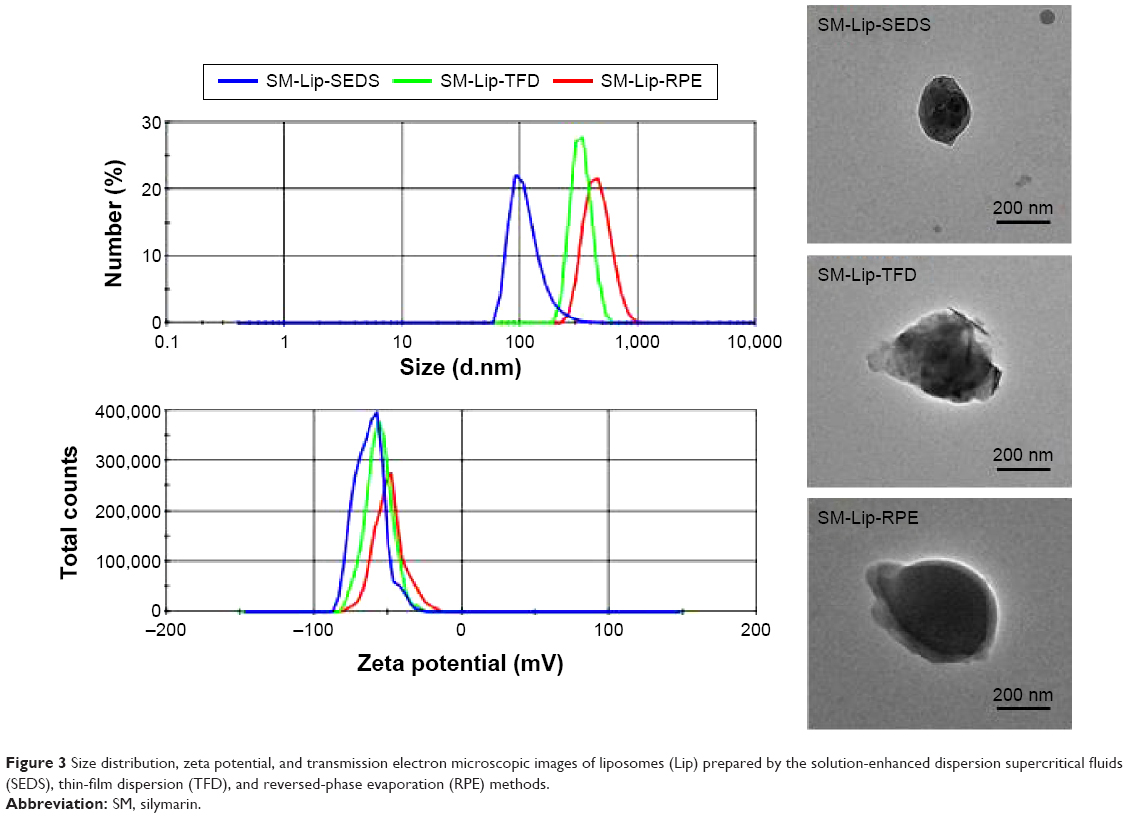 | Figure 3 Size distribution, zeta potential, and transmission electron microscopic images of liposomes (Lip) prepared by the solution-enhanced dispersion supercritical fluids (SEDS), thin-film dispersion (TFD), and reversed-phase evaporation (RPE) methods. |
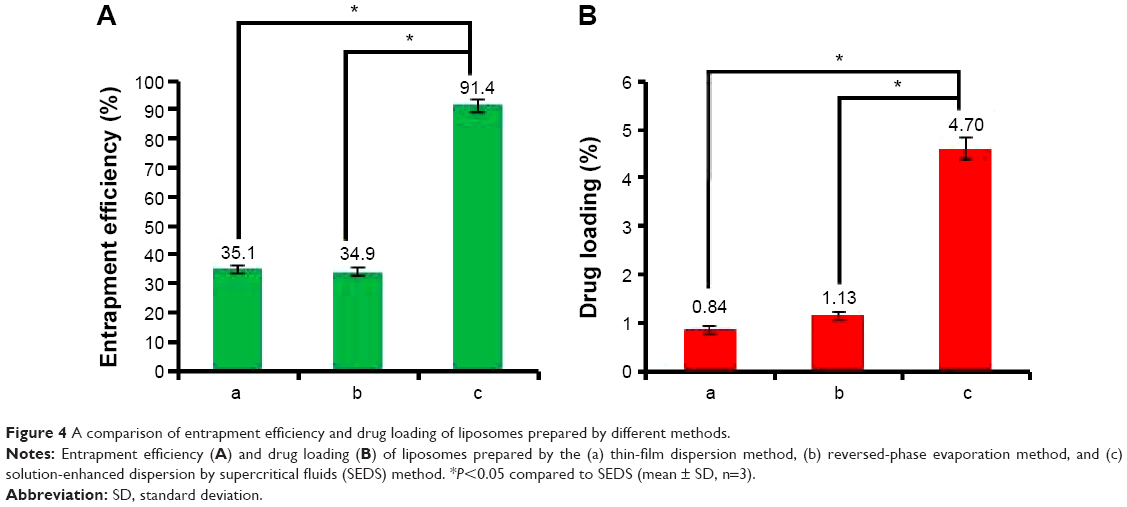 | Figure 4 A comparison of entrapment efficiency and drug loading of liposomes prepared by different methods. |
X-ray diffraction
The crystal structure of SM-Lip-SEDS and each ingredient were analyzed using X-ray diffraction (XRD) (Figure 5). SM powder and SGC showed a crystalline structure, as demonstrated by their sharp and intense diffraction peaks. The patterns for soybean phosphatidylcholine and hydrogenated soybean phosphatidylcholine exhibited no obvious diffraction peak. For the physical mixtures, the XRD pattern showed a number of different peaks that were retained from SM, SGC, and the phosphatidylcholines, suggesting that SM and SGC remained in a crystalline state when mixed together. However, the areas of these peaks were reduced compared with their original areas. This may be due to the different ratio of components in the physical mixtures. In contrast, SM liposomes did not show any strong peak at the same position, indicating that SM and SGC were in an amorphous state in liposomes.36
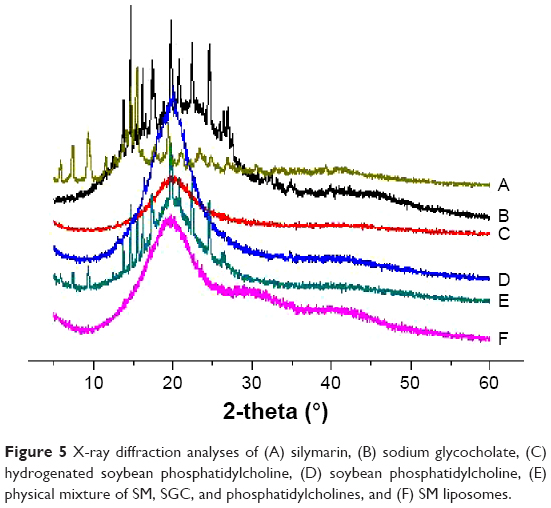 | Figure 5 X-ray diffraction analyses of (A) silymarin, (B) sodium glycocholate, (C) hydrogenated soybean phosphatidylcholine, (D) soybean phosphatidylcholine, (E) physical mixture of SM, SGC, and phosphatidylcholines, and (F) SM liposomes. |
Differential scanning calorimetry
Differential scanning calorimetry (DSC) studies were performed to investigate the physical state of the drug in the liposomes (Figure 6). SGC showed an endothermic melting peak at 175.66°C. The melting point of SM was observed in the DSC curve as an endothermic peak at 200.37°C. For both phosphatidylcholines, there was no obvious endothermic peak. Two melting peaks of SGC and SM appeared in the physical mixture with a small change, probably due to the interaction of drug and material for heat production during the mixing process, leading to decreased crystallinity of the drug. This effect has been also reported in a previous paper.37 However, these two peaks were not detected in the DSC curves obtained from liposomes. The absence of the endothermic peaks is evidence that SM and SGC existed in a noncrystalline state in liposomes. These DSC results were in agreement with the XRD results.
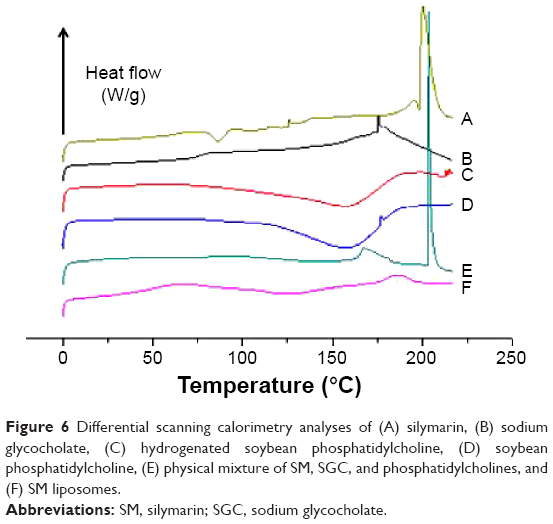 | Figure 6 Differential scanning calorimetry analyses of (A) silymarin, (B) sodium glycocholate, (C) hydrogenated soybean phosphatidylcholine, (D) soybean phosphatidylcholine, (E) physical mixture of SM, SGC, and phosphatidylcholines, and (F) SM liposomes. |
Stability study
To evaluate the stability of liposomes prepared by the three methods, sonication of the model formulation was undertaken to accelerate drug leakage (Figure 7). The drug leakage rate from all liposomal preparations increased with increasing time. For all preparations, leakage proceeded at different rates but was complete after 480 seconds. The drug leakage rate of SM-lip-SEDS increased the slowest, while the rate of increase was fastest with SM-Lip-RPE. The intactness of liposome vesicles may play a main role in their stability. Thus, liposomes prepared by SEDS existed in the most stable state compared with the other two preparation methods.
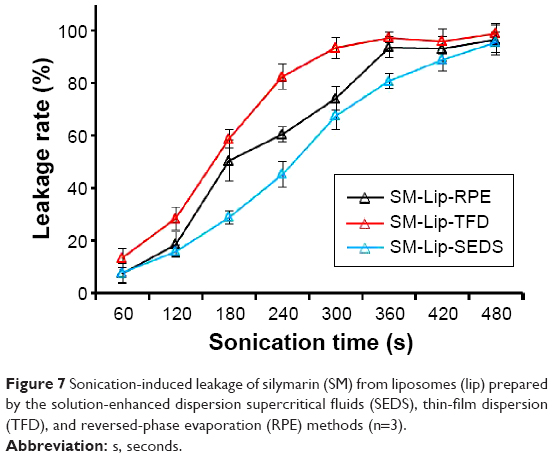 | Figure 7 Sonication-induced leakage of silymarin (SM) from liposomes (lip) prepared by the solution-enhanced dispersion supercritical fluids (SEDS), thin-film dispersion (TFD), and reversed-phase evaporation (RPE) methods (n=3). |
In vitro dissolution test
The release profiles of SM from SM powder and SM liposomes in phosphate buffer solution from three parallel experiments are shown in Figure 8. SM powder and SM liposomes have different drug release profiles. The slower rate of drug release from SM liposomes is due to the lipid bilayer that inhibits the diffusion of SM out of the liposomes. No significant burst release effect was observed in liposomes. The release profiles of SM-Lip-RPE and SM-Lip-TFD showed biphasic behavior, with the release rate during the primary phase being faster than during the steady release phase. Because SM is mainly within the bilayer lipid structure of the liposomes, the initial release is ascribed to drug detachment from the outer lamellae and drug adsorbed on or close to the surface of the particles.38 The release curve of SM from SM-Lip-SEDS was slower than other samples without biphasic behavior because of its stability and high EE. This means that SM-Lip-SEDS has an improvement in the sustained and stable release of SM compared to SM-Lip-RPE and SM-Lip-TFD.
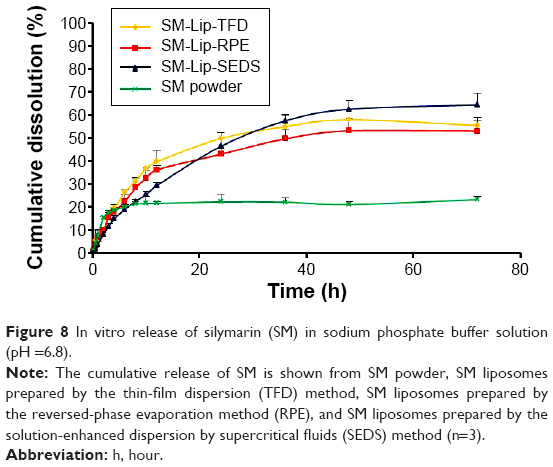 | Figure 8 In vitro release of silymarin (SM) in sodium phosphate buffer solution (pH =6.8). |
Pharmacokinetics
The plasma concentration–time profiles of SM in rats after oral administration of SM in different formulations (SM powder, commercial SM product, and SM-Lip-SEDS) are illustrated in Figure 9. The pharmacokinetic parameters are summarized in Table 3. After gavage administration of the SM suspension and commercial SM product, the absorption of SM was rapid without any significant differences (Tmax =1.42 hours and 0.92 hours for the SM suspension and commercial product, respectively). The Tmax obtained from the SM-Lip-SEDS (7.67 hours), SM-Lip-RPE (5.67 hours), and SM-Lip-TFD (5.50 hours) revealed that SM absorption took significantly longer with these formulations. This may be due to the presence of bile salts in the lipid bilayers of the liposomes making the vesicle resistant to the detrimental effects of physiological bile salts in the gastrointestinal (GI) tract, thus protecting these liposomes from enzymatic degradation.39 Moreover, the longer Tmax for SM-Lip-SEDS may be related to their better self-stability compared to SM-Lip-RPE and SM-Lip-TFD. Importantly, the pharmacokinetic parameters showed that both the commercial product and liposomal formulations improved the oral absorption of SM in rats compared to the absorption of a simple SM suspension. However, the liposome preparations were more effective in improving the bioavailability of SM. Compared to the SM powder formulation, the commercial SM product increased Cmax by 1.1-fold and the AUC by 2.0-fold, while the SM-Lip-SEDS liposome preparation increased the Cmax and AUC by approximately 2.0- and 4.8-fold, respectively.
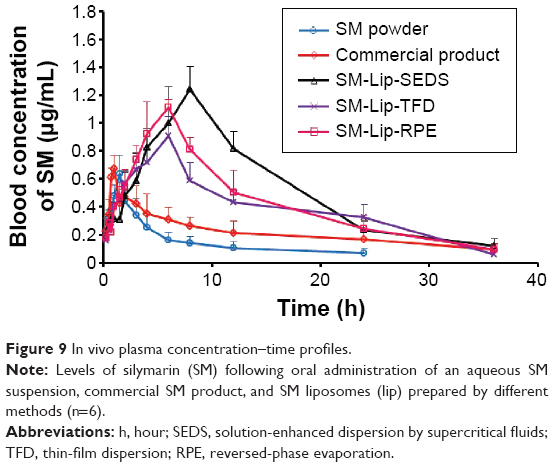 | Figure 9 In vivo plasma concentration–time profiles. |
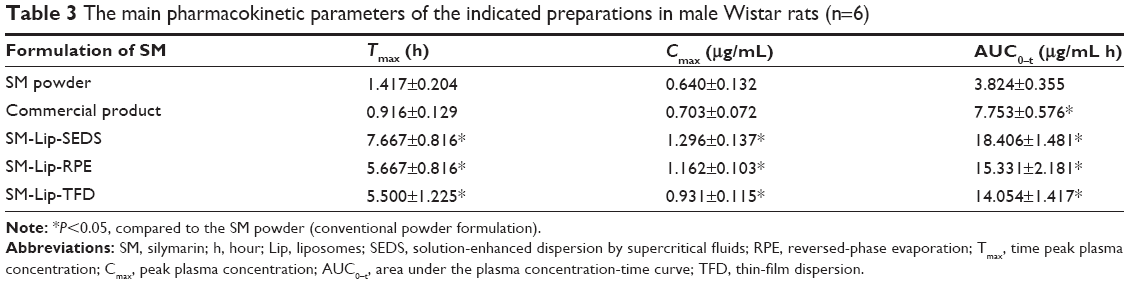 | Table 3 The main pharmacokinetic parameters of the indicated preparations in male Wistar rats (n=6) |
The improved oral bioavailability of SM with the liposome preparations may be due to the liposomes containing a bile salt that interacted with phospholipids in the GI tract to form mixed micelles that play important roles in enhancing the absorption of lipids and poorly water-soluble drugs by bile salt monomers. Likewise, bile salt monomers can penetrate into liposomal lipid bilayers and disrupt the vesicular structure, and further increases in bile salt concentrations can induce liposomes to undergo vesicle–micelle transition. The resultant mixed function as excellent vehicles for poorly water-soluble drug molecules is one of the most important mesophases before absorption. Therefore, liposomes containing bile salts (originally called transfersomes when developed for transdermal delivery) can readily transform into mixed micelles in the GI environment, thus enhancing transmembrane absorption.40–44 Furthermore, SM-Lip-SEDS has a higher bioavailability than SM-Lip-RPE and SM-Lip-TFD, possibly because of their smaller particle size and higher EE.
Overall, compared to the conventional powder formulation and commercial SM product, liposomes containing a bile salt increased the SM concentration in the plasma and prolonged drug retention. These effects may provide some therapeutic benefits by reducing the dosing frequency and improving the bioavailability of SM. Thus, the SEDS method affords a promising strategy for the preparation of therapeutically useful liposomes.
Conclusion
We have successfully prepared SM-loaded liposomes containing a bile salt (SGC) using the SEDS method. The product exhibited improved characteristics compared to liposomes prepared by the RPE or TFD methods. Moreover, SM-Lip-SEDS clearly improved the solubility of SM based on an evaluation of the in vitro drug release profiles. In vivo assays showed enhanced oral bioavailability of SM administered in liposomes containing a bile salt when compared with an aqueous SM suspension or commercial SM product. This indicates that SEDS exhibit great potential for the preparation of liposomes with improved solubility and oral bioavailability of SM.
Acknowledgments
This work was supported financially by the Subject Chief Scientist Program (10XD14303900) from the Science and Technology Commission of the Shanghai municipality, and the Specialized Research Fund for the Doctoral Program of Higher Education of China (20123107110005).
Disclosure
The authors report no conflicts of interest in this work.
References
Campodonico A, Collado E, Ricci R, Pappa H, Segall A, Pizzorno MT. Dissolution test for silymarin tablets and capsules. Drug Dev Ind Pharm. 2001;27(3):261–265. | ||
Tedesco D, Tava A, Galletti S, et al. Effects of silymarin, a natural hepatoprotector, in periparturient dairy cows. J Dairy Sci. 2004;87(7):2239–2247. | ||
Mohamed ES, Gamal ES, Tarek MI. Comparison of early treatment with low doses of nilotinib, imatinib and a clinically relevant dose of silymarin in thioacetamide-induced liver fibrosis. Eur J Pharmacol. 2001;670(2):593–600. | ||
Kvasnicka F, Biba B, Sevcik R, Voldrich M, Kratka J. Analysis of the active components of silymarin. J Chromatogr A. 2003;990(1):239–245. | ||
Wu JW, Lin LC, Hung SC, Chi CW, Tsai TH. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J Pharm Biomed Anal. 2007;45(4):635–641. | ||
Comoglio A, Tomasi A, Malandrino S, Poli G, Albano E. Scavenging effect of silipide, a new silybin-phospholipid complex, on ethanol-derived free radicals. Biochem Pharmacol. 1995;50(8):1313–1316. | ||
Wu L, Qiao YL, Wang LN. A self-microemulsifying drug delivery system (SMEDDS) for a novel medicative compound against depression: a preparation and bioavailability study in rats. AAPS PharmSciTech. Epub 2015, Feb 6. | ||
Du HH, Yong IK, Kwan HC. A novel solid dispersion system for natural product-loaded medicine: silymarin-loaded solid dispersion with enhanced oral bioavailability and hepatoprotective activity. J Microencapsul. 2014;31(7):619–626. | ||
Cao X, Fu M, Wang L. Oral bioavailability of silymarin formulated as a novel 3-day delivery system based on porous silica nanoparticles. Acta Biomater. 2012;8(6):2104–2112. | ||
Angelico R, Ceglie A, Sacco P, Colafemmina G. Phyto-liposomes as nanoshuttles for water-insoluble silybin–phospholipid complex. Int J Pharm. 2014;471(1):173–181. | ||
He J, Hou S, Lu W. Preparation, pharmacokinetics and body distribution of silymarin-loaded solid lipid nanoparticles after oral administration. J Biomed Nanotechnol. 2007;3(2):195–202. | ||
Shangguan MZ, Lu Y, Qi JP, Han J. Binary lipids-based nanostructured lipid carriers for improved oral bioavailability of silymarin. J Biomater Appl. 2014;28(6):887–896. | ||
Kumar N, Rai A, Reddy ND, Raj PV. Silymarin liposomes improves oral bioavailability of silybin besides targeting hepatocytes, and immune cells. Pharmacol Rep. 2014;66(5):788–798. | ||
Niu M, Tan Y, Guan P. Enhanced oral absorption of insulin-loaded liposomes containing bile salts: a mechanistic study. Int J Pharm. 2014;460(1):119–130. | ||
Mikov M, Fawcett JP, Kuhajda K, Kevresan S. Pharmacology of bile acids and their derivatives: absorption promoters and therapeutic agents. Eur J Drug Metab Pharmacokinet. 2006;31(3):237–251. | ||
Yu JN, Zhu Y, Wang L. Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles. Acta Pharmacol Sin. 2010;31(6):759–764. | ||
Guan PP, Lu Y, Qi JP, Niu MM. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomedicine. 2011;6(965):965–974. | ||
Chen GY, Li DH, Jin Y, et al. Deformable liposomes by reverse-phase evaporation method for an enhanced skin delivery of (+)-catechin. Drug Dev Ind Pharm. 2014;40(2):260–265. | ||
Yang KW, Joseph TD, Ulrich SS, Alfred F. Fast high-throughput screening of temoporfin-loaded liposomal formulations prepared by ethanol injection method. J Liposome Res. 2012;22(1):31–41. | ||
Yin F, Guo SY, Gan Y, Zhang XX. Preparation of redispersible liposomal dry powder using an ultrasonic spray freeze-drying technique for transdermal delivery of human epithelial growth factor. Int J Nanomedicine. 2014;9:1665–1676. | ||
Pankaj RK, Wonkyung C, Hee-Jun P, Jeong-Sook P. Characterization and stability studies of a novel liposomal cyclosporin A prepared using the supercritical fluid method: comparison with the modified conventional Bangham method. Int J Nanomedicine. 2013;8:365–377. | ||
Kim MS, Jin SJ, Kim JS. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69(2):454–465. | ||
Pasquali I, Bettini R, Giordano F. Supercritical fluid technologies: an innovative approach for manipulating the solid-state of pharmaceuticals. Adv Drug Deliv Rev. 2008;60(3):399–410. | ||
Antonio T, Eva M, Miguel A. Precipitation of tretinoin and acetaminophen with solution enhanced dispersion by supercritical fluids (SEDS). Powder Technol. 2012;217:177–188. | ||
Ragonese R, Macka M, Hughes J, Petocz P. The use of the Box-Behnken experimental design in the optimisation and robustness testing of a capillary electrophoresis method for the analysis of ethambutol hydrochloride in a pharmaceutical formulation. J Pharm Biomed Anal. 2002;27(6):995–1007. | ||
Bahia AP, Azevedo EG, Ferreira LA, Frézard F. New insights into the mode of action of ultradeformable vesicles using calcein as hydrophilic fluorescent marker. Eur J Pharm Sci. 2010;39(1):90–96. | ||
Niu M, Lu Y, Hovgaard L, Wu W. Liposomes containing glycocholate as potential oral insulin delivery systems: preparation, in vitro characterization and improved protection against enzymatic degradation. Int J Nanomedicine. 2011;6:1155–1166. | ||
Hu S, Niu M, Hu F, et al. Integrity and stability of oral liposomes containing bile salts studied in simulated and ex vivo gastrointestinal media. Int J Pharm. 2013;441(1):693–700. | ||
Liu D, Hu H, Lin Z, et al. Quercetin deformable liposome: preparation and efficacy against ultraviolet B induced skin damages in vitro and in vivo. J Photochem Photobiol B. 2013;127:8–17. | ||
Khodadoust S, Hadjmohammadi M. Determination of N-methylcarbamate insecticides in water samples using dispersive liquid–liquid microextraction and HPLC with the aid of experimental design and desirability function. Anal Chim Acta. 2011;699(1):113–119. | ||
Esfandiari N, Ghoreishi SM. Synthesis of 5-fluorouracil nanoparticles via supercritical gas antisolvent process. J Supercrit Fluids. 2013;84:205–210. | ||
Wang J, Chen J, Yang Y. Micronization of titanocene dichloride by rapid expansion of supercritical solution and its ethylene polymerization. J Supercrit Fluids. 2005;33(2):159–172. | ||
Hao J, Wang F, Wang X, et al. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur J Pharm Sci. 2012;47(2):497–505. | ||
Rahman Z, Zidan AS, Habib MJ, Khan MA. Understanding the quality of protein loaded PLGA nanoparticles variability by Plackett–Burman design. Int J Pharm. 2010;389(1):186–194. | ||
Cui FD, Tao AJ, Cun DM, Zhang LQ, Shi K. Preparation of insulin loaded PLGA-Hp55 nanoparticles for oral delivery. J Pharm Sci. 2007;96(2):421–427. | ||
García-Rodriguez JJ, de la Torre-Iglesias PM, Vegas-Sánchez MC, Torrado-Durán S, Bolás-Fernández F, Torrado-Santiago S. Changed crystallinity of mebendazole solid dispersion: improved anthelmintic activity. Int J Pharm. 2001;403(1):23–28. | ||
Semalty A, Semalty M, Singh D, Rawat MSM. Preparationand characterization of phospholipid complexes of naringenin for effective drug delivery. J Incl Phenom Macrocycl Chem. 2010;67(3–4):253–260. | ||
Haeri A, Sadeghian S, Rabbani S, Anvari MS, Boroumand MA, Dadashzadeh S. Use of remote film loading methodology to entrap sirolimus into liposomes: preparation, characterization and in vivo efficacy for treatment of restenosis. Int J Pharm. 2011;414(1):16–27. | ||
Niu M, Lu Y, Hovgaard L, et al. Hypoglycemic activity and oral bioavailability of insulin-loaded liposomes containing bile salts in rats: the effect of cholate type, particle size and administered dose. Eur J Pharm Biopharm. 2012;81(2):265–272. | ||
Dial EJ, Rooijakkers SH, Darling RL, Romero JJ, Lichtenberger LM. Role of phosphatidylcholine saturation in preventing bile salt toxicity to gastrointestinal epithelia and membranes. J Gastroenterol Hepatol. 2008;23(3):430–436. | ||
Hildebrand A, Beyer K, Neubert R, Garidel P, Blume A. Temperature dependence of the interaction of cholate and deoxycholate with fluid model membranes and their solubilization into mixed micelles. Colloids Surf B Biointerfaces. 2003;32(4):335–351. | ||
Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Andrieux K, Forte L, Lesieur S, Paternostre M, Ollivon M, Grabielle-Madelmont C. Solubilisation of dipalmitoylphosphatidylcholine bilayers by sodium taurocholate: a model to study the stability of liposomes in the gastrointestinal tract and their mechanism of interaction with a model bile salt. Eur J Pharm Biopharm. 2009;71(2):346–355. | ||
Cevc G, Vierl U, Mazgareanu S. Functional characterisation of novel analgesic product based on self-regulating drug carriers. Int J Pharm. 2008;360(1):18–28. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.