")
Back to Journals » International Journal of Nanomedicine » Volume 15
Enhanced Anti-Cancer Effect of Folate-Conjugated Olaparib Nanoparticles Combined with Radiotherapy in Cervical Carcinoma
Authors Li D, Hu C, Yang J, Liao Y , Chen Y, Fu SZ , Wu JB
Received 24 July 2020
Accepted for publication 17 November 2020
Published 10 December 2020 Volume 2020:15 Pages 10045—10058
DOI https://doi.org/10.2147/IJN.S272730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Dong Li,1,* Chuanfei Hu,1,* Juan Yang,1 Yin Liao,1 Yue Chen,2,3 Shao Zhi Fu,1 Jing Bo Wu1,3
1Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, People’s Republic of China; 2Department of Nuclear Medicine, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, People’s Republic of China; 3Nuclear Medicine and Molecular Imaging Key Laboratory of Sichuan Province, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Bo Wu
Department of Oncology, The Affiliated Hospital of Southwest Medical University, No. 25, Taiping Street, Luzhou 646000, People’s Republic of China
Email [email protected]
Shao Zhi Fu
Department of Oncology, The Affiliated Hospital of Southwest Medical University, No. 25, Taiping Street, Luzhou 646000, People’s Republic of China
Tel +86 830 316 5698
Fax +86 830 316 5690
Email [email protected]
Background: Radiotherapy (RT), one of the main treatments for cervical cancer, has tremendous potential for improvement in the efficacy. Poly (ADP-ribose) polymerase (PARP) is a key enzyme in the repair of DNA strand breaks (DSB). Olaparib (Ola) is a PARP inhibitor that is involved in preventing the release of PARP from RT-induced damaged DNA to potentiate the effect of RT. Although the basic mechanism of Ola’s radiosensitization is well known, the radiosensitization mechanism of its nanomedicine is still unclear. In addition, the lack of tumor tissue targeting is a major obstacle for the clinical success of Ola.
Materials and Methods: In this study, we developed folate-conjugated active targeting olaparib nanoparticles (ATO) and investigated the anti-tumor effect of ATO combined with radiotherapy (RT) in nude mice using cervical cancer xenograft models. We used folate (FA)-conjugated poly (ϵ-caprolactone)-poly (ethyleneglycol)-poly (e-caprolactone) (PCEC) copolymer to prepare ATO via emulsification/solvent diffusion. Further, we evaluated ATO particle size, potential, encapsulation efficiency, and in vitro release characteristics, and evaluated the shape of ATO via transmission electron microscopy (TEM). We then performed MTT and cell uptake assays to detect cytotoxicity and targeting uptake in vitro. We investigated the anti-tumor properties of ATO in vivo by apoptosis test, 18 F-FDG PET/CT, and immunohistochemical analysis. Finally, the xenografted tumor in nude mice was subjected to RT and/or ATO treatment.
Results: The results confirmed that ATO in combination with RT significantly inhibited tumor growth and prolonged survival time of tumor-bearing mice. This may be related to the inhibition of tumor proliferation and DNA damage repair and induction of cell apoptosis in vivo.
Conclusion: The ATO developed in this study may represent a novel formulation for olaparib delivery and have promising potential for treating tumors with an over-expression of folate receptors.
Keywords: olaparib, cervical cancer, folate, anti-cancer effect, active targeting
Introduction
Cervical cancer is one of the most common cancers in women worldwide,1 and RT is the main treatment for advanced cervical cancer. However, not so many effective methods to improve the efficacy of RT currently exist. Although cisplatin is traditionally widely used in combination with RT, it has severe side effects in patients. Therefore, exploring new approaches to improve the efficacy of RT has important clinical value. Research suggests that the mechanism of killing tumor cells by RT involves DNA strand breaks (DSBs).2 Poly (ADP-ribose) polymerase (PARP) is a key enzyme for DNA repair, and PARP inhibitors can selectively inhibit PARP activity, thereby inhibiting DNA strand repair and forming a large number of broken single-stranded DNAs, which eventually cause the double-stranded DNA to break at the replication fork and improves the efficiency of RT.3–5 Olaparib (Ola) is a well-known highly selective PARP inhibitor. Although current studies about the anti-tumor effects of Ola combined with RT in vitro are relatively detailed, there are few studies regarding this in vivo; thus, further research needs to be conducted.6–8 Nevertheless, with the lack of tumor tissue targeting, Ola use clinically leads to common adverse reactions such as anemia, nausea, and vomiting.9 In addition, issues such as poor water solubility, high toxicity, and poor stability have largely prevented the popularization and application of Ola in clinical use. In recent years, there are many studies about nano-delivery system for cancer therapy, which can enhance the efficacy and targeting of drugs.10–12 Therefore, developing a targeted nano-delivery system capable of solving the abovementioned issues is important.
Folate receptors have recently been confirmed to be over-expressed on the surfaces of some tumor cells such as those of cervical cancer, ovarian cancer, and nasopharyngeal cancer.13 The differential expression of folate receptors in tumor tissues and normal tissues makes them ideal targets for targeted drug delivery.14 Therefore, a new folate-conjugated nano-drug delivery system capable of targeting the folate receptor has emerged, which is considered to be a promising anti-cancer drug carrier, thus providing a feasible solution to the abovementioned problems.15,16 Our previous study had shown that Ola nanoparticles can enhance the radiosensitivity of lung cancer xenografts in nude mice.17 Furthermore, folate-conjugated honokiol nanoparticles show active-targeted inhibitory effect on nasopharyngeal carcinoma xenografts enriched with folate receptors on the cell surface.18 Therefore, in this study, we prepared folate-conjugated Ola nanoparticles that actively targeted folate receptors.
A large number of studies have reported anti-tumor effects of Ola combined with RT, including details on the drug and radiation doses.5,17 Thus, an optimal drug dose of Ola (50 mg/kg/d, 3 days) and radiation dose (10 Gy) were used for subsequent experiments. The purpose of this study was to prepare ATO and study the anti-tumor effects of ATO in combination with RT on cervical carcinoma subcutaneous xenograft model.
Materials and Methods
Materials
Ola with a purity level of 99.8% was purchased from Meilun Co., Ltd (Dalian, China). ε-Caprolactone (ε-CL), poly (ethylene glycol) (PEG, Mn=4000), and stannous octoate (Sn (Oct)2) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), N,N’-Diisopropylcarbodiimide (DIC), MTT, and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich. Further, dimethyl sulfoxide (DMSO), folate, petroleum ether, anhydrous ethanol, acetonitrile, and methanol (HPLC grade) were purchased from KeLong Co., Ltd (Chengdu, China). Tetrahydrofuran (THF) was purchased from Bioway (New York, NY). γ-H2AX, Ki-67, and CD-31 polyclonal antibodies were purchased from Bioworld Technology Co., Ltd (Nanjing, China).
Hela and A549 cell lines were provided by Zhongke Quality Inspection Biotechnology Co., Ltd (Beijing, China). BALB/c mice (female, nu/nu, 4–5 weeks old) were purchased from Chongqing TengXin biotechnology Co., Ltd (Chongqing, China).
Synthesis of Folate-Conjugated PCEC (FA-PCEC)
The synthesis route of FA-PCEC is shown in Figure 1. First, PCEC was synthesized from polyethylene glycol and ε-CL via ring-opening polymerization using Sn (Oct)2 as a catalyst, as reported previously.19 Next, the target polymer FA-PCEC was synthesized in three steps.20,21 Boc-L-Phe-terminated PCEC was obtained as follows: a nitrogen-purged flask containing 10 g of PCEC and 3.1 g of Boc-L-Phe, dissolved in 80 mL anhydrous dichloromethane, was cooled to −10°C. The mixture was then poured into a solution containing 0.8 g of DCC and 0.08 g of DAMP and incubated for 48 h at 0°C. Second, 6 g of Boc-L-Phe end-capped PCEC was added to 70 mL dry methylene dichloride; the mixture was then cooled to 0°C. Following this, 15 mL trifluoroacetic acid was added to the solution under nitrogen atmosphere for 2 h. Amino-terminated PCEC was obtained by vacuum drying at room temperature for 48 h. Finally, 806.4 mg of amino-terminated PCEC was dissolved in 60 mL DMSO, and mixed with 440 mg of folate. Subsequently, 253 mg of DIC and 230 mg of NHS were added to the solution as catalysts. The reaction was performed at 37°C for 24 h; the supernatant was collected, dialyzed for 72 h, and dried in a freeze dryer to obtain FA-PCEC. A proton NMR spectrometer was used to detect the characteristic peaks of protons to verify whether folate was attached to PCEC.
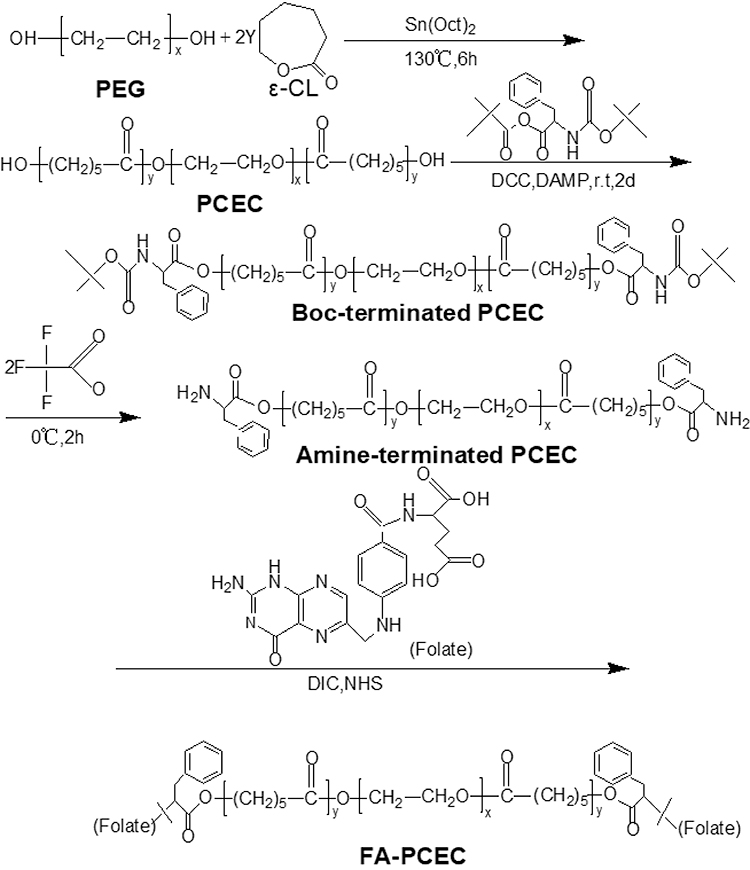 |
Figure 1 The synthesis route of FA-PCEC block copolymer. Abbreviation: PEG, polyethylene glycol. |
Preparation of ATO
ATO was prepared by emulsification/solvent diffusion method.22,23 Ola (4 mg) and FA-PCEC copolymer (46 mg) were dissolved in 2 mL DMSO to induce a diffusing phase; the solution was then added to 10 mL dispersing phase (PVA 0.5% in water) that was under a magnetic stirrer (300 rpm, 10 min) using a syringe. The nanoparticles were obtained by dialyzing against water for 48 h to remove DMSO and free Ola. A sterile filter with a 220-nm pore size was used to filter the suspension. The nanoparticles were then centrifuged at 20,000 g for 15 min, followed by several washing steps with distilled water. The orange-colored active targeting nanoparticles were then freeze-dried and stored at 4°C. Non-active targeting olaparib nanoparticles (NATO), which were not modified with folate, were prepared by a similar method, with the replacement of FA-PCEC copolymer with PCEC copolymer.
Characterization of ATO
The size distribution and Zeta potential of ATO were measured by dynamic light scattering (DLS, NanoBrook90 plus Zeta, Brookhaven, USA) at 25°C. All results were averages of three test runs. The structure of ATO was determined using a transmission electron microscope (TEM, Tecnai G2 F20, USA).
Drug Loading (DL) and Encapsulation Efficiency (EE)
The DL and EE of ATO were determined via high-performance liquid chromatography (HPLC, Agilent, Richardson, TX), and chromatographic analysis was performed using a reversed-phase C18 column (4.6 × 150 mm, 5 µm, USA) as per a previously reported method.24 The specific steps are as follows. ATO was dissolved in the mobile phase, which contained acetonitrile/water (64/36, v/v). The air from mobile phase was exhausted 30 min before use, and the mobile phase was applied with a flow rate of 1.0 mL/min through the column at 25°C. The volume of each injection was 20 µL, and the detection wavelength was set at 276 nm. DL and EE were calculated according to the following formula:
DL=Olaparib/Olaparib+Polymer
EE=Actual DL/Theoretical DL
In vitro Drug Release of Ola and ATO
The release of Ola from ATO in vitro was evaluated as follows: 1 mg Ola or ATO containing 1 mg Ola (theoretical DL, 8%) were separately dissolved in 1 mL DMSO (1 mg/mL) and 1 mL distilled water (1 mg/mL), and then separately poured into corresponding sealed dialysis bags (molecular weight cutoff, 3.5 kDa). The bags were individually incubated at 37°C in 30 mL PBS (pH = 7.4), containing Tween-80 (0.5%, w/w), with gentle stirring (100 rpm). The incubation medium was replaced with the same volume (2 mL) of freshly preheated medium (37°C) at predetermined time points. Subsequently, the collected release drug was centrifuged at 12,000 rpm for 15 min at 4°C. Following this, 1 mL of supernatant from each group was collected and stored at −20°C for further analysis. All supernatant samples were analyzed via HPLC and experiments were performed at least in triplicates.
Cytotoxicity Assay and Cellular Uptake of Nanoparticles
The cytotoxicity was evaluated via MTT assay. In brief, Hela cells were seeded in 96-well plates at a density of 5 × 103 cells/well and incubated for 24 h at 37°C; the cells were then exposed to free Ola, NATO, and ATO at different concentrations (0 to 5 nmol/L) for 24 h. Subsequently, 10 µL of MTT solution was added to the cells, and cell viability was analyzed after incubation for 4 h by dissolving the formazan precipitate in DMSO and recording the absorbance at 490 nm using a microplate reader (iMark, Bio-RAD, USA).
FITC-labeled PCEC nanoparticles and FA-PCEC nanoparticles were prepared as described previously.25 An ethanol solution of FITC was added to 1.0 mg/mL of PCEC or FA-PCEC nanoparticle solution, and the FA-PCEC nanoparticles to FITC molar ratio was controlled at 1:4. The solution was stirred (400 rpm) for 24 h at room temperature in a dark environment. The reaction product was then dialyzed against deionized water for 24 h using a dialysis membrane (MWCO: 3.5 kDa) to remove un-reacted FITC. HeLa cells and A549 cells were then seeded at a concentration of 1 × 105 cells/well in a 6-well plate and incubated for 24 h. Following this, 50 µL of medium containing 10 µg/mL FITC-PCEC nanoparticles was added to the 6-well plate with Hela cells and incubated for 6 h. The FITC-FA-PCEC nanoparticles were added to a separate 6-well plate with Hela cells and A549 cells. For comparison, free FA dissolved in DMSO was added to a fresh medium containing FITC-FA-PCEC nanoparticles at a concentration of 10 µg/mL and poured into a 6-well plate with Hela cells. After washing the cells three times with PBS (0.01 M, pH = 7.4), they were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
In vivo Evaluation of Anti-Tumor Efficacy
Animal experiments were performed according to Guidelines for the Feeding, Management and Use of Laboratory Animals and were approved by the Institutional Animal Care and Treatment Committee of Southwest Medical University (Luzhou, China). In brief, the tumor xenograft model was established by subcutaneously injecting Hela cells (1 × 106 cells in 100 µL PBS) into the right thigh of athymic BALB/c nude mice (female, 4–5 weeks old). When the average volume of the subcutaneous tumor reached 150–200 mm3 (V = a × b2/2, where a and b are the longest and shortest diameters of the tumor, respectively), the mice were randomly divided into six groups as follows (n = 12 per group): Control (0.9% normal saline [NS]); NATO; ATO; RT (a single dose of 10 Gy); NATO+RT; and ATO+RT. Subsequently, each mouse in the drug-treated group was intravenously administered 50 mg/kg of Ola standard for 3 days. RT was only conducted 30 min after the first drug administration. The long and short diameters of the tumor were measured every 2 days from the first day of drug administration using a Vernier caliper, and the tumor volume was calculated based on the tumor volume calculation formula described before. The measurement was stopped after the tumor volume reached four times the initial tumor volume. The tumor growth delay (TGD) was calculated by Ti4 – T4, where Ti4 is the time taken for the tumor volume to reach four times the initial tumor volume after treatment and T4 is the time taken for the control tumor volume to reach four times the initial tumor volume. On day 10 after drug administration, mice from each group were selected and dissected to resect the heart, liver, spleen, lung, and kidney for H&E staining for evaluating drug toxicity. The remaining mice in each group were used to observe median survival.
Small animal PET/CT was performed to explore therapy response and early metabolism of tumor tissue with different treatments. The mice were randomly selected from each group for the scan and were not allowed to drink or eat for at least 8 h before the scan. The mice were then anesthetized with 1% pentobarbital (5 mg/kg) intraperitoneal injection and injected with 100–200 µCi FDG (0.1 mL) in the tail vein. PET/CT scan imaging was performed at least 30 min after the intravenous injection. The parameters were as follows: voltage, 80 kV; current, 500 µA; section thickness, 1.5 mm, and 10 min per bed position.
Immunohistochemical Analysis
Within 48 h after the first administration, three nude mice were randomly selected from each group to obtain tumor tissues for immunohistochemical detection of γ-H2AX. Further, 10 days after drug administration, the obtained tumor tissue was used for the detection of other immunohistochemical indicators such as Ki-67. Specific steps are as follows: the tumor tissue was fixed in 10% neutral formaldehyde for at least 24 h, it was then embedded in paraffin and cut into 3–4-µm-thick sections and stained with H&E for immunohistochemical analysis. The detailed procedures for γ-H2AX and Ki-67 expression detection were according to the manufacturer’s instructions (Bioworld Technology, Nanjing, China) and pictures were taken using an optical microscope (Olympus, Tokyo, Japan). The expression levels of γ-H2AX and Ki-67 were calculated at three randomly selected areas in each tumor sample at 200× magnification as the number of positive cells/total number of cells.
Apoptosis Analysis
Ten days after drug administration, the obtained tumor tissue was used for apoptosis analysis. Cellular apoptosis was analyzed by flow cytometry (BD FACSVerse, Piscataway, NJ). The tumor tissue was shredded and added to 2 mL trypsinization buffer placed in a 37°C incubator with gentle shaking to digest. After 40 min of digestion, the serum-containing medium was added to stop the digestion. Following this, the mixture was filtered using a nylon gauze of pore size 70 µm to remove the undigested tumor tissue. The collected cell suspension was centrifuged at 1000 rpm for 2 min and washed twice with physiological saline. Subsequently, 5 µL of propidium iodide and 5 µL of fluorescein isothiocyanate were added to each test tube. Cells were incubated at room temperature in the dark for 15 min, and 400 µL of pre-cold 1× binding buffer was then added. After gentle mixing, the cell suspensions were analyzed via flow cytometry (BD FACSVerse, Piscataway, NJ).
Statistical Analysis
Data analysis was processed by SPSS 18.0 software using t-test for two-group comparisons and one-way ANOVA for comparisons of more than two groups. The value obtained is expressed as mean ± standard deviation (x ± s). P < 0.05 was considered statistically significant.
Results
Characterization of FA-PCEC Copolymer
In this study, we successfully synthesized FA-PCEC copolymer. The structure of the FA-PCEC copolymer was detected via nuclear magnetic resonance hydrogen spectroscopy. As shown in Figure 2, the copolymer shows characteristic PCL peaks at 1.4 ppm, 1.6 ppm, 2.3 ppm, and 4.1 ppm and a typical PEG characteristic peak at 3.5 ppm. The characteristic peaks of PCL and PEG appeared in the same position in the NMR hydrogen spectrum for both PCEC and FA-PCEC, but the typical peaks of folate were seen in FA-PCEC at 6.6 ppm, 7.6 ppm, and 8.6 ppm. Based on this analysis, it can be concluded that folate was successfully branched on PCEC and the target polymer FA-PCEC was successfully synthesized.
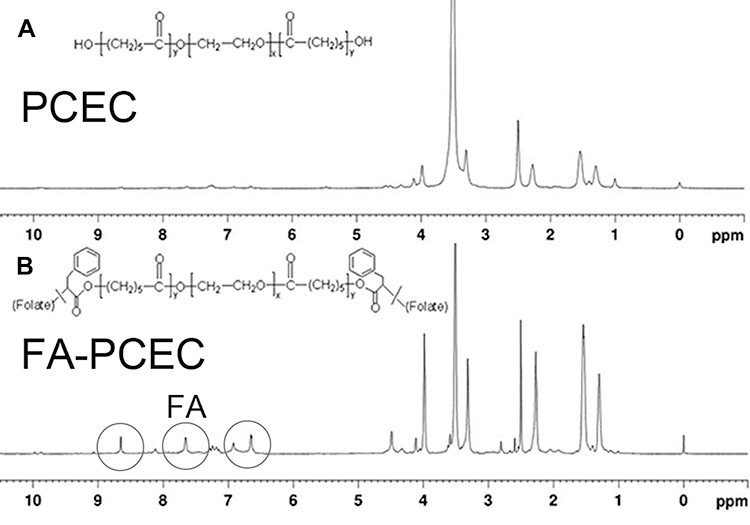 |
Figure 2 H-NMR spectra of PCEC (A) and FA-PCEC (B). The gray circle represents the characteristic peak of FA. Abbreviation: H-NMR, NMR hydrogen spectroscopy. |
In vitro Characterization of ATO
The particle size and morphology of ATO were detected by DLS and TEM. The average hydrodynamic diameter of ATO was 240.62 ± 8.79 nm (Figure 3A), furthermore, the zeta potential of ATO was −10.5 ± 1.09 mV (Figure 3C). As a comparison, the average hydrodynamic diameter of ATO was 205.43 ± 3.13 nm (Figure 3B), the zeta potential of ATO was 4.67 ± 1.17 mV (Figure 3D). The obtained ATO was mono-dispersed and stabilized in an aqueous solution; the appearance of ATO was spherical as shown in the TEM image (Figure 3E).
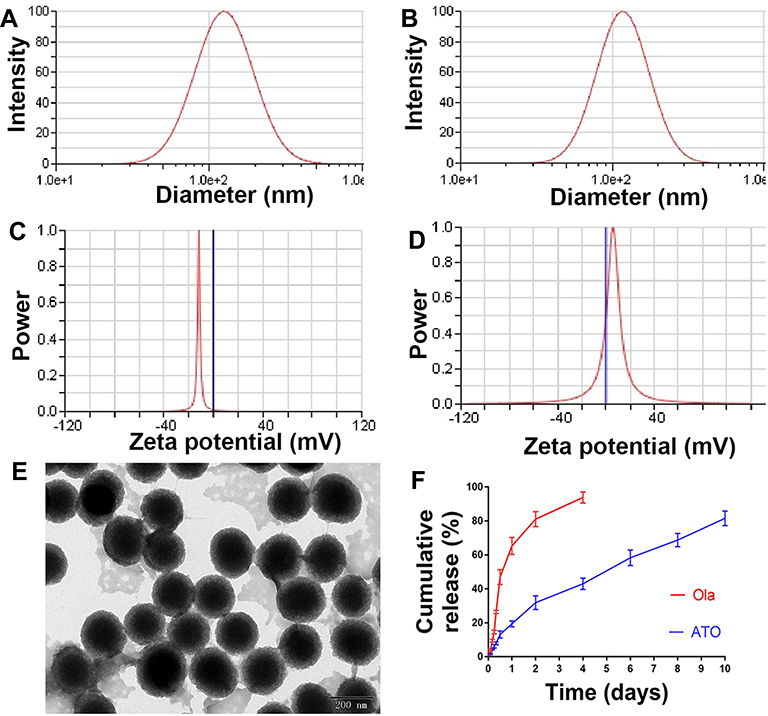 |
Figure 3 The characterization of ATO. (A) Particle size distribution of ATO, presented a mean size of 240.62 ± 8.79 nm. (B) Particles size distribution of NATO, presented a mean size of 205.43 ± 3.13 nm. (C) Zeta potential of ATO, showed a negative surface charge of −10.5 ± 1.09 mV. (D) Zeta potential of NATO, showed a positive surface charge of 4.67 ± 1.17 mV. (E) Transmission electron microscopy image of ATO (the scale is 50 nm). (F) Drug release study of Ola and ATO. |
As mentioned before, the theoretical DL of ATO was set at 8%. However, HPLC results showed that the actual DL and EE of ATO were 6.44% ± 0.05% and 80.54 ± 0.63%, respectively. As shown in Figure 3F, in vitro release of free Ola showed a rapid release trend and approximately 90% of Ola was released into the medium within 4 days. In contrast, ATO showed a slow and long-lasting release trend, and 80% of Ola was released from ATO within 10 days.
Cytotoxicity Assay and Cellular Uptake
To evaluate the cytotoxicity of ATO, MTT assay was performed using HeLa cells. The dose-dependent inhibitory effect of free Ola, NATO, and ATO on Hela cells is illustrated in Figure 4A. The cytotoxicity of ATO was superior to that of free Ola basically. Moreover, compared with free Ola and NATO, ATO showed a stronger inhibitory effect when the concentration was 200 μg/L. When the concentration was 200 μg/L, the viability of cells treated with ATO was 53.2 ± 1.5%, which was significantly lower than that of cells treated with NATO and treated with NATO, indicating that the therapeutic effect of Ola was enhanced when Ola was incorporated into nanoparticles to form ATO. Figure 4B illustrates the cytotoxic effect of FA-PCEC on HeLa cells. No cytotoxicity was detected among all the tested concentrations, indicating that the anti-tumor activity of ATOs was attributed to Ola, which released from the nanoparticles. This indicated that FA-PCEC copolymer has good cytocompatibility and can be considered a safe drug delivery vehicle.
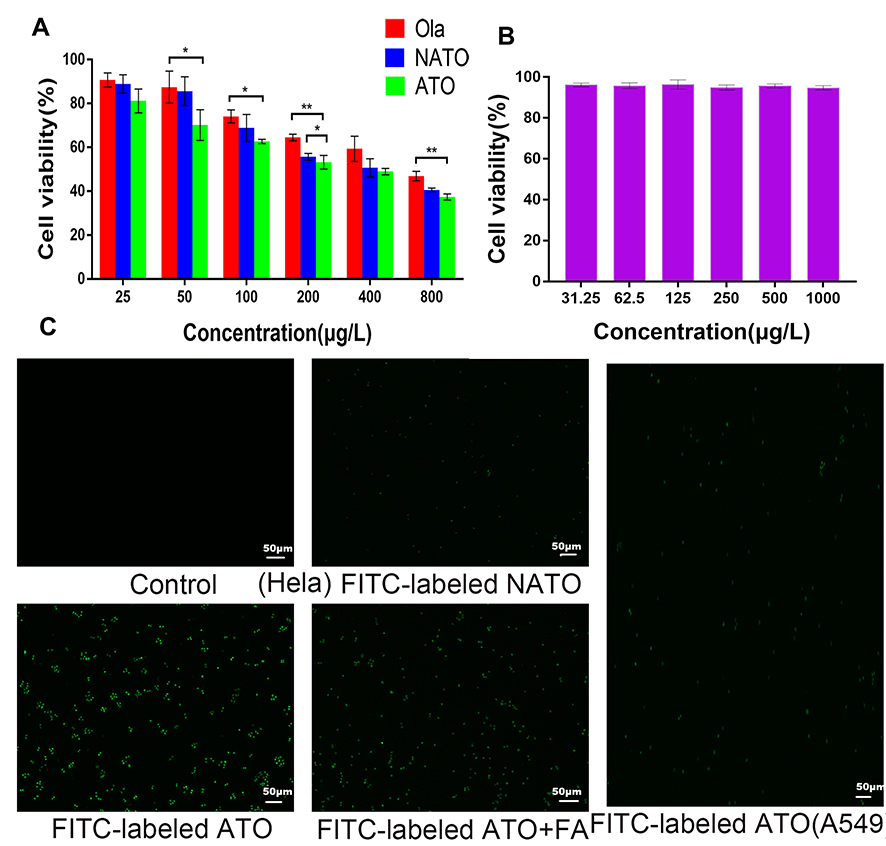 |
Figure 4 In vitro cell viability and fluorescent images. (A) Cell viability of free Ola, NATO and ATO on Hela cell after 24 h incubation. *P<0.05; **P<0.01; no statistical significance between unlabeled groups. (B) Cell viability of FA-PCEC. (C) Fluorescent images of Hela cells treated with culture medium, FITC-labeled NATO, FITC-labeled ATO and FITC-labeled ATO+FA for 2 h and fluorescent images of A549 cells treated with FITC-labeled ATO for 2 h. |
To verify the targeting ability of ATO, cell uptake experiments were conducted in Hela cells and A549 cells. Because A549 cells do not have folate receptors, thus A549 cells were chosen as negative control group. Fluorescence images obtained after incubation with FITC-labeled NATO and ATO are shown in Figure 4C. In Hela cells, no fluorescence spot was observed in the Control group, and fluorescence intensity of FITC-labeled ATO was more pronounced than that of FITC-labeled NATO, indicating that folate-conjugated nanoparticles show a better cell uptake efficiency than unconjugated nanoparticles. However, after adding free FA to the medium containing FITC-labeled ATO, a lower fluorescence intensity was observed, indicating that free FA prevents ATO from transporting into Hela cells by competitive binding to folate receptors on the cell surface. In addition, A549 cells showed relatively low fluorescence uptake on addition of FITC-labeled ATO. These results indicated that FA-conjugated nanoparticles are a potential drug delivery system that targets tumor cells through folate receptor-mediated endocytosis.
In vivo Anti-Tumor Efficacy of Radiotherapy Combined with ATO
The therapeutic efficacy of ATO+RT on Hela xenografted mice was assessed as shown in Figure 5. The PET/CT images and tumor appearance in mice in each group are shown in Figure 5A and B. According to the tumor appearance, the tumor volume was the largest in the mice in the Control group, whereas the tumor volume in the mice in the ATO+RT group was the smallest. Based on the results of PET/CT imaging (Figure 5C), the mean SUVmax of the tumor in each group was observed: 2.73 ± 0.25 (control), 2.26 ± 0.25 (NATO), 1.77 ± 0.15 (ATO), 1.63 ± 0.25 (RT), 1.33 ± 0.15 (NATO+RT), and 0.93 ± 0.05 (ATO+RT). The SUVmax of mice in the ATO+RT group was the lowest when compared to NATO+RT, RT and other groups, indicating that the tumor in this group has the lowest metabolism, and thereby implying a more powerful anti-tumor effect of ATO+RT. In addition, the tumor volume in mice in different groups after treatment was measured and plotted as shown in Figure 5D. Tumor growth was delayed by 3 days in mice treated with NATO alone (P < 0.05), whereas the ATO group and RT group showed a delay in tumor growth by 6 and 12 days, respectively (P < 0.05). The NATO+RT group showed a tumor growth delay of 18 days and the ATO+RT group exhibited a tumor growth delay of 24 days, which was longer than the other five groups mentioned above. These results indicated that ATO+RT had a more effective synergistic effect in inhibiting tumor growth compared with other therapies. At the same time, a similar effect was observed for the median survival time in mice (Figure 5E). The median survival time of mice in the ATO+RT group (50 days) was significantly longer than that of mice in the NATO+RT (42 days), RT (38 days), ATO (38 days), NATO (34 days) and Control (32 days) groups (P < 0.05).
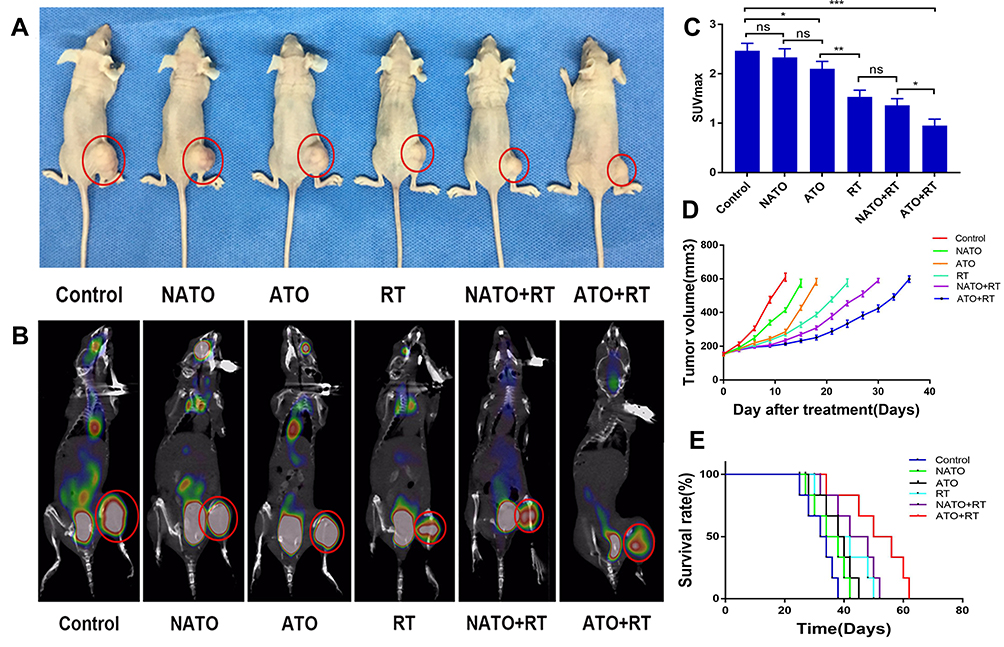 |
Figure 5 Evaluation of antitumor efficiency in vivo. (A) General view of tumor-bearing example mice with different treatments. (B) PET/CT images of example mice in each group. (C) Quantitative analysis of SUVmax of mice in each group. *P<0.05; **P<0.01; ***P<0.001; ns: no statistical significance. (D) Tumor growth curves of mice in each group. (E) The survival curves of mice in each group. |
Immunohistochemical Analysis
The immunohistochemical analysis results are demonstrated in Figure 6A. The results showed that ATO+RT significantly increased the expression of γ-H2AX and reduced the expression of Ki-67 in mice.
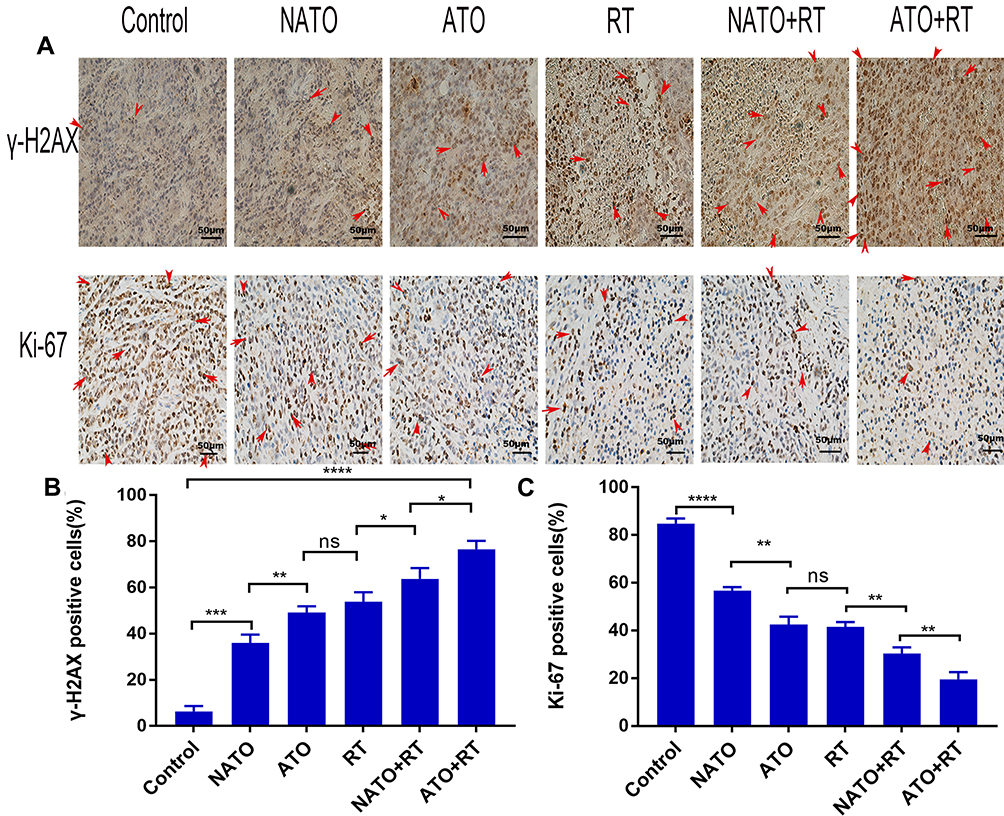 |
Figure 6 Immunohistochemical analysis. (A) Representative images of γ-H2AX and Ki-67 in each group (magnification, ×200). (B) Quantitative analysis of γ-H2AX in each group. (C) Quantitative analysis of Ki-67 in each group. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Abbreviation: ns, no statistical significance. |
The proportion of γ-H2AX-positive cells in the ATO+RT group was 76.65% ± 2.88%, which showed an increased apoptotic rate when compared to NATO+RT (63.8% ± 3.77%), RT (53.86% ± 3.32%) and other treatment groups (Figure 6B) (P<0.05).
Ki-67 is an immune indicator that reflects tumor cell proliferation. The percentage of Ki-67-positive cells in different groups is as follows (Figure 6C): Control group, 84.82% ± 2.11%; NATO group, 56.78% ± 1.41%; ATO group, 42.43% ± 3.35%; RT group, 41.55% ± 1.95%; and NATO+RT group, 30.4% ± 2.54%. In addition, there was an obvious decrease in the percentage of Ki-67-positive cells in ATO+RT group (19.51% ± 3.04%) compared with that in other groups (P<0.01).
Flow Cytometry
Flow cytometry was used to detect the apoptosis levels of tumor tissues in each group (Figure 7A). As shown in Figure 7B, there was no significant difference in apoptosis rate between ATO+RT group and NATO+RT group (P>0.05). But the rate of apoptosis in ATO+RT and NATO+RT groups were higher than that in RT (60.56% ± 4.12%), ATO (52.33% ± 3.6%) and other groups. This result indicated that a combination of ATO or NATO with RT significantly increased the apoptosis rate of tumor cells.
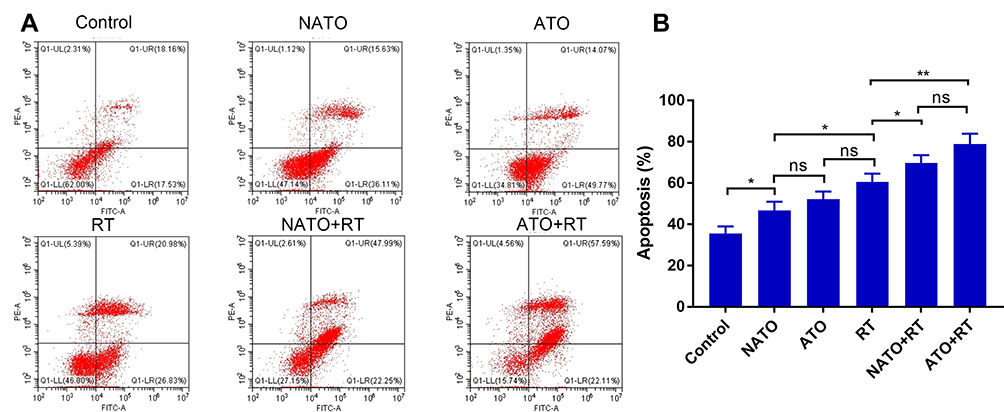 |
Figure 7 Apoptosis analysis of tumor tissues in each group. (A) Apoptosis graphs of each group. (B) Quantitative analysis of apoptosis. *P<0.05; **p<0.01. Abbreviation: ns, no statistical significance. |
Evaluation the Side Effects of ATO
In this experiment, the in vivo toxicity of different treatments was evaluated by observing the pathological changes in important organs of nude mice in each group. After the nude mice were sacrificed, the heart, liver, spleen, lung and kidney of the mice were prepared for H&E staining. As shown in Figure 8, no obvious pathological changes were found in each treatment group.
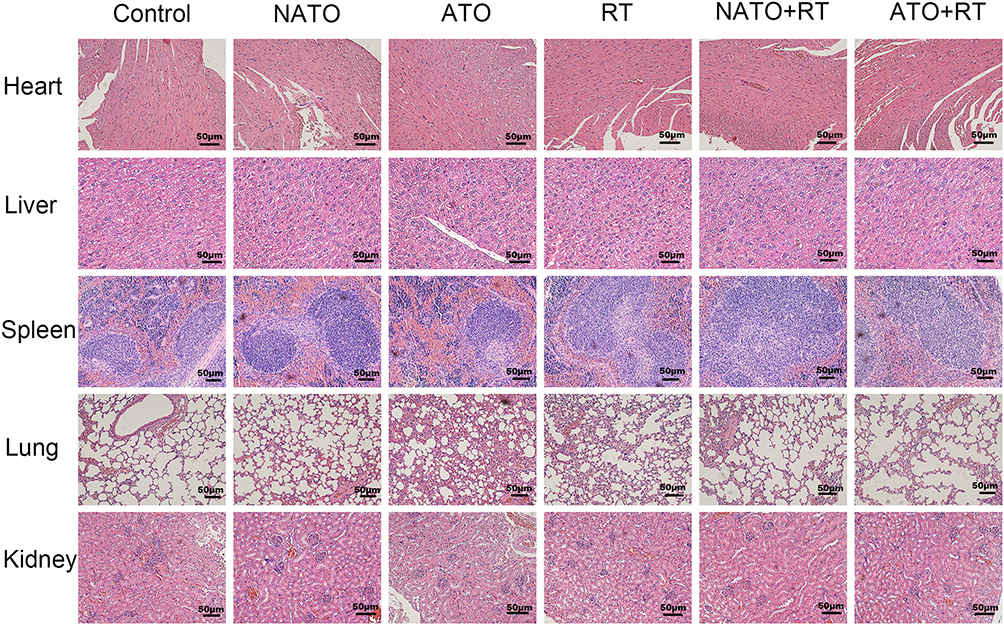 |
Figure 8 H&E staining of heart, liver, spleen, lung, and kidney of each group (magnification, ×200). |
Discussion
Cervical cancer, as a common malignant tumor that seriously threatens women’s health, has shown a high incidence in recent years.26 RT is one of the most important treatment options for cervical cancer as it not only improves tumor control but also reduces complications.27,28 The mechanism of RT is mainly to cause DNA strand breaks in tumor cells, but the self-repair function of the tumor tissue makes the tumor tissue less sensitive to radiation. Therefore, targeted inhibition of DNA repair to improve the efficacy of RT has become one of the research hotspots in recent years.2
PARP is a well-known enzyme closely associated in DNA damage repair. PARP inhibitors can precisely inhibit the activity of PARP and the repair of single-stranded DNA breaks, causing a synthetic lethal effect that leads to the death of tumor cells and enhancing the effect of RT.4 Ola is a representative PARP inhibitor, which is approved for use in the treatment of advanced ovarian cancer patients with BRCA1/2 mutation.29 Previous studies have confirmed that Ola can be used in combination with RT to enhance its anti-tumor effects.8,30,31 At the same time, our research group has previously confirmed that Ola nanoparticles combined with RT showed enhanced anti-tumor effects on lung cancer A549 xenografts in nude mice at an early stage,17 which further improves the feasibility of its clinical promotion. According to research reports, drug-loaded nanoparticles can achieve passive targeting in tumors because of enhanced permeability and retention effect (EPR).32 However, only a small fraction of nanoparticles accumulating in solid tumors through passive targeting can be absorbed.33,34 Thus, to improve the targeting efficiency, it is necessary to introduce targeting molecules (such as folic acid and monoclonal antibodies) that can recognize and bind to specific receptors on cancer cells.35–37 Furthermore, a preliminary study by our research group confirmed that folate-conjugated nanoparticles have an active targeting inhibitory effect on tumors rich in folate receptors on their cell surface.18 This work reveals that it is possible to target DNA damage to tumor cells and improve the therapeutic effect of RT.
Therefore, this study aimed to prepare ATO and explore the targeted enhancement of therapeutic effect when using ATO in combination with RT. First, an emulsification/solvent diffusion method was used to successfully prepare injectable ATO. Compared with other recently reported nanoparticles, ATO showed a stable particle size and higher DL.38,39 In addition, the sustained slow release trend of ATO may be attributed to the relatively stable structure of the folate polymer carrier. The cytotoxicity experiments suggested that ATO lead to decreased cell viability of tumor cells, which was associated with the lack of BRCA2 in cervical cancer cells and is consistent with previous research.40,41 However, FA-PCEC showed almost no cytotoxic effect. These findings indicated that the inhibitory effect of ATO was attributed to the Ola wrapped in it. In this study, the uptake of folate-conjugated nanoparticles by Hela cells was more significant. In addition, a lower fluorescence intensity was observed after adding free folate into the medium. Moreover, a lower fluorescence intensity was observed in A549 cells that are negative for folate receptor; this proves our hypothesis that the active endocytosis of ATO by Hela cells was mediated by folate receptors. Thus, these results indicated that ATO prominently inhibited cancer cell growth by delivering more Ola into Hela cells by folate-mediated endocytosis.
We subsequently established a nude mouse xenograft model using cervical cancer Hela cells to investigate the therapeutic effect of ATO+RT. A RT dose of 10 Gy has always been used in studies reporting combined use of olaparib and RT.5,42,43 In our in vivo anti-tumor study, TGD and survival duration of mice treated with ATO+RT were significantly higher than those of mice treated with other treatment measures; this further confirmed that ATO combined with RT had a better anti-tumor effect on cervical cancer xenograft model. The abovementioned results are consistent with the results of our previous study concerning the combined use of Ola and RT.17 This is probably because of the active targeted uptake of ATO by Hela cells resulting in a high concentration of Ola in the cells, which further results in a more pronounced therapeutic effect when combined with RT.18F-FDG PET/CT was used to understand the early therapeutic effects in nude mice. Previous studies have shown that18F-FDG PET/CT can reflect the level of glucose metabolism, which is closely associated with the therapeutic effect on the tumor.38 In tumor tissues, high glucose metabolism indicates a worse therapeutic effect, whereas low glucose metabolism often demonstrates a better therapeutic effect. The mice in the ATO+RT group showed the lowest glucose uptake compared with those in other groups, suggesting that ATO+RT had a better effect of inhibiting tumor growth.
In this study, immunohistochemical analysis was used to detect γ-H2AX and Ki-67 expressions in tumor tissue. The results suggested that mice in the ATO+RT group showed a higher expression level of γ-H2AX than those in other groups. Previous studies have shown that Ola combined with RT can increase DNA double-strand breaks. Therefore, the expression of γ-H2AX, a biomarker for DNA double-strand breaks, was detected to confirm the DNA damage in each group. Ki-67 is a tumor proliferation index; our results indicated that the mice in the ATO+RT group had the lowest proportion of Ki-67-positive cells among all groups. These results were similar to those reported in previous studies.2,44 The increased expression of γ-H2AX and the decreased expression of Ki-67 in the ATO+RT group may be because of the active targeting uptake of ATO in Hela cells to form high concentrations of Ola in Hela cells. The results of flow cytometry also showed that tumor tissues in ATO+RT group had the highest rate of apoptosis, which is similar to previously reported results.45 The increased sensitivity of tumor cells to apoptosis may be associated with the down-regulation of the anti-apoptotic protein.46,47
In summary, these results indicate that ATO in combination with RT has a synthetic lethal effect on cervical cancer xenografts. Although the results are encouraging, shortcomings and limitations of our research clearly exist. Therefore, the tissue distribution of nanoparticles in vivo needs to be evaluated further in subsequent experiments. Also, further in vivo studies with folate receptor-negative cell lines should be conducted in the future.
Conclusion
In this study, ATO was successfully prepared. Data showed that ATO had outstanding targeted effect on cervical cancer xenograft tumor. ATO+RT significantly inhibited the growth of cervical cancer cells and prolonged the median survival duration of mice. In addition, we explored the mechanism of synthetic lethal effect of ATO+RT and found that ATO+RT induces DSBs and increases the apoptosis of tumor cells. In summary, ATO has good application prospects for tumors with an over-expression of folate receptors.
Acknowledgments
This study was supported by the Major Project of the first Affiliated Hospital of Southwest Medical University, project number P2017001.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zuckerman AJ. IARC monographs on the evaluation of carcinogenic risks to humans. J Clin Pathol. 1995;48(7):691–a. doi:10.1136/jcp.48.7.691-a
2. Annika K, Kerstin C, Kerstin B, et al. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol Oncol. 2014;8(16):16–25.
3. Verhagen Caroline VM, de Haan R, Floor H, et al. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother Oncol. 2015;116(3):58–65.
4. Albert JM, Cao C, Kim KW, et al. Inhibition of poly (ADP-Ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13(10):33–42. doi:10.1158/1078-0432.CCR-06-2872
5. Yanyan J, Tom V, Devery Aoife M, et al. Hypoxia potentiates the radiation-sensitizing effect of olaparib in human non-small cell lung cancer xenografts by contextual synthetic lethality. Int J Radiat Oncol Biol Phys. 2016;95(7):72–81.
6. Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):52–61. doi:10.1016/S1470-2045(11)70214-5
7. Wang W, Duan B, Zeng L, et al. Effect and mechanism of radiosensitization of poly (ADP-Ribose) polymerase inhibitor on lewis cells and xenografts. Chin J Lung Cancer. 2016;19(1):16–23.
8. Senra Joana M, Telfer Brian A, Cherry Kim E, et al. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther. 2011;10(19):49–58.
9. Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):07–14. doi:10.1016/S1470-2045(14)70391-2
10. Yu X, Guoyi T, Changbing W, et al. Functionalized selenium nanoparticles for targeted siRNA delivery silence Derlin1 and promote antitumor efficacy against cervical cancer. Drug Deliv. 2020;27(1):15–25. doi:10.1080/10717544.2019.1667452
11. Yu X, Misi X, Mingqi Z, et al. Doxorubicin-loaded functionalized selenium nanoparticles for enhanced antitumor efficacy in cervical carcinoma therapy. Mater Sci Eng C Mater Biol Appl. 2020;106:110100. doi:10.1016/j.msec.2019.110100
12. Eugenia T, Catalin Z, Ariana H, et al. Impact of the magnetic field on 3T3-E1 preosteoblasts inside SMART silk fibroin-based scaffolds decorated with magnetic nanoparticles. Mater Sci Eng C Mater Biol Appl. 2020;110:110714. doi:10.1016/j.msec.2020.110714
13. Huang Y, Yang T, Zhang W, et al. A novel hydrolysis-resistant lipophilic folate derivative enables stable delivery of targeted liposomes in vivo. Int J Nanomedicine. 2014;9(45):81–95.
14. Xie M, Zhang H, Xu Y, et al. Expression of folate receptors in nasopharyngeal and laryngeal carcinoma and folate receptor-mediated endocytosis by molecular targeted nanomedicine. Int J Nanomedicine. 2013;8(24):43–51.
15. Batoul F, Carvalho MR, Prameswari K, et al. Folate-conjugated nanovehicles: strategies for cancer therapy. Mater Sci Eng C Mater Biol Appl. 2020;107(110):03–41.
16. Xue H, Jing L, Min L, et al. 9-NC-loaded folate-conjugated polymer micelles as tumor targeted drug delivery system: preparation and evaluation in vitro. Int J Pharm. 2009;372(1):25–31.
17. Min W, Jing L, ChuanFei H, et al. Olaparib nanoparticles potentiated radiosensitization effects on lung cancer. Int J Nanomedicine. 2018;13(82):61–72.
18. Bo Y, XiaoLing N, LongXia C, et al. Honokiol-loaded polymeric nanoparticles: an active targeting drug delivery system for the treatment of nasopharyngeal carcinoma. Drug Deliv. 2017;24(6):60–69.
19. MaLing G, ChangYang G, Juan Z, et al. Polymeric matrix for drug delivery: honokiol-loaded PCL-PEG-PCL nanoparticles in PEG-PCL-PEG thermosensitive hydrogel. J Biomed Mater Res A. 2010;93(2):19–26.
20. Liu P, Yu H, Sun Y, et al. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33(17):4403–4412. doi:10.1016/j.biomaterials.2012.02.041
21. Zelai HJ. Co-delivery of cisplatin and paclitaxel by folic acid conjugated amphiphilic PEG-PLGA copolymer nanoparticles for the treatment of non-small lung cancer. Oncotarget. 2015;6(421):50–68.
22. Farnaz E, Hossein GM, Nasser OS, et al. Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J Drug Target. 2008;16(4):15–23.
23. Fasehee H, Dinarvand R, Ghavamzadeh A, et al. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: in vitro and in vivo investigations. J Nanobiotechnology. 2016;14(1):32. doi:10.1186/s12951-016-0183-z
24. XiuLing Z, Bing K, MaLing G, et al. Preparation of MPEG-PLA nanoparticle for honokiol delivery in vitro. Int J Pharm. 2010;386(26):2–7.
25. Zhou YY, Du YZ, Wang L, et al. Preparation and pharmacodynamics of stearic acid and poly (lactic-co-glycolic acid) grafted chitosan oligosaccharide micelles for 10-hydroxycamptothecin. Int J Pharm. 2010;393(1–2):144–152. doi:10.1016/j.ijpharm.2010.04.025
26. Avinash B, Parshuram RV, Kirti S, et al. Prospective evaluation of the setup errors and its impact on safety margin for cervical cancer pelvic conformal radiotherapy. Rep Pract Oncol Radiother. 2020;25(2):60–65.
27. Vicharn L, Imjai C, Supatra S, et al. Concurrent mitomycin C, 5-fluorouracil, and radiotherapy in the treatment of locally advanced carcinoma of the cervix: a randomized trial. Int J Radiat Oncol Biol Phys. 2003;55(12):26–32.
28. Paulina C, Ewa B, Ewa W, et al. The effect of different segmentation methods on primary tumour metabolic volume assessed in F-FDG-PET/CT in patients with cervical cancer, for radiotherapy planning. Contemp Oncol (Pozn). 2019;23(3):183–186.
29. Zimmer AS, Gillard M, Lipkowitz S, et al. Update on PARP inhibitors in breast cancer. Curr Treat Options Oncol. 2018;19(5):21–28. doi:10.1007/s11864-018-0540-2
30. Güster JD, Weissleder SV, Busch CJ, et al. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother Oncol. 2014;113(3):345–351. doi:10.1016/j.radonc.2014.10.011
31. Polly G, James N, Emma G, et al. Radiosensitization with an inhibitor of poly (ADP-ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair (Amst). 2018;6(1):25–36.
32. Karel U, Vladimír S. Polymeric anticancer drugs with pH-controlled activation. Adv Drug Deliv Rev. 2004;56(10):23–50.
33. Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy. Adv Drug Deliv Rev. 2002;54(5):631–651. doi:10.1016/S0169-409X(02)00044-3
34. Choi KY, Chung H, Min KH, et al. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31(1):106–114. doi:10.1016/j.biomaterials.2009.09.030
35. Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Gold nanoparticles with a monolayer of doxorubicin-conjugated amphiphilic block copolymer for tumor-targeted drug delivery. Biomaterials. 2009;30(60):65–75.
36. Lin JJ, Chen JS, Huang SJ, et al. Folic acid–Pluronic F127 magnetic nanoparticle clusters for combined targeting, diagnosis, and therapy applications. Biomaterials. 2009;30(51):14–24. doi:10.1016/j.biomaterials.2009.06.004
37. Peifeng L, Zonghai L, Mingjie Z, et al. Preparation of EGFR monoclonal antibody conjugated nanoparticles and targeting to hepatocellular carcinoma. J Mater Sci Mater Med. 2010;21(55):1–6.
38. YanXin Y, Shan X, Hong Y, et al. In vivo synergistic anti-tumor effect of paclitaxel nanoparticles combined with radiotherapy on human cervical carcinoma. Drug Deliv. 2017;24(1):75–82. doi:10.1080/10717544.2016.1230902
39. Wang Y, Wu M, Gu L, et al. Effective improvement of the neuroprotective activity after spinal cord injury by synergistic effect of glucocorticoid with biodegradable amphipathic nanomicelles. Drug Deliv. 2017;24(1):391–401. doi:10.1080/10717544.2016.1256003
40. Gopeshwar N, Hugo AP, Nandula Subhadra V, et al. Promoter hypermethylation of FANCF: disruption of Fanconi Anemia-BRCA pathway in cervical cancer. Cancer Res. 2004;64:24–28.
41. Anna B, Salvatore L, Gary A, et al. PARP-1 activity (PAR) determines the sensitivity of cervical cancer to olaparib. Gynecol Oncol. 2019;155:144–150. doi:10.1016/j.ygyno.2019.08.010
42. van de Ven Anne L, Shifalika T, Paige B, et al. PTEN/TP53-nanoformulation of olaparib amplifies PARP inhibition and sensitizes deficient prostate cancer to radiation. Mol Cancer Ther. 2017;16(12):79–89.
43. Eiko N. C225 antiepidermal growth factor receptor antibody enhances the efficacy of docetaxel chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(11):63–73.
44. Cui Y, Zhang C, Li X, et al. Intravoxel incoherent motion diffusion-weighted magnetic resonance imaging for monitoring the early response to ZD6474 from nasopharyngeal carcinoma in nude mouse. Sci Rep. 2015;5(1):63–89. doi:10.1038/srep16389
45. Hongju P, Huang Q, Hongcheng Y, et al. The antitumor effect of cisplatin-loaded thermosensitive chitosan hydrogel combined with radiotherapy on nasopharyngeal carcinoma. Int J Pharm. 2019;55(6):97–105.
46. Deben C, Lardon F, Wouters A, et al. APR-246 (PRIMA-1MET) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016;375(2):313–322. doi:10.1016/j.canlet.2016.03.017
47. Kumar CG, Poornachandra Y, Chandrasekhar C. Green synthesis of bacterial mediated anti-proliferative gold nanoparticles: inducing mitotic arrest (G2/M phase) and apoptosis (intrinsic pathway). Nanoscale. 2015;7(44):18738–18750. doi:10.1039/C5NR04577K
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.