")
Back to Journals » Vascular Health and Risk Management » Volume 14
Endothelin-receptor antagonists in the management of pulmonary arterial hypertension: where do we stand?
Authors Correale M , Ferraretti A , Monaco I , Grazioli D, Di Biase M, Brunetti ND
Received 26 October 2017
Accepted for publication 23 February 2018
Published 4 October 2018 Volume 2018:14 Pages 253—264
DOI https://doi.org/10.2147/VHRM.S133921
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Takashi Kajiya
Michele Correale,1 Armando Ferraretti,2 Ilenia Monaco,3 Davide Grazioli,1 Matteo Di Biase,4 Natale Daniele Brunetti3
1Department of Cardiology, Ospedali Riuniti University Hospital, Foggia, 2Cardiology Unit, “Caduti in guerra” Hospital, Canosa di Puglia, BT, 3Department of Cardiology, University of Foggia, 4Santa Maria Hospital, GVM Care and Research, Bari, Italy
Abstract: Pulmonary arterial hypertension, a disease largely neglected until a few decades ago, is presently the object of intense studies by several research teams. Despite considerable progress, pulmonary arterial hypertension remains a major clinical problem, because it is not always easy to diagnose, treat, and prevent. The disease was considered incurable until the late 1990s, when Epoprostenol was introduced as the first tool against this illness. More recently, therapy for pulmonary arterial hypertension gained momentum after publication of the SERAPHIN and AMBITION trials, which also highlighted the importance of upfront therapy. This review also focuses on recent substudies from these trials and progress in drugs targeting the endothelin pathway. Future perspectives with regard to endothelin-receptor antagonists are also discussed.
Keywords: endothelin-receptor antagonists, pulmonary arterial hypertension, Bosentan, ambrisentan, sitaxentan, macitentan
Introduction
Pulmonary arterial hypertension (PAH), a disease largely neglected until a few decades ago, is presently the object of intense study by several research teams. Despite considerable progress, PAH remains a major clinical problem, because it is not always easy to diagnose, treat, and prevent. This disease was considered incurable until the late 1990s, when Epoprostenol (Flolan, GSK e Caripul o Velletri, Actelion) was introduced as the first tool against it.
Epoprostenol is a synthetic molecule that elicits the same effects as prostacyclin (which is produced naturally in the human body). Epoprostenol was the first drug to be approved for PAH treatment. The introduction of Epoprostenol changed therapy for PAH considerably: Epoprostenol improved exercise capacity, hemodynamic parameters, and PAH symptoms, and remarkably was the first drug able to reduce mortality due to PAH.1–3
Subsequently, another drug, Bosentan (Tracler, Actelion), showed a reduction in mortality in some forms of PAH.4 Bosentan was the first in a new class of drugs: endothelin-receptor antagonists (ERAs).5 Bosentan has a central role in PAH treatment, because it can improve exercise capacity, hemodynamics, symptoms, and right-ventricle function. Bosentan can cause an increase in the level of transaminases in ~10% of patients, but this effect is reversible upon dose reduction or discontinuation. Nevertheless, levels of transaminases should be measured every month while patients are on Bosentan therapy.6 The drug has been evaluated in several forms of PAH (idiopathic, associated with congenital heart defects, and Eisenmenger syndrome) in five studies (pilot, BREATHE-1, BREATHE-2, BREATHE-5, and EARLY) that showed improvement in exercise capacity, World Health Organization (WHO) functional class, hemodynamics, echocardiographic/Doppler variables, and time to clinical worsening.7 Bosentan improves survival in the forms of PAH associated with systemic rheumatic diseases.8 In patients with systemic sclerosis (SS), long-term treatment with Bosentan improves endothelial function and reduces the risk of PAH progression.9
More recently, other drugs have been developed. Some act on other pathways, such as phosphodiesterase 5 inhibitors (PDE5Is) guanylate-cyclase agonists, prostacyclin analogs (eg, Beraprost [Belnar, Asahi Kasei Pharma, Japan], Iloprost [Ventavis, Bayer, Germany], and Treprostinil [Remodulin, United Therapeutics, USA]), and other ERAs (eg, Ambrisentan [Volibris, GSK, UK] and Macitentan [Opsumit, Actelion, Switzerland]).7 Lately, PAH therapy has received considerable momentum after the publication of important trials, such as SERAPHIN and AMBITION.10,11 Results from the latter highlighted the importance of upfront therapy. A large part of this review focuses on new substudies of these trials. However, before evaluation of specific ERAs, we summarize the endothelin (ET) pathway. Finally, future perspectives in the field of ERAs are reported.
Methods
An extensive Internet search on PubMed until July 2017 was carried out using the following keywords alone or in combination: “pulmonary arterial hypertension”, “pulmonary hypertension”, “endothelin receptor antagonists”, “Bosentan”, “ambrisentan”, “sitaxentan”, “macitentan”. Only articles in English were selected for review, which focused on the most consistent and relevant trials, original articles, reviews, and case reports, preferentially involving humans.
Results
The ET pathway and ERAs
ET1 is a polypeptide produced mainly by vascular endothelial cells. ET1 release results in potent vasoconstriction that can induce proliferation of vascular smooth-muscle cells. In patients with PAH, high plasma levels of ET1 have been documented due to an increase in production in endothelial cells and inhibition of elimination of ET1, primarily in the lung (clearance).
The biological action of ET1 is mediated by two G-protein-coupled subtypes of receptors: ETA and ETB. ETA receptors are expressed on pulmonary smooth-muscle cells, and mediate potent vasoconstriction and promote cell proliferation. ETB receptors are expressed predominantly on the endothelial surface of vessels, and mediate vasodilatation through the production of nitric oxide and prostacyclin; they also stimulate pulmonary clearance of circulating ET1 to favor its elimination. ETB receptors not only have “protective” effects but they are also present in the muscle cells of vascular walls, where they have the same effects as ETA receptors (vasoconstriction and cell proliferation). In systemic and pulmonary hypertension, expression of ETB receptors is upregulated in the media of blood vessels, and ETA and ETB receptors contribute to the detrimental effects of ET1.12
The most efficient pharmacological mechanism for antagonizing the deleterious effects of ET1 is the use of ERAs, because they can blockade only ETA and ETB receptors. As such, ERAs can be distinguished into two types, based on the action on the ET receptor, with relative differences in pharmacologic effects: selective drugs (eg, ambrisentan) and unselective (eg, Bosentan and macitentan).13 If these drugs bind to ET1 receptors, the latter cannot be activated by ET1, which cannot exert its effects on vascular structures within the lungs. The rationale for the selective blockade of ETA receptors is the ability to maintain the potential beneficial effects mediated by ETB receptors (vasodilatation and ET1 clearance) by blocking only the adverse effects (vasoconstriction and proliferation) of ETA receptors. In fact, even ETB receptors located on muscle cells have adverse effects.13
The ETA-receptor-selective antagonist ambrisentan was approved for clinical use against PAH in 2007, followed by the more ETA-receptor-selective antagonist sitaxentan.14–16 However, in 2010 sitaxentan was withdrawn voluntarily by Thelin, Encysive Pharmaceuticals and Pfeizer, USA, because of idiosyncratic hepatitis that resulted in death from acute liver failure.17 Bosentan was the structural basis for the development of macitentan. Bosentan was approved for clinical use in 2013 and represented the next generation of antagonists, because it was more potent, with longer receptor occupancy and an active metabolite, and properties that contributed to greater pharmacodynamic and pharmacokinetic efficacy.18
Table 1 summarizes the main differences between ERAs. Table 2 details the disadvantages and advantages of ERAs. With regard to pharmacodynamic or pharmacokinetic differences, macitentan is a “dual ERA” developed by modifying the structure of Bosentan to increase efficacy and safety.19 Though it is classified as a “mixed antagonist”, macitentan tends to be more selective for the ETA receptor.20 In fact, unlike Bosentan, macitentan has higher affinity and more sustained receptor binding, and is the first ERA to show a significant reduction in risk of morbidity and mortality in PAH patients.21
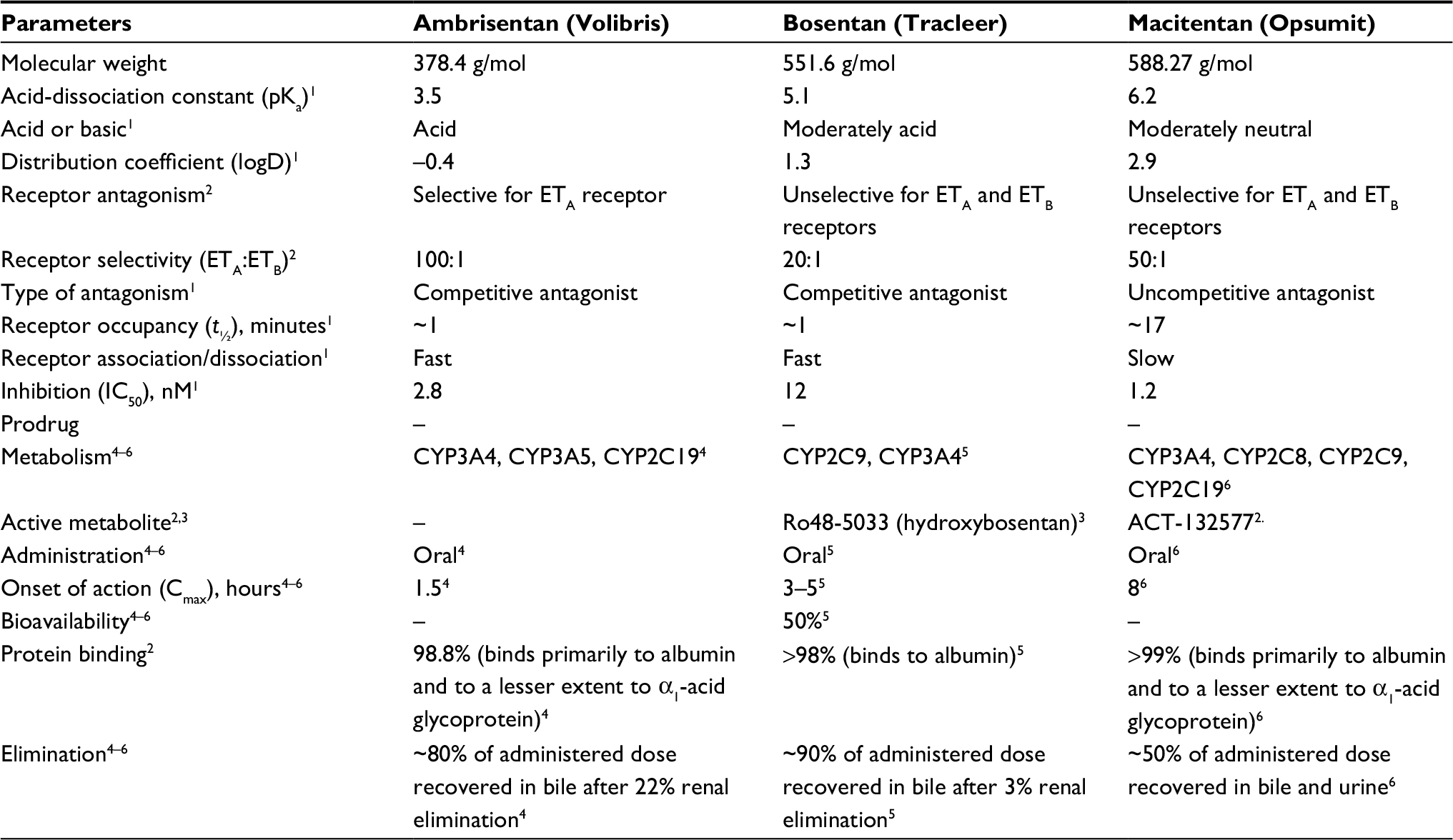 | Table 1 Pharmacodynamic or pharmacokinetic differences among endothelin (ET)-receptor antagonists
|
 | Table 2 Disadvantages and advantages of endothelin (ET)-receptor antagonists |
In a preclinical model of PAH, macitentan displayed a slow dissociation rate from receptors. The receptor occupancy half-life of macitentan (17 min) was 15-fold longer than that of Bosentan.22 The slower dissociation kinetics of macitentan have translated into an insurmountable antagonism in studies of second-messenger signaling, whereas Bosentan has shown surmountable competitive antagonism. As a result, macitentan can inhibit the ET1-induced sustained increase in intracellular levels of calcium across the whole range of ET1 concentrations tested, which is not observed with Bosentan. Macitentan is more effective than Bosentan in vivo. Bosentan has a similar mode of action (blockade of ETA and ETB receptors), but macitentan displays an improved ability to achieve more effective blockade of ET receptors via increased distribution in tissue and better receptor-binding properties. This improved efficacy may explain the unique efficacy of macitentan in reducing PAH progression, as demonstrated in SERAPHIN.23
Bosentan can be hepatotoxic, and the US Food and Drug Administration requires liver-function tests to be done every month, in addition to hematocrit monitoring every 3 months for anemia. In a study by Rubin et al, abnormal hepatic function was reported more frequently in a high-dose Bosentan group.24 In contrast, the safety profile of macitentan appears to be superior with respect to hepatic safety and edema/fluid retention than Bosentan and ambrisentan, respectively, and is similar when considering the reduction in hemoglobin concentration. Bosentan has a low propensity for drug–drug interactions, and has one circulating pharmacologically active metabolite; it does not require dose adjustments in patients with renal or hepatic impairment.25 Bosentan has a selectivity ratio for the ETA receptor of 20, macitentan a selectivity ratio of 50:1, and ambrisentan (which is ETA-receptor-selective) a selectivity ratio of 200:1.
Ambrisentan and PAH
Pharmacodynamics and pharmacokinetics of ambrisentan
Ambrisentan is an orally active propanoic acid and a potent ERA, with higher selectivity for the ETA receptor than the ETB receptor. The selectivity ratio for the ETA receptor is different among ERAs: 20:1 for Bosentan, 50:1 for macitentan, and 200:1 for ambrisentan. Ambrisentan is absorbed rapidly into the systemic circulation with a half-life of ~15 hours, which permits once-daily dosing. The pharmacokinetics are linear, and a steady state is reached after 4 days of repeated administration. The overall bioavailability of ambrisentan is 80%, and it is not affected by food. Ambrisentan is highly bound to plasma proteins (99%). Elimination is mostly through the biliary system, with most oral doses recovered in urine and feces.26,27
Ambrisentan does not affect the pharmacodynamics of concomitant medications for patients with PAH (eg, warfarin, sildenafil, tadalafil, ketoconazole) significantly. It is approved for the treatment of idiopathic, heritable PAH and PAH-associated connective-tissue disease (CTD). It has been shown to be effective in improving exercise capacity, cardiac hemodynamics, WHO functional class, and quality of life in patients with PAH. Ambrisentan displays an acceptable safety profile if used as monotherapy and in combination therapy with other vasodilators, such as a PDE5I.27
Ambrisentan is usually well tolerated, but it can lead to side effects due to systemic vasodilatation. In the ARIES-1 and ARIES-2 studies, peripheral edema, headache, and nasal congestion tended to be more frequent in patients treated with ambrisentan. Increases in serum levels of aminotransferases have been described with older ERAs (eg, Bosentan), whereas with ambrisentan the prevalence of hepatic injury due to ambrisentan is lower. ARIES-1 and ARIES-2 demonstrated a reduction in hemoglobin concentration in patients with PAH treated with ambrisentan, likely because of systemic vasodilatation and hemodilution. In AMBITION, anemia was seen more frequently in the ambrisentan–tadalafil combination than with ambrisentan or tadalafil monotherapy. ERAs are teratogenic, and should not be used during pregnancy.27
Randomized clinical trials
Several clinical studies have assessed ambrisentan use. Two randomized, double-blind, placebo-controlled, multicenter studies (ARIES-1 and -2) demonstrated a good safety profile with low risk of aminotransferase abnormalities and significant improvements in WHO functional class, 6-minute walking distance (6MWD), and time to clinical worsening relative to placebo, with a high tolerance profile. In these studies, 392 patients with PAH treated orally (per os [PO]) with placebo or ambrisentan (5 or 10 mg in ARIES-1, 2.5 or 5 mg in ARIES-2) once daily for 12 weeks were randomized. Most patients had idiopathic PAH (64%), but a certain proportion of patients had PAH related to CTD (32%), HIV infection (2.5%), or anorexigen use (1.5%). These patients were mostly in WHO functional class II (44%) and III (52%). The primary end point for each study was the change in 6MWD from baseline to week 12. 6MWD increased in all ambrisentan groups. These clinical studies moreover showed improvements in time to clinical worsening (ARIES-2), WHO functional class (ARIES-1), and brain natriuretic peptide (BNP) levels (both studies) in the absence of significant increases in aminotransferase concentrations.28
A total of 383 patients completing ARIES-1 or -2 were eligible to continue on ambrisentan for the ARIES-E extension study, which was designed to obtain additional long-term data on safety and efficacy. This study showed sustained improvements in exercise capacity and a low risk of clinical worsening and death in patients with PAH after 2 years of ambrisentan exposure. The best 6MWD results were obtained for the 5 mg (+23 m) and 10 mg (+28 m) groups, with a sustained improvement in exercise capability, dyspnea, and WHO functional class in these groups.29 More recently, ARIES-3, a long-term, multicenter, single-arm study, confirmed the results of previous placebo-controlled studies and demonstrated the tolerability and benefits of ambrisentan in patients with PAH.30
AMBITION evaluated two simultaneous strategies in PAH patients for treating drug-naïve patients with PAH: first-line combination therapy (ambrisentan and tadalafil) versus first-line monotherapy (ambrisentan or tadalafil).Patients were assigned randomly at a 2:1:1 ratio to a target daily dose of ambrisentan (10 mg) and tadalafil (40 mg). Overall, 18% of the combination-therapy group reached the primary composite end point of time to clinical failure (death, first occurrence of hospitalization for worsening PAH, disease progression, or unsatisfactory long-term clinical response) compared with 34% of the ambrisentan- and 28% of the tadalafil-monotherapy groups and 31% of the pooled-monotherapy group. The combination-therapy group demonstrated a reduction in the risk of first event of clinical failure of 50% compared with the monotherapy groups.11 In a post hoc analysis of AMBITION, Coghlan et al demonstrated that initial combination therapy reduced the risk of clinical failure compared with pooled monotherapy in each subgroup (CTD-PAH and SS-PAH).31
Ambrisentan substudies
D’Alto carried out the first prospective study evaluating hemodynamics using ambrisentan therapy in PAH in a long-term (12 months) follow-up involving 27 consecutive adult patients with PAH (idiopathic PAH, congenital heart disease [CHD]-PAH, and CTD-PAH). They showed a significant improvement in WHO functional class, 6MWD, pro-BNP level, and hemodynamics (cardiac index and pulmonary resistance).26
Combination therapy is the standard of care for patients with PAH with unsatisfactory response to monotherapy in many PAH centers.32 In the ATHENA-1 study, Shapiro et al studied the effects obtained by the addition of ambrisentan to 33 patients with PAH on a PDE5I. They showed improvements in 6MWD, N-terminal prohormone BNP (NT-pro-BNP) level, and hemodynamic parameters (mean PA pressure, systemic vascular resistance, and cardiac index) at 24 weeks.33
One observational study showed ambrisentan monotherapy to be effective and safe for the treatment of patients with portopulmonary hypertension, as evidenced by significant improvements in hemodynamic measurements (mean PA pressure, systemic vascular resistance, and cardiac output), biomarkers (BNP), and symptoms (WHO functional class), with no deterioration in systemic blood pressure, liver function, or renal function.34
A subgroup analysis of ARIES-E assessed the efficacy and safety of ambrisentan in patients with CTD. A total of 124 patients with CTD-PAH participating in ARIES-1 and ARIES-2 and their long-term extension were evaluated. Patients taking ambrisentan demonstrated fewer worsening clinical events and improved survival compared with controls.35
In a post hoc analysis of AMBITION, initial combination therapy was associated with a survival advantage compared with initial monotherapy in patients with newly diagnosed PAH.36 VOLT (a prospective, observational, multicenter study) was designed to characterize the safety profile of ambrisentan: the drug was not associated with increases in liver-function abnormalities.37 Hassoun et al demonstrated the effects of upfront combination PAH therapy (ambrisentan 10 mg and tadalafil 40 mg daily for 36 weeks) in patients with SS-PAH. They noted a significant reduction in median right-ventricle mass and pulmonary vascular resistance, as well as improvements in the median stroke volume:pulmonary pulse pressure ratio, tricuspid annular plane systolic excursion, 6MWD and serum levels of NT-pro-BNP.38 Recently, D’Alto et al showed the efficacy of 12-month upfront therapy with ambrisentan and tadalafil in improving hemodynamics in patients with incident PAH.39
Macitentan and PAH
Macitentan reduces the mean arterial pulmonary pressure in a dose-dependent manner without affecting the heart rate in animal models of PAH. Macitentan has tenfold-greater potency than Bosentan in terms of reducing mean arterial pulmonary pressure, suggesting more complete blockade of ET1 receptors under conditions of locally fluctuating ET1 levels, even if ET1 concentrations are high.19,22,25,40
Pharmacodynamics and pharmacokinetics of macitentan
Macitentan is an orally active, potent, dual ERA. It antagonizes ETA and ETB receptors, with an ETA receptor:ETB receptor-inhibitory potency ratio of 50:1. Macitentan is metabolized by cytochrome P450 enzymes (mainly CYP3A4) to its major pharmacologically active metabolite, ACT132577, which is approximately fivefold less potent than the parent drug at the ETA receptor and has an ETA receptor:ETB receptor-inhibitory potency ratio of 16:1. After oral administration, macitentan reaches its peak plasma concentration in ~8 hours in healthy volunteers, with a steady state being reached by day 3 for macitentan and by day 7 for ACT132577. The latter is responsible for ~40% of the pharmacologic activity at the steady state.41 The estimated oral bioavailability of macitentan is 74%.42 The pharmacokinetics of macitentan and ACT132577 are not altered to a clinically significant extent by food. They are highly bound to plasma proteins (>99%), mainly to albumin and to a lesser extent to α1-acid glycoprotein. They are distributed widely in tissue.43–45 Age, sex, ethnicity, renal impairment, or hepatic impairment do not affect the pharmacokinetics of macitentan or ACT132577 to a clinically relevant extent, and thus dose adjustment is not required.43,44,46–48
Macitentan and ACT132577 are not associated with clinically relevant inhibition or induction of CYP enzymes. They enter the liver by passive diffusion, and are not substrates of the OATPs 1B1, 1B3, or 2B1. At clinically relevant concentrations, macitentan and ACT132577 do not inhibit hepatic or renal uptake transporters (including OATP1B1 and OATP1B3) or hepatic or renal efflux pumps, such as Pgp and the multidrug and toxin-extrusion transporters 1 and 2K. Macitentan is not a substrate for Pgp, but can inhibit expression of the breast cancer-resistance protein. At clinically relevant concentrations, macitentan and ACT132577 do not interact with hepatic bile-salt-transport proteins, such as the bile-salt-export pump and sodium-dependent taurocholate cotransporting polypeptide.43,44,49 However, macitentan is a CYP3A4 substrate, so strong CYP3A4 inducers (eg, rifampicin) reduce macitentan exposure significantly if coadministered, so coadministration should be avoided. Instead, coadministration of macitentan with cyclosporine A (inhibitor of CYP3A4 and OATPs) in healthy volunteers does not alter the steady-state exposure to macitentan or ACT132577 significantly, so dose adjustment is not required.43,44,50 Furthermore, strong inhibitors of CYP3A4 (eg, ketoconazole, ritonavir) may increase macitentan exposure.51 Therefore, in the US, the concomitant use of macitentan and strong inhibitors of CYP3A4 (eg, antiretroviral drugs, such as ritonavir) is avoided, whereas in European countries caution is recommended if these agents are coadministered.43,44 Macitentan does not affect the pharmacokinetics of ethinylestradiol/norethisterone or the pharmacokinetics and pharmacodynamics of warfarin to a clinically significant extent.52,53
A minor but not clinically relevant pharmacokinetic interaction between macitentan and sildenafil has been observed. Based on those results, dose adjustment of either compound is not necessary during concomitant treatment with macitentan and sildenafil.54 Macitentan is excreted largely via the kidneys; the apparent elimination half-life of macitentan is ~16 hours and that of ACT132577 ~48 hours.43–45
Safety and tolerability
Macitentan improves hemodynamic parameters and functional exercise capacity in a concentration-dependent manner in patients with PAH, but no clear relationship is seen between safety parameters and macitentan concentrations.55 Macitentan (10 mg PO once daily) in general was well tolerated in SERAPHIN.10 Increases in levels of AST or ALT to more than triple the upper limit of normal (ULN) occurred in 3.4% of patients receiving macitentan (10 mg once daily) and in 4.5% of the placebo group, whereas increases in levels of AST or ALT to more than triple ULN associated with bilirubin to more than double ULN occurred in 1.7% in both groups. Massive increases in levels of liver enzymes were more frequent in macitentan patients (levels of transaminases more than eight times ULN were 2.1% in the treatment arm and 0.4% in the placebo group, which was fivefold greater).
With regard to all ERAs, the most important adverse effects are hepatotoxicity, peripheral edema, and anemia. Increases in levels of AST or ALT levels are associated with PAH and with ERA therapy. Treatment discontinuation due to hepatic adverse events occurred in 3.3% of macitentan and 1.6% of placebo arms in SERAPHIN, thereby showing a much better tolerability profile than Bosentan.10 Currently, it is recommended to monitor levels of liver enzymes before initiating macitentan treatment, and then upon clinical findings not to measure them monthly (as recommended for Bosentan). Therapy discontinuation is recommended if patients develop sustained, unexplained, clinically relevant increases in levels of aminotransferase, bilirubin above double ULN, or severe liver injury.43,44 Peripheral edema is more common and pronounced in patients on ETA-receptor-specific medications than dual RAs. In SERAPHIN, a significant difference in the incidence of peripheral edema was not observed with macitentan compared with placebo (18.2% versus 18.1%). Only a subgroup analysis on older patients showed a greater development of peripheral edema in the macitentan group (25.9% versus 18.2%).10
The third common side effect of ERAs is anemia. In general, anemia stabilizes in the first few weeks of therapy and does not necessitate treatment discontinuation. In SERAPHIN, anemia was seen in 13.2% in the 10 mg group versus 3.2% in the placebo group, with hemoglobin levels <8 mg/dL in 4.3% versus 0.4%, respectively.10 Only two patients, one in each group, discontinued therapy for severe anemia, and complete recovery was observed with cessation of the drug. It is recommended to measure the hemoglobin concentration before starting treatment with macitentan and during treatment if clinically indicated. Finally, there may be some minor respiratory and neurologic side effects. The most common is infection of the upper respiratory tract (15.3% versus 13.3%), followed by nasopharyngitis (14.0% versus 10.4%), headache (13.6% versus 8.8%), and bronchitis (11.6% versus 5.6%).
The safety profile of macitentan in the real-world postmarketing setting is now evaluated in the OPUS registry. OPUS is an ongoing (estimated completion April 2018), long-term, prospective, multicenter, observational drug registry of patients newly treated with macitentan (≤30 days of enrollment) in the US. In January 2016, OPUS included 416 patients with PAH newly treated with macitentan. The safety profile of macitentan in OPUS is similar to that reported in SERAPHIN, including hepatic adverse events related to macitentan therapy not observed until that date.56 Macitentan can be taken with or without food, and dose adjustment is not needed in those aged >65 years, but caution is advised in patients aged >75 years, because data are limited in these patients. The safety and efficacy of macitentan in the pediatric population have not been studied, and so its use is not approved in these patients.
Based on pharmacokinetic data, a dose adjustment is not required in renal impairment, but caution is recommended in severe forms, because data are lacking. It is contraindicated in those undergoing dialysis. Macitentan may cause harm to a fetus, so its use is contraindicated during pregnancy and in women of childbearing age who are not using reliable contraception. Pregnancy must be excluded before initiation, monthly during treatment, and 1 month after treatment has been discontinued. Patients must be advised to use contraception while taking macitentan, as well as 1 month after discontinuation of the drug. Macitentan is also contraindicated in women who are breastfeeding.43,44 Just two small studies (on 40 and 24 patients) have assessed the safety of switching from Bosentan to macitentan. Neither study showed a significant change in levels of liver enzymes, peripheral edema, nor anemia in patients who had switched treatment.57,58
Randomized clinical trials and long-term extension
Macitentan (10 mg PO once daily) has been approved (class I, level B) in monotherapy and combination therapy for the long-term treatment of PAH in adult patients in WHO functional classes II/III in the European Union and for the treatment of WHO group I PAH to delay disease progression and reduce hospitalization in the US. The efficacy of macitentan has been demonstrated in idiopathic PAH, hereditary PAH, CTD-PAH, and PAH related to repaired congenital systemic-to-pulmonary shunts. The efficacy of macitentan was investigated in SERAPHIN, a multicenter, double-blind, randomized, placebo-controlled, event-driven, Phase III trial.10 This trial marked a new era in the study of PAH, because of the large number of patients enrolled (742 patients randomized in three arms (1:1:1) to receive placebo, macitentan (3 mg), or macitentan (10 mg once-daily), the long duration of the trial (median duration of treatment of 115 weeks), and the composite primary end point of morbidity and mortality (death, atrial septostomy, lung transplantation), initiation of treatment with prostanoids (intravenously, subcutaneously), or worsening of PAH. SERAPHIN was unlike previous PAH trials, which had been characterized by small samples, short duration, and improvement of 6MWD as the primary end point.28,59–63
SERAPHIN enrolled patients with idiopathic PAH (55%), hereditary PAH (1.8%), or CTD-PAH (30.5%), repaired congenital systemic-to-pulmonary shunts (8.4%), HIV infection (8.4%), using drugs (1.4%), or toxin exposure (3%) in WHO functional class II/III (only 14 patients [1.9%] were in WHO functional class IV). Also, 63.7% of these patients were stably treated with oral or inhaled therapy for PAH (PDE5I in almost all cases). Patients receiving prostanoids (intravenously, subcutaneously) were excluded. SERAPHIN showed that macitentan significantly reduced the composite primary end point of morbidity and mortality among patients with PAH at a once-daily dose of 10 mg versus placebo (HR 0.55, 97.5% CI 0.39–0.76; P<0.001). The effect of macitentan on the primary end point was observed for both patients who had not received treatment previously and for both cases receiving therapy for PAH at baseline. Worsening of PAH was the most frequent primary end-point event. Also, the secondary composite end point was reached with a reduction in the prevalence of death due to PAH or hospitalization for PAH for a 10-mg dose versus placebo (HR 0.50, 97.5% CI 0.34–0.75; P<0.001), with hospitalization being the most common end-point event.
Macitentan therapy was approved only for PAH group 1, but several other randomized trials are trying to show its efficacy in other PAH groups and outside PAH. Several trials are ongoing (Table 3), but one, MERIT-1, has finished. MERIT-1 was a Phase II, double-blind, randomized, placebo-controlled trial in which macitentan (10 mg PO once daily) was assessed versus placebo in patients with inoperable chronic thromboembolic pulmonary hypertension (PAH group IV). The primary end point (pulmonary vascular resistance at rest at week 16, expressed as a percentage of baseline resting pulmonary vascular resistance) and some secondary end points were achieved. Significant improvements were also observed in other clinically relevant variables: exercise capacity, cardiac output, and NT-pro-BNP concentration. The treatment effect was consistent across the prespecified subgroups, including naïve patients and patients receiving other PAH treatments at baseline (eg, PDE5Is and oral/inhaled prostanoids).64
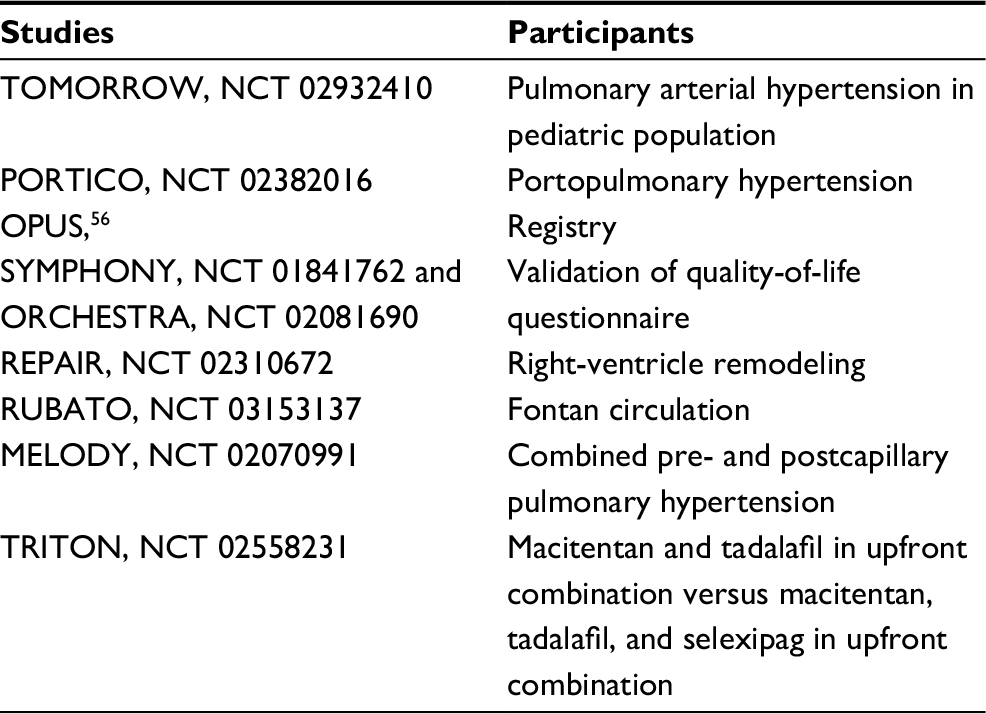 | Table 3 Ongoing studies using macitentan |
Macitentan substudies
SERAPHIN also included a hemodynamic substudy.65 It showed that patients with a cardiac index >2.5 L/min/m2, right atrial pressure <8 mmHg, or NT-pro-BNP <750 fmol/mL at month 6 had a lower risk of morbidity/mortality than those not meeting these cutoff values (HR 0.49, 95% CI 0.28–0.86; HR 0.72, 95% CI 0.42–1.22; HR 0.22, 95% CI 0.15–0.33, respectively). Hence, macitentan can increase the chance of reaching the cutoff values associated with the best prognosis for PAH patients.
In a post hoc analysis, Channick et al showed that macitentan (10 mg once daily) reduced the risk, prevalence, and number of days of hospitalization significantly. These treatment effects were driven by reductions in the risk and prevalence of PAH-related hospitalization. Macitentan reduced the risk of PAH-related hospitalization regardless of background PAH-specific therapy, but patients not receiving PAH therapy at baseline displayed a more pronounced reduction in risk.66
Another post hoc analysis was conducted on a subgroup of treatment-naïve patients enrolled in SERAPHIN. Patients allocated to placebo or macitentan groups were classified by time from the diagnosis to enrollment as “incident” (<6 months) or “prevalent” (>6 months). Macitentan (10 mg) reduced the risk of a morbidity/mortality event versus placebo by 60% in incident patients significantly (HR 0.40, 95% CI 0.20–0.79) and by 53% in prevalent patients (HR 0.47, 95% CI 0.24–0.92). Macitentan (10 mg) also reduced the risk of PAH-related death/hospitalization significantly by 77% in incident patients (HR 0.23, 95% CI 0.09–0.57) and by 62% in prevalent patients (HR 0.38, 95% CI 0.16–0.92). This analysis supports the use of macitentan as effective first-line therapy for delaying disease progression in incident and prevalent patients with PAH.67
The effect of macitentan (10 mg) versus placebo on long-term outcomes according to baseline WHO functional class has also been examined. As expected, the two subgroups differed at baseline in 6MWD and cardiac index, which were higher in patients in WHO functional class I/II than in III/IV, and pulmonary vascular resistance, which was lower. The interaction-test P-values were 0.64 and 0.60 for morbidity/mortality and death/hospitalization due to PAH, respectively. These values suggested the consistent efficacy of macitentan in reducing the risk of events irrespectively of WHO functional class at baseline.68
In another post hoc analysis of SERAPHIN, the effect of macitentan (10 mg) on the risk of morbidity and mortality was assessed in PAH patients with baseline hemodynamic values suggestive of impaired right-ventricle function. Macitentan (10 mg) reduced the risk of morbidity and mortality versus placebo significantly irrespectively of the presence or absence of right-ventricle impairment.69
Health-related quality of life was also evaluated in SERAPHIN by compiling the 36-item Short Form survey (SF36). At month 6, macitentan (10 mg) improved health-related quality of life, with significant effects observed for seven of eight SF36 domains for scores for physical component summary and mental component summary versus placebo. Furthermore, macitentan reduced the risk of a clinically meaningful deterioration in the scores for the physical component summary (HR 0.60, 95% CI 0.47–0.76; P<0.0001) and mental component summary (HR 0.76, 95% CI 0.61–0.95; P=0.0173) significantly until treatment cessation versus placebo.70
Besides SERAPHIN, there have been two small observational studies. One, by Herbert et al, assessed 15 patients with CHD-PAH (patients with complex CHD and patients with Down syndrome). Some of these patients were naïve and some shifted from Bosentan to macitentan. 6MWD increased from a median of 286 m to 360 m (P<0.05), most notably in those who were treatment-naïve.71 In another prospective observational study on a heterogeneous population of 40 adult CHD-PAH patients (including patients with Down syndrome or complex CHD) in treatment for many years with Bosentan (median 7.2 years), switching from Bosentan to macitentan showed an improvement in WHO functional class (the number of patients in WHO functional class III/IV decreased from 48% to 23%, P=0.004), NT-pro-BNP level (from 723 ng/mL to 488 ng/mL, P=0.019) and tricuspid annular plane systolic excursion (from 19 mm to 21 mm, P=0.002) at 6-month follow-up. Despite improvement in WHO functional class, 6MWD did not change, possibly due to the relatively high number of patients with Down syndrome (40%) in the cohort.57
Pizarro et al studied the effect of macitentan on right-ventricle myocardial function using speckle-tracking analysis in 16 patients with PAH. After 12 weeks of treatment with macitentan, global longitudinal (but not regional longitudinal) strain revealed significant improvement. Comparison of strain adaptation between the macitentan group and control group identified macitentan patients as experiencing a superior relative gain in global, basal septal, and basal lateral strain, whereas controls manifested deterioration of myocardial strain.72
Meta-analyses
Duo-Ji and Long carried out a network meta-analysis for multiple comparisons among PAH therapies: in 2,172 patients, all four ERAs increased mean 6MWD significantly in comparison with placebo.73 Moreover, Bosentan and ambrisentan showed a significant association with a reduction in risk of clinical worsening compared with placebo. With regard to all-cause discontinuation, ambrisentan was the only therapy associated significantly with a risk reduction compared with placebo. As such, ambrisentan could be considered the most appropriate therapy among the four ERAs for PAH patients, followed by Bosentan.
Wei et al undertook a meta-analysis on the clinical safety of ERAs. They noted that the incidence of abnormal hepatic function, peripheral edema, and anemia was significantly higher in the ERA group compared with placebo. Bosentan (but not macitentan) increased the risk of abnormal hepatic function significantly, whereas ambrisentan decreased that risk significantly. Bosentan and ambrisentan (but not macitentan) increased the risk of peripheral edema significantly. Bosentan and macitentan (but not ambrisentan) increased the risk of anemia significantly.74
Another meta-analysis from Zheng et al evaluated the efficacy and safety of oral targeted therapies in PAH and focused on overall survival. Compared with placebo, ERAs, PDE5Is, and riociguat reduced clinical worsening, ameliorated WHO functional class, and increased 6MWD significantly. However, prostanoids given via the oral route had only a mild effect on 6MWD, and did not reduce mortality or clinical worsening. ERAs and riociguat can reduce clinical worsening and ameliorate exercise capacity. Prostanoids have more significant adverse effects and weak therapeutic effects.75
Special guidelines
Current guidelines recommend the use of ambrisentan, Bosentan (recommendation I, level of evidence A) and macitentan (I, B) in patients with PAH and WHO functional class II and III. In WHO functional class IV, the first-line drug is Epoprostenol and the recommendation for ERAs is weaker (IIb, C).32 Combination therapy using two or more classes of drugs simultaneously (eg, β-blockers plus angiotensin-converting enzyme inhibitors) has been used for the treatment of systemic hypertension and heart failure. The experience with combination therapy in PAH, however, is increasing, and it may be applied sequentially or initially (upfront). In sequential therapy from monotherapy, there is addition of a second and then a third drug in cases of inadequate clinical results or in cases of deterioration. Current guidelines suggest several possible associations for sequential combination therapy,32 whereas in upfront combination therapy, the recommendation is I, B for ambrisentan plus tadalafil and IIa for ERAs plus PDE5Is.
Future developments
Several ongoing studies are assessing macitentan, the last ERA approved for PAH treatment (Table 3). Future perspectives beyond simple improvement in hemodynamics will focus on utilization of right-ventricle substrates and the effects of ERAs on right-ventricle remodeling and biventricular fibrosis.76,77 In the near future, assessment of ERA efficacy on right-ventricle remodeling will rely increasingly on cardiac magnetic resonance imaging. Moreover, new ERAs will be tested (eg, HJP272), and other ERAs will be tested in diseases other than PAH (eg, chronic kidney disease, mainly for diabetic nephropathy).78,79
Conclusion
The introduction of ERAs for PAH has changed its natural history and improved patient survival. The encouraging results arising from SERAPHIN and AMBITION with the introduction of upfront combination therapy (with PDE5Is) has shown new ways for clinical research. However, less exciting results with combination therapy (COMPASS-2, sildenafil plus Bosentan) have raised doubts about the efficacy of combination therapy, but might be linked to specific drugs, rather than to a class effect.80 Larger studies are warranted to assess the efficacy of three possible strategies with triple-combination therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol): results of a randomized trial. Ann Intern Med. 1990;112:485–491. | ||
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. | ||
Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease: a randomized, controlled trial. Ann Intern Med. 2000; 132:425–434. | ||
McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25:244–249. | ||
Chen SJ, Chen YF, Meng QC, et al. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol. 1995;79:2122–2131. | ||
Hoeper MM. Therapie der pulmonal arteriellen Hypertonie: Endothelin-Rezeptor-Antagonisten. [Treatment of pulmonary arterial hypertension: endothelin-receptor antagonists]. Dtsch Med Wochenschr. 2006;131:S308–S310. German. | ||
Pesto S, Begic Z, Prevljak S, Pecar E, Kukavica N, Begic E. Pulmonary hypertension: new trends of diagnostic and therapy. Med Arch. 2016;70:303–307. | ||
Volkov AV, Iudkina NN, Nikolaeva EV, et al. Бозентан: существенное увеличение продолжительности жизни пациентов с легочной артериальной гипертонией, ассоциированной с системными ревматическими заболеваниями. Терапевтический архив. [Bosentan: a considerable increase in the survival of patients with pulmonary hypertension associated with systemic rheumatic diseases]. Ter Arkh. 2014;86:32–39. Russian. | ||
Murdaca G, Lantieri F, Puppo F, et al. Beneficial effects of long-term treatment with bosentan on the development of pulmonary arterial hypertension in patients with systemic sclerosis. J Int Med Res. 2016;44:85–89. | ||
Pulido T, Adzerikho I, Channick R, et al. Study with an endothelin receptor antagonist in pulmonary arterial hypertension to improve clinical outcome: macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. | ||
Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. | ||
Iglarz M, Steiner P, Wanner D, Rey M, Hess P, Clozel M. Vascular effects of endothelin receptor antagonists depends on their selectivity for ETA versus ETB receptors and on the functionality of endothelial ETB receptors. J Cardiovasc Pharmacol. 2015;66:332–327. | ||
Correale M, Totaro A, Lacedonia D, et al. Novelty in treatment of pulmonary fibrosis: pulmonary hypertension drugs and others. Cardiovasc Hematol Agents Med Chem. 2013;11:169–178. | ||
Vatter H, Seifert V. Ambrisentan, a non-peptide endothelin receptor antagonist. Cardiovasc Drug Rev. 2006;24:63–76. | ||
Benza RL, Mehta S, Keogh A, Lawrence EC, Oudiz RJ, Barst RJ. Sitaxsentan treatment for patients with pulmonary arterial hypertension discontinuing bosentan. J Heart Lung Transplant. 2007;26:63–69. | ||
Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30:394–403. | ||
Don GW, Joseph F, Celermajer DS, Corte TJ. Ironic case of hepatic dysfunction following the global withdrawal of sitaxentan. Intern Med. 2012;42:1351–1354. | ||
Patel T, McKeage K. Macitentan: first global approval. Drugs. 2014;74:127–133. | ||
Bolli MH, Boss C, Binker, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N0-propylsulfamide (macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem. 2012;55:7849–7861. | ||
Davenport AP, Hyndman KA, Dhaun N, et al. Endothelin. Pharmacol Rev. 2016;68:357–418. | ||
Sidharta PN, Treiber A, Dingemanse J. Clinical pharmacokinetics and pharmacodynamics of the endothelin receptor antagonist macitentan. Clin Pharmacokinet. 2015;54:457–471. | ||
Gatfield J, Grandjean CM, Sasse T, et al. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One. 2012;7:e47662. | ||
Iglarz M, Bossu A, Wanner D, et al. Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension. Life Sci. 2014;118:333–339. | ||
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. | ||
Dingemanse J, Sidharta PN, Maddrey WC, et al. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13:391–405. | ||
D’Alto M. An update on the use of ambrisentan in pulmonary arterial hypertension. Ther Adv Respir Dis. 2012;6:331–343. | ||
Rivera-Lebron BN, Risbano MG. Ambrisentan: a review of its use in pulmonary arterial hypertension. Ther Adv Respir Dis. 2017;11:233–244. | ||
Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–3019. | ||
Oudiz RJ, Galiè N, Olschewski H, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:1971–1981. | ||
Badesch DB, Feldman J, Keogh A, et al. ARIES-3: ambrisentan therapy in a diverse population of patients with pulmonary hypertension. Cardiovasc Ther. 2012;30:93–99. | ||
Coghlan JG, Galiè N, Barberà JA, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis. 2017;76:1219–1227. | ||
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2016;37:67–119. | ||
Shapiro S, Torres F, Feldman J, et al. Clinical and hemodynamic improvements after adding ambrisentan to background PDE5i therapy in patients with pulmonary arterial hypertension exhibiting a suboptimal therapeutic response (ATHENA-1). Respir Med. 2017;126:84–92. | ||
Cartin-Ceba R, Swanson K, Iyer V, Wiesner RH, Krowka MJ. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139:109–114. | ||
Fischer A, Denton CP, Matucci-Cerinic M, et al. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): a subgroup analysis of the ARIES-E clinical trial. Respir Med. 2016;117:254–263. | ||
Hoeper MM, McLaughlin VV, Barberá JA, et al. Initial combination therapy with ambrisentan and tadalafil and mortality in patients with pulmonary arterial hypertension: a secondary analysis of the results from the randomised, controlled AMBITION study. Lancet Respir Med. 2016;4:894–901. | ||
Vachiéry JL, Hoeper MM, Peacock AJ, et al. Ambrisentan use for pulmonary arterial hypertension in a post-authorization drug registry: the Volibris tracking study. J Heart Lung Transplant. 2017;36:399–406. | ||
Hassoun PM, Zamanian RT, Damico R, et al. Ambrisentan and tadalafil up-front combination therapy in scleroderma-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:1102–1110. | ||
D’Alto M, Romeo E, Argiento P, et al. Initial tadalafil and ambrisentan combination therapy in pulmonary arterial hypertension: clinical and haemodynamic long-term efficacy (ITALY study). J Cardiovasc Med (Hagerstown). 2018;19:12–17. | ||
Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–745. | ||
Sidharta N, van Giersbergen PL, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple dose study in healthy subjects. J Clin Pharmacol. 2013;53:1131–1138. | ||
de Kanter R, Sidharta PN, Delahaye S, et al. Physiologically based pharmacokinetic modeling of macitentan: prediction of drug-drug interactions. Clin Pharmacokinet. 2016;55:369–380. | ||
Actelion Pharmaceuticals. Opsumit (macitentan) tablets, for oral use [prescribing information]. San Francisco, NC: Actelion; 2016. | ||
European Medicines Agency. Opsumit (macitentan) [summary of product characteristics]. London: EMA; 2016. | ||
Bruderer S, Hopfgartner G, Seiberling M, et al. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012;42:901–910. | ||
Bruderer S, Marjason J, Sidharta PN, et al. Pharmacokinetics of macitentan in Caucasian and Japanese subjects: the influence of ethnicity and sex. Pharmacology. 2013;91:331–338. | ||
Ahn LY, Kim SE, Yi S, et al. Pharmacokinetic–pharmacodynamic relationships of macitentan, a new endothelin receptor antagonist, after multiple dosing in healthy Korean subjects. Am J Cardiovasc Drugs. 2014;14:377–385. | ||
Sidharta PN, Lindegger N, Ulč I, Dingemanse J. Pharmacokinetics of the novel dual endothelin receptor antagonist macitentan in subjects with hepatic or renal impairment. J Clin Pharmacol. 2014;54:291–300. | ||
Treiber A, Aanismaa P, de Kanter R, et al. Macitentan does not interfere with hepatic bile salt transport. J Pharmacol Exp Ther. 2014;350:130–143. | ||
Bruderer S, Aanismaa P, Homery MC, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012;14:68–78. | ||
Atsmon J, Dingemanse J, Shaikevich D, et al. Investigation of the effects of ketoconazole on the pharmacokinetics of macitentan, a novel dual endothelin receptor antagonist, in healthy subjects. Clin Pharmacokinet. 2013;52:685–692. | ||
Hurst N, Pellek M, Dingemanse J, et al. Lack of pharmacokinetic interactions between macitentan and a combined oral contraceptive in healthy female subjects. J Clin Pharmacol. 2016;56:669–674. | ||
Sidharta PN, Dietrich H, Dingemanse J. Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clin Drug Investig. 2014;34:545–552. | ||
Sidharta PN, van Giersbergen PL, Wolzt M, Dingemanse J. Investigation of mutual pharmacokinetic interactions between macitentan, a novel endothelin receptor antagonist, and sildenafil in healthy subjects. Br J Clin Pharmacol. 2014;78:1035–1042. | ||
Zisowsky J, Sidharta PN, Krause A, Dingemanse J. Pharmacokinetic/pharmacodynamic analyses in SERAPHIN, a randomized, controlled study of macitentan in patients with pulmonary arterial hypertension. Clin Pharmacol Drug Dev. 2013;2:29. | ||
Kim NH, Chin KM, Muros-le Rouzic E, et al. Opsumit Users Registry (OPUS): insights into the safety and tolerability of Opsumit. Am J Respir Crit Care Med. 2016;193:A7396. | ||
Blok IM, van Riel AC, van Dijk AP, Mulder BJ, Bouma BJ. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: further improvement? Int J Cardiol. 2017;227:51–52. | ||
Safdar Z, Thakur A, Frost A. Tolerability of switch to macitentan from bosentan in pulmonary arterial hypertension. South Med J. 2017;110:223–228. | ||
Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–329. | ||
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. | ||
Simonneau G, Barst RJ, Galiè N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analog, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–804. | ||
Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. | ||
Galiè N, Brundage B, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–2903. | ||
Ghofrani HA, Simonneau G, D’Armini AM, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med. 2017;10:785–794. | ||
Galiè N, Jansa P, Pulido T, et al. SERAPHIN haemodynamic sub-study: the effect of the dual endothelin receptor antagonist macitentan on haemodynamic parameters and NT-proBNP levels and their association with disease progression in patients with pulmonary arterial hypertension. Eur Heart J. 2017;38:1147–1155. | ||
Channick RN, Delcroix M, Ghofrani HA, et al. Effect of macitentan on hospitalizations: results from the SERAPHIN trial. JACC Heart Fail. 2015;3:1–8. | ||
Simonneau G, Channick RN, Delcroix M, et al. Incident and prevalent cohorts with pulmonary arterial hypertension: insight from SERAPHIN. Eur Respir J. 2015;46:1711–1720. | ||
Channick RN, Delcroix M, Galie N, et al. The effect of macitentan on long-term outcomes in patients with pulmonary arterial hypertension by WHO functional class: data from the randomized controlled SERAPHIN study. Am J Respir Crit Care Med. 2014;189:A4783. | ||
Sitbon O, Channick RN, Delcroix M, et al. Macitentan reduces the risk of morbidity and mortality irrespective of the presence or absence of right ventricular (RV) impairment: results from the randomised SERAPHIN trial in pulmonary arterial hypertension (PAH). Eur Respir J. 2014;44:3419. | ||
Mehta S, Sastry B, Souza R, et al. Macitentan improves health-related quality of life for patients with pulmonary arterial hypertension: results from the randomized controlled SERAPHIN trial. Chest. 2017;151:106–118. | ||
Herbert S, Gin-Sing W, Howard L, Tulloh RM. Early experience of macitentan for pulmonary arterial hypertension in adult congenital heart disease. Heart Lung Circ. 2017;26:1113–1116. | ||
Pizarro C, Meyer-Arend JM, Schueler R, et al. Impact of macitentan on right ventricular myocardial function in pulmonary arterial hypertension. Int J Cardiol. 2016;214:438–441. | ||
Duo-Ji MM, Long ZW. Comparative efficacy and acceptability of endothelin receptor antagonists for pulmonary arterial hypertension: a network meta-analysis. Int J Cardiol. 2017;234:90–98. | ||
Wei A, Gu Z, Li J, et al. Clinical adverse effects of endothelin receptor antagonists: insights from the meta-analysis of 4894 patients from 24 randomized double-blind placebo-controlled clinical trials. J Am Heart Assoc. 2016;5:e003896. | ||
Zheng YG, Ma H, Hu EC, Liu G, Chen G, Xiong CM. Oral targeted therapies in the treatment of pulmonary arterial hypertension: a meta-analysis of clinical trials. Pulm Pharmacol Ther. 2014;29:241–249. | ||
Drozd K, Ahmadi A, Deng Y, et al. Effects of an endothelin receptor antagonist, macitentan, on right ventricular substrate utilization and function in a Sugen 5416/hypoxia rat model of severe pulmonary arterial hypertension. J Nucl Cardiol. 2016;6:1979–1989. | ||
Nielsen EA, Sun M, Honjo O, Hjortdal VE, Redington AN, Friedberg MK. Dual endothelin receptor blockade abrogates right ventricular remodeling and biventricular fibrosis in isolated elevated right ventricular afterload. PLoS One. 2016;11:e0146767. | ||
Liu X, Khadtare N, Patel H, Stephani R, Cantor J. Time-dependent effects of HJP272, an endothelin receptor antagonist, in bleomycin-induced pulmonary fibrosis. Pulm Pharmacol Ther. 2017;45:164–169. | ||
Egido J, Rojas-Rivera J, Mas S, et al. Atrasentan for the treatment of diabetic nephropathy. Expert Opin Investig Drugs. 2017;26:741–750. | ||
Actelion. Effects of the combination of bosentan and sildenafil versus sildenafil monotherapy on pulmonary arterial hypertension (PAH) (COMPASS-2). Available from: https://clinicaltrials.gov/ct2/show/NCT00303459. NLM identifier: NCT00303459. Accessed April 5, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.