")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Endoplasmic Reticulum (ER) Stress in Part Mediates Effects of Angiotensin II in Pancreatic Beta Cells
Authors Ramalingam L, Sopontammarak B, Menikdiwela KR, Moustaid-Moussa N
Received 16 April 2020
Accepted for publication 24 June 2020
Published 18 August 2020 Volume 2020:13 Pages 2843—2853
DOI https://doi.org/10.2147/DMSO.S257797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Latha Ramalingam, Boontharick Sopontammarak, Kalhara R Menikdiwela, Naima Moustaid-Moussa
Department of Nutritional Sciences, And Obesity Research Institute, Texas Tech University, Lubbock, TX 79424, USA
Correspondence: Naima Moustaid-Moussa
Texas Tech University, Department of Nutritional Sciences & Obesity Research Institute, 1301 Akron Street, Lubbock, TX 79409-1270, USA
Tel + 806-834-7946
Email [email protected]
Introduction: The renin angiotensin aldosterone system (RAAS) is a hormone system known for its role in regulating blood pressure and fluid balance. Numerous RAAS inhibitors routinely prescribed for hypertension have also beneficial effects in type 2 diabetes (T2D) prevention. RAAS components are expressed locally in many tissues, including adipose tissue and pancreas, where they exert metabolic effects through RAAS bioactive hormone angiotensin II (Ang II). Pancreatic beta cells are specialized insulin-producing cells; they have also developed endoplasmic reticulum (ER), which contributes to beta cell dysfunction, when proteins are misfolded in disease states such as T2D. However, no studies have investigated the relationship between RAAS and ER stress in beta cells as a mechanism linking pancreatic RAAS to T2D. Hence, we hypothesized that Ang II treatment of beta cells increases ER stress and inflammation leading to reduced insulin secretion.
Methods: To test this hypothesis, we treated clonal INS-1E beta cells and human islets with Ang II and assessed changes in ER stress markers. INS-1E beta cells were also used for measuring insulin secretion and for assessing the effects of various RAAS and ER stress inhibitors.
Results: We demonstrated that Ang II significantly increased the expression of ER stress genes such as Chop and Atf4 and reduced insulin secretion. Furthermore, inhibition of Ang II production with an angiotensin converting enzyme inhibitor (ACEi, captopril) significantly reduced ER stress. Moreover, the Ang II receptor blockade reduced ER stress significantly and rescued insulin secretion.
Discussion: This research provides new mechanistic insight into the role of RAAS activation via ER stress on beta cell dysfunction and provides additional evidence for protective effects of RAAS inhibition in T2D.
Keywords: beta cells, renin angiotensin aldosterone system, RAAS, type 2 diabetes, ER stress, inflammation
Introduction
Type 2 diabetes mellitus (T2D) is an emerging epidemic worldwide, with the number of people affected rising significantly over the past several decades.1 It is a complex metabolic condition that requires the simultaneous presence of two defects: insulin resistance and impaired pancreatic beta cell function.2 Pancreatic beta cells in the early stages of insulin resistance, secrete more insulin to compensate for increasing insulin resistance; however, this leads to an extra burden on beta cells, which fail at some point resulting in T2D.3
Several mechanisms have been associated with pathogenesis and the development of T2D. One of these is the renin angiotensin aldosterone system (RAAS), which is classically known to regulate blood pressure and fluid balance. While numerous studies reported contributions of RAAS to T2D,4–6 and benefits of RAAS inhibitors in T2D prevention,7 the role of this system in beta cell function is relatively less explored. Angiotensinogen (Agt) is the primary precursor in RAAS and is catalyzed into smaller angiotensin peptides, including angiotensin II (Ang II), which is the main bioactive hormone product.8 Recently, components of RAAS have been discovered locally within various tissues, including adipose tissue and pancreas, with local functions.5,9–12 Mouse and human islets treated with Ang II demonstrated significantly reduced insulin secretion, which led to beta cell dysfunction.4,5
Several clinical trials demonstrated that angiotensin receptor blockers (ARB) and angiotensin converting enzyme inhibitors (ACEi), primarily used as anti-hypertensive drugs also reduce T2D incidence by 25% in high-risk patients.7,13–15 This reduction is in part from protective actions on pancreatic islets through improved beta cell function.15–17 Furthermore, mice treated with ACEi have enhanced beta cell mass and improved beta cell proliferation markers.18,19 More importantly, pancreatic islets treated with agonists or blockers of the RAAS pathway points to a possible role of activated local RAAS in insulin secretion,5,20 but the precise mechanisms involved are not well established.
One of the downstream mechanisms through which RAAS contributes to obesity and insulin resistance is the endoplasmic reticulum (ER) stress.21,22 ER is a specialized secretory organelle, which is pivotal for protein folding. ER is especially critical in beta cells as around one million insulin molecules are produced per minute.23 When ER is overburdened due to hyperinsulinemia, immature insulin and other unfolded proteins accumulate in ER lumen, leading to activation of unfolded protein responses (UPRs).24 When UPR reaches beyond a certain capacity in cells, it results in ER stress, which then reduces insulin secretion leading to beta cell death and T2D.25 Recently, microRNAs, which are post-transcriptional gene regulators, demonstrated regulation of cell survival or cell death through ER stress mediation in beta cells.26 Specifically, miR-30 family and miR-708 were reduced or increased with ER stress, respectively.21 The detailed mechanisms by which RAAS regulates ER stress in beta cells are not completely understood. Hence, we hypothesized that activation of RAAS in beta cells led to increased ER stress and reduced insulin secretion; we further hypothesized that both of which will improve with RAAS inhibition. The results of this study will increase understanding of the role of activated RAAS in beta cells on T2D progression and may help researchers develop potential new anti-diabetic therapeutic targets.
Materials and Methods
Cell Culture and Islet Procurement
Rat pancreatic INS-1E cell lines were a kind gift from Dr. Patrick Fueger (City of hope, CA) and approved by the Texas Tech University Research Ethics Committee. Cell lines were maintained in 5% CO2 at 37°C in RPMI-1640 (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA USA), 1 mM sodium pyruvate, 10 mM HEPES, 0.05 mM 2-mercaptoethanol, and 50 U/mL penicillin and 50 g/mL streptomycin (Thermo Fisher Scientific, Waltham, MA USA). Human islets were obtained through the Integrated Islet Distribution Program (https://iidp.coh.org/). We received shipments of human islets from de identified male subjects between 20–50 years with a BMI <30. Upon arrival, islets were hand-picked for further treatments.
INS-1E cells were treated with various doses (10–100 nM) of Ang II (Sigma-Aldrich, St. Louis, MO, USA) for 24 hours and 48 hours, respectively. Pancreatic human islets were treated with various doses of Ang II for 48 hours. For other experiments, various inhibitors such as 1 mg/mL captopril (Angiotensin-converting enzyme inhibitor: ACEi), 1000 nM P-186 (Angiotensin type 2 receptor inhibitor; P) and 1 mg/mL telmisartan (Angiotensin type 1 receptor inhibitor; T), 7.5 mM 4-phenyl butyric acid (4-PBA; ER stress inhibitor; Sigma-Aldrich, St. Louis, MO, USA) were used based on previous literature.21,27
For glucose starvation treatments, cells were starved using HBSS buffer (1M NaHCO3, 1M HEPES, 1M CaCl2, 0.075M KCl, 2.5M MgSO4, 5M NaCl, 1M KH2PO4 and 1g BSA) containing 3 mM glucose for 2 hours. Cells were then treated with HBSS buffer containing 15 mM glucose along with the respective treatments. After 2 hours, media collected was used for insulin secretion experiments, while cells were used for gene expression studies.
RNA Isolation and Gene Expression
RNeasy mini kit (Qiagen, Valencia, CA, USA) was used for RNA isolation, and cDNA was reverse transcribed using iScript reverse transcription supermix (BioRad, Hercules, CA, USA). Gene expression levels were assessed by real-time quantitative polymerase chain reaction (RT-qPCR) using Sybr green master mix (BioRad, Hercules, CA, USA) and normalized to housekeeping genes (18S ribosomal RNA and GAPDH).
MicroRNA Expression
Total RNA was used for cDNA synthesis by reverse transcription using TaqMan™ Advanced microRNA cDNA Synthesis Kit (Thermo Fisher Scientific, Hereford, TX, USA). MicroRNA expression levels were assessed with real-time quantitative polymerase chain reaction (RT-qPCR) using the TaqMan™ Fast advanced master mix (Thermo Fisher Scientific, Hereford, TX, USA). The microRNA samples were normalized to housekeeping microRNAs (miR-191-5p and miR-186-5p).
Elisa
Insulin content was measured using enzyme-linked immunosorbent assay (ELISA) kits (EMD Millipore Corporation, St. Louis, Missouri, USA) according to the manufacturer’s protocol.
Statistical Analyses
Results are presented as means ± SEM where one-way ANOVA was performed with Tukey’s posthoc test (p<0.05) for experiments with more than two groups and analyzed using the Graph Pad Prism, version 8. On the contrary, Student’s t-test was used for data with two groups. qRT-PCR assay results were analyzed using the CFX Manager software provided by Bio-Rad Laboratories, Inc., using the 2-ΔΔCT method. All in vitro experiments had at least three replicates.
Results
We tested whether Ang II activates ER stress in pancreatic beta cells using dose and time-dependent studies. We treated INS-1E cells with various doses of Ang II (10–100 nM) for 24 and 48 hours, respectively, to identify the optimal concentration of Ang II for inducing ER stress. We measured markers of ER stress, such as activating transcription factor 4 (Atf4) and CCAAT-enhancer-binding protein homologous protein (Chop). The expression of these genes was no different from untreated control cells at 24 hours (Figure 1A and B). However, Atf4 mRNA levels were significantly increased by ~ 2.1 fold (p<0.05) with 50 nM Ang II treatment at 48 hours compared to untreated control cells (Figure 1C). The Chop mRNA level increased significantly with all three doses of Ang II compared to control after 48 hours (Figure 1D). Ang II at 10 nM increased Chop levels by ~ 1.5 fold, while Ang II at 50 nM increased Chop mRNA by ~ 3 fold, respectively (P<0.05) (Figure 1D). Consistent with these findings, glucose-stimulated insulin secretion was increased by ~ 8 fold (p<0.05) as expected, with 15 mM glucose compared to 3 mM glucose at 24 hours (Figure 1E). Ang II at 50 nM in the presence of 15 mM glucose reduced insulin secretion significantly by ~ 4 fold (p<0.05) compared to the 15 nM glucose at 24 hours (Figure 1E). At 48 hours, insulin secretion with 50 nM Ang II was significantly decreased by 3.7 fold compared to 15 mM glucose control, with no differences for 10 and 100 nM Ang II compared to control (Figure 1F). Based on the above findings, we chose 50 nM of Ang II to induce ER stress for subsequent experiments.
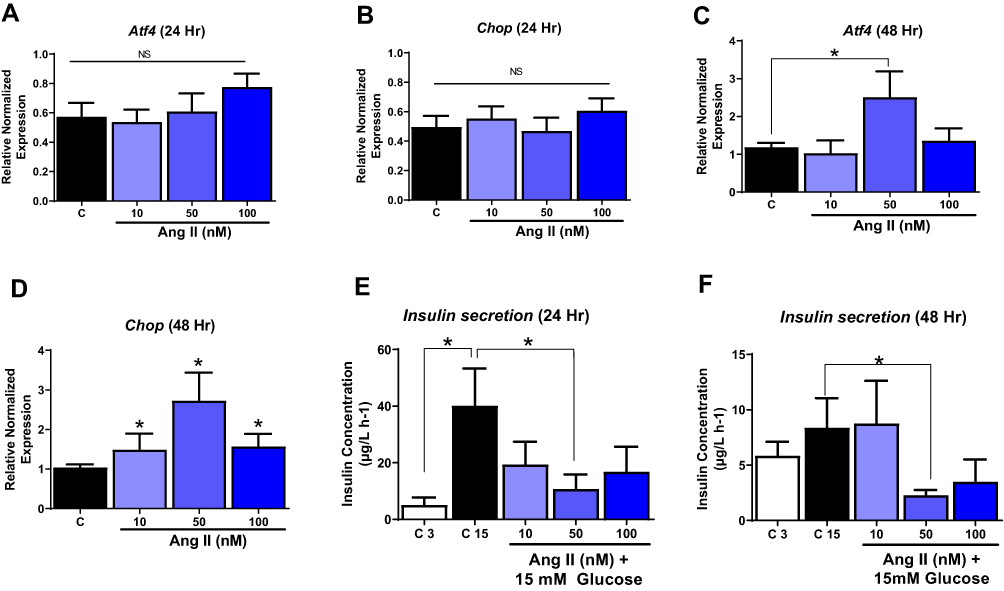 |
Figure 1 Dose and time-dependent effects of Angiotensin II. INS-1E cells were treated with different doses of Ang II to determine the optimal dose of Ang II to induce ER stress. (A and B) Ang II at all doses tested did not alter activated transcription factor 4 (Atf4) and C/EBP homologous protein (Chop) gene levels at 24 hours. (C) Ang II at 50 nM increased Atf4 gene levels, and (D) Ang II at all doses tested increased Chop gene levels at 48 hours. (E) Ang II only at 50 nm reduced insulin secretion at 24 hours. (F) Ang II at 50 nM reduced insulin secretion at 48 hours. Data are presented as mean ± SEM (n=4). *p< 0.05 compared to control. |
We validated the findings from INS-1E cells in human pancreatic islets treated with various doses of Ang II for 48 hours. As seen in rodent cells, Atf4 gene level was significantly increased with 50 nM Ang II by ~ 2 fold (Figure 2A) (p<0.05) while the Chop levels were increased by ~ 1.7 fold (p<0.05) with Ang II at both 50 and 100 nM in human pancreatic islets (Figure 2B). Due to the cost and limited availability of human islets, other experiments were performed in INS-1E cells.
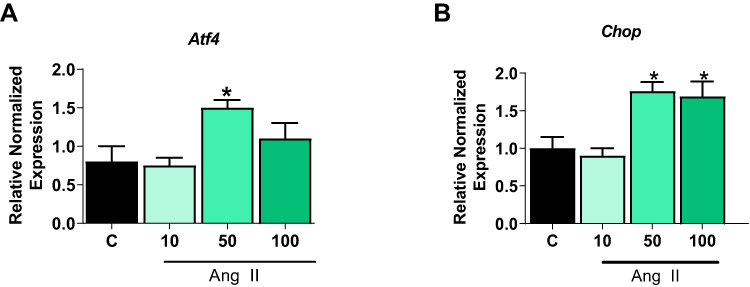 |
Figure 2 Dose and time-dependent effects of Angiotensin II on Human Pancreatic islets. Human Pancreatic islets were treated with different doses of Ang II for 48 hours to determine the optimal dose of Ang II to induce ER stress. (A) Ang II at 50 nM increased gene levels of activated transcription factor 4 (Atf4). (B) Ang II at both 50 and 100 nM increased C/EBP homologous protein (Chop) gene levels. Data are presented as mean ± SEM (n=4). *p< 0.05 compared to control. |
To further confirm the role of RAAS in ER stress, we treated INS-1E cells with captopril, an ACEi thats inhibits the production of Ang II. Pancreatic beta cells treated with captopril for 48 hours significantly lowered gene levels of Atf4 and Chop compared to control by ~ 1.5 fold (p<0.05), as seen in Figure 3A and B. Furthermore, captopril treatment increased insulin secretion by ~1.6 fold (P<0.06), which showed trending towards significance compared to control (Figure 3C).
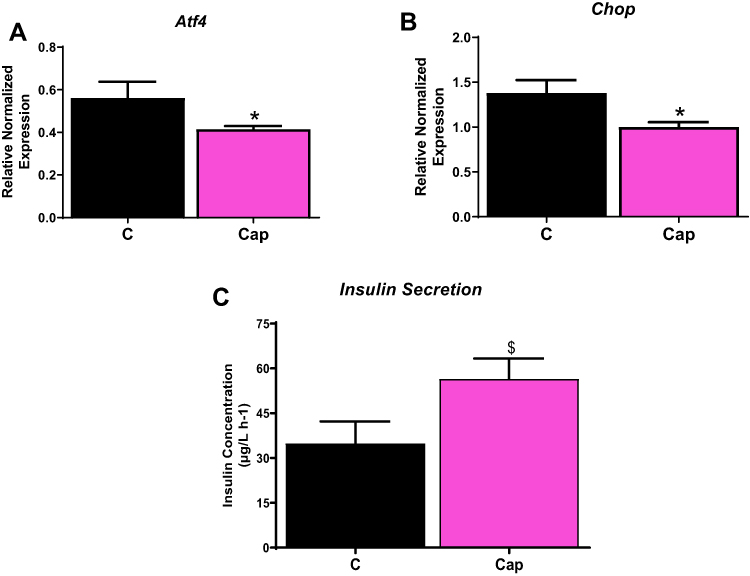 |
Figure 3 Captopril reduces ER stress. INS-1E cells treated with 100 µM captopril reduces ER stress as shown by (A) activated transcription factor 4 (Atf4) gene levels, (B) C/EBP homologous protein (Chop) expression gene levels and (C) improved insulin secretion. Data are presented as mean ± SEM (n=5 each group). *p< 0.05 compared to control. $p< 0.1. |
Ang II effects are mediated downstream through two receptors of the RAAS, namely angiotensin type 1 receptor (AT1) and angiotensin type 2 receptor (AT2). As the RAAS pathway induces ER stress, we sought to determine the Ang II receptor type mediating these effects. We treated cells with Ang II in the presence of AT1 inhibitor (telmisartan) and AT2 inhibitor (P-186), either individually or in combination. While Ang II treatment alone induced ER stress as shown by significantly increased levels of Atf4 and Chop mRNA levels by ~ 2 and 1.5 fold (p<0.05) compared to control respectively (Figure 4A and B), treatment with either P-186 or telmisartan reduced both Chop and Atf4 gene expression significantly, compared to Ang II-treated cells. Additionally, treatment with both inhibitors (telmisartan and P-186) also significantly lowered Atf4 and Chop expression levels compared to control, suggesting ER stress is equally transmitted via both the AT1 receptor and AT2 receptor as shown in Figure 4A and B (p<0.05). Lastly, insulin secretion was significantly reduced by 50 nM of Ang II by ~ 2 fold but was rescued by telmisartan, P-186 individually as well as combined receptor treatments (Figure 4C).
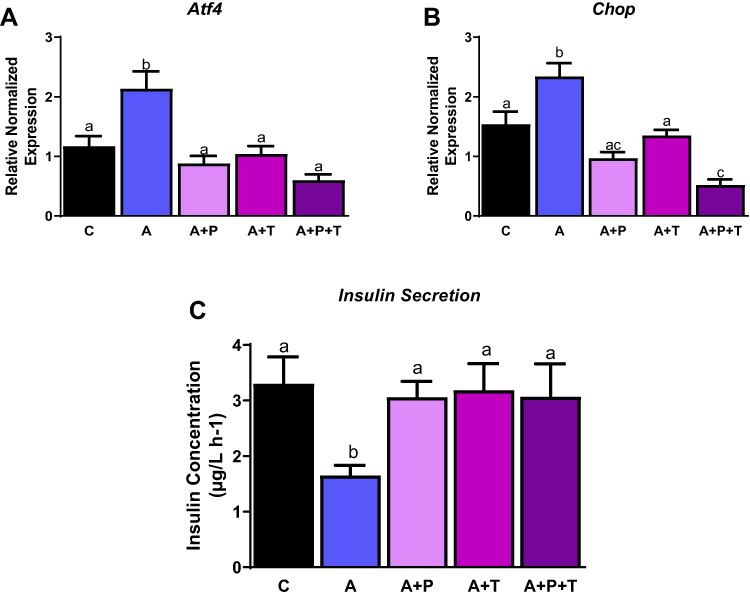 |
Figure 4 Angiotensin receptor inhibitors reduce ER stress. INS-1E cells treated with telmisartan (T; AT1 inhibitor), P186 (P; AT2 inhibitor) reduce ER stress as shown by (A) activated transcription factor 4 (Atf4) gene levels, (B) C/EBP homologous protein (Chop) gene levels and (C) improved insulin secretion. Data are presented as mean ± SEM (n=4-5 each group). Means without the same letter are different. |
To further validate Ang II effects on beta cells via ER stress, we tested whether the effects of Ang II could be reduced by ER stress inhibitor 4-PBA. INS-1E cells were treated with Ang II (50 nM) alone or in the presence of 4-PBA for 48 hours, and ER stress markers were measured. As expected, Ang II induced ER stress by 1.5 fold; however, ER stress was significantly reduced with 4-PBA along with Ang II by ~ 3 and 5 fold (p<0.05), as shown by expression levels of both Atf4 and Chop respectively in Figure 5A and B. Lastly, insulin secretion was significantly increased by ~ 1.9 fold (p<0.05) with Ang II along with 4-PBA compared to Ang II, as shown in Figure 5C.
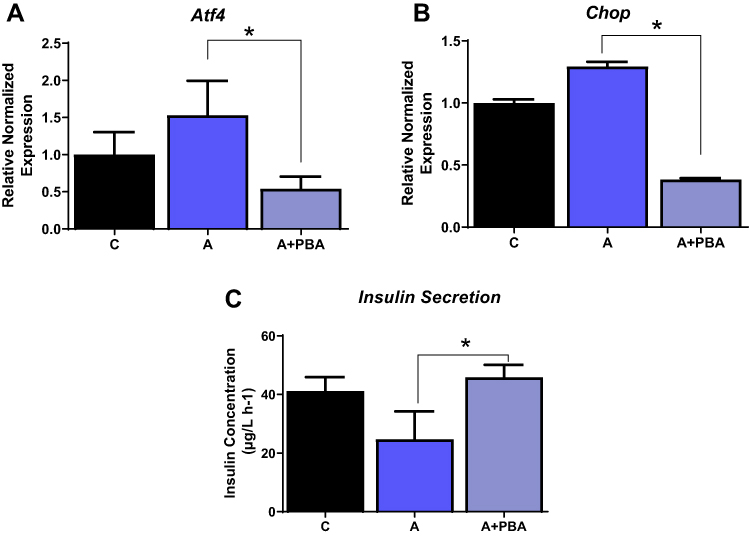 |
Figure 5 ER stress inhibitor reduces ER stress even in the presence of Angiotensin II. INS-1E cells treated with Ang II and ER stress inhibitor, 4-phenyl butyric acid reduces ER stress as shown (A) activated transcription factor 4 (Atf4), (B) C/EBP homologous protein (Chop) and (C) improved insulin secretion. Data are presented as mean ± SEM (n=5 each group). *p< 0.05 compared to Ang II. |
Since RAAS exerts proinflammatory effects in adipose tissue and adipocytes,10 we determined if activation of RAAS also led to inflammation in beta cells and, if so, whether this was mediated by Ang II receptors. We measured various inflammation markers, such as inducible nitric oxide synthase (iNos), nuclear factor-kappa B (NFfkb), and tumor necrosis factor-alpha (Tnf-α) in Ang-II treated INS-1E cells. Ang II significantly increased iNos mRNA levels by ~ 2 fold, while inhibition of AT1 and AT2 receptors individually or in combination reduced iNos mRNA levels, compared to Ang II (Figure 6A). Surprisingly, there were no differences with the above treatments for expression of Nfkb and Tnf-α genes (Figure 6B and C). In addition, we measured interleukin (Il)-6, interleukin- 1 beta (Il-1β), monocyte chemoattractant protein −1 (Mcp-1), and Il-10 mRNA levels, but their expression levels were not detectable in INS-1E cells.
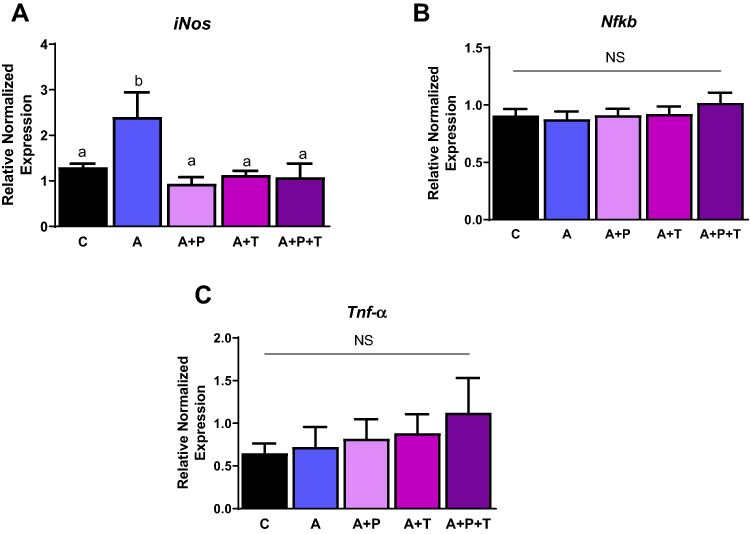 |
Figure 6 Angiotensin induces inflammation through iNos. INS-1E cells treated with telmisartan (T; AT1 inhibitor), P186 (P; AT2 inhibitor) reduce inflammation as shown by (A) inducible nitric oxide synthase (iNos) gene levels, (B) nuclear factor kappa-light-chain-enhancer of activated B cells (NfkB) gene levels and (C) tumor necrosis factor alpha (Tnf-α). Data are presented as mean ± SEM (n=5 each group). Means without the same letter are different. |
Lastly, as miRNAs have been previously reported to regulate ER stress and inflammation, and mediate Ang II effects in part in adipocytes,21 we specifically tested changes in miR-30 and miR-708-5p, negatively and positively associated with ER stress, respectively.21,28 As shown in Figure 7, Ang II treatment significantly reduced levels of miR-30c-5p by ~ 2 fold but not miR-30a-3p and miR-708-5p levels compared to control untreated cells.
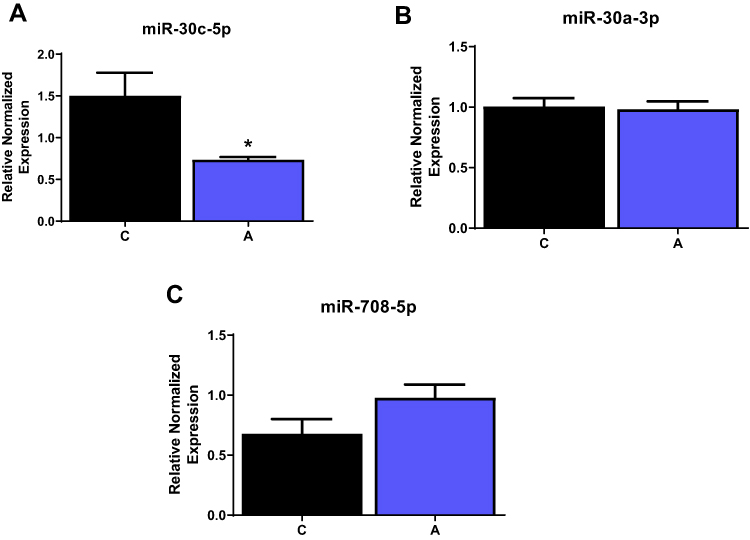 |
Figure 7 Alterations in microRNAs associated with ER stress. INS-1E cells were treated with Ang II, and levels of different miRNAs were measured. (A) miR-30c was reduced while (B) miR-30a and (C) miR-708 was unaltered in the presence of Ang II. Data are presented as mean ± SEM (n=3). *p<0.05. |
Discussion
RAAS is a critical physiological regulator of blood pressure and fluid balance. However, additional local functions of RAAS have also been identified in several metabolic tissues, including adipose tissue and the pancreas.11,29 Components of RAAS (AT1, AT2, Agt, Ang II and ACE) are expressed in murine and human pancreatic islets with a local role within the pancreas.17,30–32 RAAS regulates islet blood flow;33 however, specific non-hemodynamic functions in pancreatic beta cells are not well established.34
In our current study, Ang II treatment, which mimics RAAS overactivation, induces ER stress in pancreatic beta cells and human islets. Furthermore, Ang II reduces insulin secretion in INS-1E cells. Additionally, inhibiting the RAAS pathway reduced ER stress and improved insulin secretion in beta cells. Furthermore, increased ER stress by Ang II was mediated via both AT1 and AT2 receptors. Moreover, we have shown that one of the common miRNA regulators of ER stress, miR-30, was also reduced in our studies. These studies find in part that ER stress induced by the RAAS pathway could be a mechanistic pathway that could contribute to T2D in beta cells.
The Ang II receptor, AT1, is primarily known to mediate vasoconstriction, oxidative stress, and inflammation,35 while AT2 overall exerts beneficial properties such as vasodilation, anti-inflammatory, and anti-fibrotic effects.36 In human islets, inhibition of AT1 receptor reduced ER stress and improved its antioxidant potential and insulin secretion.37 These findings agree with our results demonstrating that ER stress is reduced when interaction of Ang II with AT1 was blocked.38 Additionally, we showed that ER stress was also, in part, mediated by the AT2 receptor, typically considered a protective receptor in other cell types.36,39 However, in rodent clonal beta cells, overexpression of AT2 receptor increased apoptosis and reduced insulin secretion.40 This suggests that AT2 receptor signaling may upregulate Chop activity, which overburdens ER, leading to increased apoptosis. This is in part consistent with our data showing that inhibition of AT2 reduced ER stress; however, whether this will trigger apoptosis needs to be further validated. Interestingly, in ob/ob mice, a model of T2D, AT1 receptor gene levels in islets were lower compared to healthy Wt mice where, AT2 receptor levels were on the other hand higher.41 The importance of such changes is specifically related to reduced ER stress and insulin secretion with AT2 blockers. In future studies, we will be using silencing approaches to specifically test the role of each RAAS component to understand detailed mechanisms linking Ang II receptor signaling to critical biological outcomes such as insulin secretion, apoptosis and other beta cell functions.
Several clinical trials with ACEi have shown reduced incidence of T2D in hypertensive patients.16,42-44 A couple of these studies have demonstrated that these beneficial effects were partly through improved insulin release.15 Additionally, increased ER stress reduces insulin biosynthesis.45 Corroborating these findings, our data reported for the first time that ACEi inhibitors reduced ER stress and improved insulin secretion. The beneficial effects of ACEi could be potentially mediated through Mas receptor,46 and worthy of future investigation.
We showed that Ang II treatment reduces insulin secretion, in agreement with few published studies.5,30,38 However, other studies have shown that activation of the RAAS pathway induced insulin secretion. This difference might be related to treatment conditions, such as culture media. Some insulin secretion experiments were conducted directly in media and did not use a starvation buffer (HBSS), which is typically used to measure glucose-stimulated insulin secretion.33,47 Also, the duration of Ang II stimulation to measure insulin secretion varied among studies. Ang II treatments could induce insulin secretion for shorter periods but could be deleterious at longer intervals. Similar effects were observed with IL-1β. Clonal beta cells treated for a short time with IL-1β potentiated insulin secretion, while longer treatment times inhibited insulin secretion.48,49 Consistent with these effects, we have previously shown that chronic RAAS overexpression in adipose tissue causes hyperinsulinemia and insulin resistance with increased adipocyte inflammation,10 while acute treatment of adipocytes with Ang II stimulated insulin signaling.50
ER stress is also known to activate inflammation, and both of these pathological factors could be induced by Ang II.4,51,52 However, the precise inflammatory pathways which are induced by Ang II are not well established yet in beta cells as well in other tissues.53,54 Our study and others have demonstrated that activation of RAAS could contribute to inflammation.21 Furthermore, increased Chop expression, an ER stress marker, induced inflammation through the NFkB pathway.55 Additionally, proinflammatory mediators under pathological conditions also trigger ER stress, further suggesting these processes are intertwined.56 Higher ER stress and inflammation, individually and in combination, could, in turn, cause insulin resistance, which could further activate RAAS leading to a vicious cycle.
Another well-established mechanism through which cellular processes are regulated is through miRNAs, also known to play a critical role in ER stress response.57 In our study, miR-30a-3p was reduced by Ang II, consistent with studies in cardiac cells and adipocytes21,28 where Ang II reduced these miRNAs. However, whether Chop is a direct target of miR-30a-3p needs further evaluation using mimic/inhibitors and luciferase reporter assays.
Current study used 10–100 nM concentration to test Ang II effects in beta cells. This is much higher than 5–50 pM circulating levels found in healthy individuals.58,59 However, the levels could rise to ~200 pM in individuals suffering from obesity.60 For example, subcutaneous adipose tissue Ang II levels are significantly higher than circulatory levels,60 Ang II levels found locally in the canine pancreas is much higher than systemic levels.61 Ang II has a dramatic effect on insulin secretion, and such effects likely stem from local Ang II production. It is important to note that it is challenging to measure the amount of Ang II accurately due to its short life and low concentrations. However, studies have demonstrated that local RAAS generates high concentrations of Ang II in isolated human pancreatic islets.30 It is also difficult to measure Ang II concentration directly in the human pancreas; hence it needs to be done in vitro using human or mouse islets.
In this original research, we primarily focused on the ATF4-CHOP pathway given its importance in beta cells.62 We also tested BiP, a key regulator of UPRs, but Ang II did not induce BiP at the doses tested (data not shown). It is possible that different doses of Ang II alter different ER stress biomarkers. Also, consistent with our findings in beta cells, Ang II induced ER stress in adipocytes by activating the ATF4-CHOP axis.21 However, studies have also demonstrated that other players like ATF3, ATF6, and IRE1α may be important as well in regulating beta cell proliferation.27 Whether Ang II regulates other branches of ER stress in beta cells or related pathways needs to be tested in future studies. Lastly, we tested ER stress markers only at gene levels, but measuring protein levels or post translational modifications will provide a more comprehensive understanding, to better explain the role of Ang II overactivation in beta cell functions.
Conclusion
In conclusion, activation of the RAAS upregulates ER stress in clonal beta cells, which is detrimental in T2D especially, and could lead to reduced insulin secretion. Our studies demonstrated that inhibition of RAAS improves insulin secretion; thus, currently used antihypertensive drugs targeting RAAS are also promising targets for insulin resistance and T2D. Our studies add significantly to currently known benefits of RAAS inhibitors in T2D prevention, by unraveling a new mechanism in the pancreas, by which these inhibitors may improve T2D.
Abbreviations
Atf4, activating transcription factor; ACEi, angiotensin converting enzyme inhibitor; Ang II, angiotensin II; ARB, angiotensin receptor blockers; AT1, angiotensin type 1 receptor; AT2, angiotensin type 2 receptor; Agt, angiotensinogen; Chop, CCAAT-enhancer-binding protein homologous protein; ER, endoplasmic reticulum; ELISA enzyme-linked immunosorbent assay; iNos, inducible nitric oxide synthase; IL, Interleukin; Mcp-1, monocyte chemoattractant protein−1; Nf-kb, nuclear factor-kappa B; 4-PBA, 4-phenyl butyric acid; RT-qPCR, real-time quantitative polymerase chain reaction; RAAS, renin angiotensin aldosterone system; Tnf-α, tumor necrosis factor-alpha; T2D, type 2 diabetes mellitus; UPR, unfolded protein response.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Diabetes; 2017. Available from: http://www.who.int/mediacentre/factsheets/fs312/en/.
2. DeFronzo RA. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia. 1992;35(4):389–397. doi:10.1007/BF00401208
3. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
4. Sauter NS, Thienel C, Plutino Y, et al. Angiotensin II induces interleukin-1 beta-mediated islet inflammation and beta-cell dysfunction independently of vasoconstrictive effects. Diabetes. 2015;64(4):1273–1283. doi:10.2337/db14-1282
5. Lau T, Carlsson PO, Leung PS. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia. 2004;47(2):240–248. doi:10.1007/s00125-003-1295-1
6. Pahlavani M, Kalupahana NS, Ramalingam L, Moustaid-Moussa N. Regulation and functions of the renin-angiotensin system in white and brown adipose tissue. Compr Physiol. 2017;7(4):1137–1150. doi:10.1002/cphy.c160031
7. Abuissa H, Jones PG, Marso SP, O’Keefe JH
8. Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obesity Rev. 2012;13(2):136–149.
9. Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369(9568):1208–1219.
10. Kalupahana NS, Massiera F, Quignard-Boulange A, et al. Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity. 2012;20(1):48–56. doi:10.1038/oby.2011.299
11. Ramalingam L, Menikdiwela K, LeMieux M, et al. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta. 2016.
12. Tahmasebi M, Puddefoot JR, Inwang ER, Vinson GP. The tissue renin-angiotensin system in human pancreas. J Endocrinol. 1999;161(2):317–322. doi:10.1677/joe.0.1610317
13. Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 2. Overview of physiological and biochemical mechanisms. Diabetes Metab. 2004;30(6):498–505. doi:10.1016/S1262-3636(07)70147-7
14. Devereux RB, Dahlof B, Kjeldsen SE, et al. Effects of losartan or atenolol in hypertensive patients without clinically evident vascular disease: a substudy of the LIFE randomized trial. Ann Intern Med. 2003;139(3):169–177.
15. van der Zijl NJ, Moors CCM, Goossens GH, Hermans MMH, Blaak EE, Diamant M. Valsartan improves beta-cell function and insulin sensitivity in subjects with impaired glucose metabolism a randomized controlled trial. Diab Care. 2011;34(4):845–851. doi:10.2337/dc10-2224
16. Group NS, McMurray JJ, Holman RR, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362(16):1477–1490. doi:10.1056/NEJMoa1001121
17. Lupi R, Del Guerra S, Bugliani M, et al. The direct effects of the angiotensin-converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets. Eur j Endocrinol/Eur Federation Endocr Soc. 2006;154(2):355–361. doi:10.1530/eje.1.02086
18. Frantz ED, Crespo-Mascarenhas C, Barreto-Vianna AR, Aguila MB, Mandarim-de-Lacerda CA. Renin-angiotensin system blockers protect pancreatic islets against diet-induced obesity and insulin resistance in mice. PLoS One. 2013;8(7):e67192. doi:10.1371/journal.pone.0067192
19. Chhabra KH, Xia H, Pedersen KB, Speth RC, Lazartigues E. Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. Am J Physiol Endocrinol Metab. 2013;304(8):E874884. doi:10.1152/ajpendo.00490.2012
20. Chu KY, Leung PS. Angiotensin II Type1 receptor antagonism mediates uncoupling protein 2-driven oxidative stress and ameliorates pancreatic islet beta-cell function in young Type 2 diabetic mice. Antioxid Redox Signal. 2007;9(7):869–878.
21. Menikdiwela KR, Ramalingam L, Allen L, Scoggin S, Kalupahana NS, Moustaid-Moussa N. Angiotensin II increases endoplasmic reticulum stress in adipose tissue and adipocytes. Sci Rep. 2019;9(1):8481. doi:10.1038/s41598-019-44834-8
22. Menikdiwela KR, Ramalingam L, Abbas MM, et al. Role of microRNA 690 in mediating angiotensin II effects on inflammation and endoplasmic reticulum stress. Cells. 2020;9(6):1327. doi:10.3390/cells9061327
23. Acosta-Montano P, Garcia-Gonzalez V. Effects of dietary fatty acids in pancreatic beta cell metabolism, implications in homeostasis. Nutrients. 2018;10:4.
24. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29(3):317–333. doi:10.1210/er.2007-0039
25. Marchetti P, Bugliani M, Lupi R, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50(12):2486–2494. doi:10.1007/s00125-007-0816-8
26. Malhi H. MicroRNAs in ER stress: divergent roles in cell fate decisions. Curr Pathobiol Rep. 2014;2(3):117–122.
27. Sharma RB, O’Donnell AC, Stamateris RE, et al. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831–3846. doi:10.1172/JCI79264
28. Chen M, Ma G, Yue Y, et al. Downregulation of the miR-30 family microRNAs contributes to endoplasmic reticulum stress in cardiac muscle and vascular smooth muscle cells. Int J Cardiol. 2014;173(1):65–73. doi:10.1016/j.ijcard.2014.02.007
29. Menikdiwela KR, Ramalingam L, Rasha F, et al. Autophagy in metabolic syndrome: breaking the wheel by targeting the renin–angiotensin system. Cell Death Dis. 2020;11(2):1–17. doi:10.1038/s41419-020-2275-9
30. Ramracheya RD, Muller DS, Wu Y, et al. Direct regulation of insulin secretion by angiotensin II in human islets of Langerhans. Diabetologia. 2006;49(2):321–331. doi:10.1007/s00125-005-0101-7
31. Chu KY, Leung PS. Angiotensin II in type 2 diabetes mellitus. Curr Protein Pept Sci. 2009;10(1):75–84. doi:10.2174/138920309787315176
32. Leung PS, Carlsson PO. Pancreatic islet renin angiotensin system: its novel roles in islet function and in diabetes mellitus. Pancreas. 2005;30(4):293–298.
33. Carlsson PO, Berne C, Jansson L. Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia. 1998;41(2):127–133.
34. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42–61. doi:10.1210/er.2007-0015
35. Ribeiro-Oliveira JA, Nogueira AI, Pereira RM, Boas WWV, Dos Santos RAS, e Silva ACS. The renin–angiotensin system and diabetes: an update. Vasc Health Risk Manag. 2008;4(4):787. doi:10.2147/VHRM.S1905
36. Yayama K, Okamoto H. Angiotensin II-induced vasodilation via type 2 receptor: role of bradykinin and nitric oxide. Int Immunopharmacol. 2008;8(2):312–318. doi:10.1016/j.intimp.2007.06.012
37. Madec AM, Cassel R, Dubois S, et al. Losartan, an angiotensin II type 1 receptor blocker, protects human islets from glucotoxicity through the phospholipase C pathway. FASEB J. 2013;27(12):5122–5130. doi:10.1096/fj.13-234104
38. Chu KY, Lau T, Carlsson PO, Leung PS. Angiotensin II type 1 receptor blockade improves beta-cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes. 2006;55(2):367–374. doi:10.2337/diabetes.55.02.06.db05-1022
39. Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II-dependent hypertension. Circ Res. 2016;119(4):532–543. doi:10.1161/CIRCRESAHA.116.308384
40. Liu M, Jing DQ, Wang Y, Liu Y, Yin SN. Overexpression of angiotensin II type 2 receptor promotes apoptosis and impairs insulin secretion in rat insulinoma cells. Mol Cell Biochem. 2015;400(12):233–244. doi:10.1007/s11010-014-2280-3
41. Keller MP, Choi Y, Wang P, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18(5):706–716. doi:10.1101/gr.074914.107
42. Hansson L. Clinical studies with candesartan. Drugs Today. 1999;35(2):117–126. doi:10.1358/dot.1999.35.2.527967
43. Dahlof B, Zanchetti A, Diez J, et al. Effects of losartan and atenolol on left ventricular mass and neurohormonal profile in patients with essential hypertension and left ventricular hypertrophy. J Hypertens. 2002;20(9):1855–1864. doi:10.1097/00004872-200209000-00032
44. van der Zijl NJ, Serne EH, Goossens GH, et al. Valsartan-induced improvement in insulin sensitivity is not paralleled by changes in microvascular function in individuals with impaired glucose metabolism. J Hypertens. 2011;29(10):1955–1962.
45. Kim MK, Kim HS, Lee IK, Park KG. Endoplasmic reticulum stress and insulin biosynthesis: a review. Exp Diabetes Res. 2012.
46. Sahr A, Wolke C, Maczewsky J, et al. The angiotensin-(17)/mas axis improves pancreatic beta-cell function in vitro and in vivo. Endocrinology. 2016;157(12):4677–4690. doi:10.1210/en.2016-1247
47. Gletsu N, Doan TN, Cole J, Sutliff RL, Bernstein KE. Angiotensin II-induced hypertension in mice caused an increase in insulin secretion. Vascul Pharmacol. 2005;42(3):83–92. doi:10.1016/j.vph.2005.01.006
48. Maedler K, Schumann DM, Sauter N, et al. Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes. 2006;55(10):2713–2722. doi:10.2337/db05-1430
49. Spinas GA, Mandrup-Poulsen T, Molvig J, et al. Low concentrations of interleukin-1 stimulate and high concentrations inhibit insulin release from isolated rat islets of Langerhans. Acta Endocrinol. 1986;113(4):551–558. doi:10.1530/acta.0.1130551
50. Kim S, Voy BH, Huang T, et al. Angiotensin II uses insulin signalling pathways in 3T3-L1 adipocytes. Adipocytes. 2005;1(4):239–248.
51. Kuno A, Yamada T, Masuda K, et al. Angiotensin-converting enzyme inhibitor attenuates pancreatic inflammation and fibrosis in male Wistar Bonn/Kobori rats. Gastroenterology. 2003;124(4):1010–1019.
52. Yamada T, Kuno A, Masuda K, et al. Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J Pharmacol Exp Ther. 2003;307(1):17–23.
53. Li TT, Jia LX, Zhang WM, et al. Endoplasmic reticulum stress in bone marrow-derived cells prevents acute cardiac inflammation and injury in response to angiotensin II. Cell Death Dis. 2016;7(6):e2258.
54. Wang J, Wen Y, Lv LL, et al. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol Sin. 2015;36(7):821–830. doi:10.1038/aps.2015.21
55. Willy JA, Young SK, Stevens JL, Masuoka HC, Wek RC. CHOP links endoplasmic reticulum stress to NF-kappaB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol Biol Cell. 2015;26(12):2190–2204. doi:10.1091/mbc.E15-01-0036
56. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398–406. doi:10.1007/s00395-008-0733-0
57. Byrd AE, Brewer JW. Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life. 2013;65(5):373–381.
58. Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310(2):H137152. doi:10.1152/ajpheart.00618.2015
59. Schulz A, Jankowski J, Zidek W, Jankowski V. Absolute quantification of endogenous angiotensin II levels in human plasma using ESI-LC-MS/MS. Clin Proteomics. 2014;11(1):37. doi:10.1186/1559-0275-11-37
60. Harte A, McTernan P, Chetty R, et al. Insulin-mediated upregulation of the renin angiotensin system in human subcutaneous adipocytes is reduced by rosiglitazone. Circulation. 2005;111(15):1954–1961. doi:10.1161/01.CIR.0000161954.17870.5D
61. Chappell MC, Millsted A, Diz DI, Brosnihan KB, Ferrario CM. Evidence for an intrinsic angiotensin system in the canine pancreas. J Hypertens. 1991;9(8):751–759. doi:10.1097/00004872-199108000-00008
62. Kitakaze K, Kurahashi K, Miyake M, Hamada Y, Oyadomari M, Oyadomari S. Targeted deletion of ATF4 in ß cells leads to the vulnerability and dedifferentiation during ER stress. Diabetes. 2018;67(Supplement 1):2104P. doi:10.2337/db18-2104-P
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.