")
Back to Journals » OncoTargets and Therapy » Volume 13
End Processing Factor APLF Promotes NHEJ Efficiency and Contributes to TMZ- and Ionizing Radiation-Resistance in Glioblastoma Cells
Authors Dong W, Li L, Teng X, Yang X, Si S, Chai J
Received 23 April 2020
Accepted for publication 2 August 2020
Published 19 October 2020 Volume 2020:13 Pages 10593—10605
DOI https://doi.org/10.2147/OTT.S254292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Wei Dong,1 Lanlan Li,2 Xuepeng Teng,1 Xinhua Yang,1 Shujing Si,3 Jie Chai1
1Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academic Sciences, Jinan, Shandong, People’s Republic of China; 2Binzhou Medical University Hospital, Binzhou, Shandong, People’s Republic of China; 3Department of Oncology, Gaoqing People’s Hospital, Zibo, Shandong, People’s Republic of China
Correspondence: Jie Chai Email [email protected]
Purpose: Glioblastoma (GBM) is the most commonly diagnosed primary brain tumor in adults. Despite a variety of advances in the understanding of GBM cancer biology during recent decades, very few of them were applied into treatment, and the survival rate of GBM patients has not been improved majorly due to the low chemosensitivity to temozolomide (TMZ) or low radiosensitivity. Therefore, it is urgent to elucidate mechanisms of TMZ- and IR-resistance and develop novel therapeutic strategies to improve GBM treatment.
Methods: TMZ- and IR-resistant cell lines were acquired by continuous exposing parental GBM cells to TMZ or IR for 3 months. Cell viability was determined by using Sulforhodamine B (SRB) assay. Protein and mRNA expression were examined by Western blotting assay and quantitative polymerase chain reaction (qPCR) assay, respectively. Homologous recombination (HR) and nonhomologous end joining (NHEJ) efficiency were measured by HR and NHEJ reporter assay. Cell apoptosis was determined by Caspase3/7 activity. Autophagy was analyzed using CYTO-ID® Autophagy detection kit. Tumor growth was examined by U87 xenograft mice model.
Results: DNA repair efficiency of non-homologous end joining (NHEJ) pathway is significantly increased in TMZ- and IR-resistant GBM cells. Importantly, APLF, which is one of the DNA end processing factors in NHEJ, is upregulated in TMZ- and IR-resistant GBM cells and patients. APLF deficiency significantly decreases NHEJ efficiency and improves cell sensitivity to TMZ and IR both in vitro and in vivo.
Conclusion: Our study provides evidence for APLF serving as a promising, novel target in GBM chemo- and radio-therapy.
Keywords: APLF, NHEJ, chemoresistance, radioresistance, GBM
Introduction
Glioblastomas (GBMs) are generally fatal, malignant brain tumors.1 It is deemed the most common and aggressive form of primary brain tumor in adults with about one year median survival and less than 10% 5-year survival rate.2 The current first-line therapy after surgical resection is a combination of radiotherapy and chemotherapy with temozolomide (TMZ).3–5 However, a majority of GBM patients developed either intrinsic or acquired chemoresistance to TMZ.6–9 Unfortunately, there are very limited alternative chemotherapeutic drugs for GBM and the TMZ-resistant patients are left without therapeutic strategies.7 Therefore, it is critical to elucidate the mechanisms of TMZ resistance in GBM and identify novel methods to improve GBM therapy. TMZ acts as an alkylating agent that methylates DNA at O6 and N7 position of guanine, and O3 position of adenine.10 O6-meG, which is the main contribution of cytotoxicity, can be directly reversed by O6-methylguanine methyl transferase (MGMT).11 The mutation of DNA triggers mismatch repair (MMR) to generate a nick and leads to DNA double stand break during second round replication.12
DNA double-strand break (DSB) is extremely toxic for cell survival and that is the strategy of variety of chemotherapeutic compounds and radiotherapy.13 Therefore, understanding the contribution of DSB repair in TMZ-resistance may provide new therapeutic targets. There are two major DSB repair pathways in human, homologous recombination (HR) and non-homologous end joining (NHEJ).14–16 HR is considered to be an error-free DSB repair pathway because it incorporates sister chromatid as template during polymerizing the gap. This dependence of template limits HR in late S to G2 phase when sister chromatids are available.17 On the other hand, NHEJ does not require templates to join DNA ends so it can be performed throughout the whole cell cycle.18 The essential factors of NHEJ include Ku70/80 heterodimer, DNA-PKcs, XRCC4, XLF and Ligase IV.18 The pathway is initiated by Ku-DNA interaction followed by recruitment of other NHEJ key factors by Ku.19 NHEJ is not a simple end-to-end joining pathway but requires ligatable DNA ends. Therefore, DNA end processing factors are important acquisitions of NHEJ, such as Artemis, PNKP, TDP1, WRN and APLF.20–25
Aprataxin‐and‐PNK‐Like Factor (APLF) was identified in 2007 as a novel component of NHEJ.26 It promotes efficient NHEJ followed by ionizing radiation, especially during the first few hours after DNA damage.27 It suggests that APLF would serve as a promising target to reduce both chemo- and radioresistance generated by NHEJ. Our study found that APLF contributes to NHEJ efficiency in GBM cell lines. Importantly, we observed that APLF deficiency can significantly sensitize GBM cells to TMZ and IR, indicating that APLF is a promising target for GBM treatments.
Methods and Materials
Cell Lines and Cell Cultures
U87 (ATCC, HTB-14) and T98G cells (ATCC, CRL-1690) were cultured at 37°C in 5% CO2 atmosphere in EMEM (ATCC 30–2003) with 10% FBS for less than 2 months. U87 and T98G TMZ-resistant cells were generated by incubating with 50 to 100 μM TMZ for 3 months.28 To create IR-resistant GBM cells, we expose U87 and T98G cells to 1 to 2 Gy IR twice a week for 2 months.
Reagents
TMZ was purchased from sigma (T2577). siAPLF was purchased from Dharmacom (J-018493-17-0002 and J-018493-17-0002). APLF mammalian expression vector (pcDNA3.1(+)) used as negative control was purchased from ThermoFisher Scientific (V79020). Autophagy detecting Cyto-ID Green dye was purchased from Enzo Life Sciences (ENZ-KIT175-0050). Crystal violate was purchased from sigma (C0775).
Cell Viability Assay
Cells were seed at 3x103 cells/well and cultured for overnight to allow adherence. Cells were incubated with drug for 72h and the cell viability was detected by using Sulforhodamine B (SRB) assay.29 Cells were fixed by 100 µL/well of 10% trichloroacetic acid at 4°C for 1 hour. Plate was washed and air dried for 1 hour at room temperature (RT). Cells were stained by 100 µL/well 0.02% SRB in 1% acetate acid for 1 hour at RT. Plates were washed for 3 times with 200 µL/well 1% acetate acid and air dried. 200 µL/well of 10 mM tris-HCl, pH 10.5 was added in each well to extract SRB with 1 hour shaking on an orbital shaker. The absorbance was measured at 510 nm by microplate reader (Thermo Scientific).
qPCR
QRT-PCR was performed according to previous studies.30 Briefly, Total RNA was extracted from cells by using RNeasy Mini Kit (QIAGEN). Complementary DNA was synthesized using TaqMan™ Reverse Transcription Reagents (ThermoFisher Scientific). Gene expression for APLF was performed using quantitative RT-PCR (qPCR) with Fast SYBR™ Green Master Mix (ThermoFisher Scientific) and human GAPDH was used as the internal normalization control. Each assay was repeated in triplicate on a Thermal Cycler Eco qPCR system (Eppendorf). APLF primers: 5ʹ-3ʹ CAAGGAAGCCCTGAAATAACC; 3ʹ-5ʹ CTGAAAGCTCTGCATTCACCT. Patients who did not exhibit significant anti-tumor effect observed by CT or MRI after 5 weeks of TMZ treatment (75mg/m2/day) were determined as TMZ-resistant patients. Studies involving patient samples were approved by the Shandong Cancer Hospital, and all patients provided informed consent, in accordance with the Declaration of Helsinki.
Knockout APLF in U87 Cells by Using CRISPR/Cas9
Cas9 along with APLF guide RNA plasmid was constructed by ligating oligonucleotide duplexes, which targets exon1 of APLF, into BbsI cut pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene #42,230). The plasmid was transfected into U87 cell line along with pcDNA3.1.puro by lipofectamine 2000 and incubated for 3 days. Cells successfully transfected with APLF KO plasmid were selected by puromycin for 3 days. Cells were harvested and seeded in 10 cm plate at concentrations of 10–100 cells/mL and incubated for 2 weeks. Induvial clones were passaged, expanded and screened for APLF expression. We randomly picked 2 APLF KO clones for further studies.
DSB Reporter Assay
NHEJ and HR reporters were previously described.31,32 Briefly, 10 μg of NHEJ reporter or HR reporter cassette were linearized by 50 U of NheI in a 50 μL reaction for 4 hours in 37°C water bath. Linearized DNA was gel purified and 1 μg of clean and linearized plasmid was transfected into U87 cells by using Lipofectamine3000 according to manufacturer’s instruction. Cells with chromosomally integrated reporter were selected by 1 mg/mL geneticin 3 days after transfection for 2 weeks. Stable transfected cells were seeded at 3x105 cells/mL in a 6-well plate and 2µg/well of I-SceI coding plasmid was transfected into the cell by lipofectamine 3000 and incubated for 48h. Cells were harvest and GFP positive cells, which indicating successful NHEJ repair, were count by flow cytometry (BD FACSCelesta™ Flow Cytometer).
Western Blot Assay
Protein samples were denatured by using SDS-PAGE sample buffer, boiled for 5 min. The samples were then loaded and separated on a 7% polyacrylamide gel (29:1) (BIO-RAD, 1,610,156) at 120 V for 1.5 hour on electrophoresis apparatus (BioRad). Separated samples were transferred to nitrocellulose membrane at 100v at 4°C for 1 hour. Membrane was blocked by 3% non-fat milk solution dilute in PBS with 0.1% Tween20 and probed by relevant antibody followed by HRP-conjugated rabbit secondary antibody. The protein signal was developed by SuperSignalTM west pico PLUS Chemiluminescent Substrate (ThermoFisher Scientific #34,580) and detected by ChemiDocTM (BioRad).33
Colony Formation Assay
Cells were seeded in 6-well plates at a concentration of 500 cells/well and cultured for overnight to allow adherence. The cells were subsequently treated with 1Gy of IR and cultured for 10 days. The colonies were washed with PBS, fixed with 4% paraformaldehyde for 10 min and stained with 0.01% crystal violate (Sigma) at room temperature for 30 min. The colonies were then washed with dH2O. Images were captured under a stereomicroscope and quantified.
Animals
Female BALB/c nude mice (5 weeks old, 18 ± 2 g) were purchased from Jiangsu ALF Biotechnology Co., LTD. (Nanjing, China). 5x106 U87-TR or U87-TR-KO cells were subcutaneously injected to generate tumor xenograft model. Treatments were given when the tumors were approximately 100 mm3 in volume. Mice were treated with vehicle or TMZ (7.5mg/kg/day) intraperitoneally for 2 weeks. Tumor volume and body weight were measured with a caliper every 3 days using the formula, volume=length x width2/2. All the animal experiments were authorized by the Laboratory Animal Care and Ethical Committee of the Shandong Cancer Hospital and Institute, and were performed following the Guide for the Care and Use of Laboratory Animals (8th edition, 2011, National Academies Press (US)).
Statistical Analysis
Data are expressed as the means ± standard deviations from 3 independent experiments. Comparisons between 2 groups and multiple groups were conducted using the Student’s t-test and one-way ANOVA. Differences were considered significant at a p value<0.05. Prism7 software was used for performing all statistical analysis (GraphPad Software, La Jolla, CA, USA).
Results
Establish TMZ- and Radioresistant GBM Cell Lines
To study the mechanism underlying chemo- and radioresistance in GBM, we first generated GBM cell lines that were resistant to TMZ or ionizing radiation (IR). As shown in Figure 1A and B, we successfully increased TMZ IC50 by 3.75- and 5.26-folds in U87 and T98G cell lines, respectively, by incubating wild type (WT) cells with 50 to 100 μM TMZ. To create IR-resistant GBM cells, we expose U87 and T98G cells to 1 to 2 Gy IR twice a week for 2 months and measured cell viability 3 days after IR. We found that U87 IR-resistant cells (U87-IRR) and T98G IR-resistant cells (T98G-IRR) had 5.07- and 4.62-folds increase of IC50 to IR as compared to their parental cell lines (Figure 1C and D). We used these cell lines for further molecular mechanism studies.
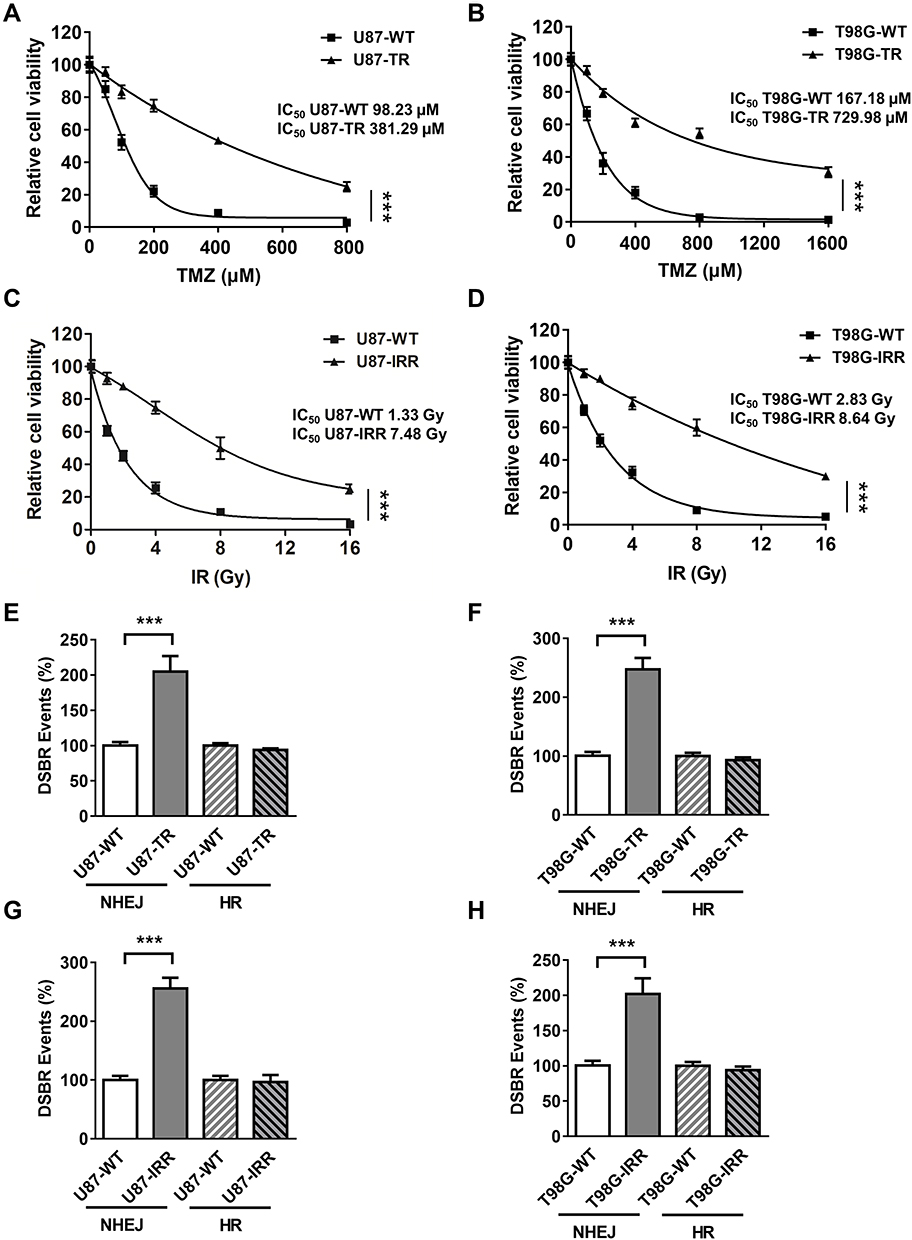 |
Figure 1 Establish TMZ-resistant and radioresistant GBM cell lines. (A) Cell viability of U87-WT and U87 TMZ resistant (U87-TR) cells. TMZ concentrations are 0 μM, 50 μM, 100 μM, 200 μM, 400 μM and 800 μM. Data are represented as mean ± standard deviations (SD) of three independent experiments. (B) Cell viability of T98G -WT and T98G TMZ-resistant (T98G-TR) cells. TMZ concentrations are 0 μM, 100 μM, 200 μM, 400 μM, 800 μM and 1600 μM. Data are represented as mean ± SD of three independent experiments. (C) Cell viability of U87-WT and U87 IR-resistant (U87-IRR) cells. IR doses are 0 Gy, 1 Gy, 2 Gy, 4 Gy, 8 Gy and 16 Gy. Data are represented as mean ± SD of three independent experiments. (D) Cell viability of T98G-WT and T98G IR-resistant (T98G -IRR) cells. IR doses are 0 Gy, 1 Gy, 2 Gy, 4 Gy, 8 Gy and 16 Gy. Data are represented as mean ± SD of three independent experiments. (E) Relative DSB repair events in U87-WT, U87 TR, (F) T98G -WT and T98G-TR cells. (G) Relative DSB repair events in U87-WT, U87 IRR, (H) T98G -WT and T98G-IRR cells. ***P<0.001. |
Both TMZ and IR induce DSBs that require HR or NHEJ to prevent cell death. To determine which DSBR pathway is activated in response to TMZ and IR treatment, we then incorporated well-established, cell-based HR and NHEJ efficiency assay that measures DSBR events on chromosomal level.34 As shown in Figure 1E and F, NHEJ was significantly upregulated in TMZ-resistant GBM cell lines as compared to the parental cell lines. Similarly, IR-resistant U87 and T98G cells exhibited increased NHEJ efficiency as compared to WT cells (Figure 1G and H). However, resistant cell lines did not exhibit difference in HR efficiency as compared to their parental cell lines (Figure 1E–H). These results suggest that NHEJ plays an important role in mechanism of TMZ- and IR-resistance.
Expression of APLF Positively Correlated to TMZ and IR Resistance
To identify which NHEJ factor is misregulated in chemo- or radioresistance GBM, we extracted RNA from TMZ sensitive- and resistant-patient samples. We next measured mRNA expression levels of NHEJ factors by using qPCR and we found that APLF was significantly increased in patients resistant to TMZ as compared to that in TMZ-sensitive patients (Figure 2A). In consistent with DSBR efficiency results, mRNA expression of RAD51 and BRCA1, which are essential factors of HR, did not change in TMZ sensitive- and resistant-patient samples (Supplementary Figure 1A and B). To further confirm whether protein expression of APLF also increases in TMZ-resistant patient sample, we used Western blotting assay to compare APLF expression and two representative results from TMZ-sensitive and TMZ-resistant samples were shown (Figure 2B). Indeed, we observed that both TMZ-resistant samples (R1 and R2) showed upregulated APLF protein expression as compared to that in TMZ-sensitive samples (S1 and S2).
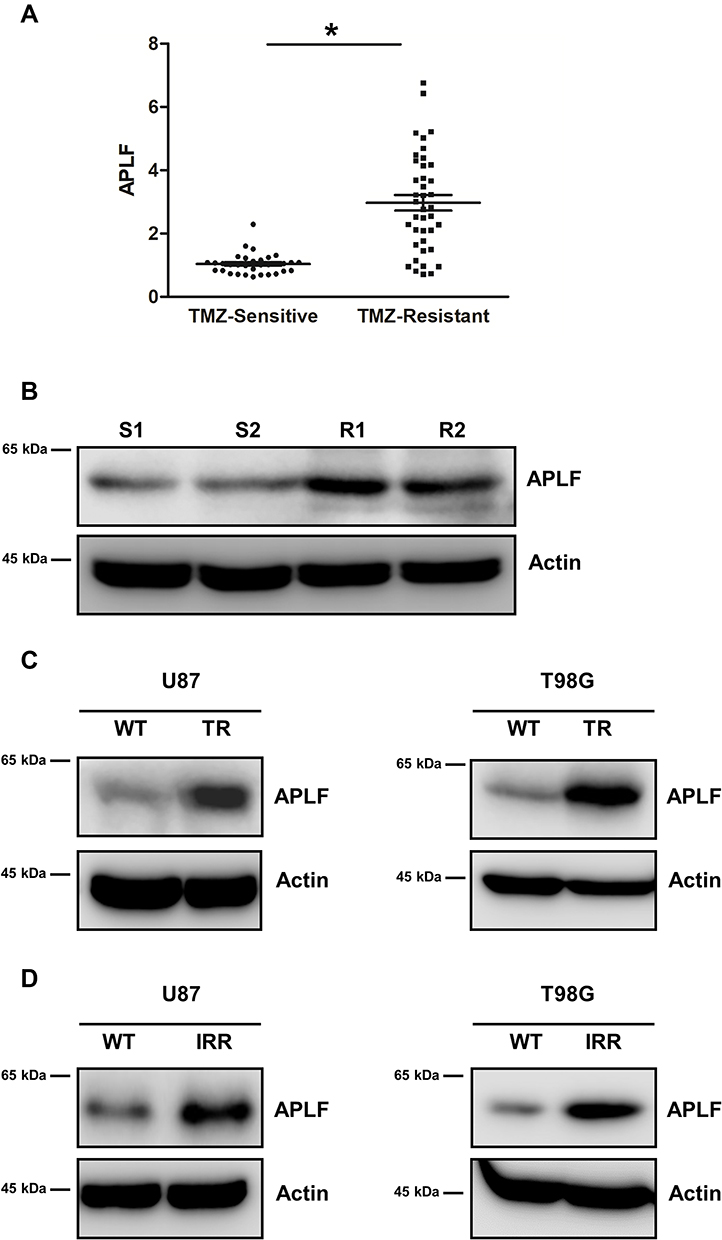 |
Figure 2 Expression of APLF positively correlated to TMZ and IR resistance. (A) APLF mRNA expression in GBM patient samples. TMZ sensitive group: n=30. TMZ-resistant group: n=40. *p<0.05. (B) Western blot analysis of APLF protein expression in 2 TMZ sensitive and 2 TMZ-resistant patient samples. (C) Western blot analysis of APLF protein expression in U87-WT, U87-TR, T98G -WT and T98G-TR cells. (D) Western blot analysis of APLF protein expression in U87-WT, U87-IRR, T98G -WT and T98G-IRR cells. |
We also used the cell lines generated above to evaluate the role of APLF in TMZ- and chemoresistance. We found that APLF was upregulated in both TMZ-resistant GBM cell lines (U87-TR and T98G-TR) as compared to that in their parental cell lines (Figure 2C, Supplementary Figure 2A and B). Similarly, we found the APLF expression was increased in IR-resistant cell lines (U87-IRR and T98G-IRR) (Figure 2D, Supplementary Figure 2C and D). In addition, as shown in Supplementary Figure 3A to D, TMZ-resistant cells were resistant to IR and vice versa. This could be explained by their shared APLF upregulation. Our results suggest that APLF is correlated with both IR- and TMZ-resistance in GBM. These data indicate that APLF contributes to radio- and chemoresistance in GBM.
APLF Contributes to NHEJ Efficiency in GBM Cells
Since APLF participates in end processing in NHEJ followed by ionizing radiation,35 we hypothesized that APLF contributes to NHEJ efficiency in resistant GBM cells. To evaluate the role of APLF in NHEJ, we generated APLF knock-out (KO) TMZ- and IR-resistant GBM cells (U87-TR-KO, U87-IRR-KO, T98G-TR-KO and T98G-IRR-KO) by using CRISPR/Cas 9 technique. As shown in Figure 3A–D, these KO cell lines had no detectable APLF protein expression. We then compared NHEJ efficiency between APLF-proficient and APLF-deficient GBM cells and found that APLF deficiency significantly impaired NHEJ efficiency in TMZ- and IR-resistant cells (p<0.001) (Figure 3E–H). It is possible that upregulation of NHEJ efficiency in TMZ-resistant cells may be regulated by other proteins rather than APLF. To confirm APLF is the primary protein contributes to NHEJ efficiency in TMZ-resistant cells, we ectopically expressed APLF in U87-TR-KO, U87-IRR-KO, T98G-TR-KO and T98G-IRR-KO cells (Figure 3A–D) and measured NHEJ efficiency subsequently. As shown in Figure 3E–H, we found that ectopic expression of APLF in KO cells restored NHEJ efficiency to a similar level as in APLF-proficient cells. Consistently, HR efficiency was not affected by APLF deficiency in resistant cell lines (Supplementary Figure 4A to D). To further confirm that APLF promotes TMZ- and IR induced DSB repair in resistant GBM cells, we examined expression of γH2AX, which indicates the presence of DSB, after TMZ or IR treatment. As shown in Supplementary Figure 5A and B, sustained γH2AX was found in APLF-deficient cells, and ectopic expression of APLF accelerated disappearance of γH2AX, indicating APLF indeed contributes to TMZ- and IR-resistance through enhancing NHEJ in GBM.
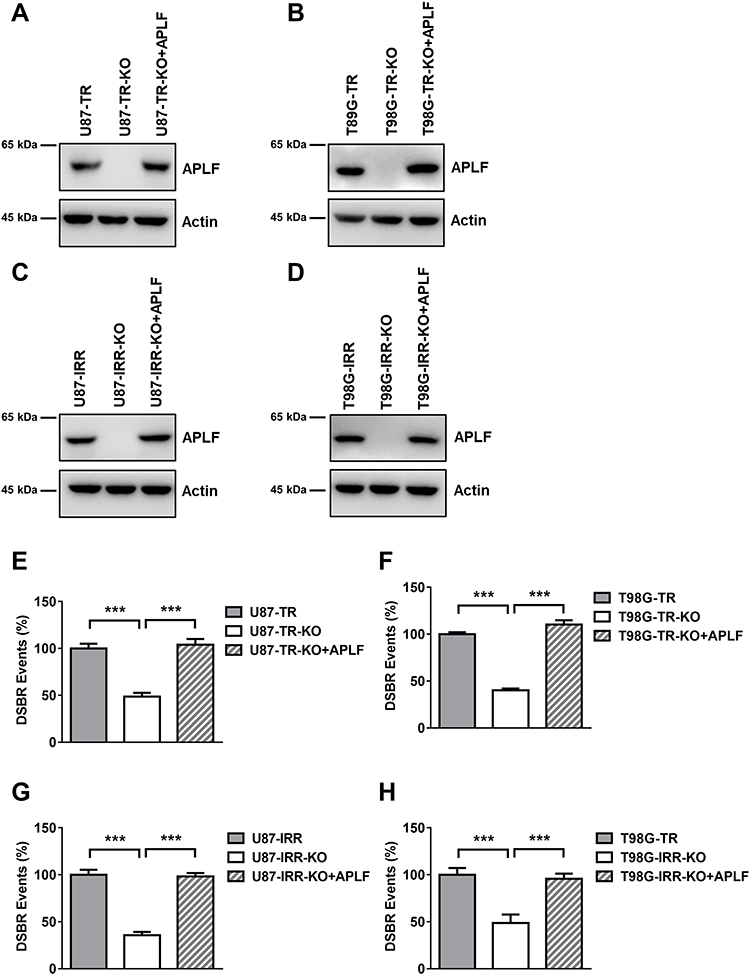 |
Figure 3 APLF contributes to NHEJ efficiency in GBM cells. (A) Western blot analysis of APLF protein expression in U87-TR, APLF-deficient U87-TR (U87-TR-KO) and U87-TR-KO with APLF ectopic expression cells (U87-TR-KO+APLF). (B) Western blot analysis of APLF protein expression in T98G-TR, APLF-deficient T98G-TR (T98G-TR-KO) and T98G-TR-KO with APLF ectopic expression cells (T98G-TR-KO+APLF). (C) Western blot analysis of APLF protein expression in U87-IRR, APLF-deficient U87-IRR (U87-IRR-KO) and U87-IRR-KO with APLF ectopic expression cells (U87-IRR-KO+APLF). (D) Western blot analysis of APLF protein expression in T98G-IRR, APLF-deficient T98G-IRR (T98G-IRR-KO) and T98G-IRR-KO with APLF ectopic expression cells (T98G-IRR-KO+APLF). (E) Quantification of GFP cells result from successful NHEJ events in U87-TR, U87-TR-KO, U87-TR-KO+APLF, (F) T98G-TR, T98G-TR-KO, T98G-TR-KO+APLF, (G) U87-IRR, U87-IRR-KO, U87-IRR-KO+APLF, (H) T98G-IRR, T98G-IRR-KO and T98G-IRR-KO+APLF cell lines. Each result represents 3 independent experiments. ***p<0.001. |
APLF is an accessory factor of NHEJ and APLF deficiency does not affect NHEJ efficiency significantly. To determine whether the APLF only contributes to NHEJ in resistant GBM cells, we knocked down APLF in U87-WT and T98G-WT cells (Supplementary Figure 6A and B) and measured NHEJ efficiency. We observed that APLF deficiency slightly decreased NHEJ efficiency in WT cells (p<0.05), and ectopic expression of APLF rescued NHEJ efficiency to the similar level of WT cells (Supplementary Figure 6C and D), suggesting APLF has a unique role in NHEJ in response to TMZ- and IR-resistance.
APLF Deficiency Improves TMZ- and IR-Sensitivity in GBM Cells
To validate whether APLF is a potential target to increase sensitivity to TMZ, we measured cell viability in response to TMZ and IR in U87-TR-KO and T98G-TR-KO cells. We found that APLF deficiency exhibited excellent synergy with TMZ, and expression of APLF restored TMZ resistance, suggesting APLF sensitizes GBM cells to TMZ (Figure 4A and B). Similarly, APLF deficiency also significantly re-sensitized U87-IRR and T98G-IRR cells to IR (Figure 4C and D). To further verify the effect of APLF deficiency on cell reproductivity to IR, we analyzed colony formation in the absence and presence of APLF. As shown in Figure 4E and F, U87-IRR-KO and T98G-IRR-KO cells are significantly more sensitive to IR as compared to their parental cell lines. Our results indicate that APLF deficiency overcomes chemo- and radioresistance in GBM.
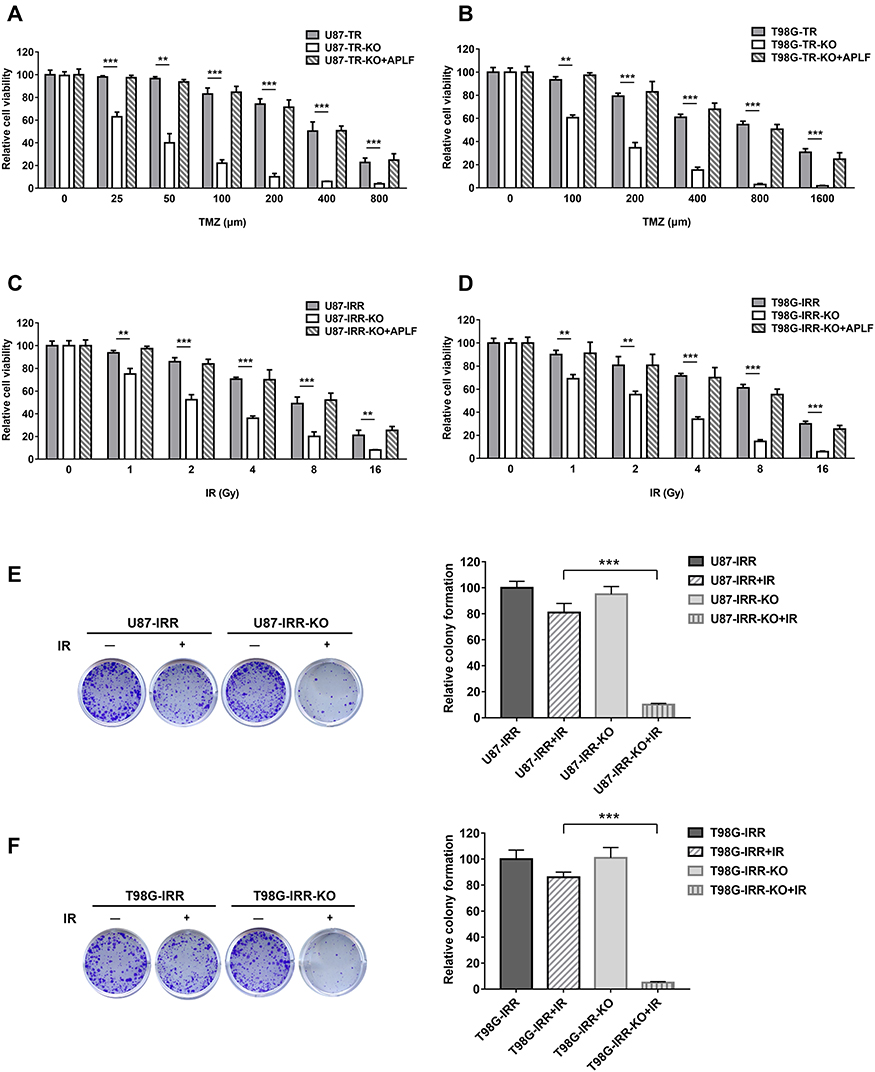 |
Figure 4 APLF deficiency improves TMZ- and IR-sensitivity in GBM cells. (A) Cell viability of U87-TR, U87-TR-KO and U87-TR-KO+APLF cells to TMZ. TMZ concentrations are 0 μM, 25 μM, 50 μM, 100 μM and 200 μM, 400 μM and 800 μM. Data are represented as mean ± SD of three independent experiments. **p < 0.01, ***p < 0.001. (B) Cell viability of T98G-TR, T98G-TR-KO and T98G-TR-KO+APLF cells to TMZ. TMZ concentrations are 0 μM, 100 μM, 200 μM, 400 μM, 800 μM and 1600 μM. Data are represented as mean ± SD of three independent experiments. **p<0.01, ***p<0.001. (C) Cell viability of U87-IRR, U87-IRR-KO and U87-IRR-KO+APLF cells to IR. IR doses are 0 Gy, 1 Gy, 2 Gy, 4 Gy and 8 Gy and 16 Gy. Data are represented as mean ± SD of three independent experiments. **p < 0.01, ***p < 0.001. (D) Cell viability of T98G-IRR, T98G-IRR-KO and T98G-IRR-KO+APLF cells to IR. IR concentrations are doses are 0 Gy, 1 Gy, 2 Gy, 4 Gy and 8 Gy and 16 Gy. Data are represented as mean ± SD of three independent experiments. **p<0.01, ***p<0.001. (E) Left, digital image showing colonies produced by U87-IRR, U87-IRR-KO (F) T98G-IRR and T98G-IRR-KO cells in response to IR. Cells were irradiated with or without 1 Gy of IR and incubated for 10 days. Right, quantification of colonies. Data are represented as mean ± SD of three independent experiments. ***p<0.001. |
APLF Deficiency Induces Apoptosis in Response to TMZ and IR
To determine the cause of increased cell death in APLF-deficient cells induced by TMZ and IR, we analyzed autophagy and apoptosis activity after TMZ or IR treatment in APLF proficient and deficient resistant GBM cells. As shown in Supplementary Figure 7A and B, autophagy induced by TMZ or IR was not further increased by APLF deficiency. To investigate whether APLF deficiency results in synergy with TMZ to inhibit cell growth via the induction of apoptosis, we measured Caspase3/7 activity after TMZ treatment in APLF proficient and deficient TMZ-resistant GBM cells. As shown in Figure 5A and B, Caspase 3/7 activity was promoted in U87-TR-KO and T98G-TR-KO cells as compared to that in U87-TR and T98G-TR cells, respectively. Similar results were observed in IR-resistant GBM cells (Figure 5C and D), indicating induced apoptosis in APLF-deficient cells after exposure to TMZ or IR. These data suggest that APLF is a promising target to overcome TMZ and IR resistance in GBM.
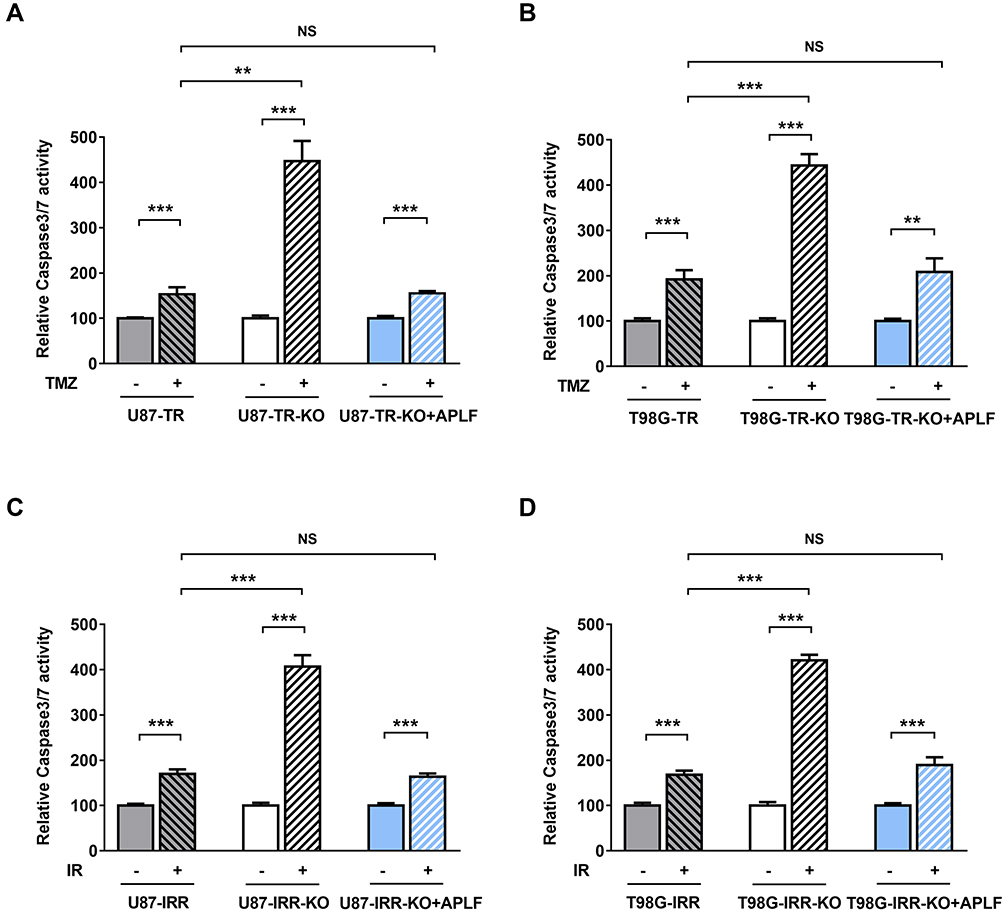 |
Figure 5 APLF deficiency induces apoptosis in response to TMZ and IR. (A) Relative Caspase 3/7 activity in U87-TR, U87-TR-KO and U87-TR-KO+APLF cells after exposure to TMZ (100 μM). **p<0.01, ***p<0.001, NS: not significant. (B) Relative Caspase 3/7 activity in T98G-TR, T98G-TR-KO and T98G-TR-KO+APLF cells after exposure to TMZ (200 μM). **p<0.01, ***p<0.001, NS: not significant. (C) Relative Caspase 3/7 activity in U87-IRR, U87-IRR-KO and U87-IRR-KO+APLF cells after exposure to IR (4 Gy). ***p<0.001, NS: not significant. (D) Relative Caspase 3/7 activity in T98G-IRR, T98G-IRR-KO and T98G-IRR-KO+APLF cells after exposure to IR (4 Gy). ***p<0.001, NS: not significant. |
APLF Deficiency Overcomes TMZ-Resistance in vivo
To further validate the effect of APLF deficiency on overcoming resistance to TMZ in GBM, we compare tumor growth inhibition in response to TMZ between U87-TR and U87-TR-KO xenograft models. The U87-TR and U87-TR-KO cells were subcutaneously implanted in female nude mice, and the TMZ treatment was initiated when the tumor volume reached 100mm3. In consistent with the in vitro cell-based assay, combination of APLF and TMZ resulted in very promising synergy in suppression of tumor growth without detectable toxicity (Figure 6A–C). We also noted that U87-TR and U87-TR-KO groups had no significant difference in tumor volume, indicating APLF deficiency alone does not affect tumor growth.
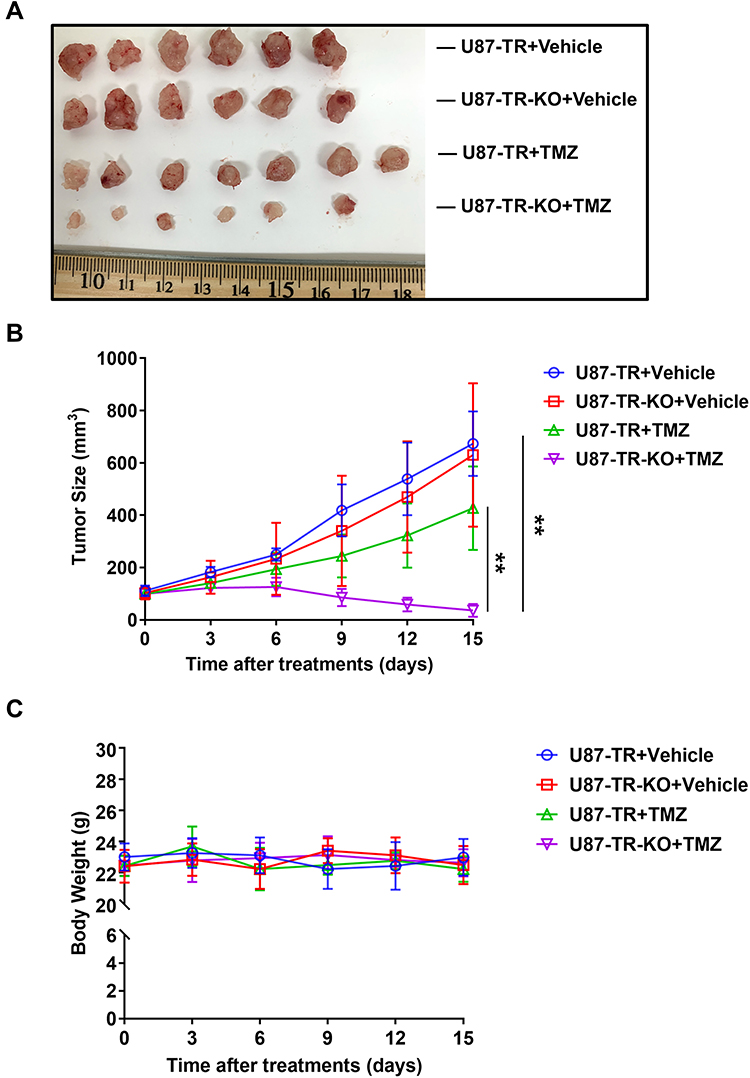 |
Figure 6 APLF deficiency overcomes TMZ-resistance in vivo. (A) Photograph of tumors dissected from mice at day 15 in U87-TR and U87-TR-KO xenograft. Female BALB/c nude mice were treated intraperitoneally with vehicle (DMSO), TMZ (7.5mg/kg/day) for 15 days. (B) Tumor size and (C) body weight of U87-TR and U87-TR-KO xenograft during treatment. **p<0.01. |
Together our data suggest that APLF is potential target and inhibition of ALPF is a promising strategy to overcome chemo- and radioresistance in GBM.
Discussion
In this study, we demonstrated the role of APLF in NHEJ in GBM cells that APLF deficiency significantly impaired NHEJ efficiency. We found that APLF expression is positively correlated with TMZ resistance in GBM patients and in laboratory GBM cell lines. Similarly, APLF is upregulated in IR-resistant GBM cells. We further showed that APLF deficiency generates great improvement of sensitivity to TMZ and IR in U87 and T98G cell lines. Our results provide evidences to demonstrate that APLF can be served as a novel target for GBM treatment after surgical resection. The limitation of this study is xenograft mice model with subcutaneous injection of human cells. We will use intracranial orthotopic model in future development of APLF inhibitors.
It has been decades that TMZ resistance is believed to be associated with MGMT and MMR36,37 because MGMT directly removes the methylation generated by TMZ and MMR is the pathway to recognize the mutant DNA. Thus, acquired TMZ resistance has been correlated with upregulated MGMT,38 In addition, loss of MMR by mutation of MMR key factors can impair TMZ treatment by lack of DNA strand breaks.6 However, they are not the mechanisms that can explain all the cases. The somatic genomic landscape of GBM showed that only about half of GBM patients express MGMT,39 and only a small portion of GBM patients showed MMR deficiency,38,40 indicating that mechanisms of TMZ resistance are still unclear and new yet undiscovered mechanisms remain to be found. DSBs can be generated as a result of MMR after TMZ treatment that require DSB repair pathways.41 In addition, the most severe DNA lesion induced by IR are DSBs, which are repaired by HR or NHEJ.42 Therefore, DSB repair pathway might play an important and frequently underestimated role in TMZ and IR-resistance mechanism in GBM.
Gil Del Alcazar et al found that HR mediates acquired TMZ resistance in GBM via inducing dissolution of Rad51 foci.43 However, specific HR efficiency was not determined in the TMZ-resistant GBM cells, and participation of NHEJ was not excluded in the study. We focused on NHEJ because it is the DNA repair pathway to join the most toxic DNA lesions in cells and it can be used regardless of cell cycle. The study of NHEJ in GBM is very limited. Liu et al found that DNA-PKcs inhibition can sensitize GBM cells to carbon ions indicating impaired NHEJ contributes to GBM treatment. Since NHEJ is so universally important, DNA-PKcs, Ku, XRCC4, Ligase IV or XLF deficiency, which leads to complete loss of NHEJ, would be toxic for GBM treatment. Therefore, our group is interested in NHEJ accessory, yet important protein: APLF. APLF deficiency results in reduced DNA ligation in human and in DT40 cells.35,44,45 Importantly, repair rate of γ ray-induced DSB is reduced at early stage in APLF-deficient human cells.44 Although neither APLF deficiency human cells nor MEFs significantly affect sensitivity to DNA damaging agents,44 APLF overexpression significantly increased resistance to IR and TMZ in resistant GBM cells. These observations suggest a specific role of APLF participating in mechanism of IR and TMZ resistance. Ku proteins are highly abundant in human cells, whereas expression of other NHEJ core factors is limited.46 To efficiently repair DSB induced by IR or TMZ in resistant cells, over expressed APLF might be recruited by Ku and fast assembles NHEJ factors to resolve DSB. This might explain elevated NHEJ efficiency in IR and TMZ-resistant GBM cells.
NHEJ key factors, such as Ku70/80, DNAPKcs and Ligase IV, are overexpressed in various types of cancer.47–49 NHEJ also plays an important role in providing chemoresistance and IR resistance in cancer cells.50,51 Therefore, NHEJ inhibitors can be used as anti-cancer drugs and sensitizers to radiations and DSB inducing agents. Most well studied NHEJ inhibitors target NHEJ factors that show enzymatic activities. For example, NU7026, AZD7648 and VX-984 are selective DNAPKcs inhibitors. Both AZD7648 and VX-984 are under evaluation in early phase of clinical trials as anti-cancer drugs (AZD7648: NCT03907969; VX-984: NCT02644278). SCR7 was developed as a Ligase IV inhibitor that impairs NHEJ by interfering Ligase IV-DNA binding.51 However, it was then demonstrated neither specific nor potent in vitro.52 Since NHEJ is one of the major DSB repair pathways, inhibition of NHEJ key factors may not generate selective obliteration of cancer cells. This prompted us to identify NHEJ factors that specifically contribute to chemo- or IR-resistance, but not deplete NHEJ.
APLF does not exhibit enzymatic activity; therefore, it is unlikely to develop APLF inhibitor using conventional substrate-product high throughput screening. The most important role of APLF in NHEJ is achieved by interacting with Ku80, XRCC4-Ligase4 and XLF, our group will use luciferase-based high throughput screening to identify small molecules that disrupt APLF interaction with Ku80, XRCC4 or XLF. Recent study found that expression of APLF is upregulated in breast cancer, and APLF knockdown induced apoptosis, inhibited cell proliferative and delayed DSB repair in MDAMB-231 cells.53 Our group, for the first time, demonstrated the role of APLF in IR- and TMZ-resistant GBM cells. Therefore, APLF inhibitors have a great potential to be used as sensitizers for IR or chemotherapeutic agents in various type of cancer.
Acknowledgment
This work was supported by funding ZR2017MH129 to Jie Chai from Shandong Provincial Natural Science Foundation.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Dunn GP, Rinne ML, Wykosky J, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi:10.1101/gad.187922.112
2. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised Phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi:10.1016/S1470-2045(09)70025-7
3. Minniti G, De Sanctis V, Muni R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma in elderly patients. J Neurooncol. 2008;88:97–103. doi:10.1007/s11060-008-9538-0
4. Komotar RJ, Otten ML, Moise G, Connolly ES. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma – a critical review. Clin Med Oncol. 2008;2:421–422.
5. Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996.
6. Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622–4629.
7. McNeill RS, Vitucci M, Wu J, Miller CR. Contemporary murine models in preclinical astrocytoma drug development. Neuro Oncol. 2015;17:12–28.
8. Kitange GJ, Mladek AC, Carlson BL, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res. 2012;18:4070–4079. doi:10.1158/1078-0432.CCR-12-0560
9. Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193.
10. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120.
11. Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–2597.
12. Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). 2007;6:1079–1099. doi:10.1016/j.dnarep.2007.03.008
13. Jekimovs C, Bolderson E, Suraweera A, et al. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Front Oncol. 2014;4:86.
14. Kakarougkas A, Jeggo PA. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br J Radiol. 2014;87:20130685. doi:10.1259/bjr.20130685
15. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi:10.1146/annurev.biochem.052308.093131
16. Jackson SP. Detecting, signalling and repairing DNA double-strand breaks. Biochem Soc Trans. 2001;29:655–661. doi:10.1042/bst0290655
17. Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113.
18. Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi:10.1074/jbc.R700039200
19. Fattah F, Lee EH, Weisensel N, et al. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi:10.1371/journal.pgen.1000855
20. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi:10.1016/S0092-8674(02)00671-2
21. Bernstein NK, Williams RS, Rakovszky ML, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell. 2005;17:657–670.
22. Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912.
23. Kanno S, Kuzuoka H, Sasao S, et al. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007;26:2094–2103. doi:10.1038/sj.emboj.7601663
24. Li J, Summerlin M, Nitiss KC, Nitiss JL, Hanakahi LA. TDP1 is required for efficient non-homologous end joining in human cells. DNA Repair (Amst). 2017;60:40–49. doi:10.1016/j.dnarep.2017.10.003
25. Heo J, Li J, Summerlin M, et al. TDP1 promotes assembly of non-homologous end joining protein complexes on DNA. DNA Repair (Amst). 2015;30:28–37. doi:10.1016/j.dnarep.2015.03.003
26. Bekker-Jensen S, Fugger K, Rendtlew Danielsen J, et al. Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J Biol Chem. 2007;282:19638–19643. doi:10.1074/jbc.C700060200
27. Rulten SL, Fisher AE, Robert I, et al. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45.
28. Yang B, Han N, Sun J, Jiang H, Xu HY. CtIP contributes to non-homologous end joining formation through interacting with ligase IV and promotion of TMZ resistance in glioma cells. Eur Rev Med Pharmacol Sci. 2019;23:2092–2102.
29. Sun J, Cai X, Yung MM, et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene. 2019;38:564–580.
30. Chen CW, Li Y, Hu S, et al. DHS (trans-4,4ʹ-dihydroxystilbene) suppresses DNA replication and tumor growth by inhibiting RRM2 (ribonucleotide reductase regulatory subunit M2). Oncogene. 2019;38:2364–2379. doi:10.1038/s41388-018-0584-6
31. Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci U S A. 2007;104:13068–13073. doi:10.1073/pnas.0702410104
32. Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst). 2008;7:1765–1771.
33. Meng Y, Chen C-W, Yung MMH, et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer Lett. 2018;428:104–116. doi:10.1016/j.canlet.2018.04.029
34. Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010. doi:10.3791/2002
35. Macrae CJ, McCulloch RD, Ylanko J, Durocher D, Koch CA. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst). 2008;7:292–302. doi:10.1016/j.dnarep.2007.10.008
36. Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi:10.1056/NEJMoa043331
37. Wiewrodt D, Nagel G, Dreimüller N, Hundsberger T, Perneczky A, Kaina B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int J Cancer. 2008;122:1391–1399.
38. Felsberg J, Thon N, Eigenbrod S, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659–670. doi:10.1002/ijc.26083
39. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477.
40. Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi:10.1038/nature07385
41. Erasimus H, Gobin M, Niclou S, Van Dyck E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat Res Rev Mutat Res. 2016;769:19–35. doi:10.1016/j.mrrev.2016.05.005
42. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi:10.1016/j.molcel.2010.09.019
43. Gil Del Alcazar CR, Todorova PK, Habib AA, Mukherjee B, Burma S. Augmented HR repair mediates acquired temozolomide resistance in glioblastoma. Mol Cancer Res. 2016;14:928–940. doi:10.1158/1541-7786.MCR-16-0125
44. Grundy GJ, Rulten SL, Zeng Z, et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013;32:112–125. doi:10.1038/emboj.2012.304
45. Hammel M, Yu Y, Radhakrishnan SK, et al. An intrinsically disordered APLF links Ku, DNA-PKcs, and XRCC4-DNA Ligase IV in an extended flexible non-homologous end joining complex. J Biol Chem. 2016;291:26987–27006. doi:10.1074/jbc.M116.751867
46. Shibata A, Jeggo P, Lobrich M. The pendulum of the Ku-Ku clock. DNA Repair (Amst). 2018;71:164–171. doi:10.1016/j.dnarep.2018.08.020
47. Pucci S, Mazzarelli P, Rabitti C, et al. Tumor specific modulation of KU70/80 DNA binding activity in breast and bladder human tumor biopsies. Oncogene. 2001;20:739–747. doi:10.1038/sj.onc.1204148
48. Shintani S, Mihara M, Li C, et al. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003;94:894–900. doi:10.1111/j.1349-7006.2003.tb01372.x
49. Kuschel B, Auranen A, McBride S, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet. 2002;11:1399–1407. doi:10.1093/hmg/11.12.1399
50. Riballo E, Critchlow SE, Teo S-H, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi:10.1016/S0960-9822(99)80311-X
51. Srivastava M, Nambiar M, Sharma S, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–1487. doi:10.1016/j.cell.2012.11.054
52. Greco GE, Matsumoto Y, Brooks RC, et al. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair (Amst). 2016;43:18–23. doi:10.1016/j.dnarep.2016.04.004
53. Majumder A, Syed KM, Mukherjee A, et al. Enhanced expression of histone chaperone APLF associate with breast cancer. Mol Cancer. 2018;17:76. doi:10.1186/s12943-018-0826-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.