")
Back to Journals » Cancer Management and Research » Volume 11
Enasidenib in acute myeloid leukemia: clinical development and perspectives on treatment
Authors Reed DR, Elsarrag RZ , Morris AL, Keng MK
Received 13 May 2019
Accepted for publication 30 July 2019
Published 30 August 2019 Volume 2019:11 Pages 8073—8080
DOI https://doi.org/10.2147/CMAR.S162784
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Daniel R Reed,1 Ramey Z Elsarrag,2 Amy L Morris,3 Michael K Keng1
1Division of Hematology/Oncology, Department of Medicine, University of Virginia, Charlottesville, VA, USA; 2Department of Medicine, University of Virginia, Charlottesville, VA, USA; 3Department of Pharmacy Services, University of Virginia, Charlottesville, VA, USA
Correspondence: Michael K Keng
Department of Medicine, Division of Hematology/Oncology, University of Virginia, 1300 Jefferson Park Avenue, West Complex, Room 6009, Charlottesville, VA 22908, USA
Tel +1 434 924 4257
Fax +1 434 244 7534
Email [email protected]
Abstract: Recently there has been a significant progression in the understanding of molecular mutations driving biochemical and cellular signaling changes leading to survival and proliferation of leukemia cells in patients with acute myeloid leukemia (AML). Preclinical studies have demonstrated a mutated enzyme in the citric acid cycle, isocitrate dehydrogenase (IDH), leads to the production of an oncogenic metabolite R-2-hydroxy-glutarate (R-2-HG). This causes the arrest in the differentiation of hematopoietic stem cells leading to the promotion of leukemia. Inhibitors of the IDH enzyme have been shown in preclinical studies to reduce the production of R-2-HG, resulting in terminal differentiation of leukemia blast cells. In recent phase I and II trials, the IDH2 inhibitor enasidenib has shown clinical activity in patients with relapsed and refractory (R/R) AML. This review will describe the preclinical and clinical developments of enasidenib and its Food and Drug Administration approval in R/R AML, treatment recommendations and management will be outlined.
Keywords: isocitrate dehydrogenase, IDH 2, relapsed/refractory, R/R acute myeloid leukemia AML, enasidenib, IDH, AML
Introduction
Acute myeloid leukemia (AML) represents 33% of all new leukemia diagnoses in 2018, yet is responsible for 44% of all leukemia-related deaths that year.1 Those with relapsed or refractory (R/R) disease, advanced age, or AML evolved from antecedent neoplasms have substantially poorer prognosis with a 2-year survival of around 20%.2 However, recent advances in the understanding of the pathophysiology and molecular changes in AML have led to the development of new agents that demonstrated improved clinical efficacy. This article will review the significance of isocitrate dehydrogenase (IDH) mutations in AML, preclinical and clinical studies, and clinical pearls for using enasidenib (IDHIFA; AG-221; Celgene Cooperation, Summit, NJ, USA) in practice.
Role of IDH, mutations, metabolites, and αKG-dependent dioxygenases in AML
Preclinical studies identified isocitrate dehydrogenase 2 (IHD2) as a commonly mutated enzyme in hematologic malignancies, represented in around 11% of the patients with AML; the products of the mutated enzymes have a pro-oncogenic effect.3–6 IDH is a homodimeric enzyme in the citric acid (TCA) cycle that catalyzes the conversion of isocitrate to a-ketoglutarate (αKG), withIDH2 acting in the mitochondria. Mutated forms of the enzyme have novel activity in reducing αKG to R-2-hydroxyglutarate (R-2-HG).6 R-2-HG is a competitive inhibitor of αKG-dependent dioxygenases, which are oxidative enzymes that have a wide variety of roles in post-translational modifications, epigenetic regulation, biosynthesis, and regulation of metabolism pictured in Figure 1.6,7 This inhibition leads to histone and DNA hypermethylation, chromatin modification, altered cellular metabolism, and an altered hypoxia response.6–8 High concentrations of R-2-HG are observed in patients with IDH2-mutated malignancies and associated with multiple roles in the malignant transformation and arrest of differentiation of hematopoietic cells both in-vitro and in-vivo.9
αKG-dependent hydroxylases play a crucial role in the regulation of the hypoxia response by catalyzing the ubiquitin-mediated degradation of hypoxia-inducible factor α (HIFa), a major regulator of the hypoxia response in humans. HIFa binds to hypoxia response elements and regulates the expression of hundreds of different genes. These genes increase oxygen supply to hypoxic tissues and shift cellular metabolism to favor aerobic glycolysis, conserving oxygen utilization.7,10 This shift is more commonly known as the Warburg effect, thought to play a significant role in the pathogenesis of many malignancies.7,11
Potentially all αKG-dependent hydroxylases also possess non-catalytic domains of great functional significance, exemplified by PHF8 and its interaction with histone H3. Two important sites of methylation on histone H3 are the lysine residues K9 and K4, where methylation at these two residues repress and activate transcription, respectively. The catalytic domain of PHF8 is guided to its substrate H3 dimethyl-lysine 9, demethylation it (activating transcription), through the interaction of the noncatalytic plant homeobox domain with histone H3 by binding to the H3 trimethyl-lysine 4. This additional interaction increases the PHF8’s affinity for its substrate, additionally promoting activation of transcription.8,12,13 There is a mounting body of evidence supporting this and other αKG-dependent enzymes play crucial roles in regulating a wide range of critical cellular functions, whose inhibition by R-2-HG would have profound and far-reaching effects on the cell.
IDH mutations represent an excellent therapeutic target due to the powerful effects of R-2-HG and presence of IDH mutations in hematologic malignancies. Kats et al demonstrate that the introduction of IDH mutations and FMS-like tyrosine kinase 3 (FLT3) mutations can drive the development and maintenance of leukemia in transgenic mice, but inhibition of these mutant enzymes can reduce proliferation and improve differentiation of leukemic blasts.14
All mutations in IDH2 cause a single amino acid substitution at either Arg140, substituted to Lys, Met, Gly or Trp or Arg172, substituted to either Gln or Trp. These residues, and the substitutions caused by the mutations, occur in the active site of IDH2 and are of great catalytic importance, as it allows for the novel reducing the activity of the mutated enzyme. As such the Arg140 site is most commonly mutated to Gln and the Arg172 site to Lys to allow for this novel activity. Investigations by K Yen et al reveal that enasidenib has a nearly 50 times lower in-vitro IC50 for the Arg140Gln mutant over the Arg172Lys mutant. This effect is notable in the cell lines treated with enasidenib, explaining the superior R-2-HG suppression of enasidenib in the setting of Arg140Gln mutations.15
Role of enasidenib in AML
Enasidenib is a novel, first in class, oral, small molecule inhibitor of IHD2.16 It is a non-cytotoxic differentiating agent that functions by inducing the maturation of malignant cells. Similar in class to all-trans retinoic acid (ATRA) for the treatment of acute promyelocytic leukemia (APML), enasidenib induces terminal differentiation of leukemic myeloblasts and restore normal tri-lineage hematopoiesis in responding patients without evidence of apoptosis.2 This unique effect of differentiation therapy may provide an additional advantage over traditional cytotoxic therapy by aiding in the reconstitution of the immune system and restoration of normal function.
The Food and Drug Administration (FDA) approved enasidenib in August 2017 for the treatment of IDH2 mutated R/R AML.
Pre-clinical studies
Enasidenib was a compound identified through “hit to lead” high-throughput drug screen looking for small molecule inhibitors of IDH2 mutants given the known importance of IDH mutations in oncogenesis. Through protein chemistry and X-ray crystallography, enasidenib was found to inhibit the enzyme IDH2 by binding to the allosteric site of the enzyme and stabilizing the homodimer conformation preventing conformation change of the enzyme required for the production of the R-2-HG.15 Further experiments identified dose-dependent decreases of R-2-HG, causing differentiation in cell culture of TF1 erythroleukemia cells to erythrocytes without apoptosis. Mature granulocyte markers were observed after treatment of xenograft AML blast cells, demonstrating the effectiveness of IDH inhibition in promoting differentiation of these cells. Enasidenib did not induce cellular death as demonstrated with myeloblasts differentiation into mature and functional neutrophils while continuing to express the mutant IDH2 allele. Enasidenib demonstrated a dose-dependent survival benefit in xenograft mice compared to mice treated with cytarabine and demonstrated dose-dependent reduction in R-2-HG, reduction in bone marrow blasts, as well as differentiation into mature granulocytes without impacting the IDH2 allele frequency.15
Clinical trials
The landmark trial for enasidenib was published by Stein et al in 2017.16 This was a Phase I/II, multicenter, multinational clinical trial, conducted from September 2013 to April 2016, that assessed enasidenib in humans for the first time. Patients were eligible for the trial if they were greater than 18 years of age with confirmed diagnosis of AML with a mutated IDH2 by next-generation sequencing (NGS). The study was designed with an initial dose escalation to determine the maximum tolerated dose (MTD) and then a subsequent dose expansion to evaluate the efficacy of enasidenib in terms of types of response, duration of response, and transfusion independence. One hundred thirteen patients were evaluated in the dose escalation phase and no MTD was identified up to the maximum dose of 650 mg daily. The dose chosen for the expansion phase was 100 mg daily based on its pharmacokinetic and pharmacodynamic analyses. One-hundred nine patients with R/R AML received enasidenib at 100 mg daily. The median age was 67 years old (range: 19–100); 58% of the patients were female (n=63). Twenty-five percent (n=27) of the patients had AML with myelodysplastic-related change and 36% (n=29) of the patients had poor risk cytogenetics. The majority of patients were either refractory to initial induction (32% (n=35)) or relapsed within 1 year of treatment (23% (n=25)). Twelve patients (11%) had prior stem cell transplant. Both IDH2 mutation locations were represented in the trial; however, the majority (76%) of patients had R140 mutations. The overall response rate (ORR) of R/R AML defined as complete remission (CR)+complete remission with incomplete hematologic recovery (CRi)+complete remission with incomplete platelet recovery (CRp) and partial response (PR)+morphologic leukemia-free state was 40.3% (95% CI, 33–48%) including 34 patients who achieved a CR. The majority of patients achieved a response by cycle 5 (87.3%). Patients with R172 mutations had an improved ORR compared to patients with R140 mutations, 53.3% vs 35.4%, respectively. However, only a slightly higher number of patients with R172 mutations achieved a CR compared to patients with R140, 24.2% vs 17.7%. The median duration of response was 5.8 months and median overall survival (OS) in patients with R/R AML was 9.3 months (8.2–10.9 months) with an estimated one-year survival of 39%. In patients who achieved a CR, the median OS was 19.7 months. There were 10% of the patients who were bridged to stem cell transplant using enasidenib. Most common grade 3 or 4 adverse events were hyperbilirubinemia, thrombocytopenia, anemia, and IDH differentiation syndrome (DS). This trial led to the FDA accelerated approval for enasidenib for IDH2 mutated R/R AML.
Through extensive correlative studies done on samples obtained during this trial, there were several findings.17,18 First, patients were found to have persistent IDH2 mutant alleles despite months of treatment. This was confirmed after peripheral blood mature neutrophils were found to have same allele burden compared to the stem cell leukemia clone, supporting that enasidenib in vivo does not induce apoptosis rather promotes differentiation. Correlative studies with this trial confirmed the mechanism of action of enasidenib, showing a reduction in R-2-HG levels with a median suppression of 90.6%. As shown in previous preclinical trials, enasidenib appears to be more efficacious at reducing R-2-HG levels in patients with the R140 mutant compared to the R172 mutant. Unfortunately, the degree or presence of suppression of R-2-HG was not correlated with response to enasidenib and therefore is not a useful biomarker. Variant allele frequently (VAF) burden in patients with IDH2 mutation did not correlate with clinical response and therefore should not be used for selection criteria for treatment with enasidenib. Only mutations occurring in the RAS and MAPK pathways were associated with decreased clinical response to enasidenib. In addition, patients with more than six mutations found with IDH2 were less likely to achieve a response with enasidenib.17
An update on Stein et al trial was recently published which described the treatment of 214 patients with R/R AML.18 All patients were treated with 100 mg of enasidenib daily. The CR rate was 19.6% (n=42) and ORR was 38.8% (n=83). The median OS was 8.8 months (95% CI, 7.7–9.6) and median time to best response was 3.7 months (95% CI, 0.6–14.7). The updated median OS for the 42 R/R AML patients who achieved a CR was 23 months; those who did not have an OS for 5.6 months. Enasidenib was used successfully as a bridge to transplant in patients leading to a median OS of 23.6 months. Patients with less than 3 co-mutations had improved ORR compared to those with high mutation burden (>6) at 31.3 vs 54.8% (p=0.06). In the R/R AML cohort, NRAS mutations were associated with decreased CR rate at 3% vs 19% (p=0.025) and the presence of FLT3 – internal tandem duplication (ITD) or – tyrosine kinase domain (TKD) was associated with nonresponse (p=0.014).18 Other populations are undergoing an evaluation to determine the application of enasidenib treatment. Patients who were not candidates for intensive induction chemotherapy with the IDH2 mutation (n=39) were eligible on the phase I trial to receive enasidenib and outcomes from this population were recently published.19 The median age was 77 (range 58–87) and the majority of patients (n=25) were classified as adverse risk according to ELN leukemia criteria. ORR in this population was found to be 30.8% (17–47.6) with a median overall survival of 11.3 months. Out of this cohort of patients, 18% (n=7) achieved a complete remission and went on to have overall survival of 19.8 months. In this elderly population of patients, 49% (n=19) of patients experiencing grade 3–4 adverse events. The three most common adverse events experienced were anemia (n=5, 13%), hyperbilirubinemia (n=5, 13%), and differentiation syndrome (DS) (n=4, 10%). Unfortunately, 21% (n=8) of the patients experienced serious adverse events including tumor lysis syndrome (TLS) and other life-threatening conditions. Studies are currently investigating the use of enasidenib monotherapy in patients with IDH2-mutant myelodysplastic syndrome (MDS) (NCT03383575), and as maintenance therapy after salvage induction chemotherapy (NCT03881735) or post allogeneic hematopoietic stem cell transplantation in patients harboring the IDH2 mutation (NCT03515512, NCT03728335). In addition, enasidenib is being studied combined with chemotherapy agents in patients with AML and MDS (NCT03839771, NCT03825796, NCT03683433, NCT02677922, NCT02632708, NCT03013998). These trials are listed in Table 1.
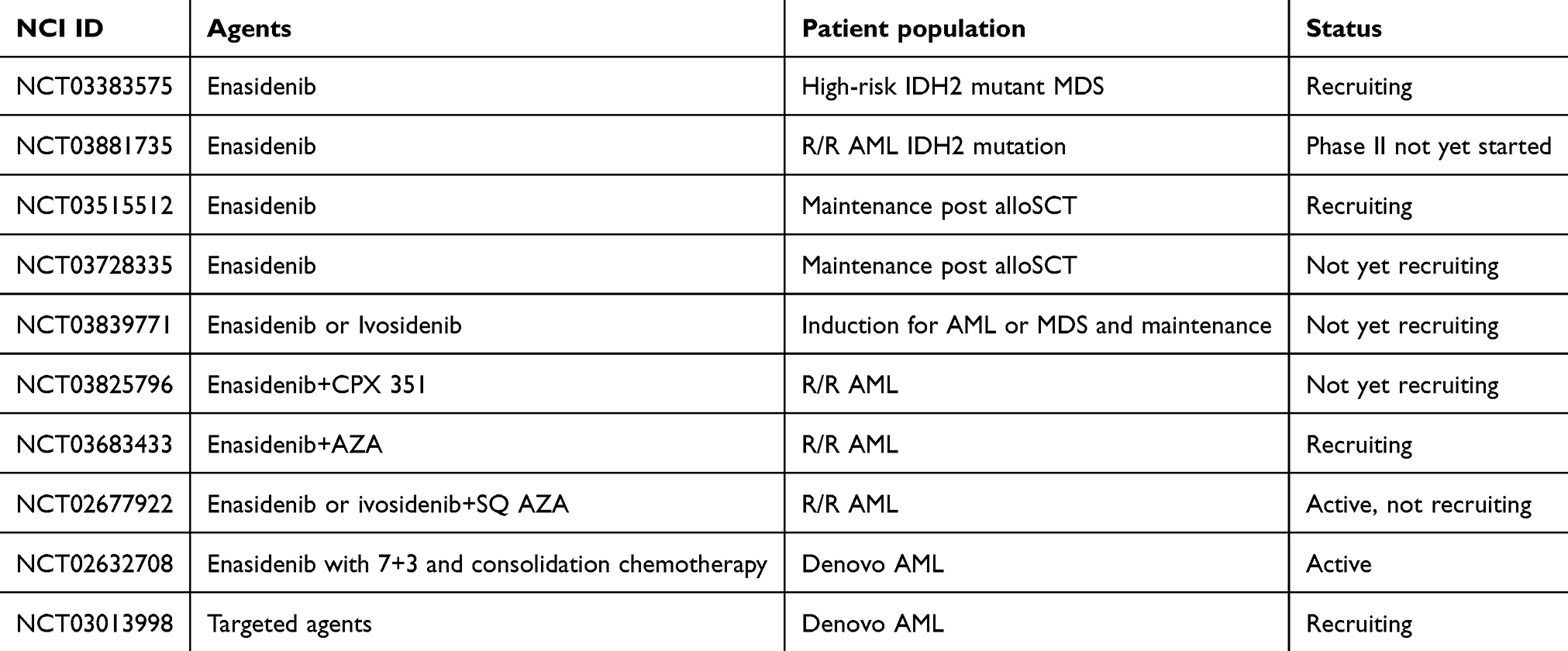 |
Table 1 Current clinical trials with enasidenib |
Resistance to enasidenib
Recent proposed mechanisms of resistance to IDH inhibitors have been described. Intlekofer et al investigated two patients with IDH mutations at R140Q that developed resistance to enasidenib. Both patients initially had a response with reductions in R-2-HG and differentiation identified. The first patient developed resistance after 9 months of treatment and was found to have relapse. The second patient developed resistance to treatment after 6 months. These two patients were found to have a second IDH2 mutation in trans which led to resistance to treatment with enasidenib.20 Quek et al did not find second site mutations leading to resistance however did establish clonal evolution of multiple different cellular signaling pathways inhibiting differentiation and leading to resistance to treatment with enasidenib. This suggests a more complicated mechanism of resistance leading to mutated clones that do not appear to be dependent on the IDH2 mutation.21
Management of patients on enasidenib
At the University of Virginia Medical Center, NGS is sent on all bone marrow biopsies to assess for targetable mutations at both initial diagnosis and relapse. If positive for an IDH2 mutation, patients with R/R AML after standard induction chemotherapy are offered reinduction with cytotoxic chemotherapy or enasidenib monotherapy. For patients with AML who are not candidates for frontline intensive chemotherapy, an individualized approach is completed. Patients are considered for treatment with combination therapy with hypomethylating agent with either venetoclax or cytarabine or the combination of glasdegib with cytarabine.22–24 Monotherapy with hypomethylating agent is also a consideration for treatment in this setting, however, because of the minimal evidence and lack of FDA approval monotherapy enasidenib is not considered front line over these agents.
When starting enasidenib, patients are counseled to expect adverse events such as nausea, vomiting, diarrhea, decreased appetite, and dysgeusia. In addition, clinical pharmacists review for potential drug interactions and assist with counseling and monitoring for enasidenib-specific adverse events including DS, non-infectious leukocytosis, TLS, and indirect hyperbilirubinemia.16,25,26 Interestingly, patients who responded to enasidenib typically do not exhibit bone marrow aplasia as a result of therapy.26
Enasidenib carries a black box warning for risk of DS. By targeting mutant IDH2 enzymes, enasidenib promotes myeloid differentiation thereby causing increased cytokine release, inflammation, and tissue damage in some patients.26 Left untreated, DS can be fatal. In a review of patients from the phase I/II trial, 11.7% of the patients experienced DS (n=33); however, a recent study by Norsworthy et al identified an incidence rate of 19% (41/214).27 In the phase I/II trial, patients with DS were significantly more likely to have had a lesser median number of previous therapies (P=0.05) and less likely to have had less than 20% bone marrow blasts at baseline (P=0.04). No other predictive factors were identified.26 Enasidenib-associated DS occurs much later than ATRA associated DS; median time to onset of enasidenib associated DS was 19–30 days and ranged from 1 to 219 days after starting therapy.26,27 This creates additional challenges for managing, as patients will likely be in the outpatient setting if and when DS occurs. Patients should be educated on the signs and symptoms of DS including unexplained fever, weight gain, and respiratory symptoms such as new onset shortness of breath and/or cough. The manufacturer website provides a way to print wallet cards for patients which include the medication name, prescribing oncologist’s contact information, and most importantly a QR code which emergency medical providers can scan to quickly obtain information on the diagnosis and treatment of DS. Treatment of DS involves initiating corticosteroids (eg, dexamethasone 10 mg IV BID) and hemodynamic monitoring. If patients require intubation or ventilator support, and/or renal dysfunction persists for more than 48 hrs after initiation of corticosteroids, enasidenib therapy should be interrupted. Clinicians should be aware that due to the extremely long half-life, enasidenib and its metabolites can remain in the system for days to weeks after discontinuation.28 Enasidenib may be reinitiated at the original dose once DS signs and symptoms improve to Grade 2 (moderate) or lower.25,26
White blood cell counts should be monitored frequently upon initiation of enasidenib as rapid myeloid proliferation may cause non-infectious leukocytosis, typically with the first 60 days of treatment.16 Fifteen patients (6%) in the phase I/II trial experienced treatment-related non-infectious leukocytosis events, which was not always accompanied by DS; six patients were classified as having grade 3–4 leukocytosis per Common Terminology Criteria for Adverse Events (CTCAE) guidelines. Leukocytosis led to dose interruption for six patients and permanent discontinuation for one patient.16 If patients experience leukocytosis, cytoreduction with hydroxyurea is recommended per institutional standard and enasidenib should be continued. Enasidenib may be interrupted if the WBC does not respond to increasing doses of hydroxyurea; however, due to a long half-life, holding doses may not impact the WBC for many days.28 If enasidenib must be held, it may be reinitiated at 100 mg daily once the WBC<30 k/µL.25,26
TLS may occur upon initiation of enasidenib, defined by elevations of uric acid, potassium, and/or phosphate, decrease in calcium, and/or increase in serum creatinine. Three percent of patients in the phase I/II trial had a tumor lysis defined as serious treatment-related emergent adverse event.16 Patients should have uric acid, electrolytes, and renal function monitored closely in the first several weeks of therapy, and if experiencing non-infectious leukocytosis it may be prudent to watch these parameters more closely. Supportive care medications may be indicated, including, rasburicase, phosphate binders, and potassium removal medications. Patients should start allopurinol upon initiation of enasidenib for prophylaxis of tumor lysis syndrome.
Indirect hyperbilirubinemia occurred in 38% of the patients in the phase I/II study but was not linked to liver toxicity, as no meaningful increases in alanine aminotransferase or aspartate aminotransferase were observed.16 Hyperbilirubinemia may occur via inhibition of UGT1A1, an enzyme responsible for bilirubin metabolism. Bilirubin elevation greater than 3 times the upper limit of normal (ULN) sustained for greater than 2 weeks warrants a dose reduction to 50 mg daily until bilirubin resolves to less than 2 times ULN, then 100 mg daily can be resumed.25
Gastrointestinal disturbances may occur in patients taking enasidenib. Nausea occurred in over 20% of the patients in the phase I/II study, categorizing enasidenib as a moderate to high emetic risk oral anticancer agent per National Comprehensive Cancer Network (NCCN) guidelines. According to the NCCN antiemesis guideline, routine premedication is not required for continuously dosed oral chemotherapy, but clinicians should follow up with patients after starting therapy to ensure nausea and/or vomiting are not hindering compliance and decreasing quality of life. In our experience, nausea has been severe in several patients. If patients experience nausea and vomiting, an oral 5-HT3 receptor antagonist such as ondansetron or granisetron should be administered daily prior to taking enasidenib. Other treatment-related gastrointestinal toxicities reported in the trial included decreased appetite (18%), diarrhea (15%), vomiting (14%), and dysgeusia (10%).16
Enasidenib pharmacokinetics support convenient once-daily dosing of 100 mg in subjects with relapsed and refractory AML. The absolute bioavailability after one 100 mg oral dose is approximately 57%, and median time to Cmax is 4 hrs.29 Absorption is not meaningfully affected by food intake.25 Enasidenib plasma protein binding is 98.5% with a mean volume of distribution of 55.8 L.29 Enasidenib has a terminal half-life of 137 hrs, reaching steady stateby day 29 of therapy. If enasidenib is held for toxicity, clinicians should be aware that enasidenib presence in the body will persist for an extended period following the last dose. Elimination occurs via the feces (89%) and urine (11%). Pharmacokinetics are not altered by age, race, mild hepatic impairment, renal impairment, sex, body weight, or body surface area.25
Enasidenib and its metabolite AGI-16903 are extensively metabolized by multiple hepatic enzymes, including CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and UGT1A1, UGT1A3, UGT1A4, UGT1A9, UGT2B7, and UGT2B15. Despite this extensive hepatic metabolism, enasidenib is not known to be a substrate of any hepatic or gastrointestinal enzymes and drug transport proteins, and therefore does not require dose adjustments when given concurrently with medications that inhibit or induce these enzymes.18 In-vitro studies suggest enasidenib inhibits multiple hepatic and gastrointestinal enzymes and drug transport proteins, including CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, P-gp, BCRP, OAT1, OATP1B1, OATP1B3, and OCT2.25 Narrow therapeutic index medications which are substrates of these enzymes will likely be affected by enasidenib (eg, phenytoin, warfarin, digoxin); patients on these medications should be switched to alternative agents if possible or should be monitored closely for either side effects or decreased efficacy of the concomitant medication if an alternative is not available. A pharmacokinetic study investigating the effects of enasidenib on other medications is currently being conducted (NCT03720366).
Conclusion
Enasidenib is a promising FDA approved agent in the treatment of R/R AML. Through preclinical and clinical evaluation, enasidenib has demonstrated remarkable activity in patients with mutated IDH2. Although generally well tolerated, enasidenib does require careful monitoring throughout its treatment course. Enasidenib is currently being evaluated in a multicenter, randomized Phase III clinical trial (IDHENTIFY trial, NCT 02577406) to confirm the Phase I/II clinical trial results, as well as various combinations in both the frontline and R/R settings for AML and other hematologic malignancies.
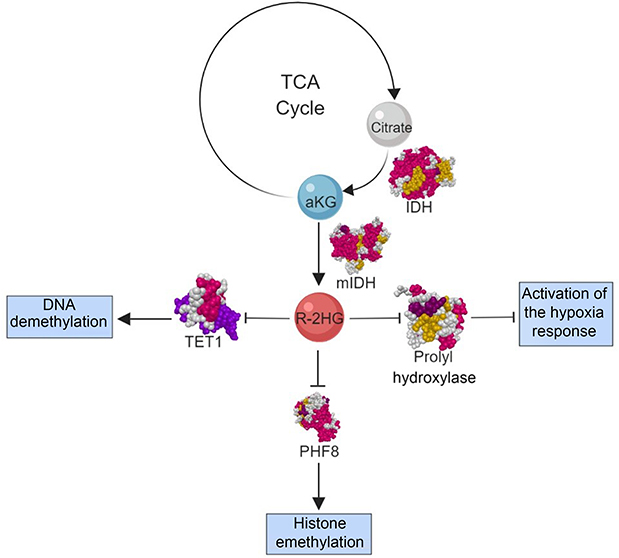 |
Figure 1 R-2-HG mechanism of action. Abbreviations: IDH, isocitrate dehydrogenase; mIDH, mutated isocitrate dehydrogenase; TCA, citric acid cycle; aKG, a-ketoglutarate; R-2-HG, R-2-hydroxyglutarate; TET1, ten-eleven translocation−1; PHF8, PHD-finger protein 8. |
Disclosure
Michael Keng has participated in a Physician Advisory Board Meeting with Agios. The authors report no other conflicts of interest in this work.
References
1. 2001–2015 database: national program of cancer registries and surveillance, epidemiology, and end results SEER*Stat database: NPCR and SEER incidence – USCS 2001–2015 public use research database. Available from: www.cdc.gov/cancer/uscs/public-use.
2. Tchekmedyian R, Elson P, Gerds AT, et al. Analysis of outcomes of patients with relapsed/refractory acute myeloid leukemia treated in randomized clinical trials. Blood. 2016;128(22):4000.
3. Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122–2126. doi:10.1182/blood-2009-11-250878
4. DiNardo CD, Jabbour E, Ravandi F, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia. 2016;30(4):980–984. doi:10.1038/leu.2015.211
5. Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28(14):2348–2355. doi:10.1200/JCO.2009.27.3730
6. Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–281. doi:10.1038/leu.2016.275
7. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi:10.1016/j.ccr.2010.12.014
8. Markolovic S, Wilkins SE, Schofield CJ. Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenases. J Biol Chem. 2015;290(34):20712–20722. doi:10.1074/jbc.R115.662627
9. Fathi AT, Sadrzadeh H, Borger DR, et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood. 2012;120(23):4649–4652. doi:10.1182/blood-2012-06-438267
10. Ziello JE, Jovin IS, Huang Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80(2):51–60.
11. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001
12. Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–727. doi:10.1038/nrg1945
13. Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol. 2010;17(1):38–43. doi:10.1038/nsmb.1753
14. Kats LM, Reschke M, Taulli R, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14(3):329–341. doi:10.1016/j.stem.2013.12.016
15. Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493. doi:10.1158/2159-8290.CD-16-1034
16. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi:10.1182/blood-2017-04-779405
17. Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–741. doi:10.1182/blood-2017-04-779447
18. Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–687. doi:10.1182/blood-2018-08-869008
19. Pollyea DA, Tallman MS, de Botton S, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019. doi:10.1038/s41375-019-0472-2
20. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559:125–129.
21. Quek L, David MD, Kennedy A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med. 2018;24(8):1167–1177. doi:10.1038/s41591-018-0115-6
22. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2018:
23. Wei AH, Strickland SA
24. Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379–389. doi:10.1038/s41375-018-0312-9
25. IDHIFA {Package Insert}. Summit, NJ: Celgene; 2017.
26. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. 2018;4(8):1106–1110. doi:10.1001/jamaoncol.2017.4695
27. Norsworthy KJ, Mulkey F, Ward AF, et al. Incidence of differentiation syndrome with ivosidenib (IVO) and enasidenib (ENA) for treatment of patients with relapsed or refractory (R/R) isocitrate dehydrogenase (IDH)1- or IDH2-mutated acute myeloid leukemia (AML): a systematic analysis by the U.S. Food and Drug Administration (FDA). Blood. 2018;132(Suppl1):288LP. doi:10.1182/blood-2018-99-117426
28. Fan B, Chen Y, Wang F, et al. Evaluation of Pharmacokinetic-Pharmacodynamic (PKPD) relationship of an oral, selective, first-in-class, potent IDH2 inhibitor, AG-221, from a Phase 1 trial in patients with advanced IDH2 mutant positive hematologic malignancies. Blood. 2014;124(21):3737LP. Available from: http://www.bloodjournal.org/content/124/21/3737.abstract.
29. Tong Z, Atsriku C, Yerramilli U, et al. Absorption, distribution, metabolism and excretion of an isocitrate dehydrogenase-2 inhibitor enasidenib in rats and humans. Xenobiotica. 2019;49(2):200–210. doi:10.1080/00498254.2018.1425511
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.