")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Empagliflozin Protects Against Proximal Renal Tubular Cell Injury Induced by High Glucose via Regulation of Hypoxia-Inducible Factor 1-Alpha
Authors Ndibalema AR , Kabuye D, Wen S , Li L, Li X, Fan Q
Received 20 December 2019
Accepted for publication 13 May 2020
Published 12 June 2020 Volume 2020:13 Pages 1953—1967
DOI https://doi.org/10.2147/DMSO.S243170
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Muthuswamy Balasubramanyam
Angelamellisy Revelian Ndibalema,1,2 Deo Kabuye,3,4 Si Wen,1 Lulu Li,1 Xin Li,1 Qiuling Fan1
1Department of Nephrology, The First Hospital of China Medical University, Shenyang 110001, People’s Republic of China; 2Kairuki Hospital, Dar es Salaam, Tanzania; 3Department of Laboratory Medicine, The First Affiliated Hospital of China Medical University, Shenyang, 110001, People’s Republic of China; 4Kalisizo Hospital Uganda, Kalisizo, Uganda
Correspondence: Qiuling Fan Tel +86 13904012680
Email [email protected]
Background: Evidence from both animal and human studies clearly supports the renal beneficial effects of empagliflozin (emp), a sodium glucose co-transporter 2 (SGLT2) inhibitor, but the mechanism in which it exerts its effect is not well understood. In this study, we investigated the capability of emp on reducing hyperglycemia-induced renal proximal tubular epithelial cells injury and we evaluated if the renoprotective effect of emp associates with hypoxia-inducible factor-1α (HIF-1α).
Materials and Methods: Human kidney cell lines (HK-2 cells) were incubated in normoxia, high glucose with or without emp treatment for 72 hours to evaluate the induction of HIF-1α, glucose transporter-1, SGLT2, the fibrosis signal pathway and epithelial–mesenchymal transition (EMT) markers.
Results: High glucose (HG) increased expression of Collagen IV, Fibronectin, transforming growth factor-beta1 (TGF-β 1). However, emp treatment remarkably decreased expression of TGF-β 1, accumulation of extracellular matrix proteins (Fibronectin, Collagen IV), as well as (phosphorylated-smad3) P-smad3. HG increased SGLT2 protein expression compared to normal glucose (NG) while emp significantly decreased SGLT2 expression. Furthermore, emp decreased high glucose-induced alpha-smooth muscle actin (α-SMA) expression and reversed epithelial marker (E-catherin) suppression induced by high glucose. In addition, emp treatment for 72 h increased expression of HIF-1α protein (95% CI: -0.5918 to – 0.002338, at 100nM, P < 0.05, 95% CI – 0.6631 to – 0.07367 at 500nM, P < 0.05) in hyperglycemic normoxic HK-2 cells. Furthermore, we observed increased expression of GLUT-1 protein after emp treatment and remarkably decreased cell proliferation.
Conclusion: Emp treatment protected proximal renal tubular cells injury induced by high glucose. Induction of HIF-1α expression by emp may play an essential role in the protection of high glucose-induced proximal renal tubular epithelial cells injury.
Keywords: humans, animals, transforming growth factor-beta1, empagliflozin, diabetic nephropathies, glucose
The Plain Language Summary
High blood glucose can initiate multiple structural and functional changes in the kidney, resulting in protein loss in urine and decline of kidney function.
Kidney disease related to high blood glucose level is the leading cause of end-stage kidney disease, once the disease progress cannot be reversed. Since the disease is irreversible, more research has been done to find ways to prevent disease development or delay disease progression.
Accumulated evidence supports the kidney protective effects of empagliflozin (emp), a new class of glucose-lowering oral agents by blocking a Sodium-glucose co-transporter 2 in the upper segment of the kidney tubular cells, and promoting glucose loss in urine. However, the way in which emp prevents kidney damage is not clear.
In our study, we tested whether emp can reduce the kidney tubular cells damage caused by high glucose exposure. We also tested the effect of emp on the induction of a key protein responsible for the regulation of tissue reaction to low oxygen tension (HIF-1α)
Human kidney cell lines were incubated under normal air condition. Treated with normal glucose, high glucose with or without emp treatment to detect specific proteins and cells multiplication.
Emp increased HIF-1α production and reduced human kidney tubular cell damage caused by high glucose exposure; therefore, HIF-1α may play a contribution in the kidney protective benefits of emp.
More research on emp drug in the treatment of high blood glucose is necessary to interrupt the development and progression of kidney disease and other complications of high blood glucose.
Introduction
Although there numerous risk factors for diabetic kidney disease like hypertension, dyslipidemia, smoking, obesity, ethnic familial and genetic predisposition.1,2 Hyperglycemia, however, has been considered as a major risk factor in the pathogenesis of renal diseases.2 Diabetes is a worldwide escalating public health problem with an estimate of 8–10% of which 20–40% develop diabetic kidney disease,2–4 in 2017 there was an estimate of 8.8% among adults aged 20–79 years with diabetes and expected to rise to 9.9% by 2045.4,5 Furthermore, it is revealed that diabetes increases the risk of microvascular and macrovascular complications.6,7 Due to the rise of diabetes epidemic global level, Diabetic kidney disease has surfaced as a leading cause of end-stage renal disease (ESRD).4,6 However, early and intensive diagnosis and management of hyperglycemia and high blood pressure may slow down the development and progression of diabetic kidney disease.8,9
Hyperglycemia causes multiple changes in the kidney functional units by inducing the constriction and dilatation of the efferent and afferent arterioles, respectively, resulting in glomerular capillary hypertension and hyperfiltration that has been reported as a physiopathological mechanism in the early development of diabetic kidney disease.6,10 Moreover, thickening of glomerular basement membrane and damage to podocytes together with increased mesangial cells and matrix leads to increased glomerular abnormalities.6,7 It is also postulated that growth factors such as transforming growth factor-beta 1(TGF-β1) are responsible for laying down extracellular matrix in diabetic kidney disease.3 Likewise, in vivo study revealed that high glucose-induced expression of TGF-β1 which influenced EMT (epithelial–mesenchymal transition) in rats tubular epithelial cell.11 Similarly, HK-2 cells (human kidney cell line) exposed to high glucose increased TGF-β1 expression.3
The sodium-glucose co-transporters (SGLT) are the sodium-dependent transporters expressed apically in the epithelial cells of the proximal convoluted tubule3,12–14 responsible for the simultaneous transportation of glucose against the concentration gradient with sodium being transported downhill following the gradient.13,14 SGLT2 accomplishes reabsorption of about 90% of filtered glucose in the proximal tubular cells with 1:1 sodium: glucose coupling ratio. Then, complete reabsorption occurs via SGLT1 with a high affinity to a low capacity for sodium and glucose with a coupling ratio of 2:1, respectively.14 Recent discoveries demonstrated localized expression of SGLT2 and SGLT1 proteins in the brush border membrane of the early and late segments of the proximal tubules, respectively.9 On the other hand, glucose is also passively transported through the basolateral membrane towards the plasma compartment by glucose transporters (GLUT).
In diabetes, SGLT2 activation increases oxygen consumption caused by high intracellular glucose level in the proximal tubular cells,12,13 and reduces sodium delivery to the juxtaglomerular apparatus thereby inducing glomerular hyperfiltration. Alternatively, hypoxic tubular cells activate the adaptive process for the survival against tissue hypoxia.15 Hypoxia-inducible factor−1alpha (HIF-1α) is a key protein that plays a major role in modulating the reaction of tissue to hypoxia, HIF-1 has a heterodimeric structure consists of oxygen-sensitive HIF-1alpha subunit bound to constitutively expressed Beta (β) subunit that subsequently activates the HIF-response element (HRE) pro-survival genes that enhance oxygen delivery in hypoxic organs such as brain, heart, liver and kidney,15–17 but its function is depressed in diabetes.18,19 Although studies have shown that overexpression of HIF-1α in tubular epithelial cells contributes to renal fibrosis, its inhibition prevented the progression of renal fibrosis and attenuated diabetic kidney disease.20,21 In particular, it is also reported that stabilization of HIF-1α by inhibition of prolyl hydroxylase domain (PHD) attenuated Ischemic kidney injury,22 and HIF-1α deficiency accelerated diabetic kidney disease progression in HIF-Iα deficient mice.15 Therefore, it is not clear whether HIF-1α expression has a protective or harmful effect on the development and progression of diabetic kidney disease. Apart from hypoxia, HIF-1α can also be regulated by hyperglycemia, TGF-β1, reactive oxygen species, cobalt chloride, nitric oxide and angiotensin II.23
Empagliflozin, a highly selective SGLT2 inhibitor, is a new class of glucose-lowering oral agents for type 2 diabetes mellitus that blocks SGLT2 transporter in the proximal tubular cells.1,24 The mechanism of action increases glucose excretion in urine leading to glycosuria and hence improving glycemic control.25,26 Evidence from both animal and human studies have shown that SGLT2 inhibition exerts renoprotective effects by reducing glomerular hyperfiltration, SGLT2 inhibitors reduce proximal tubular sodium reabsorption with subsequent increase in sodium delivery to and transport in the cells of the macula densa in the distal convoluted tubule, and thus stimulates tubuloglomerular feedback (TGF) and increases afferent arteriolar tone, leading to decrease in glomerular filtration and intraglomerular pressure with a transient lowering of glomerular filtration rate (GFR).1 Dapagliflozin an SGLT2 inhibitor induces HIF-1α expression whereas Luseogliflozin another SGLT2 inhibitor reported to reduce HIF-1α expression in renal tubular epithelial cells.27,28 CREDENCE (Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation), which showed that treatment with canagliflozin was associated with a significant 30% decreased risk of the trial’s primary composite outcome of end-stage renal disease (ESRD), a doubling of serum creatinine level, or death from renal or cardiovascular (CV) causes compared with placebo among patients with type 2 diabetes and kidney disease. Investigators presented the findings at the 2019 World Congress of Nephrology in Melbourne, Australia, on April 15. The presentation coincided with the online publication of the findings in The New England Journal of Medicine.29 Ultimately, it remains unclear whether SGLT2 inhibitor regulates HIF-1α. Therefore, in this study, we investigated whether emp Protects Proximal Renal Tubular Cells Injury Induced by High Glucose via Regulation of Hypoxia-inducible factor 1-Alpha.
Materials and Methods
Cell Culture and Treatment of Proximal Renal Tubular Epithelial Cells
HK-2 cells, immortalized proximal tubule epithelial cell line, were purchased from China Centre for Type Culture Collection (CCTCC, Wuhan, Hubei, China) and cultured. Briefly, cells were passed every 3–4 days in 6-well plates using Dulbecco`s Modified Eagles Medium-F12 (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, Waltham, Massachusetts, USA), 1% antibiotic penicillin (100 u/mL) and streptomycin (100ug/mL)). For experimental use, these cells were incubated in a humidified atmosphere of 5% CO2, at 37°C and subcultured. The medium was changed every 48 h. When 80% confluent reached, cells were seeded into 6-well plates with medium containing 5.5mM glucose (normal glucose) and 25mM glucose (high glucose, Gibco Glucose solution contains D-glucose at a concentration of 200g/L, Cat. No. A2494004) and 20mM mannitol then incubated in a humidified atmosphere of 5% Co2, at 37°C for 24 h and then harvested. For treatment with the drug, Emp an SGLT2 inhibitor purchased from (MedchemExpress.com.Shangai China. Cat.No.HY-15,409/cs-0940) was used. When 50–60% confluent reached HK-2 cells were exposed in the medium containing either 5.5mM glucose (normal glucose level) or 25mM glucose (high glucose level) with or without emp treatment (50nM, 100nM, 500nM) and cultured for 72 h in a humidified atmosphere of 5% Co2, at 37°C. Cells were collected at 72 h post treatment. Experiments were repeated three times.
Western Blot
Western blot was performed for protein analysis. Cells were collected at 97% confluent and the cell pellet was resuspended in RIPA (radio immunoprecipitation assay) lysis buffer with 1% protease and phosphatase inhibitors for 30 min. All reagents and lysates were kept on cold ice. After scraping adherent cells off the dish, suspension transferred to different corresponding EP tubules and sonicated 5 times for 3 s to complete cell lysis and shear DNA to reduce sample viscosity. Cell lysate was spun at 1200 rpm and 4°C for 20 min and stored at −80°C. Protein quantification as an integral part of any experimental work involving protein extraction, purification, labeling or analysis was carried out by using BCA (Bicinchoninic acid assay) (Beyotime-Haimen, Jiangsu China). Cell lysate assayed to determine protein concentration. A 30 ug of total protein was mixed with 5x Laemmli buffer containing (mercaptoethanol) and heated at 100°C for 10 min. A 30 ug of total protein was loaded into the gel in each lane containing 8% separating gel and 5% stacking gel, then analyzed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) with a biotinylated molecular weight standard marker for 80 min. Then, the sample was wet transferred to nitrocellulose membrane (Merck Millipore Ltd Tullagreen Carrigtwohill Country Cork Ireland) and electroblotted for 120 min. Membranes were blocked for 2 h with 5% non-fat dry milk in TBST buffer (Tris-HCl (PH 8.0), 0.8 NaCl and 0.05%Tween20, Beyotime, Haimen Jiangsu, China) with shaking. Membranes were washed with TBS-T buffer 4 times (5 min each) and incubated overnight at 4°C on the shaker with the following primary antibodies: SGLT2 1:500 (Proteintech, Wuhan Hubei China), HIF-1a 1:1000 (Rabbit Polyclonal antibody, Cat. No. 20,960-1-AP. Proteintech, China), calculated molecular weight 93 kDa but the modified protein HIF-1α is about 110–120 kDa (PMID:11,698,256, PMID:7,539,918). GLUT-1 1:500 (Proteintech, China), TGF-b1 1:1000 (Proteintech, China), SMAD3 1:1000 (Proteintech, China), P-SMAD3 1:1000 (Proteintech, China), Collagen IV 1:2000 (Proteintech, China), Fibronectin 1:2000 (Proteintech, China), E-Cadherin 1.2000, a-SMA 1:300. Beta-Actin 1:5000 (Proteintech, China) with shaking. Membranes were washed again with TBST 4 times (5 minutes each) and incubated with 1:8000 secondary anti-rabbit IgG-Horse Radish Peridoxidase-linked antibody (Cat.01.A21020.Abbkine Scientific-Wuhan Hubei, China) for 2 hours at room temperature on the shaker. The bands were detected after wash with TBS-T buffer using chemiluminescence HRP substrate (Millipore Corporation, Billerica, MA01821, USA) and exposed to films. We used the electrochemiluminescent (ECL) Bio Imaging System (MF ChemiBIS 3.2) to detect antibody binding. All membranes were reported with beta-actin and results were corrected for actin as loading control. The optical density for quantification was obtained using Image J software.
Measurement of Cell Proliferation
Cell Counting Kit-8 (CCK-8) purchased from (Abbkine, Inc. Wuhan Hubei China, KTCO11001), was used to assess cell proliferation according to the manufacturer’s protocol. HK −2 cell lines were seeded into 96-well plates with five duplicate wells in each group. Cells were treated with 5.5mM glucose, 25 mM glucose, and 25 mM glucose plus different concentrations of Emp (50nM, 100 nM, and 500 nM) and incubated for 24 h in a humidified atmosphere of 5% CO2, 95% air, at 37°C incubation and subcultured. After 24 h, CCK-8 solution with serum-free medium 110 ul was added to each well followed by a further 2-h incubation under humidified atmosphere of 5% CO2, 95% air at 37°C. Absorbance was detected at 450 nm by a microplate reader (BioTek China). Absorbance was measured at 24 h, 48 h, and 72 h. Each experiment was repeated 3 times.
Statistical Analysis
Data are reported as mean ± Standard Deviation (SD) of at least three independent experiments. Multiple comparisons among groups were performed by using one-way analysis of variance (ANOVA) with a post –hoc Tukey’s test (GraphPad Prism. Version 8.1.2 (33)). P value <0.05 was considered to indicate statistical significance.
Results
Effect of Empagliflozin on Expression of HIF-1α and GLUT-1
HIF-1α is a cellular transcriptional protein which responds to reduced oxygen levels and responsible for prosurvival genes activation. We examined whether the expression of HIF-1α was altered by HG exposure in HK-2 cultured cells under normoxia. Cells were cultured in the medium containing either 5.5 mM (normal glucose) or 25 mM glucose (high glucose) with or without emp treatment (50 nM, 100 nM, 500 nM) for 72 h. The rate of glucose consumption on incubation in the medium containing 5.5 mM or 25 mM was not detected, although this did not affect the results as the experiment was controlled three times. The results showed that exposure of HK-2 cells to high glucose (HG) under normoxia did not significantly increase expression of HIF-1α protein level compared with NG (HG: 0.56 ± 0.10 vs. NG: 0.33 ± 0.0.08, P > 0.05 95% CI −0.5222 to 0.06726) as demonstrated in Figure 1A, B and D). Addition of emp for 72 h revealed significant increase of HIF-1α protein expression in dose-dependent manner (Emp100nM: 0.64 ± 0.16 vs. HG: 0.56 ± 0.10, P < 0.05, 95% CI −0.5918 to −0.002338) (Emp500nM: 0.86 ± 0.05 vs. 0.56 ± 0.10, P < 0.05, 95% CI, −0.6631 to −0.07367) compared to untreated hyperglycemic HK-2 cells as shown in (Figure 1A, B and D). Based on these results we tested whether expression of GLUT-1 a HIF-1α mediated protein is altered by HG in HK-2 cells. We found that HG exposed HK-2 cells faintly expressed GLUT-1 protein. However, emp treatment for 72 h at 100nM significantly increased GLUT-1 protein expression in hyperglycemic HK-2 cells compared to HG group (Emp100nM: 1.2 ± 0.27 vs. HG: 0.4 ± 0.07, P < 0.05, 95% CI −1.382 to - 0.1471), surprisingly, emp at 500nM did not significantly increase GLUT-1 protein expression as illustrated in (Figure 1A, C and E)
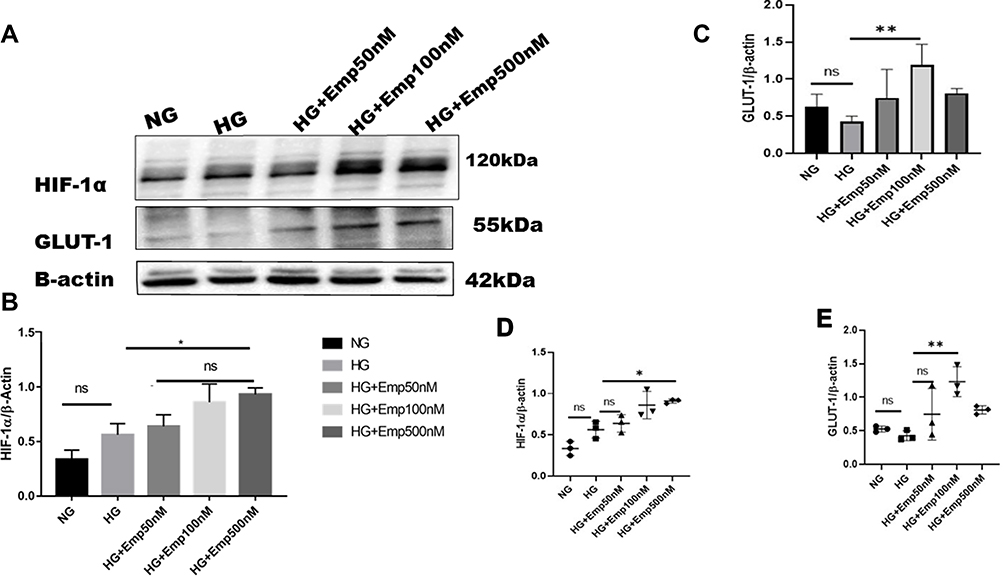 |
Figure 1 Effect of empagliflozin on expression of HIF-1α pathway in HK-2 cell line. HK-2 cells were planted in 5.5mM glucose and 25mM glucose medium with or without empagliflozin treatment under normoxia condition for 72 h. (A) Western blotting was performed using anti-HIF-1α, anti-GLUT-1 and anti-β-actin antibodies. β-actin was used as a loading control. (B, C) Quantification of the Western blot data. (D–E) Dot plots scatter represented individual measurement. The data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. high glucose. Abbreviation: ns, non-significant. |
Effect of Empagliflozin on Sodium Glucose Co-Transporter2
The blockade of SGLT2 the principal glucose transporter in the proximal tubular cells improved glycemic control and prevented hyperglycemia complications. Here we based on the impact of emp on renal proximal tubular cells (HK-2 cell lines). Cells were cultured in the medium containing either 5.5mM (normal glucose) or 25mM (high glucose) with or without emp (50nM, 100nM, and 500nM) for 72 h. We evaluated whether the expression of SGLT2 was affected by high glucose in HK-2 cells. We found that expression of SGLT2 protein level was significantly increased in response to HG compared with NG (HG:1.22 ± 0.03 vs. 0.57 ± 0.13, P < 0.05, 95% CI −1.07 to −0.2156). However, emp treatment for 72 h significantly decreased SGLT2 protein expression in dose-dependent manner (Emp 50nM: 0.74 ± 0.08 vs. HG:1.22 ± 0.03, 95% CI 0.04860 to 0.9110, Emp100nM: 0.49 ± 0.02 vs. HG: 1.22 ± 0.03, 95% CI 0.3045 to 1.167, Emp500nM: 0.46 ± 0.05 vs. HG:1.22 ± 0.03, 95% CI 0.3224 to 1.185, P < 0.05) as demonstrated in (Figure 2A–C).
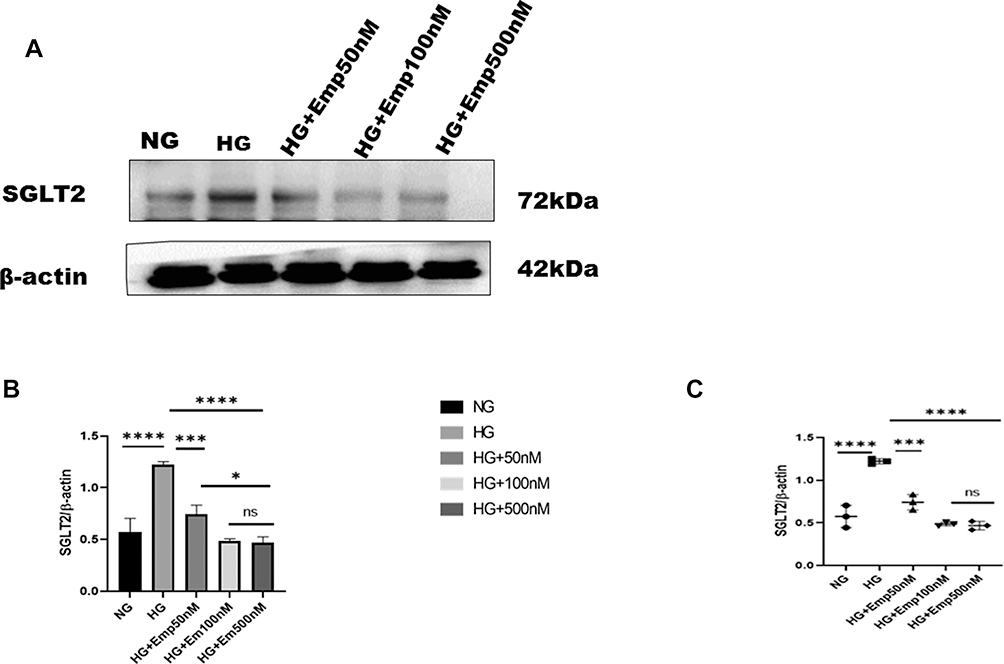 |
Figure 2 Effect of empagliflozin on SGLT2 expression in HK-2 cells. (A) Representative of Western blots illustrating SGLT2 and β-actin expression. HK-2 cells were planted in 5.5mM glucose and 25mM glucose medium with or without empagliflozin treatment for 72 h. Western blotting was performed using anti- SGLT2 and anti-β-actin antibodies. β-actin used as a loading control. (B) Quantification of Western blot data in HK-2 cells. (C) Dot plots scatter represented individual measurement. The data represent the mean ± SD of three independent experiments.*P < 0.05, ***P < 0.001, ****P < 0.0001 vs. high glucose. Abbreviation: ns, non-significant. |
Anti-Fibrotic Effect of Empagliflozin
Overexpression of extracellular matrix proteins is a hallmark of diabetic kidney disease and leads to interstitial fibrosis in HK-2 cells exposed to HG in an attempt to mimic diabetic milieu. The exposure of HK-2 cells to high glucose for 24 h significantly increased the accumulation of extracellular matrix proteins (FN, Col IV), as well as a key profibrotic cytokine TGF-β1. HG exposed HK-2 cells significantly increased expression of extracellular matrix components such as fibronectin (HG:3.23 ± 0.11 vs. NG: 0.4 ± 0.10, P < 0.05) as shown in (Figure 3A, B and E), and Col IV protein level compared to NG (HG: 2.9 ± 0.02 vs. NG:0.5 ± 0.09, P < 0.05) (Figure 3A, C and F). Furthermore, HG exposed HK-2 cells for 24 h revealed marked elevation of TGF-β1 protein expression compared to NG (HG:3.4 ± 0.05 vs. NG: 0.3 ± 0.18, P < 0. 05) as demonstrated in (Figure 3A, D and G). These findings were confirmed using Mannitol (MA) an osmotic agent and a free radical scavenger to mimic hyperglycemia. Mannitol has been reported as a renal protective agent from acute kidney injury. Exposure of HK-2 cells to MA for 24 h did not significantly alter the expression of Col IV neither TGF-β1 as shown in (Figure 3A, C, F and A, D, G) respectively. However, MA exposured HK-2 cells significantly induced expression of FN compared to NG (MA: 0.7 ± 0.05 vs. NG: 0.4 ± 0.10, P < 0.05) as illustrated in (Figure 3A, B and E). Based on these results, we investigated whether the expression of extracellular matrix proteins (Col IV, FN), TGF-β1, as well as P-smad3, was altered by blocking SGLT2 the principal transporter of glucose in the proximal tubular cells. The results showed that HG exposed HK-2 cells for 72 h significantly increased Fibronectin (FN) protein level expression in HG group compared to NG (HG: 1.02 ± 0.22 vs. NG: 0.4 ± 0.09, P < 0.05). Addition of emp treatment for 72 h remarkably decreased expression of FN in hyperglycemic HK-2 cells in dose-dependent manner (Emp50: 0.49 ± 0.16 vs. HG:1.02 ± 0.22, Emp100: 0.47 ± 0.02 vs. HG:1.02 ± 0.22, Emp500: 0.43 ± 0.07 vs. HG:1.02 ± 0.22, P < 0.05).(Figure 4A, B and D). TGF- β1 protein expression was significantly increased in hyperglycemic HK-2 cells compared to NG (HG: 1.4 ± 0.07 vs. NG:0.7 ± 0.07, 95% CI - 0.9395 to - 0.2057, P < 0.05). Emp pretreatment for 72 h significantly decreased expression of TGF-β1 protein induced by HG in HK-2 cells in dose-dependent manner (Emp50nM: 0.83 ± 0.04 vs. HG; 1.36 ± 0.07, Emp100nM: 0.57 ± 0.03 vs. HG:1.36 ± 0.07, Emp500nM: 0.2 ± 0.01 vs. HG: 1.36 ± 0.07, 95% CI 0.2144 to 0.9482, P < 0.05) as illustrated in (Figure 4A, C and E).
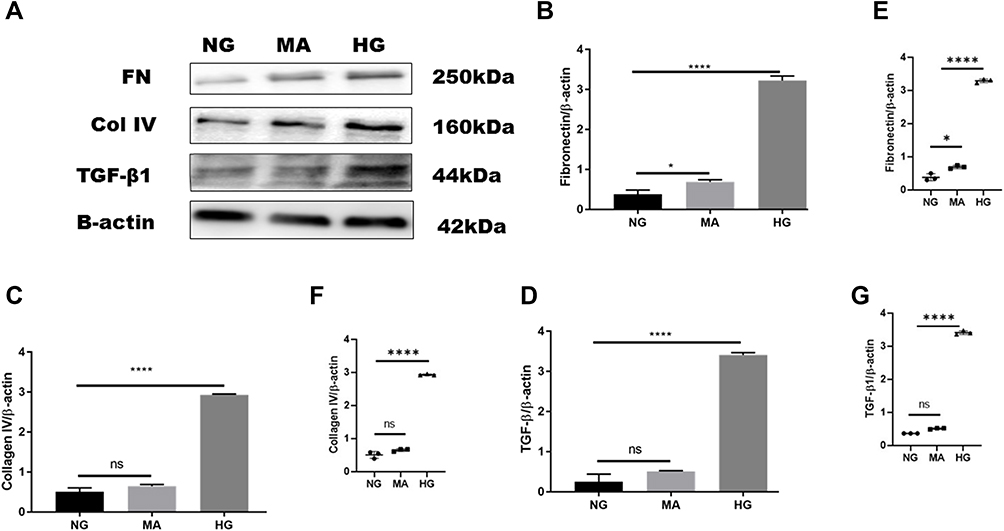 |
Figure 3 Effect of high glucose on expression of proteins that affect fibrosis in HK-2 cell line. HK-2 cells were planted in 5.5mM glucose, 5.5mM glucose plus 20mM mannitol and 25mM glucose for 24 h. (A) Western blotting was performed using anti-fibronectin, anti-collagen IV, anti-TGF-β1, and anti-β-actin antibodies. β-actin used as a loading control. (B–D) Quantification of the Western blot data in HK-2 cells. (E–G) Scatter dot plots represented individual measurement. The data represents the mean ± SEM of three independent experiments. *P < 0.05, ****P < 0.0001 vs. high glucose. Abbreviation: ns, non-significant. |
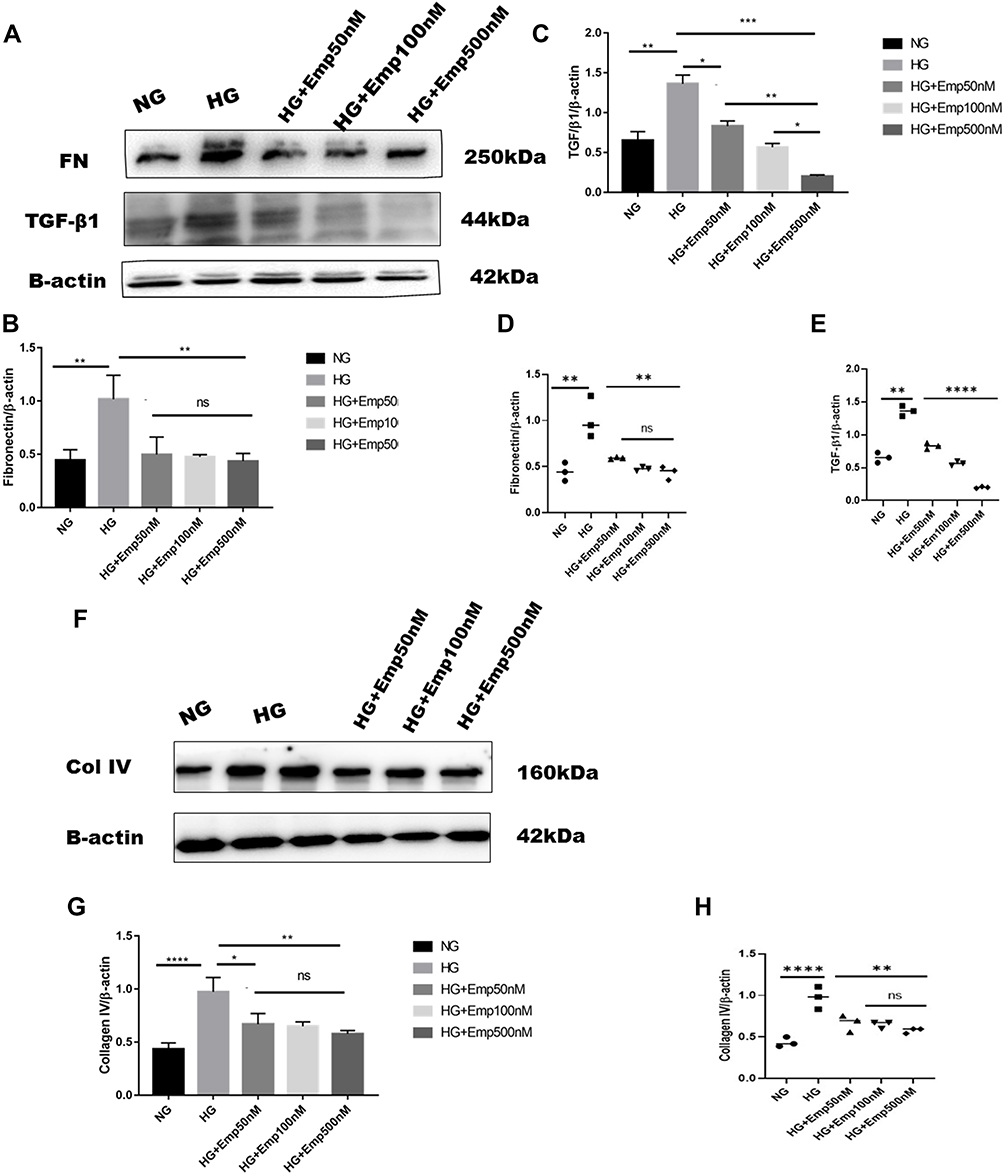 |
Figure 4 Continued. |
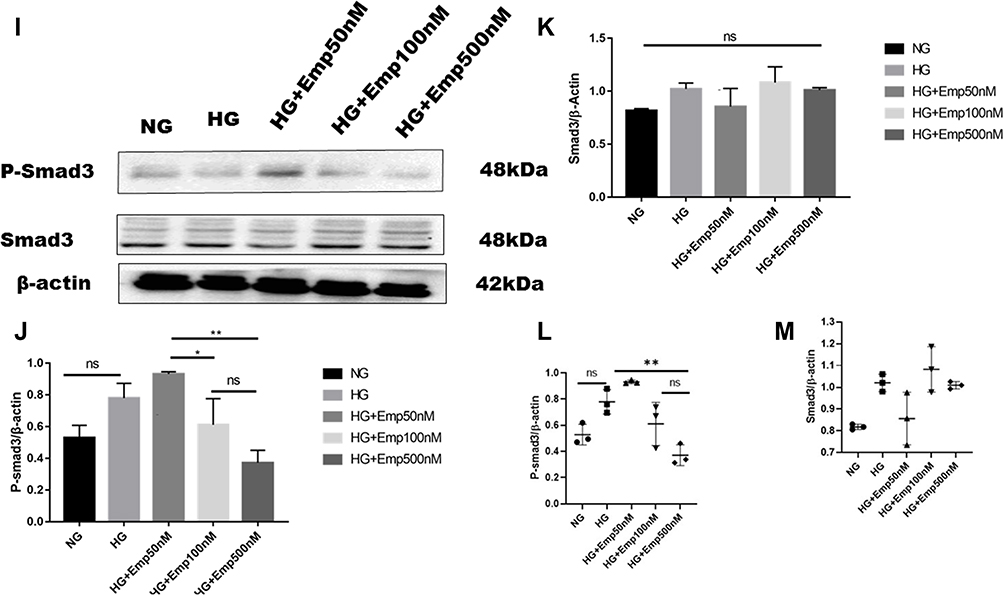 |
Figure 4 Effect of Empagliflozin on the expression of proteins that affects fibrosis in HK-2 cell line. HK-2 cells were planted in 5.5mM glucose and 25mM glucose with or without empagliflozin treatment for 72 h. (A, F, L) Representative of Western blots illustrating fibronectin, TGF-β1, Col IV, P-smad3, Smad3, and β-actin protein expression in HK-2 cells. (B, C, G, J, K) Quantification of Western blot data in HK-2 cells. (D, E, H, L, M) Individual measurement represented as dot plot scatter. β-actin used as a loading control. The data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. high glucose. Abbreviation: ns, non-significant. |
HG increased expression of Collagen IV (Col IV) in HK2 cells compared to NG (HG: 0.97 ± 0.13 vs. NG: 0.44 ± 0.05, P < 0.05). However, empagliflozin pretreatment reduced Col IV protein level expression in dose dependent manner (Emp50nM: 0.7 ± 0.10 vs. HG:0.97 ± 0.13, Emp100nM: 0.6 ± 0.04 vs. HG:0.97 ± 0.13, Emp500nM: 0.57 ± 0.03 vs. HG:0.97 ± 0.13, P < 0.05) as shown in (Figure 4F– H).
Activation of smad3 pathway plays a central role in TGF-β1- induced EMT, and leads to renal fibrosis. Even though the results showed that emp at 50 nM concentration auguments expression of P-smad3 induced by high glucose compared to NG (Emp50nM: 0.83 ± 0.11 vs.NG 0.5 ± 0.07, P < 0.05). However, emp treatment for 72 h at 500 nM concentration significantly decreased expression of P-smad3 protein level induced by high glucose (Emp500nM: 0.4 ± 0.07 vs. HG 0. 8 ± 0.09, Emp100nM: 0.5 ± 0.16 vs.HG: 0.8 ± 0.09, P < 0.05). (Figure 4I, J and L). HG did not show any effect on the expression of Smad3 in HK-2 cells compared to NG (HG: 1.02 ± 0.04 vs. NG: 0.8 ± 0.01, P > 0.05). Morever, the addition of emp did not affect the expression of Smad3 in hyperglycemic HK-2 cells (Emp50nM: 0.9 ± 0.12 vs. HG:1.02 ± 0.04, Emp100: 1.1 ± 0.10 vs. HG:1.02 ± 0.04, Emp500nM: 1.0 ± 0.01 vs. HG:1.02 ± 0.04, P > 0.05) as demonstrated in (Figure 4I, K and M).
Anti-Epithelial–Mesenchymal Transition (EMT) Effect of Empagliflozin
The effect of emp on HK-2 cells exposed to high glucose in an attempt to mimic the diabetic milieu. Exposure of HK-2 cells to HG-induced cellular changes that are characteristic to the epithelial–mesenchymal transition marker, such as induction of α- SMA (alpha-smooth muscle actin) protein expression compared with that of NG (HG:1.12 ± 0.04 vs. NG:0.6 ± 0.00, P < 0.05, 95% CI −1.017 to −0.06735). However, treatment with emp for 72 h remarkably decreased the expression of α-SMA protein level induced by high glucose in HK-2 cells (Emp500nM: 0.6 ± 0.09 vs. HG 1.12 ± 0.04, P < 0.05, 95% CI 0.02286 to 0.9729), as shown in (Figure 5A, B and E) but reversed the decreased expression of epithelial E-cadherin in dose-dependent manner (Emp100nM: 0.80 ± 0.05 vs. HG:0.36 ± 0.06, Emp500nM: 0.96 ± 0.16 vs. HG:0.36 ± 0.06, 95% CI −0.8489 to −0.3667, P < 0.05).(Figure 5A–D)
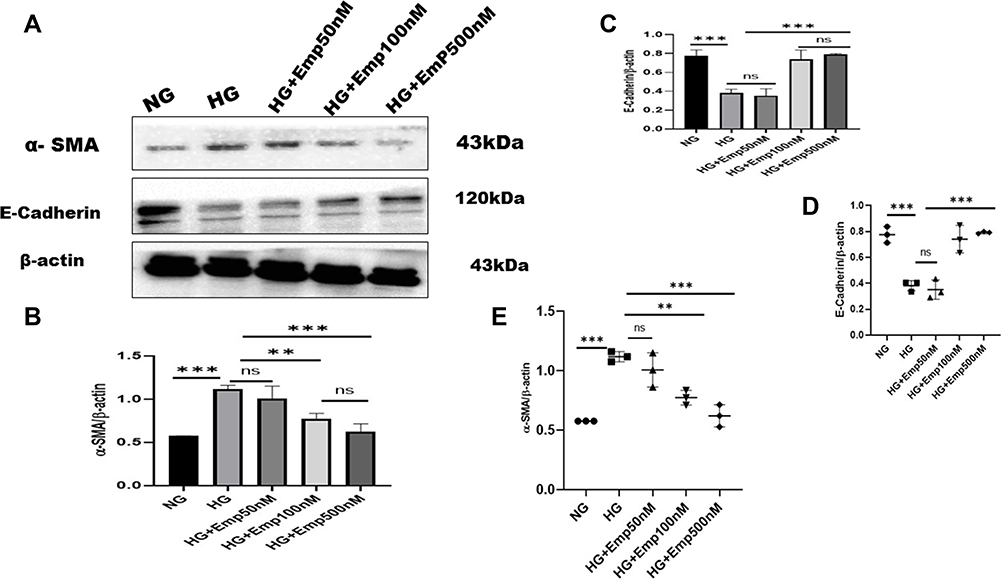 |
Figure 5 Effect of empagliflozin on the expression of proteins related to EMT in HK-2 cell line. HK-2 cells were planted in 5.5mM glucose and 25mM glucose with or without empagliflozin treatment under normoxia for 72 h. (A) Western blotting was performed using anti-α-SMA, anti-E-cadherin, and anti-β-actin antibodies. β-actin used as a loading control. (B-C) Quantification of Western blot data. (D–E) Individual measurements represented as dot plots. The data represent the mean ± SD of three independent experiments., **P < 0.01, ***P < 0.001, vs. high glucose. Abbreviation: ns, non-significant. |
The Anti-Proliferative Effect of Empagliflozin on HK-2 Cell Line
To evaluate whether the renal protective effect of emp is associated with cell hyperproliferation, we assessed cell proliferation by CCK-8 assay. The proliferation of cells was significantly increased in HG exposed HK-2 cells compared to NG (HG:1.23 ± 0.02 vs. NG: 0.53 ± 0.01, P < 0.05) at 24 h, 48 h, and 72 h. However, emp treatment significantly decreased cell proliferation in dose-dependent manner (Emp50nM: 0.78 ± 0.02, Emp100nM: 0.68 ± 0.04 and Emp500nM: 0.48 ± 0.07, P < 0.05) as demonstrated in (Figure 6A). Furthermore, we assessed whether the effect of high glucose on HK-2 cells correlates with time exposure. The results showed that HG significantly increased cell proliferation at 24 h, 48 h, and 72 h but was not significantly increased in a time-dependent manner. Emp treatment significantly decreased cell proliferation at 24 h, 48 h, and 72 h in dose-dependent manner, but was not significantly decreased in time-dependent manner as shown in (Figure 6B).
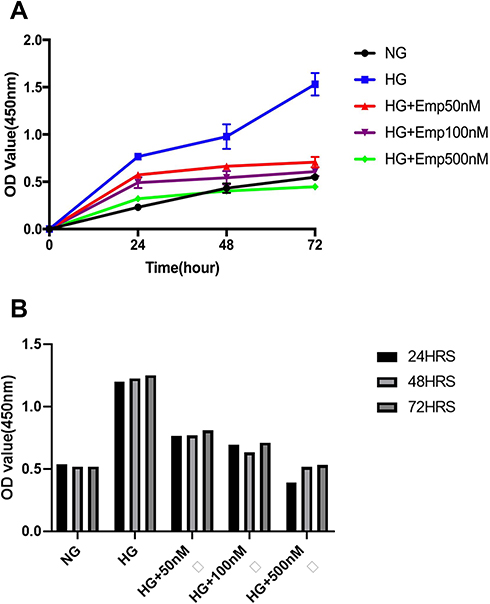 |
Figure 6 The anti-proliferative effect of empagliflozin on HK-2 cell line. Empagliflozin treatment significantly decreased cell proliferation induced by high glucose (P <0.0001). (A) HK-2 cells were planted in 5.5mM glucose and 25mM glucose with or without Empagliflozin treatment for 72 h. CCK-8 assay solution was added to detect cell proliferation. OD values were detected at 24 h, 48 h, and 72 h. (A, B). The data represents the mean ± SD of three independent experiments. |
Discussion
This study aimed to investigate the renoprotective role of emp, an SGLT2 inhibitor on high glucose-induced proximal tubular cell injury. The results clearly showed that emp reduced expression of SGLT2 protein, EMT and fibrosis markers, and increased E-cadherin expression in cultured hyperglycemic HK-2 cells. In addition, emp induced HIF-1α expression and its regulated protein GLUT-1 in normoxia cultured hyperglycemic HK-2 cells. These findings suggest that emp has a direct renoprotective effect on proximal renal tubular cell injury.
HIF-1α is a key protein that plays a major role in modulating the reaction of tissues to hypoxia in many body organs,17,22 but its function reported to be depressed in normoxia diabetic milieu in epithelial tubular cells,16,19 whereas, HIF-1α reported to be induced by high glucose under normoxia in mouse mesangial cells.19 Therefore, stabilization of HIF-1α in hyperglycemia is not clear.19 Our results showed that hyperglycemic HK-2 cells did not increase HIF-1α protein expression compared with normal glucose group which is in agreement with Bessho et al who found faintly expressed HIF-1α protein under normoxia conditions in human renal proximal tubular cells (HRPTECs),27 and we observed the decreased of GLUT-1 protein expression in HK-2 cells exposed to high glucose under normoxia condition. These findings suggest that in spite of profound renal hypoxia, HIF-1α activation in diabetes is of substandard. Although several reports have found that overexpression of HIF-1α protein in tubular epithelial cells contributed to renal fibrosis but its inhibition prevented the progression of renal fibrosis and attenuated diabetic kidney disease,22 Therefore, it is not clear whether HIF-1α play a beneficial or harmful role in the progression of diabetic kidney diseases. A recent study on renal Ischaemia reperfusion injury, dapagliflozin an SGLT2 inhibitor increased expression of HIF-1α protein level, and this was associated with attenuation of ischemic reperfusion kidney injury in proximal tubular cells.28 Luseogliflozin SGLT2 inhibitor treatment reduced expression of HIF-1α in HRPECs exposed to hypoxia which is contrary to our study results was emp significantly increased HIF-1α protein level expression in normoxia cultured hyperglycemic HK-2 cells.27 Although different factors are known to regulate HIF-1, the mechanism underlying emp-induced HIF-1α expression in hyperglycemia is not clear. In addition, our results revealed that emp reduced the expression of extracellular matrix proteins (Col IV, FN), and a key profibrotic cytokine (TGF-β1) that consequently leads to the pathogenesis and development of renal fibrosis of hyperglycemic HK-2 cells in a dose-dependent manner. These results correlate with canagliflozin treatment which showed a significant decrease of TGF-β1 and fibronectin expression in mesangial cells induced by high glucose.30 These findings suggest that emp causes an adaptive reprogramming of stressed cells in a manner that promotes homeostasis and survival. Increased SGLT2 expression increases proximal tubular sodium and glucose reabsorption in early diabetic kidney disease, the increased proximal tubular sodium reabsorption contributes to glomerular hyperfiltration,31 and consequently oxygen deprivation. Hypoxia-inducible factors (HIF-1α and HIF-2α) are lower-energy sensors that may play a crucial role in renal tubular autophagy and thereby attenuates tubulointerstitial damage.32,33 This could explain why the pharmacological induction of HIF signaling can prevent kidney injury and thus a suggestion that the increase of HIF-1α expression contributes to the renoprotective effect of emp. More so, Dimethyloxalyglycine (DMOG) 4 week treatment in rats induced activation of HIF-1α protein, reduced progression of proteinuria, prevented podocyte injury in the remnant kidney model,34 Similarly, activation of HIF-1α by Cobalt chloride prevented diabetic kidney disease by attenuating proteinuria and tubulointerstitial damage.22
Although Luseogliflozin treatment attenuated tubulointerstitial fibrosis, improved cortical tubular oxygenation, and increased AMPK (activated protein kinase) phosphorylation. However, its treatment inhibited HIF-1α expression in HRPTCs and in type 2 diabetic db/db mice,27 thus distinguishing its actions from other SGLT2 inhibitors. This effect may explain why Luseogliflozin at 8 week treatment increased urinary albumin, body weight and blood pressure in diabetic db/db mice.27 In other words, these effects might negate the renoprotective effect of SGLT2 inhibitors as well as the cardiovascular benefits of SGLT2 inhibitors. This could be due to difference in chemical structure and molecular weight of these compounds.
Empagliflozin is an oral hypoglycemic agent that inhibits the SGLT2, a principal transporter in the proximal tubular cells, thus prevent glucose reabsorption and subsequently induces the excretion of glucose via urine.9 Therefore, we tested whether high glucose alters the expression of SGLT2 protein in HK-2 cells. However, it has been reported that no regulation of SGLT2 was found in response to high glucose (30mM D-glucose)3 which could be due to cytotoxicity of hyperglycemia that induced diabetic state. However, our results demonstrated that normoxic hyperglycemic HK-2 cells increased SGLT2 protein level expression. Our results correlate with the findings in kidneys of diabetic rats where emp treatment significantly reduced expression of SGLT2 in dose-dependent manner,35 also canagliflozin, another SGLT2 inhibitor reduced expression of SGLT2 induced by high glucose in both mouse mesangial and renal proximal tubular cells.30 In a similar development, Dapagliflozin an SGLT2 inhibitor reduced proteinuria and ameliorates podocyte dysfunction and loss in mice with protein-over load proteinuria.36 In general, Sglt2 knockout in STZ-diabetic mice reduced glomerular filtration of glucose thereby preventing glomerular hyperfiltration.31 These findings suggest that by blocking SGLT2 a principal transporter as a therapeutic agent improves glycemic control and subsequently reverses hyperglycemia complications. The bulk of filtered glucose is reabsorbed by SGLT2 in the proximal tubular segmental cells. Therefore, we investigated further whether SGLT2 blockade alters the expression of other glucose transporters. From other studies, emp treatment reported to have no alteration on the expression of sodium-glucose co-transporter1 (SGLT1) and glucose transporter-2 (GLUT-2) in the renal proximal tubular epithelial cells3 which are contrary to our study results were emp treatment at 100nM significantly increased expression of GLUT-1 protein, a basal lateral glucose transporter in hyperglycemic HK-2 cells. Also, it is emphasized that emp treatment increased SGLT1 expression in proximal tubular epithelial cells.35 These findings suggest that the decrease of SGLT2 expression may shift transport to downstream segments further from the proximal segment as a compensatory in response to SGLT2 blockade. In this study, emp increased the expression of GLUT-1 protein in hyperglycemic HK-2 cells at 100nM. Emp treatment at 500nM did not significantly increase GLUT-1 expression in HK-2 cells, this could be due to lack of early proximal glucose reabsorption via SGLT2 blockade, indicating the reduction in filtered glucose balanced the inhibition of glucose reabsorption. The lack of SGLT2 enhances luminal glucose delivery to the distal nephron in the absence of hyperglycemia which increases kidney weight that could negate the renoprotective effect of SGLT2 inhibitors.
Apart from SGLT2 inhibitors, HIF-1α induction by hypoxia also has been reported to reduce SGLT2 expression in proximal tubular cells.37 However, it is controversial whether SGLT2 inhibition regulates HIF-1α. From our findings emp significantly increased HIF-1α protein level expression in normoxia hyperglycemic HK-2 cells which is in compliance with another study on dapagliflozin an SGLT2 inhibitor which induced expression of HIF-1α in proximal tubular cells and that was associated with attenuation of renal ischemia reperfusion injury.28 In contrast to other studies on Luseoglifozin treatment which revealed significant decreases of HIF-1α expression in both in vivo and in vitro which was also associated with amelioration of tubulointerstitial fibrosis.27 Considering these outcomes, it seems likely that the increase or decrease of HIF-1α expression by SGLT2 inhibitors does not affect the renoprotective effect of SGLT2 inhibitors. Thus, more studies need to be done to evaluate this issue. Our study findings revealed that emp increased the expression of GLUT-1 a target protein of HIF-1α in hyperglycemic HK-2 cells at 100nM. These findings suggest that emp exerts its renoprotective role through HIF-1α pathway regulation but, the mechanism under which emp regulates HIF-1α still undetermined. SGLT2 inhibition was reported to aggravate renal medulla hypoxia in rat nephron.38 Even though not evaluated in our study, this may be possible, since the bulk of filtered glucose is reabsorbed by SGLT2 in the early segment of the proximal tubular cells. Therefore, SGLT2 inhibition shifts transport to segments further downstream of the proximal tubule. In the outer medulla, there is baseline reduced oxygen tension; therefore, there is a possibility of exacerbating renal medullary tubular hypoxia and subsequently induction of HIF-1α in response to hypoxia.38 However, further analysis needs to be done to determine whether the persistence of HIF-1α expression in response to SGLT2 inhibition has a contribution to the renoprotective effect of SGLT2 inhibitors. Furthermore, it has been reported that dapagliflozin an SGLT2 inhibitor increases HIF-1α expression in normoxic normoglycemic HK-2 cells,23 although not done in our study, it may be possible since SGLT2 inhibition increases ATP consumption,38 which leads to hypoxia and subsequent HIF-1α induction.
Smad3 signaling pathway plays a central role in TGF-β1-induced EMT, its reduced expression and activation improve diabetic nephropathy in diabetic mice. Therefore, we investigated whether there is a possible involvement of SGLT2 inhibitor in Smad3 signaling in the proximal tubular cells. Our results demonstrated that HG exposed HK-2 cells did not increase the expression of P-smad3 to a significant level. Unexpectedly, emp pretreatment at a low concentration of Emp50nM showed to augment the expression of P-smad3 in hyperglycemic HK-2 cells. However, emp treatment at a high concentration of 500nM significantly reduced expression of P-smad3 induced by high glucose so we may hypothesize that at low drug concentration receptors are maximumly activated as compared to high dose. This may explain why high glucose did not significantly increase the expression of P-smad3 in HG exposed HK-2 cells. Our results are in consensus with findings on mesangial cells in diabetic mice where high glucose exposed mesangial cells did not show significant phosphorylation of smad3 compares to Advanced Glycated-End products (AGE),39 also a similar case for podocytic cells in diabetic mice.40 These results suggest that the phosphorylation of smad3 in diabetic state is glucotoxicity dependent. Total smad3 was not regulated by high glucose and its pretreatment with emp in our study, similar to other studies which reported that Smad3 expression between control and db/db mice was nonsignificant.39 These results suggest that changes in smad3 phosphorylation (P-smad3) may not be associated with changes in the total smad3 protein level. Basing on our findings, emp treatment could target SGLT2 and then affect HIF-1α mediated TGF-β1/smad3 phosphorylation thereby attenuating tubulointerstitial fibrosis.
In addition, emp exerted an impact on epithelial–mesenchymal transition (EMT) markers. Our results showed that HG exposed HK-2 cells increased expression of α-SMA, however, emp pretreatment significantly reduced expression α-SMA protein level induced by high glucose in HK-2 cells at 500nM, not only that but also emp treatment significantly reversed the decreased expression of epithelial marker E-catherin in dose-dependent manner. These results were similar with the results found in diabetic mice heart which shows that treatment with emp significantly decreased marked upregulation of α-SMA in heart tissue of diabetic mice,40 dapagliflozin treatment on both in vivo and in vitro studies effectively reduced high glucose-induced over-expression of α-SMA and reversed the downregulation of E-cadherin expression induced by high glucose.41 These findings suggest that Emp may reverse epithelial–mesenchymal transition effect induced by high glucose.
Diabetic kidney disease progression has been attributed to hyperglycemia-induced abnormal growth and function of glomerular and tubular cells. Therefore, we tested whether the renal protective effect of Emp is related to cell proliferation. Emp treatment revealed a significant decrease in cell proliferation induced by high glucose in HK-2 cells at 24 h, 48 h and 72 h. Similarly to Canagliflozin which exerts an antiproliferative effect on mesangial cells in diabetic mice,42 our results correlate with findings on diabetic rats that high glucose induced cell proliferation in renal tubular epithelial cells.43 No significant variation observed among time groups, it seems likely that transient glucotoxicity has a direct effect on cellular function as a response to cell injury. Thus, this result suggests that the renal protective effect of Emp may be related with the control of cell growth and proliferation in Diabetic Kidney disease, as a hallmark feature of the disease progression. Recently, SGLT2 inhibition reported to have a potential protective effect and capability to reduce cancer growth and this might be due to its anti-proliferative effect.44
In recent clinical control trial, emp reduced cardiovascular-associated deaths in type 2 diabetic patients.45 It is possible that the cardiovascular benefit of empagliflozin is via activation of HIF-1α pathway. There might be additional and synergistic direct protective renal effects beyond glucose-lowering effects. Despite the fact that HIF-1α activation plays a major role in the early response to prevent hyperglycemia-induced tissue injury, its deficiency results in a faster progression of diabetic kidney disease15 but prolonged HIF-1α activation may induce tissue fibrosis and the development and progression of chronic kidney disease,21 however, this effect may be counteracted by the antihyperglycemic effect of SGLT2 inhibitors in type 2 diabetic patients.
Limitations of This Study
Our experiment did not validate the role of HIF-1α on the effect of emp. This can be improved through blocking HIF-1α pathway by small interference RNA (siRNA). Also, the expression of HIF-1α mRNA in normal glucose and high glucose condition was not determined. This was due to the epidemic outbreak since January 24, 2020, till now whereby we could not access laboratory. HIF-1α is expressed in both proximal and distal tubule, cortex and medulla but we only assessed the proximal tubule cells to explain the renoprotective effect of SGLT2 inhibitors. Our ongoing study will be done on other cells.
Sample size was small, we propose this study to be done with a larger sample size to validate the role of HIF-1α on the renal protective effect of sodium-glucose transporter2 inhibitors.
Conclusion
In summary, we found that emp treatment reduced SGLT2 expression, induced HIF-1α expression and increased GLUT-1 expression in HK-2 cells cultured in high glucose and also emp reduced expression of TGF-β1, P-Smad3, α-SMA, reversed the downregulation of E-cadherin, inhibited HK-2 cells extracellular matrix proteins (FN, Col IV) accumulation induced by high glucose. Therefore, the induction of HIF-1α by emp might play a role in the protection of hyperglycemia-induced proximal renal tubular cells injury and emp treatment might also be particularly useful in reducing tubulointerstitial fibrosis in diabetes patients. Although our study reports the reno-protective effect of empagliflozin mediated by HIF-1α induction, one should be cautious considering the “dual-edge sword” nature of HIF-1α and its downstream signaling. Therefore, we suggest that future in-depth studies should investigate the causal role of HIF-1α in the experimental design with the application of small interference RNA siRNA/HIF-1α mimic tools.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China (No. 81770724), Chinese National Key Technology R and D Program, Ministry of Science and Technology (Nos. 2017YFC0907601, 2017YFC0907602, and 2017YFC0807603), and Shenyang Science and Technology Bureau (No. RC170172).
Author Contributions
All authors made a substantial contribution to conception and design, acquisition of data, analysis and interpretation of data; took part in drafting the article and revising it; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fioretto P, Zambon A, Rossato M, Busetto L, Vettor R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care. 2016;39(Suppl 2):S165–S171. doi:10.2337/dcS15-3006
2. Gheith O, Farouk N, Nampoory N, Halim MA, Al-Otaibi T. Diabetic kidney disease: world wide difference of prevalence and risk factors. J Nephropharmacol. 2016;5(1):49–56.
3. Panchapakesan U, Pegg K, Gross S, et al. Effects of SGLT2 inhibition in human kidney proximal tubular cells–renoprotection in diabetic nephropathy? PLoS One. 2013;8(2):e54442. doi:10.1371/journal.pone.0054442
4. Cho NH, Shaw JE, Karuranga S, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271–281. doi:10.1016/j.diabres.2018.02.023
5. Ogurtsova K, da Rocha Fernandes JD, Huang Y, et al. IDF diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50. doi:10.1016/j.diabres.2017.03.024
6. Toth-Manikowski S, Atta MG. Diabetic kidney disease: pathophysiology and therapeutic targets. J Diabetes Res. 2015;2015:697010. doi:10.1155/2015/697010
7. Lim A. Diabetic nephropathy - complications and treatment. Int J Nephrol Renovasc Dis. 2014;7:361–381. doi:10.2147/IJNRD.S40172
8. American Diabetes A. Standards of medical care in diabetes–2014. Diabetes Care. 2014;37(Suppl 1):S14–S80.
9. Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia. 2017;60(2):215–225. doi:10.1007/s00125-016-4157-3
10. Tonneijck L, Muskiet MH, Smits MM, et al. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol. 2017;28(4):1023–1039. doi:10.1681/ASN.2016060666
11. Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010;31(1):68–74. doi:10.1159/000256659
12. Layton AT, Vallon V, Edwards A. Modeling oxygen consumption in the proximal tubule: effects of NHE and SGLT2 inhibition. Am J Physiol Renal Physiol. 2015;308(12):F1343–F1357. doi:10.1152/ajprenal.00007.2015
13. Ito M, Tanaka T. The anticipated renoprotective effects of sodium-glucose cotransporter 2 inhibitors. Internal Medx. 2018;57(15):2105–2114. doi:10.2169/internalmedicine.9842-17
14. Ghezzi C, Loo DDF, Wright EM. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia. 2018;61(10):2087–2097. doi:10.1007/s00125-018-4656-5
15. Jiao Y, Jiang H, Lu H, et al. Deficiency of hypoxia inducible factor-1alpha promoted progression of diabetic nephropathy with hypertension. Exp Ther Med. 2018;16(4):3658–3662. doi:10.3892/etm.2018.6621
16. Katavetin P, Miyata T, Inagi R, et al. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J Am Soc Nephrol. 2006;17(5):1405–1413. doi:10.1681/ASN.2005090918
17. Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2(3):336–361. doi:10.1002/wsbm.69
18. Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. 2011;54(8):1946–1956. doi:10.1007/s00125-011-2191-8
19. Isoe T, Makino Y, Mizumoto K, et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 2010;78(1):48–59. doi:10.1038/ki.2010.99
20. Nayak BK, Shanmugasundaram K, Friedrichs WE, et al. HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes. 2016;65(5):1387–1397. doi:10.2337/db15-0519
21. Kabei K, Tateishi Y, Nozaki M, et al. Role of hypoxia-inducible factor-1 in the development of renal fibrosis in mouse obstructed kidney: special references to HIF-1 dependent gene expression of profibrogenic molecules. J Pharmacol Sci. 2018;136(1):31–38. doi:10.1016/j.jphs.2017.12.004
22. Nordquist L, Friederich-Persson M, Fasching A, et al. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J Am Soc Nephrol. 2015;26(2):328–338. doi:10.1681/ASN.2013090990
23. Chang YK, Choi H, Jeong JY, et al. Correction: dapagliflozin, SGLT2 inhibitor, attenuates renal ischemia-reperfusion injury. PLoS One. 2016;11(7):e0160478. doi:10.1371/journal.pone.0160478
24. Zhang YJ, Han SL, Sun XF, et al. Efficacy and safety of empagliflozin for type 2 diabetes mellitus: meta-analysis of randomized controlled trials. Medicine. 2018;97(43):e12843. doi:10.1097/MD.0000000000012843
25. Kalra S. Sodium glucose co-transporter-2 (SGLT2) inhibitors: a review of their basic and clinical pharmacology. Diabetes Ther. 2014;5(2):355–366. doi:10.1007/s13300-014-0089-4
26. Vallon V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med. 2015;66:255–270. doi:10.1146/annurev-med-051013-110046
27. Bessho R, Takiyama Y, Takiyama T, et al. Hypoxia-inducible factor-1alpha is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep. 2019;9(1):14754. doi:10.1038/s41598-019-51343-1
28. Chang Y-K, Choi H, Jeong JY, et al. Dapagliflozin, SGLT2 inhibitor, attenuates renal ischemia-reperfusion injury. PLoS One. 2016;11(7):e0158810. doi:10.1371/journal.pone.0158810
29. Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–2306. doi:10.1056/NEJMoa1811744
30. Maki T, Maeno S, Maeda Y, et al. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: a possible role of SGLT2 in mesangial cells. Sci Rep. 2019;9(1):4703. doi:10.1038/s41598-019-41253-7
31. Vallon V, Gerasimova M, Rose MA, et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic akita mice. Am J Physiol Renal Physiol. 2014;306(2):F194–F204. doi:10.1152/ajprenal.00520.2013
32. Li H, Satriano J, Thomas JL, et al. Interactions between HIF-1alpha and AMPK in the regulation of cellular hypoxia adaptation in chronic kidney disease. Am J Physiol Renal Physiol. 2015;309(5):F414–F428. doi:10.1152/ajprenal.00463.2014
33. Schonenberger MJ, Kovacs WJ. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol. 2015;3:42. doi:10.3389/fcell.2015.00042
34. Song YR, You SJ, Lee YM, et al. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant. 2010;25(1):77–85. doi:10.1093/ndt/gfp454
35. Jin J, Jin L, Luo K, Lim SW, Chung BH, Yang CW. Effect of empagliflozin on tacrolimus-induced pancreas islet dysfunction and renal injury. Am J Transplant. 2017;17(10):2601–2616. doi:10.1111/ajt.14316
36. Cassis P, Locatelli M, Cerullo D, et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight. 2018;3(15). doi:10.1172/jci.insight.98720
37. Zapata-Morales JR, Galicia-Cruz OG, Franco M, Martinez YMF. Hypoxia-inducible factor-1alpha (HIF-1alpha) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J Biol Chem. 2014;289(1):346–357. doi:10.1074/jbc.M113.526814
38. Layton AT, Vallon V, Edwards A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Renal Physiol. 2016;310(11):F1269–1283. doi:10.1152/ajprenal.00543.2015
39. Ono H, Abe H, Sakurai A, et al. Novel interplay between Smad1 and Smad3 phosphorylation via AGE regulates the progression of diabetic nephropathy. Sci Rep. 2018;8(1):10548. doi:10.1038/s41598-018-28439-1
40. Li C, Zhang J, Xue M, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18(1):15. doi:10.1186/s12933-019-0816-2
41. Huang F, Zhao Y, Wang Q, et al. Dapagliflozin attenuates renal tubulointerstitial fibrosis associated with type 1 diabetes by regulating STAT1/TGFbeta1 signaling. Front Endocrinol (Lausanne). 2019;10:441. doi:10.3389/fendo.2019.00441
42. Hou S, Zheng F, Li Y, Gao L, Zhang J. The protective effect of glycyrrhizic acid on renal tubular epithelial cell injury induced by high glucose. Int J Mol Sci. 2014;15(9):15026–15043. doi:10.3390/ijms150915026
43. Inoue MK, Matsunaga Y, Nakatsu Y, et al. Possible involvement of normalized Pin1 expression level and AMPK activation in the molecular mechanisms underlying renal protective effects of SGLT2 inhibitors in mice. Diabetol Metab Syndr. 2019;11:57. doi:10.1186/s13098-019-0454-6
44. Kaji K, Nishimura N, Seki K, et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int J Cancer. 2018;142(8):1712–1722. doi:10.1002/ijc.31193
45. Fitchett D, Inzucchi SE, Cannon CP, et al. Empagliflozin reduced mortality and hospitalization for heart failure across the spectrum of cardiovascular risk in the EMPA-REG OUTCOME trial. Circulation. 2019;139(11):1384–1395. doi:10.1161/CIRCULATIONAHA.118.037778
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.