")
Back to Journals » OncoTargets and Therapy » Volume 13
Emodin Reverses Gemcitabine Resistance of Pancreatic Cancer Cell Lines Through Inhibition of IKKβ/NF-κB Signaling Pathway
Authors Tong H, Huang Z , Chen H, Zhou B, Liao Y , Wang Z
Received 20 March 2020
Accepted for publication 4 August 2020
Published 2 October 2020 Volume 2020:13 Pages 9839—9848
DOI https://doi.org/10.2147/OTT.S253691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Hongfei Tong,1,* Zhen Huang,2,* Hui Chen,1 Bin Zhou,1 Yi Liao,1 Zhaohong Wang1
1Department of Hepatobiliary Surgery, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou City, Zhejiang Province 325027, People’s Republic of China; 2Department of Pediatric Hematology, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou City, Zhejiang Province 325027, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhaohong Wang Department of Hepatobiliary Surgery
The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, 109 Xueyuan West Road, Lucheng District, Wenzhou City, Zhejiang Province 325027, People’s Republic of China
Tel +86-577-88002710
Email [email protected]
Background: Pancreatic cancer is one of the most malignant tumors, and gemcitabine has been considered as the standard treatment and been widely utilized as a first-line drug for advanced pancreatic cancer, but gemcitabine-resistance always occurs after a short period of treatment.
Methods: Two pancreatic cancer cell lines Panc-1 and MIA-PaCa-2 were used as the study subject and their gemcitabine-resistant cells were established. Both drug-resistant cells were divided into four groups: blank, emodin, gemcitabine, and emodin+gemcitabine. Cell viability was detected by MTT assay. Flow cytometry was performed to detect cell apoptosis rate and P-gp function. Quantitative real-time polymerase chain reaction and Western blotting were used to detect Survivin, XIAP, Caspase-9/3, NF-κB p65, IKKβ and IκB-α mRNA/protein expressions, respectively. Electrophoretic mobility shift assay (EMSA) was performed to detect NF-κB binding activity. Rhodamine 123 efflux assay was used to detect P-gp function.
Results: Emodin could inhibit cell activity in all cell lines. Both emodin and gemcitabine can significantly increase the apoptosis rate, and the combination of the two drugs can further significantly increase the apoptosis rate in normal pancreatic cancer cell lines. In both drug-resistant pancreatic cancer cell lines, it can be observed that although gemcitabine can increase the apoptosis rate, the effect of promoting apoptosis is significantly lower than that of emodin; the drug combination can still significantly increase the apoptosis rate on the basis of emodin alone. Emodin can significantly reduce the mRNA and protein expression levels of Survivin, XIAP, NF-κB, and IKKβ, and significantly increase the mRNA and protein expression levels of Caspase-3/9 and IκB-α. Emodin significantly reduced NF-κB activity and emodin significantly promoted P-gp fluorescence intensity from Rhodamine 123 efflux assay.
Conclusion: Emodin inhibits the expression of IKKβ, thereby inhibiting the expression and activity of downstream NF-κB, and inhibits P-gp function at the same time, ultimately achieving the purpose of reversing the drug-resistance of pancreatic cancer cell lines.
Keywords: emodin, pancreatic cancer, gemcitabine, NF-κB, IκB-α
Introduction
Pancreatic cancer is one of the most malignant tumors, and East Asian countries, especially the People's Republic of China, have been the regions with the highest incidence of pancreatic cancer. According to statistics, in 2015, there were 48,960 new pancreatic cancer patients in the People's Republic of China, and the number of deaths was as high as 40,560.1 Pancreatic cancer has a poor prognosis, with a median survival time of less than 6 months and a 5-year survival rate of only 5%.2,3 Although in recent years, the level of pancreatic cancer-related surgery has greatly improved, 80% of patients with pancreatic cancer are diagnosed at an advanced stage that cannot be treated surgically, due to the lack of early clinical symptoms of pancreatic cancer and the high metastatic potential of pancreatic cancer. Most of them are accompanied by distant metastases, so less than 20% of patients are suitable for surgical resection.4,5 Chemotherapy is another important method to treat advanced pancreatic cancer and prolong the survival of patients.6 In the last decade, gemcitabine has been the first-line chemotherapy drug of choice for clinical treatment of advanced pancreatic cancer,7 two combination regimens for metastatic pancreatic cancer have turned out to be gold standards in recent years: 5-fluorouracil (5-FU)/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX), and gemcitabine with nab-paclitaxel since 2011.8 Gemcitabine has become a blockbuster drug for the treatment of advanced pancreatic cancer since it was launched in 1996.9
Unfortunately, patients with pancreatic cancer develop resistance to gemcitabine after a few weeks of administration. The emergence of this acquired resistance has greatly limited the clinical efficacy of gemcitabine.10 More and more evidence indicates that NF-κB may be the most important regulatory molecule among them, not only because of the high expression of NF-κB in most gemcitabine-resistant cell lines, but also because NF-κB, as a widely acting transcription factor, also plays an important transcriptional regulatory role on gemcitabine resistance-related proteins including ENT, HIF1-α.11–13
As the dominant subunit of regulatory kinases in the classic NF-κB signaling pathway, IKKβ plays a huge role in regulating cell proliferation, apoptosis, and metastasis. Therefore, the IKKβ/NF-κB signaling pathway has been one of the most popular anti-tumor targets, and its over-activation has been found in almost all solid tumor cells.14,15
In summary, IKKβ/NF-κB signaling pathway inhibitors have great potential in reversing gemcitabine resistance and are expected to become potential drugs to overcome the clinical problem of gemcitabine resistance. Unfortunately, the related research in this area has not yet attracted enough attention. In addition, currently known inhibitors of IKKβ/NF-κB signaling pathways, such as BMS−345,541, ML−120B, TPCA−1, etc., although they have good inhibitory activity, are also accompanied by strong cytotoxic effects.16–18 Curcumin and its analogs EF24 and EF31 are also toxic, and there are huge defects in drug metabolism.19–21 Therefore, finding a new type of IKKβ/NF-κB inhibitor with high efficiency and low toxicity, and exploring if it can reverse the resistance of gemcitabine has become our main research goal.
Our previous research screened a large number of active monomers from natural products, and successfully found that the natural product emodin had good anti-pancreatic cancer activity. Numerous papers have also reported that emodin had a significant inhibitory effect on a variety of solid tumors including gastric, breast, lung, liver, and pancreatic cancer.22–24 Unfortunately, the molecular target of emodin has not been confirmed, which greatly limits the clinical application prospect of using emodin as an antitumor drug.
In previous studies, we found that emodin showed synergistic antitumor effects with gemcitabine in vivo and in vitro. However, the specific effect and detailed mechanism of its resistance reversal need to be further studied. Based on the results reported in the existing literature and our preliminary basis, we propose the hypothesis that the reversal effect of emodin on gemcitabine resistance in pancreatic cancer cells is exerted by inhibiting IKKβ, then we will further verify the correlation between abnormal activation of IKKβ and gemcitabine resistance in pancreatic cancer.
Materials and Methods
Reagents and Drugs
Emodin (Sigma, USA) was dissolved in DMSO (dimethylsulfoxide), the concentration of the solution was 0.2 mmol/L, and it was stored at −20°C. Gemcitabine (Ely Lilly, Germany) was dissolved in sterile saline, the concentration of the solution was 50 g/L. FBS (fetal bovine serum) and DMEM were purchased from Gibco (USA).
Cell Culture
Panc-1 and MIA-PaCa-2 (human pancreatic cancer cell lines) were obtained from ATCC (USA). Cells were cultured in RPMI-1640 purchased from Gibco, then they were supplemented with 10% FBS (Gibco, USA), and were kept in 5% CO2 + 37°C incubator.
Resistant Cell Lines Panc-1/Gem and MIA-PaCa-2/Gem Establishment
Gemcitabine-resistant cell lines Panc-1/Gem and MIA-PaCa-2/Gem were derived from the human pancreatic cancer cell lines Panc-1 and MIA-PaCa-2 by exposing the cells to intermittently increasing concentrations of gemcitabine. After a week, cell death was observed, therefore, median lethal dose (LD80) was chosen as the initial concentration to cultivate the resistant cell line for 72 hours. Cells were passaged twice and cultivated with a double LD80 concentration of gemcitabine when cells entered the logarithmic growth phase. The process lasted 10 months, there were a total of nine concentration gradients.
Cell Grouping
Both cell lines were divided into four groups, namely the blank group, the emodin group, the gemcitabine group, and the emodin + gemcitabine group. The blank group was treated with normal saline, the emodin group was treated with 40 μM concentration of emodin, the gemcitabine group was treated with 20 μM concentration of gemcitabine, and the emodin + gemcitabine group was the combination of two drugs.
Inhibition Effect of Emodin at Different Concentrations on Cell Proliferation
Cells in logarithmic phase were cultured in 96-well plates (4x103/well) in an incubator with 5% CO2 at 37°C overnight. Emodin with a concentration of 10, 20, 40, 80 and 160 μM was administered to cells for 72 hours (n = 6). In addition, a group of series was set: emodin of 40 μM alone, gemcitabine of 20 μM alone, emodin + gemcitabine (combination) and a control group (blank) with only normal saline (n = 6). incubation was continued at 37°C for 72 hours, then 20μL MTT solution (5g/L) was added to each well, and followed by incubating in a 37°C 5% CO2 incubator for 4 hours. Next 200 μL of standard concentration of DMSO was added to each well, mixed by shaking for 15 minutes, and then the absorbance value was measurd with a microplate reader at 490 nm wavelength. Cell viability = Absorbance of test group/Absorbance of Control group × 100%.
Flow Cytometry
The cells in each group were counted after resuspending in medium. About 1×106 cells were moved into a centrifuge tube and centrifuged at 1000 r/min for 5 minutes before discarding the supernatant, adding 1 mL of PBS solution, and gently shaking the tubes to suspend the cells. The cells were then centrifuged at 1000r/min for 5 minutes before discarding the supernatant and repeating 2 times. Next, 500 μL of binding buffer was added to the cells, and gently shaken to suspend the cells, before adding 5 μL of Annexin V-FITC and mixing gently. The cells were kept at room temperature for 15 minutes in the dark, after which 300 μL of binding buffer and 10 μL of PI were added and mixed gently. After incubating the cells at room temperature in the dark for 5 minutes, the data was obtained through Cell Quest software.
Western Blot
The cells in each group were extracted according to the instructions of the protein extraction kit (Beyotime, the People's Republic of China), and the total protein was measured using the BCA kit. The extracted protein samples were electrophoresed in SDS-PAGE, and transferred to nitrocellulose membrane, then samples were closed with 5% skimmed milk for 2 hours. Add the appropriate concentration of primary antibody anti-survivin, anti-XIAP, anti-Caspase-9, anti-Caspase-3, anti- IKKβ, anti-NF-κB p65, anti- IκB-α primary antibodies (all purchased from Abcam) overnight at 4°C, the internal reference was β-actin. After washing the membrane, the secondary antibody was incubated for 2 hours. After color exposure, protein bands were obtained. Image Lab software was used to analyze the gray value of the protein bands and the relative expression of the protein. The experiment was repeated 3 times.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, USA). Applied Biosystems 7300 Fast Real-Time PCR system was used. Primers were specifically designed by Applied Biosystems Primer Express 3.0 (Thermo Fisher Scientific, USA). Specific primers were fixed using the BLAST program. Each 20 μL reaction contained 1×SYBR® Premix Ex Taq™ II, 10 μM forward and reverse primers, 0.4 μL ROX reference dye and 2 μL cDNA. The primer sequences are provided in Table 1. ABI 7300 sequencer reaction conditions are: 94°C 5 minutes, 94°C 40 seconds × 40 cycles, 60°C 40 seconds. The relative quantification (RQ) of target gene expression is calculated by the 2−ΔΔCT method. GAPDH was the internal reference. The PCR reaction was repeated 3 times for each sample.
 |
Table 1 Primer Sequences for RT-PCR |
Electrophoretic Mobility Shift Assay (EMSA)
The DNA probes used in this experiment were amplified and recovered by PCR technology. The amplification primer was introduced with 5ʹbiotin label during synthesis for subsequent detection. The operation of the EMSA test was based on the instructions of the LightShift® Chemiluminescent EMSA Kit (Thermo Fisher Scientific). The protein, probe and reaction buffer were completed in a 20 μL reaction system. During the test, the reaction system was incubated at room temperature for 20 minutes. Then staining agent was added to it and it was placed in 6.5% non-denaturing gel, and run in 0.5×TBE buffer for 1 hour. After electrophoresis was completed, nylon membrane was used for transfer, cross-linking, and color development. The chemiluminescence imager was used to observe the binding of NF-κB protein to the target gene probe.
Rhodamine123 Efflux Assay of P-gp Function in Panc-1/Gem and MIA-PaCa-2/Gem Cells (Flow Cytometry)
Rhodamine 123 staining solution with 10 μL/mL (Changchun Keygen Biological Products Co., Ltd., the People's Republic of China) was added to the culture media, then cells were cultured in 5% CO2 at 37°C for 30 minutes and then centrifuged at 2000 r/min for 5 minutes and washed twice, resuspended and incubated for 2 hours. Finally, the cells were centrifuged twice, washed twice with PBS and the fluorescence intensity was measured by flow cytometry at 488/530 nm.
Synergy Assessment
Growth inhibition following combinations of gemcitabine with and without emodin was performed (emodin at 10, 20, 40, 80 and 160 μM, gemcitabine 20 μM) in Panc-1 and MIA-PaCa-2 cell lines. After 72 hours, cell viability was quantified using CellTiter-Glo Luminescent Cell Viability Assay (Promega). Growth was normalized to wells treated with DMSO vehicle (100% viability) and bortezomib (0% viability). Synergy was scored using Bliss independence modelling as described below.
Statistical Analysis
Data analysis were performed by SPSS 21.0 software. Data were expressed as mean ± standard deviation. The t-test was performed for comparisons between the two groups, One-way analysis of variance was applied for comparisons among multiple groups. Statistical significance was assumed as P < 0.05.
Bliss independence between the drug responses to combinations of the two drugs equalled the sum of the two fractional responses of individual drugs minus their product, where (Fa + Fb) – (Fa × Fb). ΔBliss is zero for a given dose combination when there is no overall synergy or antagonism, while negative ΔBliss values indicate synergy at that combination. Overall synergy score for a drug combination was calculated by ΣΔBliss.
Results
Emodin Inhibits Pancreatic Cancer Cell Growth
Stable gemcitabine-resistant cell lines Panc-1/Gem and MIA-PaCa-2/Gem were generated from their parental cell lines Panc-1 and MIA-PaCa-2 by exposing them to intermittently increasing concentrations of gemcitabine for 10 months. Their IC50 for gemcitabine increased significantly compared to their parental cell lines (Panc-1: 0.201 μM vs Panc-1/Gem: 359.9 μM; MIA-PaCa-2: 0.087 μM vs MIA-PaCa-2/Gem: 570.6 μM, Figure 1A and B). Different concentrations of emodin were applied to Panc-1, Panc-1/Gem, MIA-PaCa-2, and MIA-PaCa-2/Gem cell lines. The results are shown in Figure 1. Emodin could inhibit cell viability in all cell lines, and the higher the dose, the stronger the inhibitory effect (Figure 1C and D). Emodin’s inhibitory effect was statistically significant starting from a concentration of 40 μM, P<0.05, so our subsequent experiments were carried out at a concentration of 40 μM. Furthermore, drug effect was detected on gemcitabine-resistant cell lines and their parental cell lines, results (Figure 1E) showed that emodin alone could significantly reduce cell viability compared to the blank group after 72 hours of drug exposure, P< 0.05, while gemcitabine had no significant effect on gemcitabine-resistant cells, P>0.05. Growth inhibition over 72 hours was quantified after gemcitabine alone and in combination with emodin at different concentrations by CellTiter-Glo, the ΔBliss analysis was −0.647, demonstrating that emodin had a synergic effect with gemcitabine.
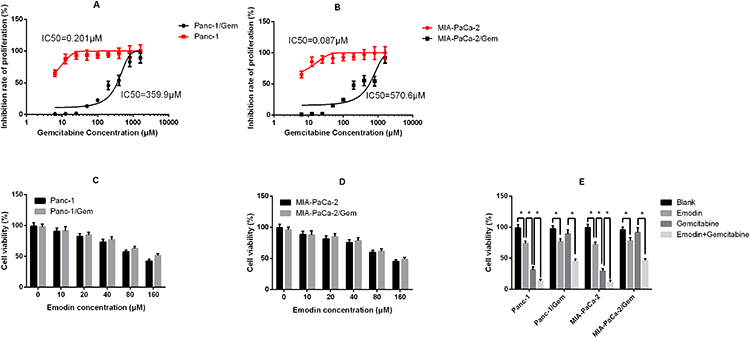 |
Figure 1 (A) Inhibition rate of proliferation of Panc-1 (parental cell line) and Panc-1/Gem (gemcitabine-.resistant cell line); (B) Inhibition rate of proliferation of MIA-PaCa-2 (parental cell line) and MIA-PaCa-2/Gem (gemcitabine-resistant cell line). (C) Cell viability of Panc-1 and Panc-1/Gem cells after 72 hours of emodin application at different concentrations detected by MTT assay. (D) Cell viability of MIA-PaCa-2 and MIA-PaCa-2/Gem cells after 72 hours of emodin application at different concentrations detected by MTT assay. (E) Cell viability of all cell lines after 72 hours of treatment from all groups (emodin 40 μM, gemcitabine 20 μM). *P< 0.05. |
Emodin Promotes Cell Apoptosis of Pancreatic Cancer Cell Lines
It can be observed in two normal pancreatic cancer cell lines (Figure 2) that: compared to the blank group, both emodin and gemcitabine can significantly increase the apoptosis rate, and the combination of the two drugs can further significantly increase the apoptosis rate after 72 hours of drug exposure, all P< 0.05. There was no statistically significant difference in the apoptosis rate between emodin and gemcitabine groups, P>0.05;.On the other hand, in both drug-resistant pancreatic cancer cell lines, it can be observed that although gemcitabine can increase the apoptosis rate, the effect of promoting apoptosis is significantly lower than that of emodin, P<0.05. The drug combination can still significantly increase the apoptosis rate on the basis of emodin alone, P<0.05.
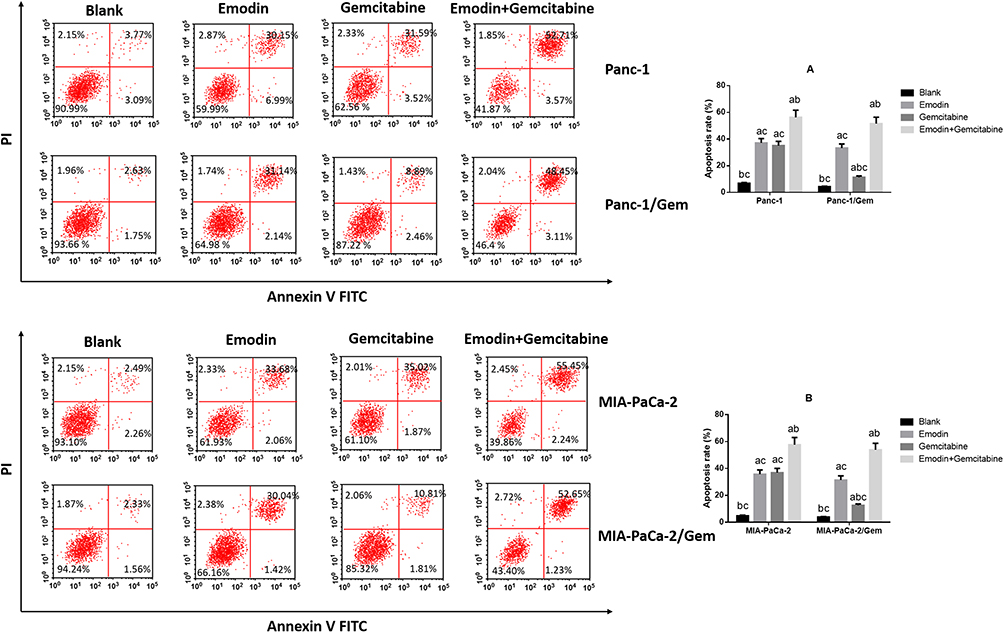 |
Figure 2 Cell apoptosis was detected by Flow cytometry. (A) Emodin’s effect on cell apoptosis of Panc-1 and Panc-1/Gem cells. (B) Emodin’s effect on cell apoptosis of MIA-PaCa-2 and MIA-PaCa-2/Gem cells. aP<0.05 compared to the blank group, bP<0.05 compared to emodin group, cP<0.05 compared to the gemcitabine group. |
Emodin Reduces NF-κB and IKKβ Expressions
The qRT-PCR result showed that NF-κB p65 and IKKβ mRNA expressions were significantly up-regulated in parental cell lines (Panc-1 and MIA-PaCa-2), while IκB-α mRNA expression was significantly down-regulated (Supplementary figure 2E and F), suggesting that NF-κB pathway was more active in gemcitabine-resistant cell lines. It was observed in two drug-resistant pancreatic cancer cell lines (Figure 3 and Supplementary figure 2) that after 72 hours of drug exposure, the use of gemcitabine did not significantly change the mRNA and protein expression levels of survivin, XIAP, NF-κB, IKKβ and IκB-α, all P>0.05, while emodin can significantly reduce the mRNA and protein expression levels of survivin, XIAP, NF-κB and IKKβ, and significantly increase the mRNA and protein expression levels of caspase-3/9 and IκB-α, all P<0.05; The combination of drugs can significantly expand this change on the basis of emodin alone: the mRNA and protein expression levels of survivin, XIAP, NF-κB and IKKβ were significantly reduced, and the mRNA and protein expression levels of caspase-3/9 and IκB-α were significantly increased, all P<0.05. Meanwhile, the NF-κB, IKKβ and IκB-α expressions in the parental cell lines shared a similar tendency (Supplementary figure 1A–D), showing that emodin can have an effect in both parental and drug-resistant cell lines.
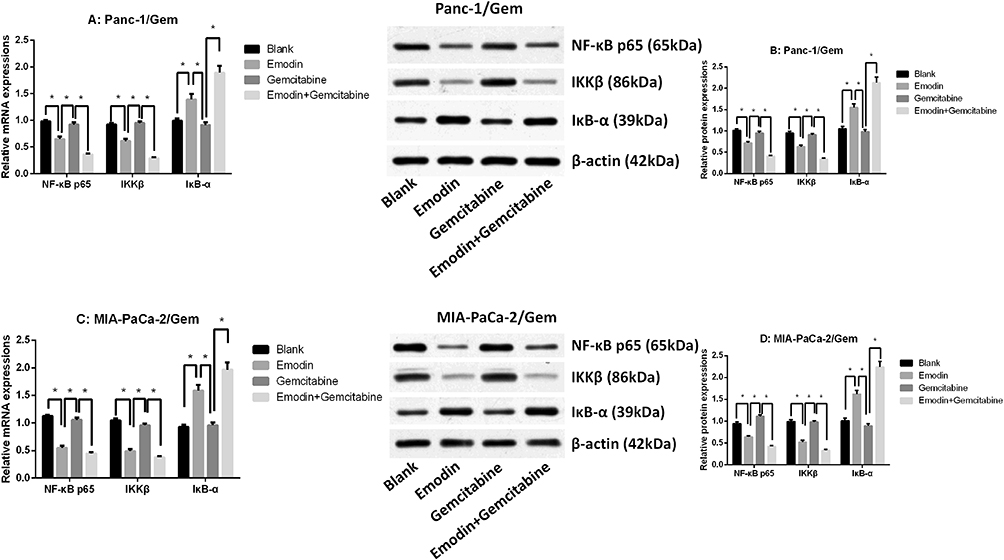 |
Figure 3 (A) mRNA expressions of NF-κB p65, IKKβ and IκB-α of Panc-1/Gem cell. (B) mRNA expressions of NF-κB p65, IKKβ and IκB-α of MIA-PaCa-2/Gem cell. (C) Protein expressions of NF-κB p65, IKKβ and IκB-α of Panc-1/Gem cell. (D) Protein expressions of NF-κB p65, IKKβ and IκB-α of MIA-PaCa-2/Gem cell. *P<0.05. |
Emodin Reduces NF-κB Activity
NF-κB DNA binding activity of nuclear extracts were detected by EMSA. As shown in Figure 4, in both Panc-1/Gem and MIA-PaCa-2/Gem cell lines, compared with the blank group, emodin significantly reduced NF-κB activity, P<0.05, while gemcitabine significantly increased NF-κB activity. Additionally, emodin combined with gemcitabine could still significantly decrease NF-κB DNA-binding activity on the basis of emodin alone, P<0.05.
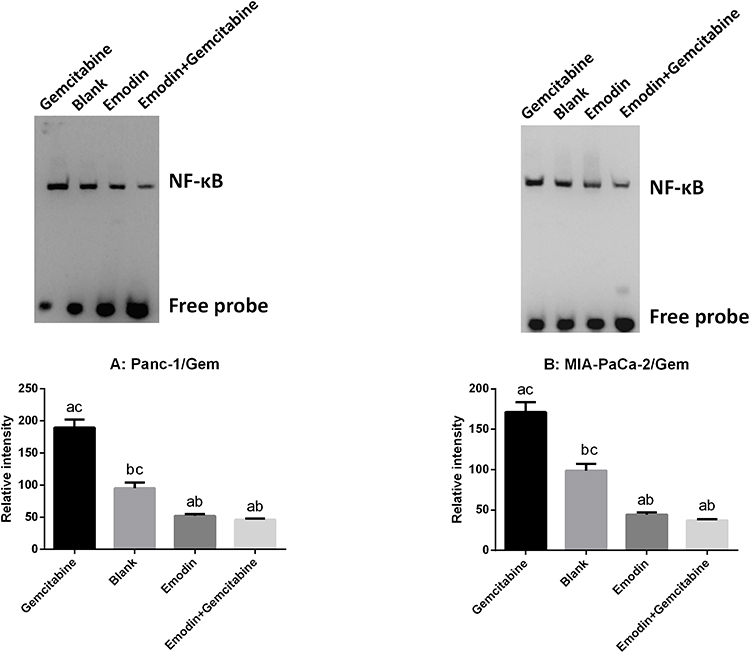 |
Figure 4 (A) Emodin’s effect on NF-κB and its downstream signaling protein expressions in Panc-1/Gem cell detected by Western Blot. (B) Emodin’s effect on NF-κB and its downstream signaling protein expressions in MIA-PaCa-2/Gem cell detected by Western Blot. aP<0.05 compared to the blank group, bP<0.05 compared to the emodin group, cP<0.05 compared to the gemcitabine group, dP<0.05 compared to the emodin+gemcitabine group. |
Emodin Reduces P-gp Function
P-glycoprotein (P-gp), also called ABCB1 (ATP-ABC subfamily B member 1) or MDR1 (for multiple drug resistance), plays an important role in development of drug resistance. Rhodamine 123 (Rh123) is a fluorescent dye which locates in the mitochondria of cells. It is a substrate for P-glycoprotein (P-gp) and can, therefore, be used as a molecular probe in studies of the multidrug resistance (MDR) phenotype. Flow cytometry was used to measure rhodamine 123 efflux in drug-resistant pancreatic cancer cell lines. As shown in Figure 5, after 72 hours of drug exposure, in both Panc-1/Gem and MIA-PaCa-2/Gem cell lines, compared with the blank group, emodin significantly promoted fluorescence intensity, P<0.05, while gemcitabine had no significant effect on it, P>0.05. Emodin combined with gemcitabine could also significantly increase the fluorescence intensity, P<0.05, but compared to emodin alone, the difference was not significant, P>0.05.
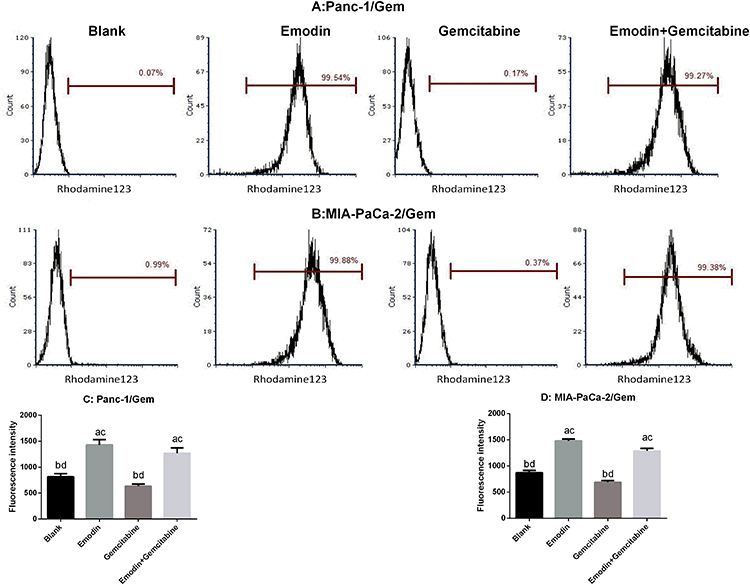 |
Figure 5 (A and C) Emodin’s effect on P-gp function (presented as fluorescence intensity) in Panc-1/Gem cell detected by flow cytometry. (B and D) Emodin’s effect on P-gp function (presented as fluorescence intensity) in MIA-PaCa-2/Gem cell detected by flow cytometry. aP<0.05 compared to the blank group, bP<0.05 compared to the emodin group, cP<0.05 compared to the gemcitabine group, dP<0.05 compared to the emodin+gemcitabine group. |
Discussion
Resistance to anticancer drugs is the primary cause of the failure of chemotherapy for malignant tumors. It includes both intrinsic resistance and acquired resistance. Acquired resistance is the result of natural selection by anticancer drugs, it consists of a gradually developed resistance in an originally sensitive cancer during anticancer drug therapy, and it occurs in almost half of all cancer patients.25 Gemcitabine-resistant pancreatic cancer is one of the acquired resistances occurring in pancreatic cancer patients.
Emodin is a polyhydroxyanthraquinone compound extracted from the dried rhizomes and roots of Polygonum cuspidatum and rhizomes of Rhubarb palmatum.26 Pharmacological researches show that emodin not only has various pharmacological effects such as antibacterial and fungal infections, anti-inflammatory, antioxidant, anti-allergic, etc, but also can regulate the expression of various cancer-related proteins such as Akt/mTOR, NF-κB, HIF1α, and STAT3 to achieve the effects of inhibiting cell proliferation and migration, promoting cell apoptosis, and inhibiting multidrug resistance in a variety of malignant tumors.23,27,28 In 2013, Zhang et al29 found that the combined use of emodin and gemcitabine significantly reduced the expression of NF-κB, XIAP and survivin in pancreatic cancer cells, thereby making BxPC-3 cells that were originally resistant to gemcitabine have restored sensitivity to gemcitabine.29 Unfortunately, the molecular target of emodin has not been confirmed, which greatly limits the clinical application prospect of emodin as an antitumor drug. In order to determine the target of emodin, we performed computer target prediction experiments on emodin. Through reverse virtual screening of the drugable site database (sc-PDB), which allowed one-to-one docking of emodin with the protein in the data by molecular docking technology, and sorting by docking to select the most potential target. We found that IKKβ, which is extremely important in the NF-κB signaling pathway, had a higher order. Further structural analysis showed that emodin bound to the allosteric site of the junction of the IKKβ kinase C-terminal and ubiquitination domain. Based on this finding, we conducted this study, confirming our original hypothesis that emodin down-regulated the expression of NF-κB in drug-resistant cells most likely caused by inhibiting the activity of its upstream kinase IKKβ.
The IκB kinase (IKK) complex includes the catalytic subunits IKKα and IKKβ and the regulatory subunits NEMO/IKKγ. IKK plays an important role in the activation of the NF-κB signaling pathway.30 IKKβ is the main catalytic subunit of the IKK complex and is required for inflammatory mediators to activate NF-κB. When exposed to external stimuli such as TNF-α, inflammatory factors, ultraviolet light, and lipopolysaccharide, cytokines bind to TNF receptors on the surface of cell membranes, and then TNF receptors multimerize and interact with TRADD in the cytoplasm.31 TRADD recruits TRAF and RIP, which passes the signal to IKK. IKK activation phosphorylates Ser32 and Ser36 residues of the alpha subunit of IκB and Ser19 and Ser23 residues of the beta subunit. IκB was then released from the p50/p65/IκB heterotrimer, which was degraded by proteasome after ubiquitination. Furthermore, NF-κB was able to expose its nuclear localization signals (NLS), quickly enter the nucleus from the cytoplasm, and combine with specific sequences on nuclear DNA to initiate or enhance the transcription of related genes and participate in the regulation of inflammation, infection and immune response.32
IκB is an inhibitor of NF-κB, including p100, p105, IκBα, IκBβ, IκBγ, IκBε, Bcl-3, and IκB-R. Most IκBs contain 3–8 ankyrin repeat motifs at the C-terminus and bind to NF-κB through this motif, covering the nuclear localization sequences (NLS) of the Rel homology domain, inhibiting NF- κB activity.33 The N-terminus of IκB is a signal response region, which contains serine phosphorylation and ubiquitination sites, and plays an important role in the induced degradation of IκB. Normally, NF-κB p65, p50 in the cytoplasm and inhibitory protein IκB combine to form a trimer complex, and NF-κB exists in a latent state.32,34 In this study, we found that expression of NF-κB was down-regulated in pancreatic cancer-resistant cell lines under the action of emodin, which is consistent with previous studies. At the same time, we also found that the expression level of IKKβ was also significantly down-regulated, which was consistent with the trend of NF-κB. In contrast, IκB-α expression was up-regulated. These results further confirmed that emodin regulates IKKβ to achieve the purpose of regulating NF-κB, thereby restoring the chemotherapy sensitivity of pancreatic cancer cells. Furthermore, we are still curious to know if emodin can act on cell lines which also have high levels of NF-κB (such as many other cancer cell lines).
Studies have confirmed that chemotherapy resistance of cancer cells may also be caused by multi-drug resistance gene (MDR-1) and its encoded protein P-gp.35 Our previous studies have also confirmed that emodin affected the expression of MRP and P-gp expression levels. P-gp, encoded by MDR, is a transmembrane protein and belongs to the ATP-binding cassette transporter protein superfamily.36 It acts as an ATP-dependent drug efflux pump and has been shown to reduce the concentration of intracellular chemotherapeutic agents. MDR-1/P-gp plays a very important role in the process of tumor chemotherapy resistance.35,37 In this study, rhodamine 123 efflux experiments demonstrated that P-gp function was significantly reduced under the action of emodin.
Considering that the relationship between the expression level of apoptosis-related proteins and emodin has been investigated in our previous research, we did not expand the apoptosis part in this study. Our future research will focus on exploring the specific molecular mechanism by which emodin affects the proliferation and apoptosis of pancreatic cancer cells, and try to knock out or overexpress the IKKβ gene through gene editing technology to observe the effect of this operation on chemotherapy resistance of pancreatic cancer.
In summary, our research confirms that emodin inhibits the expression of IKKβ, thereby inhibiting the expression and activity of downstream NF-κB, and inhibits P-gp function at the same time, ultimately achieving the purpose of reversing the drug-resistance of pancreatic cancer cell lines. The combination of gemcitabine and emodin may improve the efficacy of pancreatic cancer patients, reduce the possibility of drug resistance, and further extend the survival of patients with pancreatic cancer.
Acknowledgments
This work was supported by the Natural Science Foundation of Zhejiang project (LQ18H290003 and Y19H290009) and Wenzhou Science and Technology Bureau planning project (Y20190199, Y20160143).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21(26):7988–8003. doi:10.3748/wjg.v21.i26.7988
2. Liu Y, Shi S, Hua J, et al. Differentiation of solid-pseudopapillary tumors of the pancreas from pancreatic neuroendocrine tumors by using endoscopic ultrasound. Clin Res Hepatol Gastroenterol. 2020. doi:10.1016/j.clinre.2020.02.002
3. Moore A, Donahue T. Pancreatic cancer. JAMA. 2019;322(14):1426. doi:10.1001/jama.2019.14699.
4. Fan Z, Luo G, Gong Y, et al. Prognostic value of the C-Reactive protein/lymphocyte ratio in pancreatic cancer. Ann Surg Oncol. 2020. doi:10.1245/s10434-020-08301-3
5. van der Sijde F, Vietsch EE, Mustafa DAM, Li Y, van Eijck CHJ. Serum miR-338-3p and miR-199b-5p are associated with the absolute neutrophil count in patients with resectable pancreatic cancer. Clin Chim Acta. 2020;505:183–189. doi:10.1016/j.cca.2020.03.005
6. Bulle A, Dekervel J, Deschuttere L, et al. Gemcitabine recruits M2-type tumor-associated macrophages into the stroma of pancreatic cancer. Transl Oncol. 2020;13(3):100743. doi:10.1016/j.tranon.2020.01.004
7. Kim SY, Choi M, Hwang HK, Rho SY, Lee WJ, Kang CM. Intraoperative transfusion is independently associated with a worse prognosis in resected pancreatic cancer-a retrospective cohort analysis. J Clin Med. 2020;9:3. doi:10.3390/jcm9030689
8. Zeng Z, Pottler P, Lan L, Grutzmann G, Pilarsky P, Yang Y. Chemoresistance in pancreatic cancer.Int J Mol Sci. 2019;20(18):4504. doi:10.3390/ijms20184504
9. Okuyama H, Ikeda M, Okusaka T, et al. A Phase II trial of everolimus in patients with advanced pancreatic neuroendocrine carcinoma refractory or intolerant to platinum-containing chemotherapy (NECTOR trial). Neuroendocrinology. 2020. doi:10.1159/000505550
10. Thompson BJ. YAP/TAZ: drivers of tumor growth, metastasis, and resistance to therapy. Bioessays. 2020;42(5):e1900162. doi:10.1002/bies.201900162
11. Gamble C, McIntosh K, Scott R, Ho KH, Plevin R, Paul A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br J Pharmacol. 2012;165(4):802–819. doi:10.1111/j.1476-5381.2011.01608.x
12. Yu C, Chen S, Guo Y, Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-kappaB signaling pathway. Theranostics. 2018;8(12):3224–3236. doi:10.7150/thno.23259
13. Pramanik KC, Makena MR, Bhowmick K, Pandey MK. Advancement of NF-kappaB signaling pathway: a novel target in pancreatic cancer. Int J Mol Sci. 2018;19:12. doi:10.3390/ijms19123890
14. Liao J, Yang Z, Carter-Cooper B, et al. Suppression of migration, invasion, and metastasis of cisplatin-resistant head and neck squamous cell carcinoma through IKKbeta inhibition. Clin Exp Metastasis. 2020;37(2):283–292. doi:10.1007/s10585-020-10021-7
15. Page A, Bravo A, Suarez-Cabrera C, et al. IKKbeta overexpression together with a lack of tumour suppressor genes causes ameloblastic odontomas in mice. Int J Oral Sci. 2020;12(1):1. doi:10.1038/s41368-019-0067-9
16. Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 targets inhibitor of kappaB kinase and induces apoptosis in melanoma: involvement of nuclear factor kappaB and mitochondria pathways. Clin Cancer Res. 2006;12(3 Pt 1):950–960. doi:10.1158/1078-0432.CCR-05-1220
17. Al-Katib A, Arnold AA, Aboukameel A, et al. I-kappa-kinase-2 (IKK-2) inhibition potentiates vincristine cytotoxicity in non-Hodgkin’s lymphoma. Mol Cancer. 2010;9:228. doi:10.1186/1476-4598-9-228
18. Gaddipati S, Lu Q, Kasetti RB, et al. IKK2 inhibition using TPCA-1-loaded PLGA microparticles attenuates laser-induced choroidal neovascularization and macrophage recruitment. PLoS One. 2015;10(3):e0121185. doi:10.1371/journal.pone.0121185
19. Shrestha S, Zhu J, Wang Q, et al. Melatonin potentiates the antitumor effect of curcumin by inhibiting IKKβ/NF-κB/COX-2 signaling pathway. Int J Oncol. 2017;51(4):1249–1260. doi:10.3892/ijo.2017.4097
20. He Y, Li W, Hu G, Sun H, Kong Q. Bioactivities of EF24, a novel curcumin analog: a review. Front Oncol. 2018;8:614. doi:10.3389/fonc.2018.00614
21. Xie X, Tu J, You H, Hu B. Design, synthesis, and biological evaluation of novel EF24 and EF31 analogs as potential IkappaB kinase beta inhibitors for the treatment of pancreatic cancer. Drug Des Devel Ther. 2017;11:1439–1451. doi:10.2147/DDDT.S133172
22. Sanders B, Ray AM, Goldberg S, et al. Anti-cancer effects of aloe-emodin: a systematic review. J Clin Transl Res. 2018;3(3):283–296.
23. Su J, Yan Y, Qu J, Xue X, Liu Z, Cai H. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway. Oncol Rep. 2017;37(3):1565–1572. doi:10.3892/or.2017.5428
24. Shrimali D, Shanmugam MK, Kumar AP, et al. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013;341(2):139–149. doi:10.1016/j.canlet.2013.08.023
25. Yoneyama H, Takizawa-Hashimoto A, Takeuchi O, et al. Acquired resistance to gemcitabine and cross-resistance in human pancreatic cancer clones. Anticancer Drugs. 2015;26(1):90–100. doi:10.1097/CAD.0000000000000165
26. Dong X, Fu J, Yin X, et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;30(8):1207–1218. doi:10.1002/ptr.5631
27. Kim Y-S, Lee Y-M, Oh T-I, et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism.Int J Mol Sci. 2018;19(10):10. doi:10.3390/ijms19103127
28. Li N, Wang C, Zhang P, You S. Emodin inhibits pancreatic cancer EMT and invasion by upregulating microRNA1271. Mol Med Rep. 2018;18(3):3366–3374. doi:10.3892/mmr.2018.9304
29. Zhang W, Chen H, Liu DL, et al. Emodin sensitizes the gemcitabine-resistant cell line Bxpc-3/Gem to gemcitabine via downregulation of NF-kappaB and its regulated targets. Int J Oncol. 2013;42(4):1189–1196. doi:10.3892/ijo.2013.1839
30. Durand JK, Baldwin AS. Targeting IKK and NF-kappaB for therapy. Adv Protein Chem Struct Biol. 2017;107:77–115. doi:10.1016/bs.apcsb.2016.11.006
31. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. 2017;277(1):113–127. doi:10.1111/imr.12550
32. Mulero MC, Huxford T, Ghosh G. NF-kappaB, IkappaB, and IKK: integral components of immune system signaling. Adv Exp Med Biol. 2019;1172:207–226. doi:10.1007/978-981-13-9367-9_10
33. Liu B, Sun L, Liu Q, et al. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27(3):370–381. doi:10.1016/j.ccell.2015.02.004
34. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi:10.1016/j.cell.2008.01.020
35. Tang H, Zeng L, Wang J, et al. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and down-regulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget. 2017;8(47):82842–82853. doi:10.18632/oncotarget.20666
36. Tsujimura S, Saito K, Nakayamada S, Tanaka Y. Relevance of multidrug resistance 1 and P-glycoprotein to drug resistance in patients with systemic lupus erythematosus. Histol Histopathol. 2007;22(4):465–468. doi:10.14670/HH-22.465
37. Xu HB, Fu J, Huang F, Yu J. Guggulsterone sensitized drug-resistant human hepatocarcinoma cells to doxorubicin through a Cox-2/P-gp dependent pathway. Eur J Pharmacol. 2017;803:57–64. doi:10.1016/j.ejphar.2017.03.045
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.