")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Emodin induces apoptosis and autophagy of fibroblasts obtained from patient with ankylosing spondylitis
Authors Ma C, Wen B, Zhang Q, Shao P, Gu W, Qu K, Shi Y, Wang B
Received 31 July 2018
Accepted for publication 27 November 2018
Published 11 February 2019 Volume 2019:13 Pages 601—609
DOI https://doi.org/10.2147/DDDT.S182087
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Cristiana Tanase
Cong Ma, Bo Wen, Qin Zhang, Pei-Pei Shao, Wen Gu, Kun Qu, Yang Shi, Bei Wang
Department of Rheumatology and Immunology, Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, Beijing 100010, People’s Republic of China
Background: Ankylosing spondylitis (AS) is a type of rheumatoid disease, which has been reported to be associated with the excessive proliferation of fibroblasts recently. Emodin, a single component from a traditional Chinese medicine Rheum palmatum, exerts anti-inflammation and antirheumatic arthritis activities. However, could emodin be used to treat AS remains unclear? Thus, this study aimed to investigate the effect of emodin on AS.
Methods: Fibroblasts obtained from patients with AS were used in the current study. In addition, multiple cellular and molecular biology techniques such as Cell Counting Kit-8, Western blotting, flow cytometry, monodansylcadaverine staining, and immunofluorescence assay were applied as well.
Results: Emodin-induced apoptosis of fibroblasts obtained from patient with AS via increasing active caspase-9, active caspase-3, and Bax levels and downregulating Bcl-2. Meanwhile, emodin enhanced autophagy in fibroblasts via upregulation of the expression of Atg12, Atg5, and Beclin 1, which was further confirmed by monodansylcadaverine staining. As expected, autophagy inhibitor 3-methyladenine (3MA) completely reversed emodin-induced autophagy in fibroblasts. Moreover, 3MA significantly increased emodin-induced apoptosis of fibroblasts obtained from patient with AS by increasing the levels of γH2AX, active caspase-9, active caspase-3, and cleaved poly ADP-ribose polymerase.
Conclusion: Our results indicated that emodin effectively induced apoptosis and autophagy of fibroblasts obtained from patient with AS. In addition, suppression of autophagy enhanced emodin-induced apoptosis in fibroblasts. Therefore, we proposed that combination of emodin with autophagy inhibitor might be a potent strategy for improving the symptoms of AS in the future.
Keywords: emodin, apoptosis, autophagy, 3MA, ankylosing spondylitis
Introduction
Ankylosing spondylitis (AS) is a sort of spinal arthritic, which can make the people debilitating.1 The clinical manifestations are that the spine become painful and gradually stiffening. Males are more likely to suffer from this disease, and the onset is in late adolescence or early adulthood universally.2 The symptom characteristics of AS include acute arthritis, anterior uveitis, persistent back pain, and enthesopathy.3 In enthesis or ligament tissue, the largest number of connective tissue cells is fibroblasts.4,5 Recent researches have shown that fibroblasts play an important role in ligament tissue ossification and ankylosis.6,7 Heterotopic ossification of the entheses is one of the main features in AS, while fibroblasts are potential target cells for heterotopic ossification.8 In patients with AS, the unusual proliferation of fibroblasts is always connected with the formation of new bones by heterotopic ossification.8,9
Emodin (1,3,8-trihydroxy-6-methyl-anthraquinone), extracted from the rhizome of Rheum palmatum, is a natural active anthraquinone compound.10,11 Emodin has been revealed to have multiple biological functions, including anti-inflammation, anticancer, immunosuppression, and antivirus properties.12,13 In addition, emodin has a role in the treatment of rheumatic arthritis, which exerted anti-inflammatory effects via inhibition of the nuclear factor-κB pathway.14 Cui et al suggested that emodin induced hepatic cell apoptosis through directly affecting the mitochondria.15 Moreover, emodin induced human T cells and lung cancer cell A549 apoptosis via downregulation of Bcl-2 and upregulation of Bax and exerted immunosuppressive and antitumor actions.16,17
Autophagy is a pathway of cellular lysosomal degradation that is important for the regulation of cellular homeostasis.18,19 Depending on different circumstances, autophagy could play either a prosurvival or a death role.18,19 Recent studies revealed that autophagy performed an important cytoprotective role under physiological conditions.20,21 The relationship between apoptosis and autophagy is close and complex.
In spite of many reports on the biological activities of emodin, could emodin be used to treat AS remains unclear? Therefore, we aimed to explore the underlying mechanisms of emodin in the treatment of patients with AS.
Materials and methods
Isolation and culture of fibroblasts from patients with AS
Forty patients with AS in Beijing Traditional Chinese Medicine Hospital were enrolled between April 2017 and May 2018, which consisted of 25 males and 15 females. Patients with AS were confirmed pathologically, and the primary fibroblast cells were obtained during surgery. Patients aged from 25 to 45 years and the diagnostic criteria conformed to New York criteria.22 Clinical and pathological data of these patients were collected with their written informed consents. This study was approved by the Ethics Committee of Beijing Traditional Chinese Medicine Hospital (Beijing, People’s Republic of China). In addition, the study was carried out in accordance with the principles of the Declaration of Helsinki. The isolation and culture methods of fibroblasts from patients with AS were the same as those by Yang et al.23 Human fibroblasts CCD-18Co were provided with American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in Eagle’s Minimum Essential Medium with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA).
Cell Counting Kit-8 (CCK-8) assay of cell viability
Fibroblasts obtained from patients with AS (5×103 cells/well) were seeded into 96-well plate and treated with different concentrations of emodin (0, 2, 5, 10, or 20 μM) for 24, 48, and 72 hours. Then, cell viability was evaluated with CCK-8 (Beyotime, Shanghai, People’s Republic of China) according to the manufacturer’s protocols. The absorbance was determined at 450 nm using a Thermo Multiskan FC microplate reader (Thermo Fisher Scientific). Emodin standard product was purchased from Sigma (#30269, Sigma-Aldrich, St Louis, MO, USA).
Flow cytometric analysis of cell apoptosis
Fibroblasts obtained from patients with AS were incubated overnight in 6-well plates and treated with 10 μM emodin for another 72 hours; 0.2% dimethyl sulfoxide (DMSO; Sigma-Aldrich) was used as control. Afterward, pelleted cells were stained with 10 μL Annexin V and 5 μL propidium iodide (PI) for 15 minutes at room temperature in the dark according to the manufacturer’s protocols (Thermo Fisher Scientific). The Annexin V/PI-positive cells were then measured by flow cytometer (FCM; BD Biosciences, San Jose, CA, USA).
Western blot analysis
Fibroblasts obtained from patients with AS were exposed to 10 μM emodin for 72 hours. Then, cells were collected and washed three times with PBS in cell lysis buffer. Bradford Protein Assay Kit (Beyotime) was used to determine the protein concentration. After electrophoresis with 10% SDS polyacrylamide gel, proteins were transported into polyvinylidene fluoride membranes (PVDF, Thermo Fisher Scientific) in 2 hours. The membranes were blocked with 5% skim milk in Tris-Buffered Saline with Tween 20 (TBST) for 1 hour at room temperature. Then, membranes were washed in TBST three times and incubated with primary antibodies: anti-Bax (Abcam, Cambridge, MA, USA; ab32503) (1:1,000), anti-Bcl-2 (Abcam; ab32124) (1:1,000), antiactive caspase-9 (Abcam; ab32539) (1:1,000), antiactive caspase-3 (Abcam; ab2302) (1:1,000), anti-β-actin (Abcam; ab8227) (1:1,000), anti-Beclin 1 (Abcam; ab207612) (1:1,000), anti-Atg5 (Abcam; ab228668) (1:1,000), anti-Atg12 (Abcam ab155598) (1:1,000), anti-γH2AX (Abcam; ab2893) (1:1,000), anti-cleaved PRAP (Abcam; ab32561) (1:1,000). After washing, the PVDF membrane was incubated with secondary antibody antirabbit (Abcam; ab7090) (1:5,000) before being determined by chemiluminescence. Finally, the PVDF membranes were incubated with enhanced chemiluminescent reagent (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) to detect the blots. The density of blots for targets was normalized to β-actin.
Monodansylcadaverine (MDC) staining
Fibroblasts obtained from patients with AS were exposed to DMSO, 10 μM emodin with or without 5 mM 3-methyladenine (3MA) at 37°C for 72 hours. Then, cells were stained with 0.05 mM MDC (#D4008, Sigma-Aldrich) at 37°C for 30 minutes. Fibroblast cells were washed with PBS three times to wipe off redundant MDC. Fluorescent cells were instantly observed and counted with a Hitachi F-2000 Fluorescence Spectrophotometer (Olympus Corporation, Tokyo, Japan).
Immunofluorescence
Fibroblasts obtained from patients with AS were exposed to DMSO, 10 μM with or without 5 mM 3MA at 37°C for 72 hours. Immunofluorescence assay was performed according to the previously described method.24 Briefly, cultured cells were washed in PBS two times, prefixed with 4% paraformaldehyde for 15 minutes at room temperature, then fixed in ice-cold methanol (100%) for 10 minutes at −20°C. Next, cells were cultured with primary antibodies for anti-γH2AX (Abcam; ab2893) (1:1,000), DAPI (ab104139), and LC3 (ab48394) at 4°C overnight. After washing with PBS, cells were incubated with secondary antibodies (Abcam; ab150080) (1:5,000) at 37°C for 1 hour. Then, the samples were immediately observed by fluorescence microscope (Olympus CX23).
Statistical analysis
Each group were executed at least three independent experiments, and all data were expressed as mean ± SD. The comparison between two groups was analyzed by Student’s t-test. The comparisons among multiple groups were made with one-way ANOVA followed by Dunnett’s test. P<0.05 or P<0.01 was considered to indicate a statistically significant difference (*P<0.05, **P<0.01).
Results
Emodin suppressed the proliferation of fibroblasts isolated from patients with AS
The chemical structure of emodin is shown in Figure 1A. CCK-8 assay was used to determine the effect on the viability of fibroblasts isolated from patients with AS following treatment with emodin (0, 2, 5, 10, and 20 μM) for 24, 48, and 72 hours. As shown in Figure 1B and C, both 10 and 20 μM emodin markedly decreased the cell viability of fibroblasts. Since the number of cells was significantly decreased by >50% following treatment with 20 μM emodin, therefore, 10 μM emodin was chosen for use in the following experiments. In addition, emodin had very limited effect on human normal fibroblasts CCD-18Co (Figure 1D). These results suggested that emodin could suppress the proliferation of AS-associated fibroblast cells.
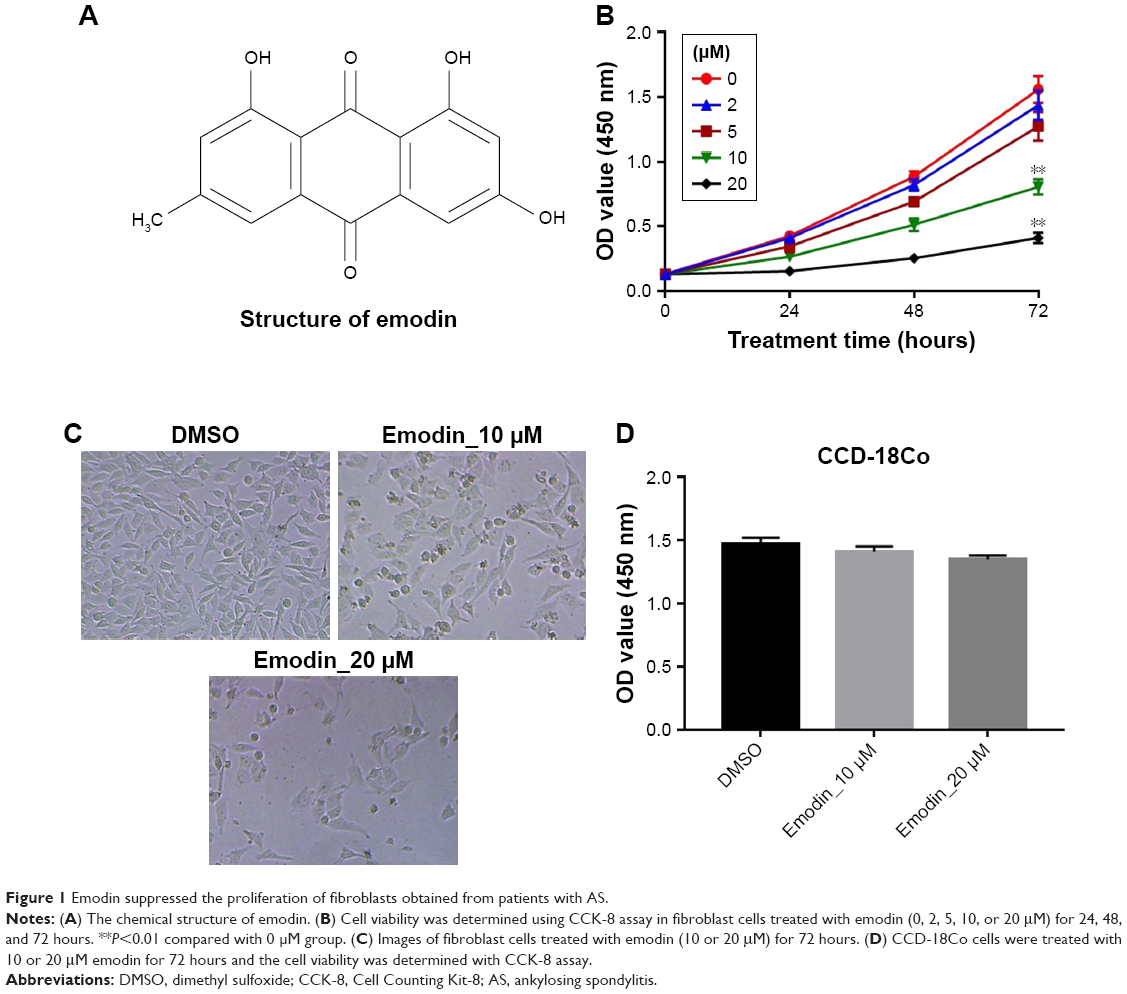 | Figure 1 Emodin suppressed the proliferation of fibroblasts obtained from patients with AS. |
Emodin induced the apoptosis of fibroblasts isolated from patients with AS
To further determine the effect of emodin on the apoptosis of fibroblasts obtained from patients with AS, cells were treated with 10 μM emodin for 72 hours. Then, the apoptosis of cells were detected with flow cytometry. As indicated in Figure 2A and B, the apoptotic rate of fibroblast cells was significantly increased by emodin interference compared with the DMSO group (P<0.01). In addition, apoptosis-related proteins Bax, Bcl-2, active caspase-9, and active caspase-3 in cells were detected by Western blotting. As shown in Figure 2C–G, the expressions of Bax, active caspase-9, and active caspase-3 were dramatically increased, whereas Bcl-2 were significantly decreased in the emodin-treated group compared with the DMSO group (P<0.01). These data indicated that emodin induced the apoptosis of fibroblasts isolated from patients with AS.
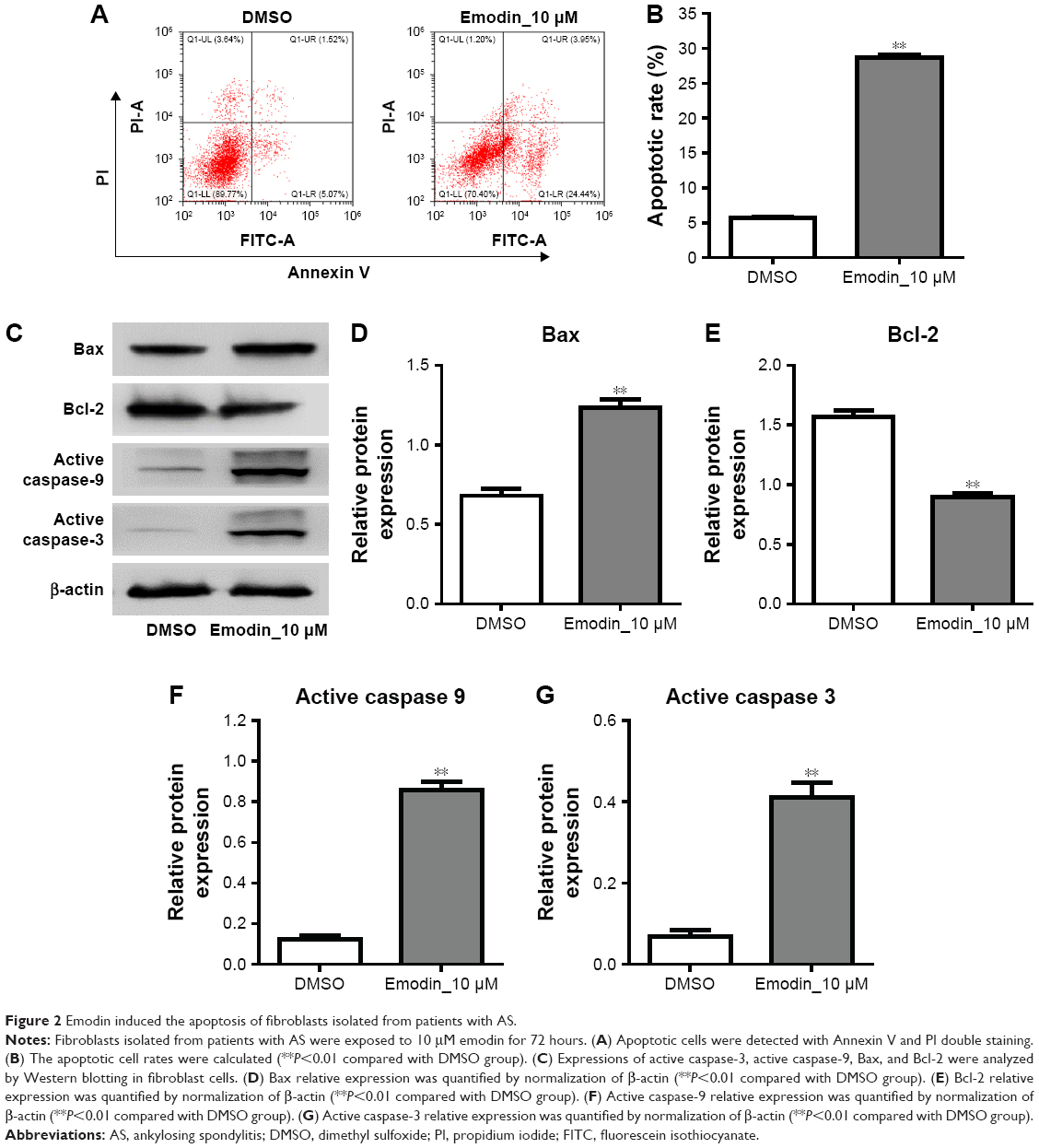 | Figure 2 Emodin induced the apoptosis of fibroblasts isolated from patients with AS. |
Emodin induced the autophagy of fibroblasts isolated from patients with AS
We next assessed if emodin could induce autophagy in fibroblasts isolated from patients with AS. MDC assay and immunofluorescence assay were performed to investigate the effect of emodin on autophagy in fibroblast cells. In addition, autophagy inhibitor 3MA was used to further testify the effect of emodin on cells. The MDC staining data indicated that the autophagic vacuoles and autophagosome were dramatically increased in the emodin-treated group compared with the DMSO group (Figure 3A and B; P<0.01). As expected, the autophagic vacuoles and autophagosome were significantly decreased in the presence of 3MA treatment compared with the emodin-treated group (P<0.01) (Figure 3A and B). Additionally, the immunofluorescence data showed that the levels of LC3-II were notably increased by emodin, which was correlated with autophagosome formation (Figure 3C).
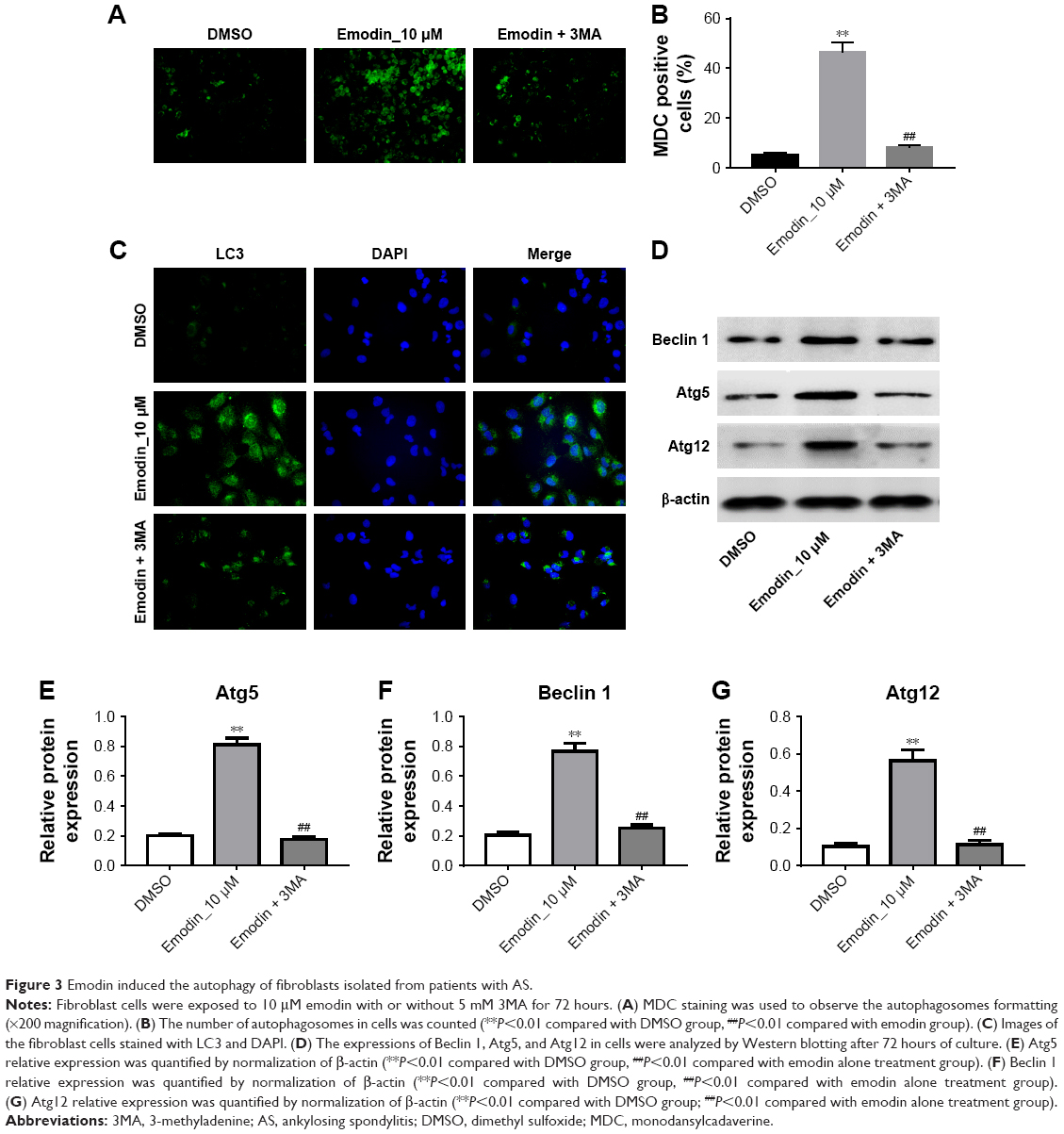 | Figure 3 Emodin induced the autophagy of fibroblasts isolated from patients with AS. |
We next used Western blotting to detect the expression of autophagy-related proteins Beclin 1, Atg5, and Atg12 in cells. As shown in Figure 3D–G, compared with the DMSO group, the levels of Beclin 1, Atg5, and Atg12 were dramatically increased in emodin-treated fibroblast cells (P<0.01). In addition, emodin-induced Beclin 1, Atg5, and Atg12 protein upregulation was significantly reversed by 3MA treatment (P<0.01) (Figure 3D–G). All these data suggested that emodin induced the autophagy of fibroblasts isolated from patients with AS, which could be markedly reversed by 3MA.
Inhibition of autophagy enhanced emodin-induced apoptosis in fibroblasts isolated from patients with AS
The autophagy inhibitor 3MA was then used to investigate the association between apoptosis and autophagy in fibroblasts isolated from patients with AS induced by emodin. Compared with the DMSO group, the cell viability was markedly decreased, whereas apoptotic rate was significantly increased in emodin-treated fibroblasts cells (Figure 4A–C; P<0.01). Besides, these effects were further enhanced by 3MA treatment group compared with the emodin-treated group (Figure 4A–C; P<0.01). These data demonstrated that inhibition of autophagy enhanced emodin-induced apoptosis in fibroblasts isolated from patients with AS.
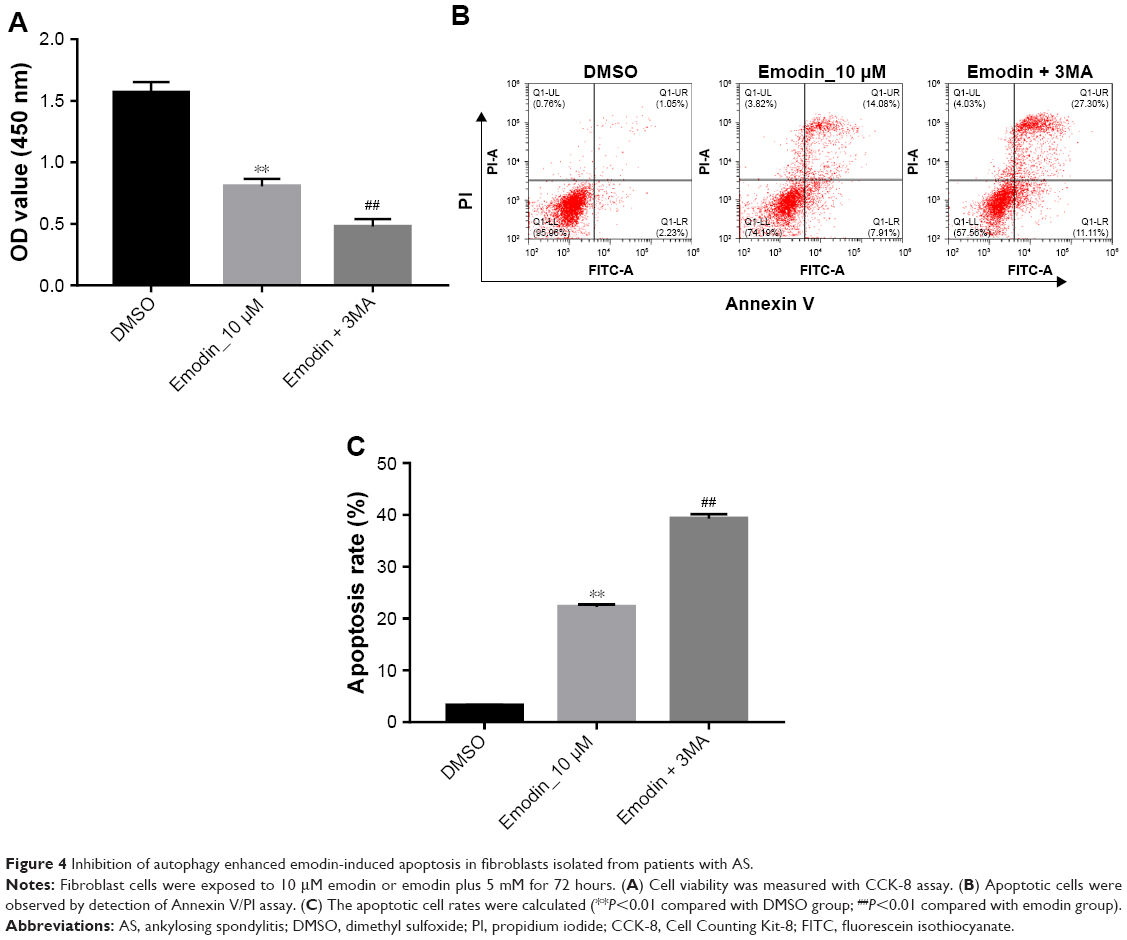 | Figure 4 Inhibition of autophagy enhanced emodin-induced apoptosis in fibroblasts isolated from patients with AS. |
Inhibition of autophagy enhanced emodin-induced DNA damage in fibroblasts isolated from patients with AS
As we know, apoptosis is an important path of cell inactivation, which is caused by DNA damage. Then, immunofluorescence assay and Western blotting were used to further detect the effect of emodin on DNA damages in fibroblasts. The mark of DNA double-strand break was the formation of γH2AX. As indicated in Figure 5A, the count of DNA double-strand break was dramatically increased in emodin-treated group compared with the DMSO group (P<0.01). As expected, the count of DNA double-strand break in the cells was further increased following treatment with 3MA compared with emodin group (Figure 5A; P<0.01). In addition, as shown in Figure 5B–F, the levels of γH2AX, active caspase-3, and cleaved poly ADP-ribose polymerase (PARP) in fibroblasts were significantly increased by emodin (P<0.01). Moreover, the expression of γH2AX, active caspase-9, active caspase-3, and cleaved PARP were further increased following treatment with 3MA compared with the emodin group (P<0.01). All these results evidenced that inhibition of autophagy enhanced emodin-induced DNA damage in fibroblasts isolated from patients with AS.
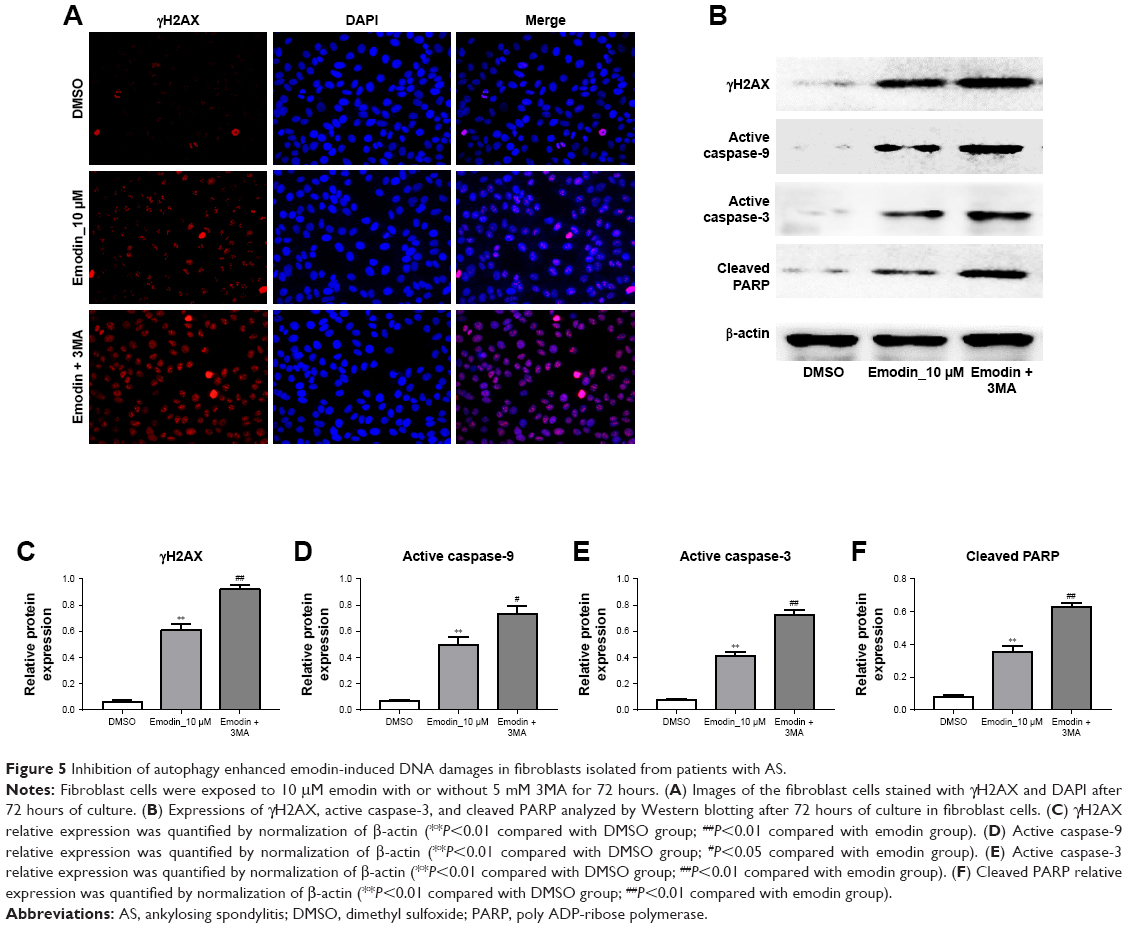 | Figure 5 Inhibition of autophagy enhanced emodin-induced DNA damages in fibroblasts isolated from patients with AS. |
Discussion
Recent studies indicated that the development of human AS was partly due to excessive proliferation of fibroblast cells.6,7 In this study, it was demonstrated that emodin inhibited the proliferation of fibroblasts obtained from patients with AS via inducing apoptosis and autophagy. In addition, emodin upregulated active caspase-9, active caspase-3, and Bax levels and downregulated Bcl-2 levels in fibroblast cells. Meanwhile, emodin increased the autophagy of fibroblasts via increasing the expressions of Beclin 1, Atg12, and Atg5. These findings are consistent with previous observations that emodin significantly inhibited proliferation of rheumatoid arthritis synoviocytes, which was stimulated by IL-1β and lipopolysaccharide under hypoxic condition;25 consistently, emodin inhibited the osteoclast differentiation by macrophage colony-stimulating factor induced in bone marrow macrophages.14 In addition, a study by Wei et al found that fluoride induced apoptosis and autophagy in MC3T3–E1 cells.26 Besides, emodin inhibited colon cancer cell viability via induction of apoptosis and autophagy.27 Our findings were evidenced by these reports. Therefore, we proposed that emodin may improve the symptoms of AS via inducing apoptosis and autophagy in fibroblasts obtained from patients with AS.
Autophagy is a process of selective degradation of cellular ingredients. The connection between apoptosis and autophagy is complex.28 Accumulating data indicated that autophagy performs a primarily cytoprotective role in physiologically related conditions.29 In the current study, we found that 3MA enhanced the antiproliferation effect of emodin on fibroblasts via increasing apoptosis, which was further evidenced by the upregulation of apoptosis-related proteins. These results indicated that inhibition of autophagy possibly aggravated DNA damage in fibroblasts obtained from patients with AS, sequentially leading to fibroblast cell death. Furthermore, these finding were consistent with previous studies that inhibition of autophagy by 3MA markedly promoted the apoptotic rate in human osteosarcoma and hepatocellular carcinoma cell lines.30,31 Similarly, the inhibition of autophagy significantly increased the apoptosis induced by doxorubicin through upregulation of active caspase-3 in osteosarcoma.32 In addition, paclitaxel increased autophagic activity and through suppressing autophagy enhances paclitaxel-induced apoptosis in osteosarcoma cells.33 All these reports further confirmed our findings that inhibition of autophagy could enhance emodin-induced growth inhibition and apoptosis in fibroblasts obtained from patients with AS.
In this study, we indicated that emodin significantly inhibited the proliferation of fibroblasts obtained from patients with AS via inducing apoptosis and autophagy. In addition, inhibition of autophagy notably enhanced emodin-induced growth inhibition and apoptosis in fibroblast cells. In conclusion, we suggested that combination of emodin and autophagy inhibitor could be a potent method for improving the symptoms AS in the future.
Acknowledgments
Funding was received from the Beijing Municipal Administration of Hospitals Incubating Program (PZ2016017), the Foundation for Young Scientists of Gongyan Xu and Shouren Xia (XX-201708), and the Beijing Traditional Chinese Medicine Eternal Flame Heritage Project “3+3”, The Master Laboratory of Weilan Wang (2014-SZ-A-30). Yanjing School Innovatively Inherits “Fist” project - Rheumatology Department.
Disclosure
The authors report no conflicts of interest in this work.
References
Reveille JD, Weisman MH. The epidemiology of back pain, axial spondyloarthritis and HLA-B27 in the United States. Am J Med Sci. 2013;345(6):431–436. | ||
Ersözlü-Bozkirli ED, Keşkek SO, Bozkirli E, Yücel AE. The effect of infliximab on depressive symptoms in patients with ankylosing spondylitis. Acta Reumatol Port. 2015;40(3):262–267. | ||
Hellmann DB, Imboden JB, McPhee SJ. Blood disorders. In: Papadakis MA, McPhee SJ, editors. Current Medical Diagnosis and Treatment. New York: Mc Graw Hill; 2014:822–823. | ||
Zhang HY, Liu R, Xing YJ, Xu P, Li Y, Li CJ. Effects of hypoxia on the proliferation, mineralization and ultrastructure of human periodontal ligament fibroblasts in vitro. Exp Ther Med. 2013;6(6):1553–1559. | ||
Yang HS, Lu XH, Chen DY, et al. Upregulated expression of connexin43 in spinal ligament fibroblasts derived from patients presenting ossification of the posterior longitudinal ligament. Spine. 2011;36(26):2267–2274. | ||
Hamdi W, Chelli-Bouaziz M, Ahmed MS, et al. Correlations among clinical, radiographic, and sonographic scores for enthesitis in ankylosing spondylitis. Joint Bone Spine. 2011;78(3):270–274. | ||
Li DH, He CR, Liu FP, et al. Annexin A2, up-regulated by IL-6, promotes the ossification of ligament fibroblasts from ankylosing spondylitis patients. Biomed Pharmacother. 2016;84:674–679. | ||
Zou YC, Yang XW, Yuan SG, Zhang P, Ye YL, Li YK. Downregulation of dickkopf-1 enhances the proliferation and osteogenic potential of fibroblasts isolated from ankylosing spondylitis patients via the Wnt/β-catenin signaling pathway in vitro. Connect Tissue Res. 2016;57(3):200–211. | ||
Zou YC, Yang XW, Yuan SG, Zhang P, Li YK. Celastrol inhibits prostaglandin E2-induced proliferation and osteogenic differentiation of fibroblasts isolated from ankylosing spondylitis hip tissues in vitro. Drug Des Devel Ther. 2016;10:933–948. | ||
Shi YQ, Fukai T, Sakagami H, et al. Cytotoxic and DNA damage-inducing activities of low molecular weight phenols from rhubarb. Anticancer Res. 2001;21(4A):2847–2853. | ||
Tsai T, Chen C. Ultraviolet spectrum identification of emodin in rabbit plasma by HPLC and its pharmacokinetics application. Asia Pac J Pharm. 1992;7:53–56. | ||
Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P. Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent. Med Res Rev. 2007;27(5):591–608. | ||
Kuo YC, Meng HC, Tsai WJ. Regulation of cell proliferation, inflammatory cytokine production and calcium mobilization in primary human T lymphocytes by emodin from Polygonum hypoleucum Ohwi. Inflamm Res. 2001;50(2):73–82. | ||
Hwang JK, Noh EM, Moon SJ, et al. Emodin suppresses inflammatory responses and joint destruction in collagen-induced arthritic mice. Rheumatology. 2013;52(9):1583–1591. | ||
Cui YT, Liu B, Xie J, Xu P, Habte-Tsion HM, Zhang YY. The effect of emodin on cytotoxicity, apoptosis and antioxidant capacity in the hepatic cells of grass carp (Ctenopharyngodon idellus). Fish Shellfish Immunol. 2014;38(1):74–79. | ||
Qu K, Shen NY, Xu XS, et al. Emodin induces human T cell apoptosis in vitro by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction. Acta Pharmacol Sin. 2013;34(9):1217–1228. | ||
Li WY, Ng YF, Zhang H, et al. Emodin elicits cytotoxicity in human lung adenocarcinoma A549 cells through inducing apoptosis. Inflammopharmacology. 2014;22(2):127–134. | ||
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. | ||
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. | ||
Bhogal RH, Weston CJ, Curbishley SM, Adams DH, Afford SC. Autophagy: a cyto-protective mechanism which prevents primary human hepatocyte apoptosis during oxidative stress. Autophagy. 2012;8(4):545–558. | ||
Hui B, Shi YH, Ding ZB, et al. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer. 2012;118(22):5560–5571. | ||
van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27(4):361–368. | ||
Yang M, Yuan H, Miao M, Xu W. The osteogenic potential of ligament fibroblasts is greater in ankylosing spondylitis patients than in patients with osteoarthritis. Z Rheumatol. 2015;74(4):340–345. | ||
Chiu SJ, Chao JI, Lee YJ, Hsu TS. Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells. Toxicol Lett. 2008;179(2):63–70. | ||
Ha MK, Song YH, Jeong SJ, et al. Emodin inhibits proinflammatory responses and inactivates histone deacetylase 1 in hypoxic rheumatoid synoviocytes. Biol Pharm Bull. 2011;34(9):1432–1437. | ||
Wei M, Duan D, Liu Y, Wang Z, Li Z. Autophagy may protect MC3T3-E1 cells from fluoride-induced apoptosis. Mol Med Rep. 2014;9(6):2309–2315. | ||
Wang Y, Luo Q, He X. Emodin induces apoptosis of colon cancer cells via induction of autophagy in a ROS-dependent manner. Oncol Res. 2017;26(6):889–899. | ||
Zhang T, Li Y, Park KA, et al. Cucurbitacin induces autophagy through mitochondrial ROS production which counteracts to limit caspase-dependent apoptosis. Autophagy. 2012;8(4):559–576. | ||
Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3(1):17–27. | ||
Tu P, Huang Q, Ou Y, et al. Aloe-emodin-mediated photodynamic therapy induces autophagy and apoptosis in human osteosarcoma cell line MG-63 through the ROS/JNK signaling pathway. Oncol Rep. 2016;35(6):3209–3215. | ||
Chang Z, Shi G, Jin J, et al. Dual PI3K/mTOR inhibitor NVP-BEZ235-induced apoptosis of hepatocellular carcinoma cell lines is enhanced by inhibitors of autophagy. Int J Mol Med. 2013;31(6):1449–1456. | ||
Zhao D, Yuan H, Yi F, Meng C, Zhu Q. Autophagy prevents doxorubicin-induced apoptosis in osteosarcoma. Mol Med Rep. 2014;9(5):1975–1981. | ||
Kim HJ, Lee SG, Kim YJ, et al. Cytoprotective role of autophagy during paclitaxel-induced apoptosis in Saos-2 osteosarcoma cells. Int J Oncol. 2013;42(6):1985–1992. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.