")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes
Authors Ye B, Chen X, Dai S , Han J, Liang X, Lin S, Cai X, Huang Z, Huang W
Received 21 November 2018
Accepted for publication 12 February 2019
Published 25 March 2019 Volume 2019:13 Pages 975—990
DOI https://doi.org/10.2147/DDDT.S195412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Bozhi Ye,1,* Xudong Chen,1,* Shanshan Dai,2 Jibo Han,3 Xiaohe Liang,1 Shuang Lin,1 Xueli Cai,1 Zhouqing Huang,1 Weijian Huang1
1Department of Cardiology, The Key Lab of Cardiovascular Disease of Wenzhou, The First Affiliated Hospital of WenZhou Medical University, WenZhou, ZheJiang, People’s Republic of China; 2Department of Emergency, The First Affiliated Hospital of WenZhou Medical University, WenZhou, ZheJiang, People’s Republic of China; 3Department of Cardiology, The Second Affiliated Hospital of Jiaxing University, Jiaxing, ZheJiang, People’s Republic of China
*These authors contributed equally to this work
Background: Emodin has recently been reported to have a powerful antiinflammatory effect, protecting the myocardium against ischemia/reperfusion (I/R) injury. Pyroptosis is a proinflammatory programmed cell death that is related to many diseases. The present study investigated the effect of emodin on pyroptosis in cardiomyocytes.
Materials and methods: Sprague Dawley rats were randomly divided into sham, I/R, and I/R+Emodin groups. I/R model was subjected to 30 minutes’ ligation of left anterior descending coronary artery, followed by 2 hours of reperfusion. Cardiomyocytes were exposed to hypoxic conditions for 1 hour and normoxic conditions for 2 hours. The level of the pyroptosis was detected by Western blot, real-time PCR analysis, and ELISA.
Results: The level of gasdermin D-N domains was upregulated in cardiomyocytes during I/R or hypoxia/reoxygenation (H/R) treatment. Moreover, emodin increased the rate of cell survival in vitro and decreased the myocardial infarct size in vivo via suppressing the levels of I/R-induced pyroptosis. Additionally, the expression of TLR4, MyD88, phospho-IκBα, phospho-NF-κB, and the NLRP3 inflammasome was significantly upregulated in cardiomyocytes subjected to H/R treatment, while emodin suppressed the expression of these proteins.
Conclusion: This study confirms that emodin treatment was able to alleviate myocardial I/R injury and inhibit pyroptosis in vivo and in vitro. The inhibitory effect of emodin on pyroptosis was mediated by suppressing the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway. Therefore, emodin may provide an alternative treatment for myocardial I/R injury.
Keywords: emodin, ischemia/reperfusion injury, gasdermin D, pyroptosis, heart
Introduction
The number of patients with acute myocardial infarction (AMI) has gradually increased, and AMI has become one of the major causes of death worldwide.1 Timely restoration of blood flow to ischemic myocardium could limit infarct size and reduce mortality.2 However, this treatment could cause an injury to the myocardium known as ischemia/reperfusion (I/R) injury.3 Reactive oxygen species (ROS) and calcium (Ca2+) overload have been involved in the mechanism of I/R injury.4,5 In recent years, an increasing number of studies have shown that inflammation plays an irreplaceable role in myocardial I/R injury.6 I/R increases the expression of inflammasomes and subsequently leads to the cleavage of IL-1β, causing inflammatory cell infiltration and increasing the expression of cytokines in the heart.7
Pyroptosis is a proinflammatory programmed cell death, which was proposed by Cookson and Brennan.8 Pyroptosis shares some characteristics with apoptosis, including DNA fragmentation, nuclear condensation, caspase dependence, and positive Annexin V staining. However, pyroptosis is programmed by an inflammatory caspase.9 Activated inflammatory caspase assembles a pore in the plasma membrane, causing cell swelling and extensive release of proinflammatory substances.10 Recent studies have verified that the gasdermin D (GSDMD) protein is the executioner of pyroptotic cell death, which is cleaved by caspase-1/4/5/11.11 Then, the gasdermin D-N domains (GSDMD-N) are moved to the plasma membrane to form a pore with an internal diameter of 10–14 nm. This pore allows the leakage of mature IL-1β and IL-18.12 Ions and water then rush into the cell, causing it to swell and dissolve, resulting in the release of cytoplasmic contents.10
Excessive neutrophil infiltration in the infarcted site is thought to be harmful to cardiomyocyte survival.13 Inflammation increases myocardial infarct size (INF),14 whereas knocking out TLR4 reduces the INF after I/R injury,15 making inflammation an important therapeutic target for improving outcomes following I/R. IL-1 receptor expression is upregulated in myocardial infarction, which increases the mortality in AMI.16 Blocking IL-1 could significantly reduce INF and attenuate cardiomyocyte injury.17 Therefore, if an inhibitor can be found to reduce the myocardial inflammation, it will effectively inhibit INF and protect the myocardium from I/R injury.
Emodin is an anthraquinone derivative from the rhizome of Rheum palmatum, which is widely used as a laxative in traditional Chinese medicine.18 Emodin has been reported to have anti-inflammatory activities.19,20 Du and Ko found that emodin pretreatment protected the myocardium against I/R injury by decreasing serum lactate dehydrogenase (LDH) and improving cardiac function.21 However, despite these important functions, the potential role of GSDMD and its mechanism of action in myocardial I/R injury remain unknown. In addition, there is currently no evidence that intervention of GSDMD-mediated pyroptosis can reduce myocardial I/R injury. The present study was designed to explore the molecular target of emodin in I/R injury-associated signal transduction pathways in myocardial cells in rats.
Materials and methods
Separation of rat primary cardiomyocytes
Cardiomyocytes were isolated from 1- to 3-day-old neonatal Sprague Dawley (SD) rats. Hearts were washed in HEPES-buffered saline solution after dissection from the rats. HEPES-buffered saline solution contained 0.08% trypsin, 0.06% collagenase type IV, 136 mM NaCl, 4.2 mM NaHCO3, 5.5 mM glucose, 4 mM KCl, and 8.3 mM HEPES. After the buffer was removed, the tissue was finely minced, immersed in 10 mL of dissociation medium, and agitated slowly for 8 minutes at 37°C. The supernatant containing cell debris and blood cells was discarded. The tissue pieces were immersed in the dissociating medium and stirred for another 8 minutes at 37°C to release cells from the tissue fragments, and this process was repeated 15 times. Lysates were collected and centrifuged at 175× g for 10 minutes at room temperature. Precipitated cells were resuspended in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (Thermo Fisher Scientific) supplemented with 1% penicillin/streptomycin (pen/strep, 10,000 U/mL each; Thermo Fisher Scientific). The cells were incubated at 37°C for 1 hour. Non-adherent cells were primary cardiomyocytes, which were gathered and cultivated for the experiment.
Treatment and culture of cardiomyocytes
Primary cardiomyocytes were separated and maintained in DMEM containing 4.5 g/L of glucose and supplemented with 10% (v/v) FBS, and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2 incubator. The cardiomyocytes were pretreated with 2.5, 5, and 10 μM emodin (E106693; Aladdin, Shanghai, China), 5 μM Bay-117082 (NF-κB pathway inhibitor) (S2913; Selleck, Shanghai, China), 10 μM NLRP3 inflammasome inhibitor (S3680; Selleck), or N-acetylcysteine (NAC, 1 mM, S0077; Beyotime, Shanghai, China) for 1 hour before the cardiomyocytes were stimulated with hypoxia/reoxygenation (H/R).
H/R with cardiomyocytes
The cells were cultured in FBS and glucose-deprived DMEM, and were first exposed to hypoxic conditions (InvivO2 hypoxia cabinet; 1% O2, 5% CO2, and 94% N2) for 1 hour and normoxic conditions for 2 hours. Meanwhile, the cells in the control group (without FBS and glucose-deprived DMEM) were maintained under normoxic conditions (21% O2, 5% CO2, and 74% N2) for 3 hours.
Animals
Six-week-old male SD rats were purchased from the Animal Center of Wenzhou Medical University and housed under specific pathogen-free conditions. The Wenzhou Medical University Animal Policy and Welfare Committee approved these experiments. Animals were kept under a 12-hour/12-hour light–dark cycle and were allowed free access to food and water. All operations were conducted in accordance with the National Institutes of Health Guide for Care and Use of Animals.
Experimental protocol and induction of I/R
Twenty-four SD rats (age, 7–8 weeks; body weight, 260–280 g) on standard diet were randomly divided into three groups: sham group (sham, n=8), I/R group (I/R, n=8), and I/R plus 20 mg/kg emodin group (I/R+Emodin [20 mg/kg], n=8). Rats were injected intraperitoneally with emodin 1 hour before I/R treatment. Surgery was performed as previously described.22 Briefly, rats were anesthetized by 2% pentobarbital sodium (50 mg/kg intraperitoneally) and ventilated artificially using a rodent respirator (HX-101E; Taimeng, Chengdu, China) in volume-controlled mode at 80 strokes per minute. Then, the left axilla was exposed through a left thoracotomy from the left sternal border at the fourth intercostal space. The left anterior descending (LAD) coronary artery was ligated ~2 mm from its origin using a 6–0 surgical suture (Surgical Specialties Corporation). Occlusion was confirmed by observing a pale myocardium in the left ventricle below the suture. Animals in the sham group were also anesthetized, and a suture was passed under the LAD without occlusion. I/R and I/R+Emodin (20 mg/kg) rats were subjected to 30 minutes of LAD ligation, followed by 2 hours of reperfusion.
Measurements of infarct size and area at risk (AAR)
AAR/left ventricles (LV) reflect the extent of myocardial ischemia, while infarct size (INF)/AAR reflects the level of dead myocardium. INF was identified as previously described.23 Briefly, the suture underneath the LAD was reoccluded after completion of I/R procedure. To identify the AAR, the animals were perfused with 2 mL of 2% Evans Blue (Sigma-Aldrich Co., St Louis, MO, USA) through the inferior vena cava. The heart was excised, rinsed with PBS solution, and frozen at −20°C. Then, the hearts were cut into five 1 mm slices, from the apex to the base, and incubated with 1% solution of 2,3,5-triphenyltetrazolium chloride in PBS solution at 37°C for 15 minutes. Then, the hearts were fixed in 10% formalin for 2 hours. The non-ischemic myocardium was stained by using Evans Blue, and the INF appeared pale after staining. The images were analyzed using ImageJ software, and the INF was calculated as a percentage of the AAR using a weight-based method.
Assessment of cell viability with CCK-8 assay
Cell viability was assessed with the CCK-8 assay according to the manufacturer’s instruction (C0038; Beyotime). The kit contains a water-soluble tetrazolium salt (WST-8), a substrate for the mitochondrial succinate dehydrogenases that converts WST-8 into formazan (orange-colored). The activities of these enzymes are proportional to the living cells. Primary cardiomyocytes were seeded into 96-well plates at a concentration of 5,000 cells/well and exposed to various culture conditions, including control, H/R, and H/R+emodin. At the end of each treatment, the culture medium was replaced with 100 μL of CCK-8 solution (containing 90 μL of serum-free DMEM with 10 μL of CCK-8 reagent). Absorbance at 450 nm was measured in each well by visualization of color intensity development.
Western blot analysis
Heart tissue samples (50–100 mg) and cardiomyocytes samples were lysed, the samples were centrifuged at 12,000× g for 15 minutes, and then the supernatants were collected. Protein samples (20 μg) were added, separated by SDS-PAGE gel (10% separation gel), and transferred to a PVDF membrane (Merck & Co., Inc., Whitehouse Station, NJ, USA, Germany). The membrane was blocked with 5% fat-free milk solution for 1 hour at room temperature and subsequently incubated overnight at 4°C with primary antibodies: TLR4 (ab22048, 1:1,000; Abcam, Cambridge, UK), IκBα (#4814, 1:1,000; Cell Signaling Technology, Danvers, MA, USA), phospho-IκBα (p-IκBα, #9246, 1:1,000; Cell Signaling Technology), NF-κB (p65, #8242, 1:1,000; Cell Signaling Technology), phospho-NF-κB (p-p65, #3033, 1:1,000; Cell Signaling Technology), MyD88 (#4283, 1:1,000; Cell Signaling Technology), ASC/TMS1 (#13833, 1:1,000; Cell Signaling Technology), NLRP3 (NBP2-12446, 1:1,000; Novus Biologicals, Littleton, CO, USA), caspase-1 (sc-56036, 1:200; Santa Cruz Biotechnology, Dallas, TX, USA), GSDMD (sc-393581, 1:200; Santa Cruz Biotechnology). After washing three times, immunoreactive bands were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (A0208, 1:2,000; Beyotime) and goat anti-mouse secondary antibody (A0216, 1:2,000; Beyotime). Proteins were detected with the ECL procedure (Bio-Rad). P-p65 was standardized by p65, and p-IκBα was standardized by IκBα. The expression of other proteins was standardized by GAPDH (#5174, 1:1,000; Cell Signaling Technology).
Real-time PCR analysis
Hearts from the SD rats were used to prepare total RNA using TRIzol Reagent according to the manufacturer’s protocol (Thermo Fisher Scientific). One microgram of total RNA from each sample was used to generate cDNAs using the RevertAidTM First Strand cDNA Synthesis Kit (#K1622; Thermo Fisher Scientific). The resultant cDNA was amplified by SYBR (#RR037A; Takara Biotechnology, Dalian, China). The PCR reaction was directly monitored by the ABI7500 platform. All results were normalized against GAPDH (B661204; Sangon Biotech, Shanghai, China).
The real-time PCR used the following primers:
GSDMD: Forward primer 5′-CCAACATCTCAGGGCCCCAT-3′-
Reverse primer 5′-TGGCAAGTTTCTGCCCTGGA-3′-
IL-1β: Forward primer 5′-CACCTCTCAAGCAGAGCACAG-3′-
Reverse primer 5′-GGGTTCCATGGTGAAGTCAAC-3′-
GAPDH: Forward primer 5′-GACATGCCGCCTGGAGAAAC-3′-
Reverse primer 5′-AGCCCAGGATGCCCTTTAGT-3′.
H&E staining
Heart tissue specimens were fixed in 4% paraformaldehyde, processed in graded alcohol, processed in xylene, and then embedded in paraffin. The paraffin-embedded tissue specimen of each sample was cut into 5 μm thick sections. After rehydration, the sections were stained with H&E. The stained sections were then viewed under a microscope (Olympus Corporation, Tokyo, Japan).
ELISA
After various treatments, the concentration of IL-1β in supernatants or in serum was measured by ELISA kit (R&D Systems). Assays were performed according to the protocols included with the kit. Absorbance at 450 nm was measured in each well by visualization of color intensity development.
Immunohistochemistry
Fresh tissue was fixed in 4% paraformaldehyde and embedded in paraffin. Five-micron sections were obtained, deparaffinized, and rehydrated as previously described. After antigen retrieval, endogenous peroxidase was blocked using 3% hydrogen peroxide at room temperature for 10 minutes. Sections were blocked with 5% BSA and then incubated with primary antibody (GSDMD, 1:50) in a humid chamber at 4°C overnight, followed by incubation with an HRP-conjugated secondary antibody (1:200) at room temperature for 1 hour. After color development through incubation with diaminobenzidine, the sections were counterstained with hematoxylin. The developed tissue sections were visualized under a microscope (Olympus Corporation).
Measurement of ROS production
Dihydroethidium (DHE, S0063; Beyotime) was used to measure ROS production in cardiomyocytes according to the manufacturer’s instruction. Cardiomyocytes were incubated in 2.5 μM DHE for 30 minutes at 37°C in a dark environment, and then washed with PBS for three times. Fluorescence was detected by fluorescence microscope (Olympus Corporation). Malondialdehyde (MDA, S0131; Beyotime) and superoxide dismutase (SOD, S0101; Beyotime) assay was also used to measure ROS production according to the manufacturer’s instruction.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (pH 7.4) at room temperature for 10 minutes and washed with PBS 3×5 minutes. Subsequently, cells were permeabilized with 0.25% Triton X-100 in PBS for 10 minutes at room temperature. Cells were then washed with PBS 3×5 minutes, blocked with 1% BSA for 30 minutes, and incubated for 1 hour at room temperature in 1% BSA containing Ki67 (ab15580, 1 μg/mL; Abcam). Cells were then washed for 3×5 minutes with PBS, followed by incubation with secondary antibody (ab6718, 1:500; Abcam) at room temperature for 1 hour, and washed for 3×5 minutes with PBS. Cell nucleus was stained with DAPI (C1006; Beyotime) served for cell localization.
Statistical analysis
All values were presented as the mean ± SD, and statistical analyses were performed using GraphPad Prism 7 (GraphPad, San Diego, CA, USA). The assumptions of normality were checked using Shapiro–Wilks test and equal variance was checked using Levene’s test. The unpaired two-tailed t-test was used to compare variables between two groups. A one-way ANOVA followed by a multiple comparisons test with a Tukey’s correction was employed to analyze the differences within multiple groups. A P-value <0.05 was considered significant.
Results
GSDMD mRNA levels and its pyroptosis-inducing fragment GSDMD-N were upregulated in the heart after I/R injury
To investigate the relationship between pyroptosis and I/R, we first evaluated the expression of GSDMD and IL-1β in the heart between the sham group (n=8) and I/R group (n=8).
Both GSDMD and IL-1β mRNA levels were upregulated in the I/R group compared with those in the sham group (Figure 1A and B). Although the protein expression of GSDMD-full length (FL) has not changed, the GSDMD-N was increased at the protein level (Figure 1C and D), and the increase in serum IL-1β was verified by ELISA (Figure 1E), suggesting that pyroptosis was increased in the myocardial I/R injury model. In addition, the cellular viability was decreased (Figure 1F) and the expression of GSDMD-N was increased when cardiomyocytes were treated with H/R (Figure 1G and H).
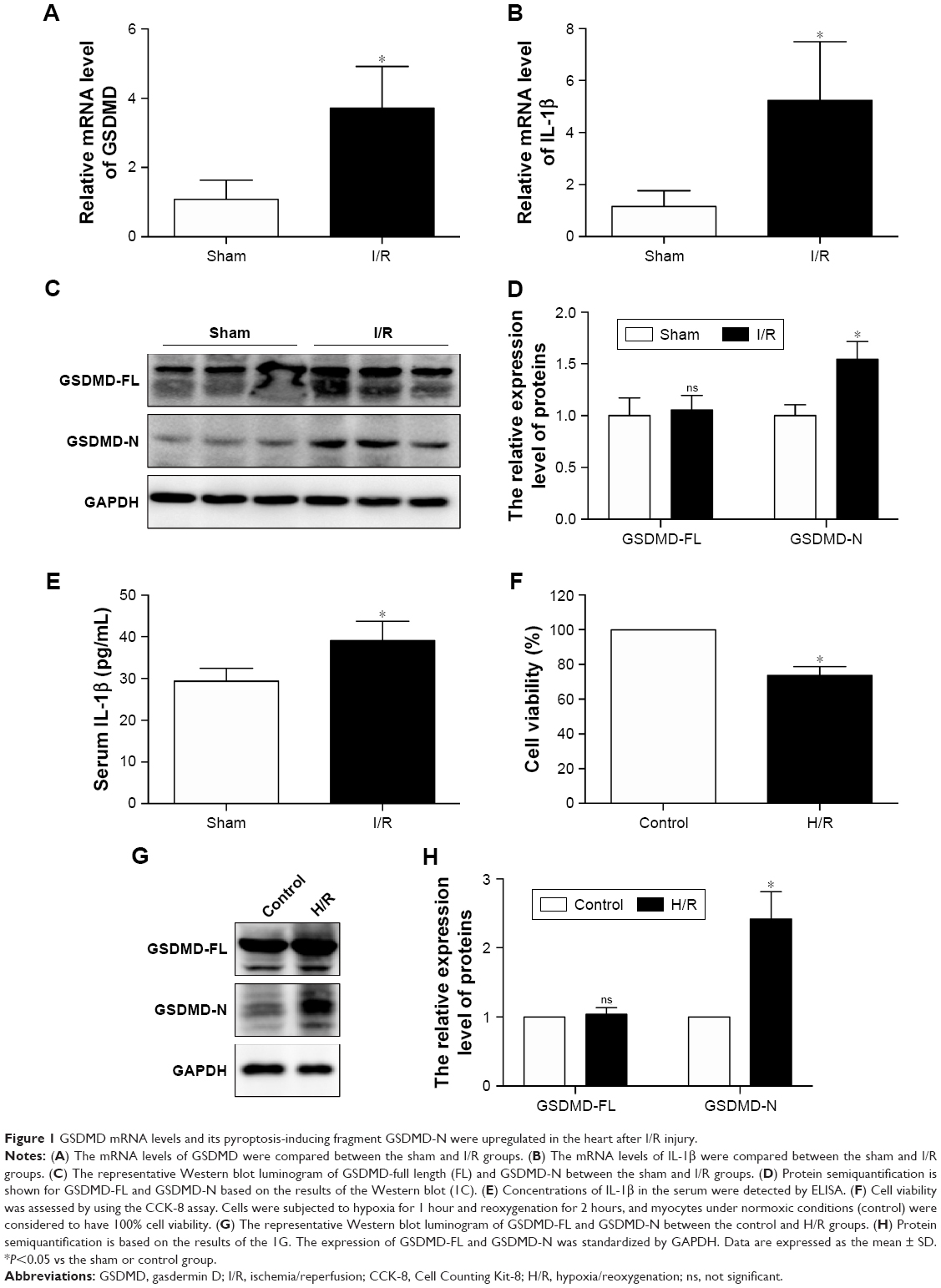 | Figure 1 GSDMD mRNA levels and its pyroptosis-inducing fragment GSDMD-N were upregulated in the heart after I/R injury. |
Emodin reduced infract size and ameliorated cardiomyocyte cell morphology
The structure of emodin used in this study is shown in Figure 2A. To evaluate the protective effect of emodin on myocardial I/R injury, the INF was measured. The AAR/LV ratio did not differ between I/R and I/R+Emodin (20 mg/kg) group. However, compared with the I/R group, the I/R+Emodin (20 mg/kg) group had a significantly decreased INF/AAR ratio, indicating that emodin protected rats from myocardial I/R injury (Figure 2B–D). H&E staining was used to examine the changes of cardiomyocyte cell morphology. Myocardial cells in the sham group were intact and arranged in order. However, in the I/R group, myocardial cells were disordered and swollen, myofibrils were contractured, and sarcolemma was disrupted. In the I/R+Emodin (20 mg/kg) group, the number of cells that were swollen was much lower than that in the I/R group (Figure 2E and F).
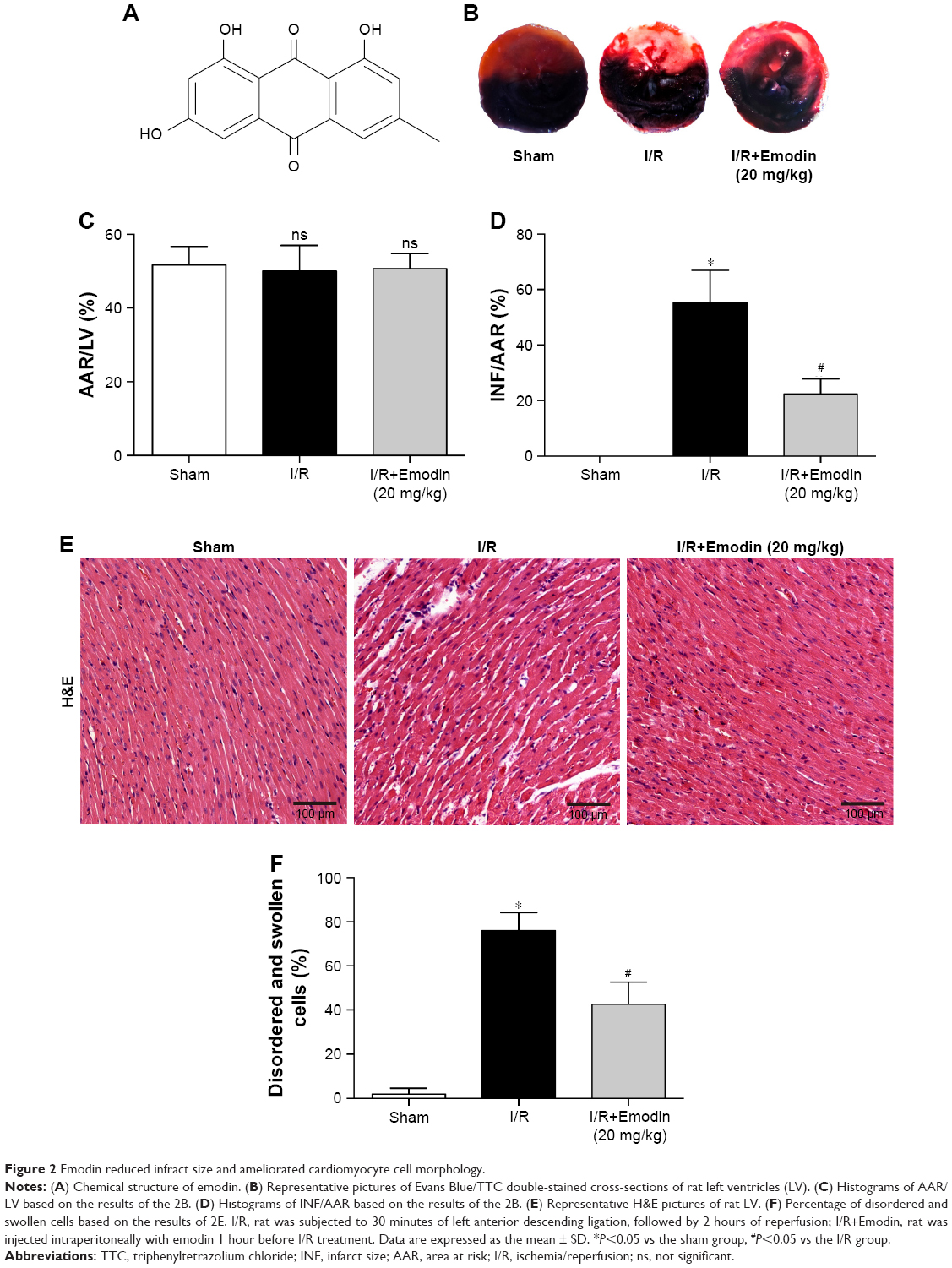 | Figure 2 Emodin reduced infract size and ameliorated cardiomyocyte cell morphology. |
Emodin protected cardiomyocytes from H/R-induced pyroptosis
The concentration ranging from 0 to 80 μM for 1 hour was used to find the optimal concentration of emodin, which is not cytotoxic (Figure 3A). Although 20 μM of emodin has no statistical significance in cell viability, it began to show a tendency toward decreased cell viability. So, we finally chose emodin at a concentration of 2.5, 5, and 10 μM for the next experiment. Meanwhile, emodin enhanced the survival of primary cardiomyocytes exposed to H/R (Figure 3B). LDH measurement confirmed these results (Figure 3C and D).
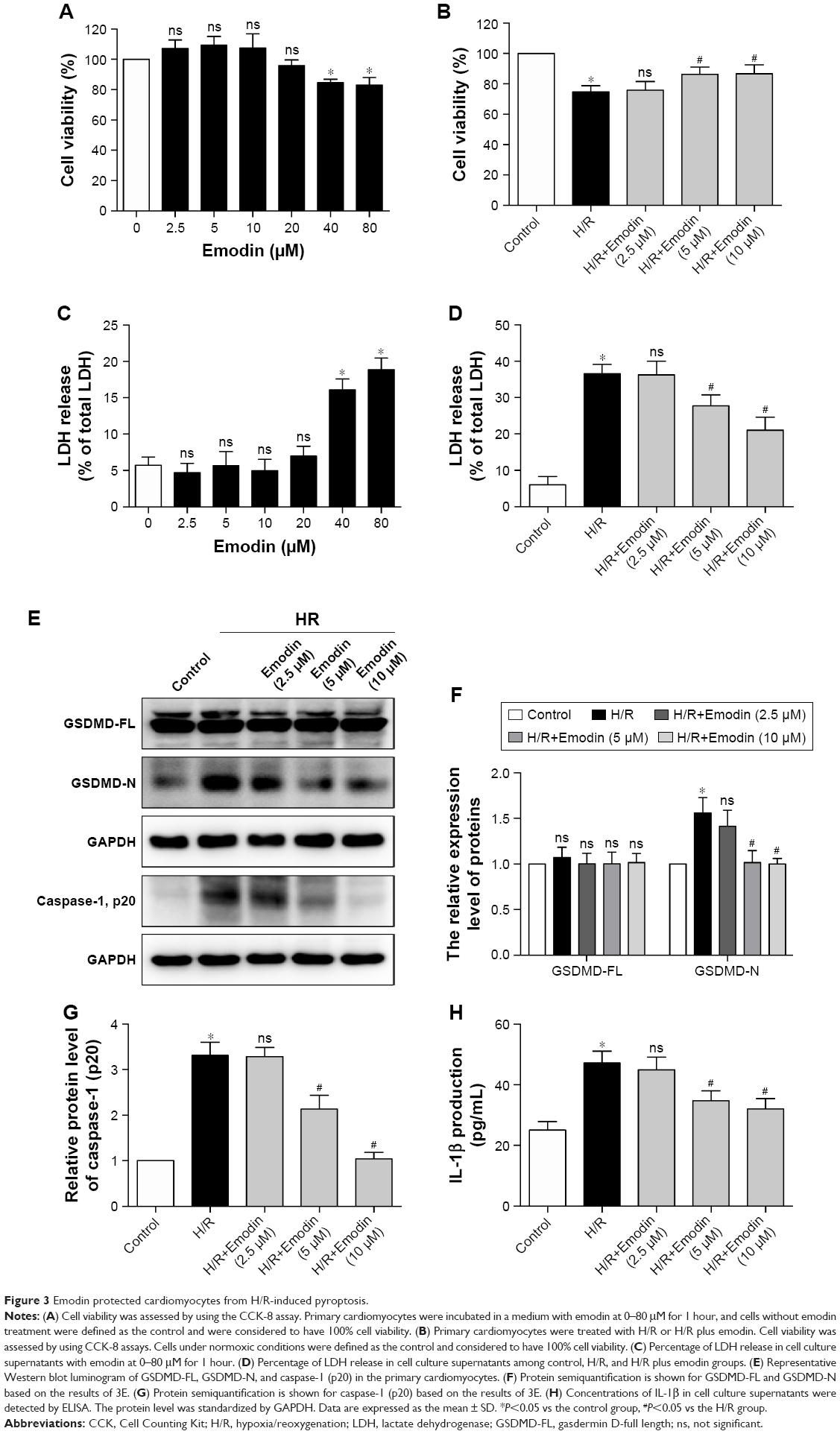 | Figure 3 Emodin protected cardiomyocytes from H/R-induced pyroptosis. |
GSDMD-N and caspase-1 (p20) in the primary cardiomyocytes and the concentration of IL-1β in the supernatant were increased after H/R exposure. Pretreatment with emodin significantly reduced the expression of GSDMD-N and caspase-1 (p20) in the primary cardiomyocytes in a concentration-dependent manner (Figure 3E–G). Pretreatment with emodin also decreased the concentration of IL-1β in the supernatant (Figure 3H). However, no significant changes occurred with the expression of GSDMD-FL (Figure 3E and F). Ki67 is a well-known proliferation marker for the evaluation of cell proliferation. Mammalian cardiomyocytes, unlike H9c2 myoblast, have only limited proliferative capacity. Ki67 can hardly be detected in primary cardiomyocytes (Figure S1).
Emodin treatment attenuated I/R-induced myocardial pyroptosis in vivo
Since we have shown that emodin treatment enhanced the survival of primary cardiomyocytes exposed to H/R by suppressing excessive pyroptosis, we wondered whether this mechanism is relevant during myocardial I/R in vivo. The expression of GSDMD-N was greatly induced in the left ventricle tissue of rats after I/R injury (Figure 4A and B), suggesting that pyroptosis was activated in the cardiac myocytes exposed to I/R. Pretreatment with emodin in the I/R group significantly reduced INF (Figure 2B–D) and decreased serum IL-1β levels (Figure 4C), suggesting that pyroptosis plays an adverse role during I/R and leads to cardiomyocyte death. Emodin (20 mg/kg) treatment repressed the induction of GSDMD-N; however, emodin did not affect the expression of GSDMD-FL (Figure 4A and B). The immunohistochemistry analysis of cardiac tissues (Figure 4D) was consistent with Western blot analysis. These findings were consistent with results from our in vitro experiments (Figure 3E–H).
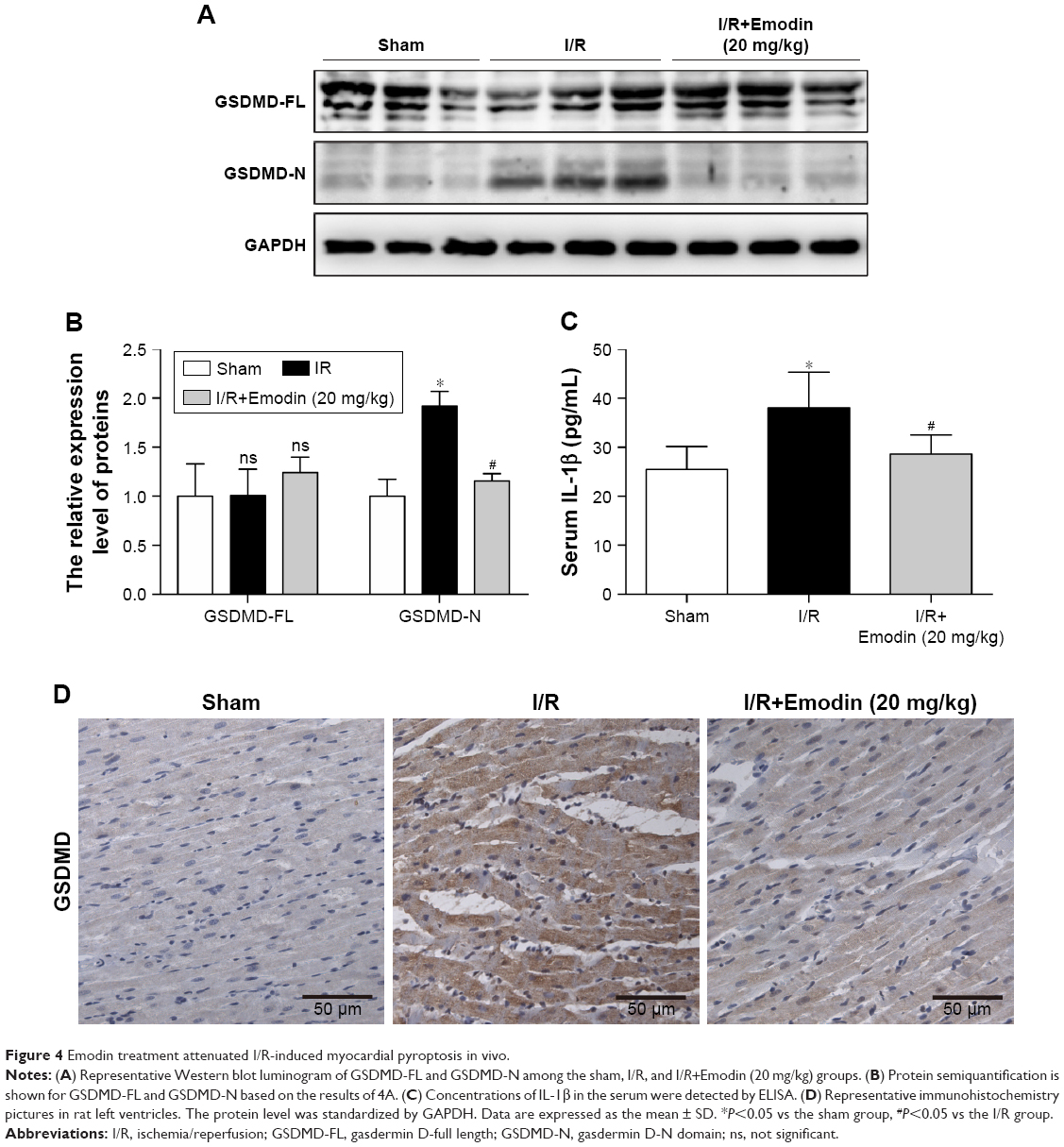 | Figure 4 Emodin treatment attenuated I/R-induced myocardial pyroptosis in vivo. |
Taken together, these findings suggest that pyroptosis is highly induced in rat cardiac myocytes after I/R injury, and that emodin treatment reduces pyroptosis activation in vivo.
Inhibition of pyroptosis by emodin involves the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway
TLR4/MyD88/NF-κB participate in NLRP3 inflammasome activation.24 The NLRP3 inflammasome is involved in the activation of caspase-1, and activated caspase-1 cleaves GSDMD to generate N-terminal fragments.25 Thus, we monitored the changes in TLR4 expression in H/R-induced cardiomyocytes and noticed that H/R stimulation increased the expression of TLR4. The expression level of TLR4 was reduced in a concentration-dependent manner in the emodin treatment group (Figure 5A and B).
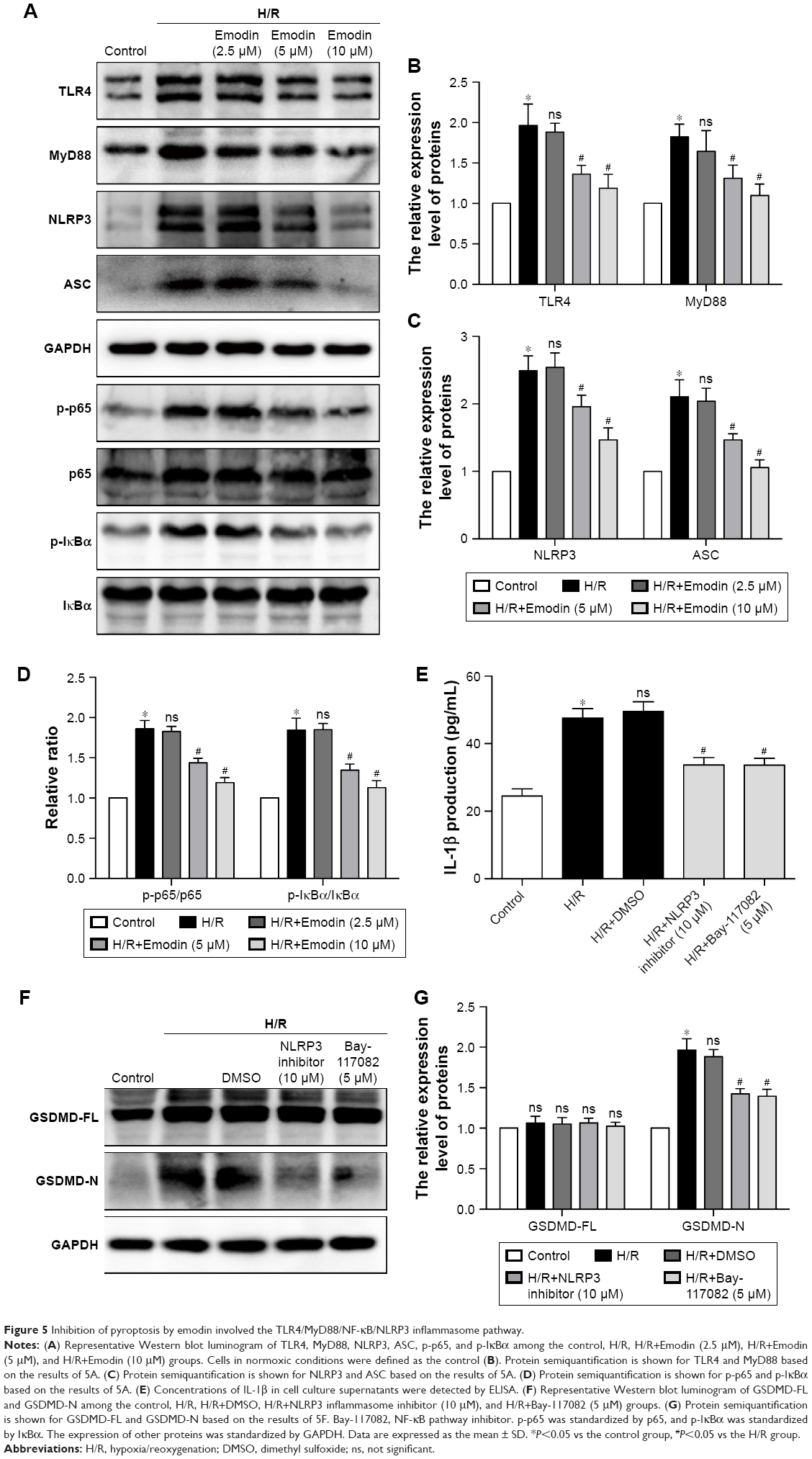 | Figure 5 Inhibition of pyroptosis by emodin involved the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway. |
TLRs recruit the intracellular TIR-domain-containing adaptor protein MyD88, triggering downstream activation of NF-κB.26 Therefore, we examined the expression of these associated downstream signaling molecules in cardiomyocytes by Western blot analysis. H/R increased the expression of MyD88, p-IκBα, p-p65, the NLRP3 inflammasome, and ASC in primary cardiomyocytes. Emodin treatment dramatically downregulated the H/R-induced expression of MyD88, p-IκBα, p-p65, the NLRP3 inflammasome, and ASC in primary cardiomyocytes in a concentration-dependent manner (Figure 5A–D).
As mentioned earlier, the NF-κB/NLRP3 inflammasome is involved in the activation of caspase-1; therefore, we wanted to know whether inhibiting the NF-κB/NLRP3 inflammasome could reduce pyroptosis. The primary cardiomyocytes were pretreated with 10 μM of NLRP3 inflammasome inhibitor or 5 μM of Bay-117082 (NF-κB pathway inhibitor) for 1 hour before the H/R stimulation. When cells were cultured in the presence of the NLRP3 inflammasome inhibitor or Bay-117082, partial inhibition of the expression of GSDMD-N and IL-1β was observed (Figure 5E–G). This result suggests that the inhibition of the NF-κB/NLRP3 inflammasome signaling pathway by emodin attenuates pyroptosis in H/R-induced cardiomyocytes.
Emodin inhibited the expression of TLR4 by decreasing ROS production
Although emodin reduces both the mRNA and protein expression of TLR4 in lipopolysaccharide (LPS)-induced acute liver injury,27 the potential mechanism still needs to be clarified. Previous studies demonstrated that NAC (a ROS scavenger) reduced high glucose-induced TLR4 expression in H9c2 cardiomyocytes28 and mouse podocytes.29 So we hypothesized that emodin reduced the expression of TLR4 by attenuating production of ROS. We found that pretreatment with emodin (10 μM) decreased MDA level induced by H/R (Figure 6A) and increased SOD level (Figure 6B). DHE fluorescence also confirmed the antioxidant activity of emodin (Figure 6C and D). Pretreatment with NAC for 1 hour reduced H/R-induced TLR4 expression (Figure 6E and F).
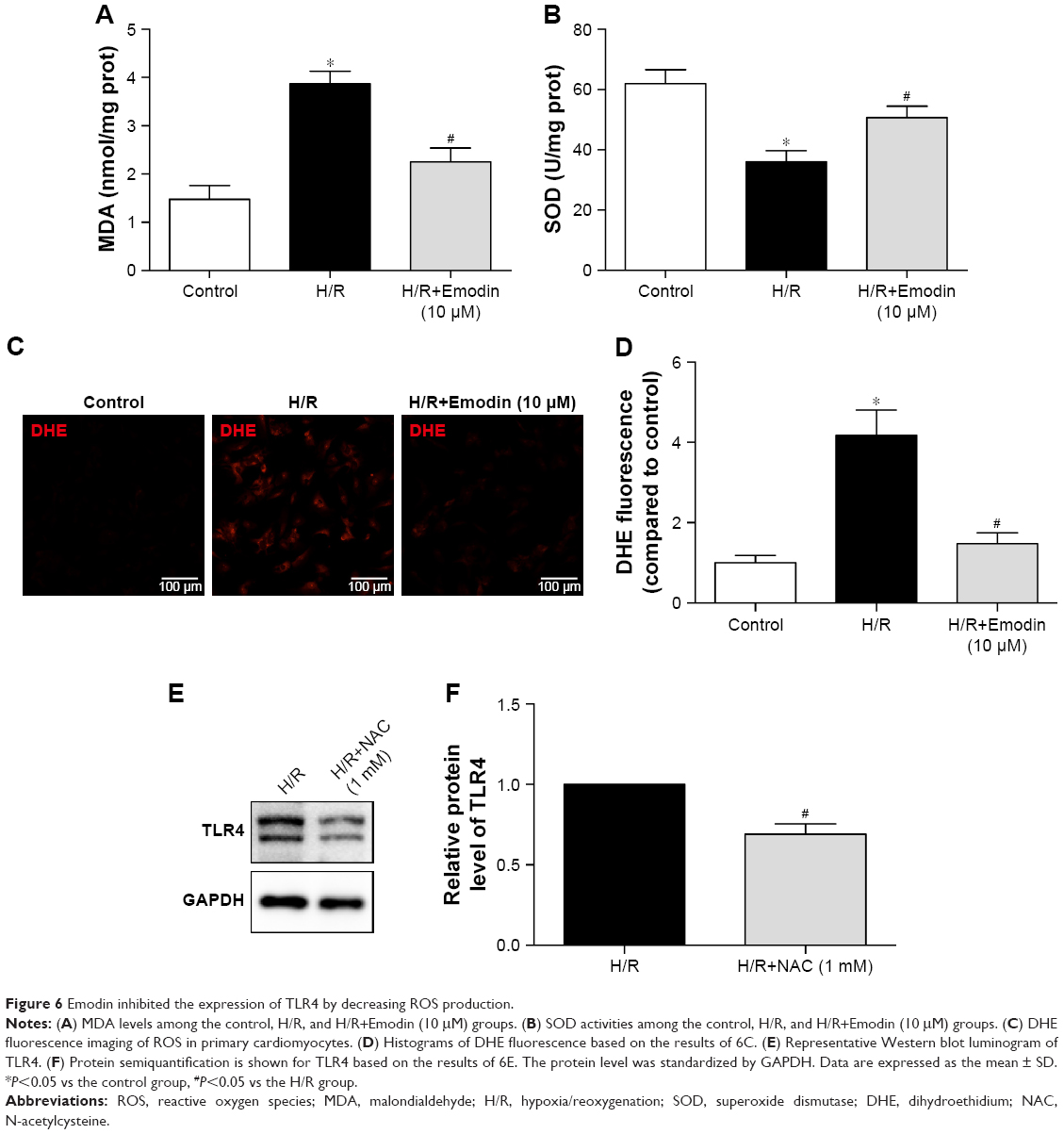 | Figure 6 Emodin inhibited the expression of TLR4 by decreasing ROS production. |
All these findings suggest that emodin inhibited the expression of TLR4, at least partly, by decreasing ROS production.
Discussion
This study shows that GSDMD-induced pyroptosis is increased in myocardial I/R injury in rats, and emodin treatment partially inhibits pyroptosis in rats in vivo and in vitro. The inhibitory effect of emodin on pyroptosis is mediated by suppressing the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway. Therefore, these results suggest a novel therapeutic target for emodin to ameliorate myocardial I/R injury.
Shortening the door-to-balloon time reduces ischemic injury, but mortality is still very high. Therefore, we should pay more attention to reperfusion injury.2 ROS, intracellular Ca2+ overload, and rapidly changing pH are involved in reperfusion injury;6 however, treatment is limited. Finding a new therapeutic target to suppress I/R injury is imperative.
An increasing amount of research has revealed that inflammation plays a critical role in myocardial I/R injury. Attenuating inflammation protects cardiomyocytes against I/R injury.30,31 GSDMD has been reported to perform proinflammatory programmed cell death and control the release of proinflammatory cytokines IL-1β and IL-18.12
Toll-like receptors recruit MyD88, leading to activation of NF-κB.26 NF-κB increases synthesis of NLRP3 and pro-IL-1β.32 At the same time, the NLRP3 inflammasome can be activated by many factors, such as K+ efflux,33 cell volume regulation,34 and oxidized mitochondrial DNA.35 The activated NLRP3 inflammasome cleaves pro-caspase-1 to produce activated caspase-1.36 GSDMD-FL could be cleaved by activated caspase-1 (canonical inflammasomes) or activated caspase-11 (non-canonical inflammasomes).9 Then, the GSDMD-N forms a large pore in the plasma membrane37 and controls IL-1β release.11 Subsequently, water enters the cell, causing cell swelling and eventual lysis.38 Therefore, GSDMD-N is the executioner of pyroptotic cell death. In our current study, H/R and emodin treatment did not affect the expression of GSDMD-FL protein; however, emodin treatment reduced the GSDMD-N expression and alleviated myocardial I/R injury.
Pyroptosis is closely correlated with many diseases. For example, pyroptosis facilitates cytokine secretion and lipogenesis, causing non-alcoholic steatohepatitis.39 Endothelial pyroptosis is increased in endotoxemia-induced lung injury.40 Nicotine promotes atherosclerosis through the pyroptosis of endothelial cells.41 However, whether pyroptosis participates in I/R injury is still unknown. These observations suggest that pyroptosis is activated and accompanied by an inflammatory response in myocardial I/R injury. We demonstrated that myocardial I/R injury increases the expression of GSDMD-N and promotes IL-1β release. Therefore, GSDMD could be used as a molecular target to protect the myocardium from I/R injury.
Traditional Chinese medicines are widely distributed throughout nature, and they are abundant and easily accessible. Emodin has recently been proven to decrease the release of inflammatory mediators (TNF-α, IL-1β, IL-6) and inhibit LPS-induced pulmonary inflammation, pulmonary edema, and MCP-1 expression in mice.42 In the current study, emodin treatment could alleviate cardiomyocyte I/R injury and reduce the expression of GSDMD-N and IL-1β.
Previous studies have shown that melatonin could alleviate inflammasome-induced pyroptosis by blocking NF-κB/GSDMD signaling in mouse adipose tissue.43 In addition, the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway is essential for activation of inflammation. Therefore, we examined whether NF-κB and NLRP3 inflammasome participate in the activation of pyroptosis in H/R-induced cardiomyocytes. In our study, H/R-activated pyroptosis, and NF-κB or the NLRP3 inflammasome inhibitor reduced the expression of GSDMD-N, decreasing the expression of IL-1β. In this report, H/R treatment increases the expression of TLR4, MyD88, p-IκBα, p-p65, and the NLRP3 inflammasome, which confirms the observations of previous investigators.23,44 Although TLR4 plays an important role in bacterial infection and sepsis, TLR4 can also be activated by ROS.45 These data demonstrated that I/R injury could activate pyroptosis, which are at least partially mediated by the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway. Emodin inhibited the expression of TLR4 by decreasing ROS production and reduced the expression of GSDMD-N by decreasing the expression of TLR4, MyD88, NF-κB, and the NLRP3 inflammasome (Figure 7).
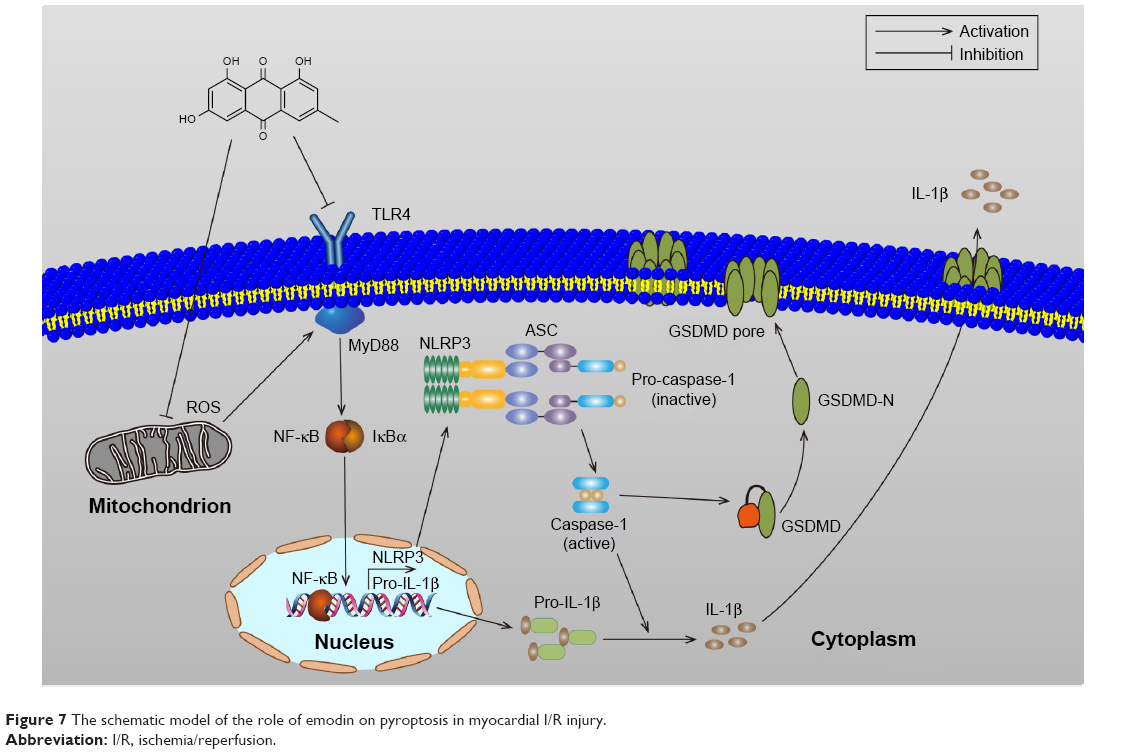 | Figure 7 The schematic model of the role of emodin on pyroptosis in myocardial I/R injury. |
Conclusion
Our current study provides strong evidence that emodin treatment is able to alleviate cardiomyocyte I/R injury and inhibit pyroptosis in vivo and in vitro. The inhibitory effect of emodin on pyroptosis is mediated by suppressing the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway. Therefore, emodin may be a potential therapeutic drug for treating myocardial I/R injury. Moreover, the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway may contain important therapeutic targets for myocardial I/R injury.
Acknowledgments
This study was funded by the Cardiac Rehabilitation and Metabolic Therapy Research Fund, National Natural Science Foundation of China (Grant No 81670227), the Natural Science Foundation of Zhejiang Province (LQ19H020004), the Traditional Chinese Medicine Administration of Zhejiang Province (Grant No 2016ZA137), and Wenzhou Science and Technology Bureau (Grant Nos Y20170045, Y20160030, and Y20180105). Xia Hong provided technical help in immunofluorescence and ROS production measurement.
Disclosure
The authors report no conflicts of interest in this work.
References
Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet. 2017;389(10065):197–210. | ||
Ibáñez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65(14):1454–1471. | ||
Hausenloy DJ, Yellon DM. Time to take myocardial reperfusion injury seriously. N Engl J Med. 2008;359(5):518–520. | ||
Oshiro Y, Shimabukuro M, Takasu N, Asahi T, Komiya I, Yoshida H. Triiodothyronine concomitantly inhibits calcium overload and postischemic myocardial stunning in diabetic rats. Life Sci. 2001;69(16):1907–1918. | ||
Maczewski M, Beresewicz A. The role of endothelin, protein kinase C and free radicals in the mechanism of the post-ischemic endothelial dysfunction in guinea-pig hearts. J Mol Cell Cardiol. 2000;32(2):297–310. | ||
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–1135. | ||
Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604. | ||
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114. | ||
Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–142. | ||
Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular Microbiol. 2006;8(11):1812–1825. | ||
Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. | ||
Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. | ||
Woodall MC, Woodall BP, Gao E, Yuan A, Koch WJ. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res. 2016;119(10):1116–1127. | ||
Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87. | ||
Oyama J, Blais C Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in Toll-like receptor 4-deficient mice. Circulation. 2004;109(6):784–789. | ||
Shibata M, Endo S, Inada K, et al. Elevated plasma levels of interleukin-1 receptor antagonist and interleukin-10 in patients with acute myocardial infarction. J Interferon Cytokine Res. 1997;17(3):145–150. | ||
Suzuki K, Murtuza B, Smolenski RT, et al. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104(suppl 1):I-308–I-313. | ||
Wu Z, Chen Q, Ke D, Li G, Deng W. Emodin protects against diabetic cardiomyopathy by regulating the Akt/GSK-3β signaling pathway in the rat model. Molecules. 2014;19(9):14782–14793. | ||
Li H, Yang T, Zhou H, Du J, Zhu B, Sun Z. Emodin combined with Nanosilver inhibited sepsis by anti-inflammatory protection. Front Pharmacol. 2016;7:536. | ||
Xue J, Chen F, Wang J, et al. Emodin protects against concanavalin A-induced hepatitis in mice through inhibiting activation of the p38 MAPK-NF-kappaB signaling pathway. Cell Physiol Biochem. 2015;35(4):1557–1570. | ||
Du Y, Ko KM. Effects of pharmacological preconditioning by emodin/oleanolic acid treatment and/or ischemic preconditioning on mitochondrial antioxidant components as well as the susceptibility to ischemia-reperfusion injury in rat hearts. Mol Cell Biochem. 2006;288(1–2):135–142. | ||
de Couto G, Gallet R, Cambier L, et al. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation. 2017;136(2):200–214. | ||
Izumi T, Saito Y, Kishimoto I, et al. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J Clin Invest. 2001;108(2):203–213. | ||
Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. | ||
Man SM, Kanneganti TD. Gasdermin D: the long-awaited executioner of pyroptosis. Cell Research. 2015;25(11):1183–1184. | ||
Ve T, Vajjhala PR, Hedger A, et al. Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat Struct Mol Biol. 2017;24(9):743–751. | ||
Ding Y, Liu P, Chen ZL, et al. Emodin attenuates lipopolysaccharide-induced acute liver injury via inhibiting the TLR4 signaling pathway in vitro and in vivo. Front Pharmacol. 2018;9:962. | ||
Liang W, Chen M, Zheng D, et al. The opening of ATP-sensitive K+ channels protects H9c2 cardiac cells against the high glucose-induced injury and inflammation by inhibiting the ROS-TLR4-necroptosis pathway. Cell Physiol Biochem. 2017;41(3):1020–1034. | ||
Wei M, Li Z, Xiao L, Yang Z. Effects of ROS-relative NF-κB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol Immunol. 2015;68(2 Pt A):261–271. | ||
Yang Z, Day YJ, Toufektsian MC, et al. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111(17):2190–2197. | ||
De Meyer SF, Savchenko AS, Haas MS, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5217–5223. | ||
Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol. 2017;18(8):861–869. | ||
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. | ||
Compan V, Baroja-Mazo A, López-Castejón G, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity. 2012;37(3):487–500. | ||
Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. | ||
Ives A, Nomura J, Martinon F, et al. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6(1):6555. | ||
Sborgi L, Rühl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35(16):1766–1778. | ||
Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. | ||
Xu B, Jiang M, Chu Y, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2018;68(4):773–782. | ||
Cheng KT, Xiong S, Ye Z, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124–4135. | ||
Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. | ||
Xiao M, Zhu T, Zhang W, et al. Emodin ameliorates LPS-induced acute lung injury, involving the inactivation of NF-κB in mice. Int J Mol Sci. 2014;15(11):19355–19368. | ||
Liu Z, Gan L, Xu Y, et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J Pineal Res. 2017;63(1):e12414. | ||
Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates I B kinase-beta, giving insight into hypoxia-induced NF B activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. | ||
Lorne E, Dupont H, Abraham E. Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Intensive Care Med. 2010;36(11):1826–1835. |
Supplementary material
 | Figure S1 Emodin has no effect on Ki67 expression in the H/R model. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.