")
Back to Journals » Psoriasis: Targets and Therapy » Volume 9
Emerging treatment options for the treatment of moderate to severe plaque psoriasis and psoriatic arthritis: evaluating bimekizumab and its therapeutic potential
Authors Chiricozzi A , De Simone C , Fossati B , Peris K
Received 14 February 2019
Accepted for publication 1 May 2019
Published 24 May 2019 Volume 2019:9 Pages 29—35
DOI https://doi.org/10.2147/PTT.S179283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Uwe Wollina
Andrea Chiricozzi,1,2 Clara De Simone,1,2 Barbara Fossati,2 Ketty Peris1,2
1Institute of Dermatology, Catholic University, Rome, Italy; 2Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
Abstract: Plaque psoriasis (PsO) is a chronic inflammatory skin disorder that may be associated with several comorbidities, including arthritis. The increasing knowledge of psoriasis pathogenesis led to the identification of novel targeted therapeutic interventions. Among them, anti-IL-17A and anti-IL-17F antibodies are currently being investigated for the treatment of PsO and/or psoriatic arthritis (PsA). Bimekizumab is one of these agents, capable ofsimultaneously neutralizing both IL-17A and IL-17F cytokines. In this review we included preclinical and clinical data related to this highly promising agent that shows a peculiar molecular structure differing from other bispecific agents.
Keywords: psoriasis, psoriatic arthritis, bimekizumab, bispecific agent, dual specific agent
Corrigendum for this paper has been published
Introduction
Psoriasis is a chronic inflammatory skin disorder that is associated with a large number of comorbid diseases, including psoriatic arthritis (PsA), that is reported in at least 20% of psoriasis cases.1 Although skin and joint inflammation is driven by a wide array of immune pathways, mounting evidence supported the central role of the interleukin (IL)-23/IL-17A axis in the psoriasis pathogenic model.2 The increased expression of IL-23 and IL-17, as detected in psoriatic patients, reflects the activation of both immune cells and tissue cells producing IL-23 which in turn potently stimulates different T cell subsets and innate lymphoid cells to release IL-17A, IL-17F, IL-17A/F, IL-22, and other inflammatory cytokines. Additional cells contributing to the increased IL-17A expression in psoriasis are represented by neutrophils and mast cells, though neutrophils appear to be IL-17A-storing cells and not IL-17A producers, while mast cells are considered a minor source of IL-17A.3 In addition to IL-23 and IL-17A, a variety of pro-inflammatory and/or proproliferative mediators including cytokines, chemokines, and antimicrobial peptides (AMPs) contribute to the development of the psoriatic phenotype.2 Among them, IL-17F, a homodimeric cytokine belonging to the IL-17 cytokine family, shows similarities with IL-17A, having both greater than 50% structural homology and overlapping biological functions.4 Likewise IL-17A, IL-17F is mainly produced by T helper (Th) 17 cells upon stimulation with IL-23, and is thought to potentiate IL-17A activity as it induces the expression of keratinocyte genes, though 30-fold less biologically active than IL-17A.5,6 IL-17A and IL-17F heterodimers can also be produced and they are supposed to have an intermediate biological potency compared to IL-17A and IL-17F homodimers. Increased expression of both IL-17A and IL-17F was found in psoriatic lesional and nonlesional skin, and in inflamed PsA synovia.7–12 In vitro studies assessing the effects of IL-17 family cytokines in monolayer keratinocytes, confirmed the overlapping effects of IL-17A and IL17F in stimulating the expression of AMPs (ie, DEFB4, LCN2, S100A9, S100A7A), chemokines (ie, CCL20, CXCL8), and cytokines (ie, IL-36γ).13 The in vitro keratinocyte transcriptomes induced by IL-17A and IL-17F significantly correlated with the psoriasis transcriptome, suggesting their contribution to psoriasis phenotype.13 In particular, IL-17A creates inflammatory reverberating loops, acting in synergism with TNF-α and IL-22, that are crucial in maintaining skin inflammation.14–16
Along with IL-17A, IL-17F can also cooperate with tumor necrosis factor-α (TNF-α) in inducing the production of key pro-inflammatory keratinocyte mediators.9–11,17 Primary normal human dermal fibroblasts and synoviocytes obtained from PsA patients, stimulated in vitro with recombinant TNF, IL17A and IL-17F, showed the contribution of IL-17F to chronic tissue inflammation. IL-17F was able to induce the expression of pro-inflammatory genes such as IL-6 and IL-8, though to a lesser extent compared to IL-17A.9–11,17 Additionally, a complex in vitro model explored the effects of the dual neutralization of IL-17A and IL-17F in suppressing inflammation, compared with the blocakde of IL-17A alone.10,11,17 Synoviocytes from PsA patients and dermal fibroblasts were treated with pro-inflammatory mediators derived from supernatant of sorted Th17 cells, and subsequently treated with anti-IL17A monoclonal antibodies, anti-IL17F monoclonal antibodies, or bimekizumab, a humanized monoclonal IgG1 antibody that can simultaneously neutralize both IL-17A and IL-17F.10,11,17 Dual inhibition induced by bimekizumab led to greater reduction of IL-6, IL-8, and other inflammatory gene (ie, CXCL1, CXCL2, CXCL3, and IL-15RA) expressions, compared to the reduced expression obtained by the IL-17A blockade.9,10,16 This study also assessed chemotactic potential of neutrophils towards Th17-stimulated dermal fibroblasts, implementing this in vitro model with neutrophils derived from whole blood of healthy donors. Greater suppression of neutrophil migration through transwell permeable membrane was detected using bimekizumab, compared to IL-17A or IL-17F blockade.9,10,16 These data provided the rationale to the clinical development of bimekizumab in both PsO and PsA. Conversely to other therapeutic monoclonal antibodies classified as bispecific agents neutralizing two different cytokines through two distinct binding sites, bimekizumab shows one binding site that simultaneously neutralizes IL-17A and IL-17F cytokines. Bispecific agents neutralizing both IL-17A and IL-17F include afasevikumab (also known as NI-1401), and ALX-0761 (also known as MSB0010841), a trivalent anti-IL-17A/F nanobody, consisting of an N-terminal IL-17F specific moiety, a-C terminal moiety that binds both IL-17A and IL-17F, and a central portion binding human serum albumin.17 Because bimekizumab shows a peculiar structure differing from these bispecific agents, it could be defined as dual specific agent (Figure 1).
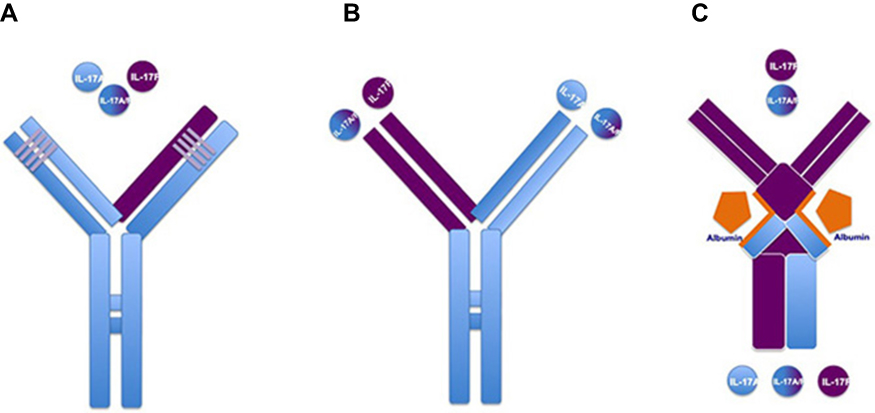 | Figure 1 Different molecular structures among therapeutic antibodies targeting both IL-17A and IL-17F. (A) Bimekizumab shows a unique binding site neutralizing IL-17A, IL-17F, and their heterodimers (dual specificity). (B) Afasevikumab (also known as NI-1401) shows two distinct binding sites neutralizing IL-17A (light blue chain) and IL-17F (purple chain) (bispecificity). (C) ALX-0761 (also known as MSB0010841) represents a trivalent anti-IL-17A/F nanobody, consisting of an N-terminal IL-17F specific moiety, a-C terminal moiety that binds both IL-17A and IL-17F, and a central portion binding human serum albumin which stabilizes the molecule for plasma half-life extension. |
Material and methods
We carried out a search of the English-language literature regarding the pathogenic role of IL-17A and IL-17F in psoriasis and PsA, in addition to publications reporting preclinical and clinical data on bimekizumab for treatment of psoriasis and PsA utilizing the following databases: PubMed, Embase, Google Scholar, ResearchGate, and Scopus. Key words used were: “‘psoriasis’”, “psoriatic arthritis”, “psoriasis pathogenesis”, ‘“IL-17Aʹ”, “IL-17”, ”IL-17F”, “bimekizumab”, “psoriasis pipeline”. All published articles plus data from recent international meetings were reviewed.
Clinical studies testing bimekizumab for the treatment of psoriasis
The first-in-human, double-blind, placebo-controlled, single-dose, dose-escalating Phase I study (NCT02529956) randomized 39 subjects with mild-to-moderate plaque psoriasis to receive escalating intravenous doses of bimekizumab (8 mg, 40 mg, 160 mg, 480 mg, and 640 mg) or placebo.19 Patients had psoriasis involvement lower than 5% of whole body surface area, mean lesion severity score (LSS) ranging from 4.2 to 5.0 across all bimekizumab and placebo groups, median Psoriasis Area and Severity Index (PASI) ranging from 2.60 to 3.75 across all bimekizumab and placebo groups. All subjects treated at baseline with a single bimekizumab dose of 8 mg, 40 mg, 160 mg, 480 mg, or 640 mg were followed for 20 weeks and completed the study with the exception of two subjects who withdrew the study because of adverse events (AE). Treatment-emergent AEs (TEAEs) were observed in 84.6% and 76.9% of subjects treated with bimekizumab and placebo, respectively.
The majority of TEAEs were of mild intensity (61.5% in all bimekizumab groups vs 53.8% in the placebo group). Only one serious AE occurred, which was, however, not classified as treatment-related.19 Commonly reported TEAEs occurring in ≥10% of all subjects receiving bimekizumab were headache, oropharyngeal pain, nasopharyngitis, and ECG alterations (not clinically meaningful). No infusion-site reactions, study discontinuations due to TEAEs, severe TEAEs, or deaths were reported. Mean neutrophil counts were similar between treatment groups and no subject experienced clinically meaningful changes in neutrophil count.19 Bimekizumab pharmacokinetics linearly increased with dose. Half-life ranged from 17 days (bimekizumab 40 mg) to 22 days (bimekizumab 160 mg) across the treatment groups.19 Notably, antibimekizumab antibodies were detected and confirmed in one subject prior to dose administration while five subjects (5/26, 19.2%) developed antibimekizumab antibodies during the follow-up period. Nevertheless, the detection of antibimekizumab antibodies did not affect pharmacokinetic parameters.
Skin amelioration was observed in patients treated with higher bimekizumab doses (160 mg, 480 mg, and 640 mg) by week 2, reaching the maximal improvement between weeks 4 and 6, that was maintained through 16–20 weeks.19 Fast onset of response, evaluated as mean reduction of >80% from baseline LSS as early as week 2, was detected in patients group treated with 480 mg and 460 mg bimekizumab.19 Maximal improvement of baseline PGA score consisting of 75%, 100%, and 94% reduction, was measured in the 160 mg, 480 mg, and 640 mg bimekizumab group, respectively. Maximal PASI score reduction was >85% across the three highest bimekizumab dose groups. This Phase I study was followed by a multicenter, randomized, double-blinded, placebo-controlled, parallel-group, dose-ranging, Phase IIb study (named BE ABLE 1 trial: NCT02905006; extension study: NCT03010527) investigating efficacy and safety of bimekizumab vs placebo in adult patients affected by moderate to severe chronic plaque psoriasis with mean baseline PASI score ranging from 18.4 to 20.6.20 The BE ABLE 1 study consisted of 12-week treatment period followed by an extension phase, and included 250 psoriasis patients who were randomized 1:1:1:1:1:1 to receive subcutaneous bimekizumab every 4 weeks at doses of 64 mg, 160 mg, 160 mg with 320 mg loading dose, 320 mg, 480 mg, or placebo.20 Primary endpoint of this study was the achievement of PASI90 response at week 12. Secondary endpoint included the reduction of at least two categories or the achievement of clear or almost clear skin condition as defined by Investigator’s Global Assessment (IGA) at week 8 and at week 12, PASI90 response at week 8, PASI75 response at week 12, and PASI100 response at week 12.20 Most randomized patients, 224/250 subjects (89.6%), completed the 12-week study whereas 26 interrupted treatment due to AEs (N=5); lack of efficacy (N=1); protocol violations (N=2); lost to follow-up (N=2); withdrew consent (N=3), and for other reasons (N=13). Rapid and marked PASI score reduction was observed since week 4. A significantly higher percentage of bimekizumab-treated patients achieved both primary and secondary endpoints compared to the placebo group. In detail, PASI90 (primary endpoint) was achieved by a significantly higher proportion of patients treated with each bimekizumab dose compared with placebo (41.0–86.0% vs 0%; P<0.0001, all comparisons) at week 8.20
A significantly higher rate of patients in all bimekizumab-treatment groups obtained PASI75, PASI90, and PASI100 response at week 12 (Figure 2).20 In particular, PASI75 and PASI90 response was more frequently observed in the bimekizumab 320 mg group (93% and 79.1%, respectively) compared to placebo (4.8% and 0%, respectively), while the highest PASI100 response (60%) was detected in the bimekizumab 160 mg with loading dose of 320 mg (vs 0% placebo).
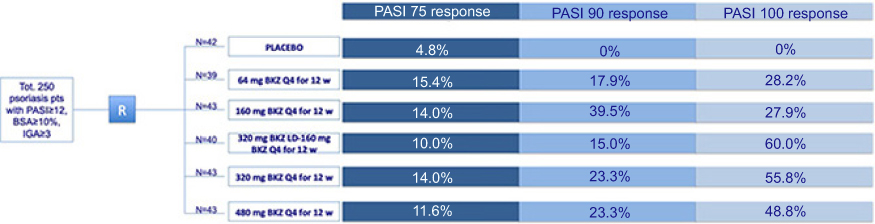 | Figure 2 Bimekizumab efficacy in treating plaque psoriasis patients, assessed in a phase II trial. Clinical outcomes in psoriasis patients after 12-week therapy with placebo or different bimekizumab dosages. Abbreviations: BKZ, bimekizumab; BSA, body surface area; IGA, investigator global assessment; PASI, psoriasis area severity index; pts, patients; Q4, every 4 weeks; R, randomization; tot, total; w, weeks. |
Bimekizumab efficacy also resulted significantly higher compared to placebo in terms of both absolute PASI values and IGA score reduction, over time.20
Safety data set showed treatment-emergent adverse events (TEAEs) in 61% of bimekizumab-treated patients versus 36% of placebo-treated patients. The most commonly reported TEAEs (>5% patients in any group) at week 12 were nasopharyngitis, upper respiratory tract infections, arthralgia, γ-glutamyltransferase increase, neutropenia, rhinitis, tonsillitis, hypertension, oral candidiasis, headache, leukopenia, and vomiting.20 Fungal infections including oral candidiasis, vulvovaginal mycotic infection, and tinea pedis were reported in 9 (4.3%) of bimekizumab-treated patients. Such AEs could be expected because of the protective role of both IL-17 cytokines against fungal infections. However, they led to treatment discontinuation in 4.8% of bimekizumab-treated patients and in 2.4% of the placebo group.20 Main limitations of this Phase II trial are linked to the short study duration. Defining the primary endpoint at week 12, the clinical response assessed at this timepoint was based on two bimekizumab injections only, thereby, any further beneficial effect related to drug exposure has not been explored and we cannot assess whether the maximum efficacy is achieved after a 12-week-observation period.
Clinical studies testing bimekizumab for the treatment of psoriatic arthritis
A phase Ib, double-blind, placebo-controlled, proof-of-concept trial randomized patients with PsA to bimekizumab (N=39) or placebo (N=14) for 20 weeks.11 Patients were included if tender joints and swollen joints ≥3, and either erythrocyte sedimentation rate was ≥28 mm/hour or high sensitivity C reactive protein was >3 mg/L; inadequate response to ≥1 nonbiological disease-modifying antirheumatic drug (DMARD, including methotrexate) and/or one approved biological DMARD. Different intravenous bimekizumab dose regimens were tested concomitantly with the use of anti-inflammatory and antirheumatic products, folic acid, analgesics, and DMARDs. Particularly, all patients reported the concomitant use of anti-inflammatory and antirheumatic products, while 84.6% also used DMARDs. Baseline characteristics included mean swollen joint count ranging from 10.3 to 14.0, across all groups, while tender joint count ranged from 20.5 to 33.4, across all groups. Disease severity was assessed by the American College of Rheumatology (ACR) score. Patients receiving bimekizumab had greater joint improvement obtaining higher rates of ACR20 (at least 20% improvement of baseline ACR score), ACR50, and ACR70 responses, compared with placebo.11 Primary efficacy endpoint (ACR20 at week 8) was obtained in 80% of patients, while ACR50 was observed in 40%, and ACR70 in 23% of bimekizumab-treated patients.11 Maximal ACR20 response rate (80% of bimekizumab-treated patients) was observed at week 8, while the highest rate of ACR50 and ACR70 response (achieved in 57% and 37% of patients, respectively) was observed at week 12 and 16, respectively. Notably, clinical response was evident as early as week 2 and it was maintained to week 20. In patients with skin involvement ≥3% body surface area, week 8 response rates for PASI75 and PASI100 were 100% and 87%, respectively.11 No warning signals related to the bimekizumab safety profile were detected, with the majority of TEAEs classified as mild or moderate, while serious AEs were not related to the study drug.11 A more recent multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging, Phase IIb (BE ACTIVE) study enrolled 206 patients with PsA.21 Patients were randomized to receive subcutaneous bimekizumab 16 mg, 160 mg, 160 mg with 320 mg loading dose, 320 mg, or placebo every 4 weeks, during the 12-week double-blind treatment period, followed by re-randomization to the different dose-blind bimekizumab treatment groups (160 mg or 320 mg) for 36 weeks for those patients who received placebo or 16-mg bimekizumab.21 All other patients continued on their previous dose. The overall treatment duration was 48 weeks. ACR50 (primary endpoint) response at week 12 was detected in up to 46% of bimekizumab-treated patients, compared to 7% of placebo-treated patients (Figure 3).21 At the same timepoint, ACR 20 was observed in 51.2–70.7% of patients and ACR70 in 12.2–31.7% of patients across all bimekizumab groups. Resolution of enthesitis, as defined by MASES index, was reported in 26.3–59.1% across all bimekizumab-treated patients, and in 28.6% of placebo-treated patients. Joint improvement was progressively greater achieving higher rates of ACR20,50,70 and the resolution of enthesitis at week 24 and 48.
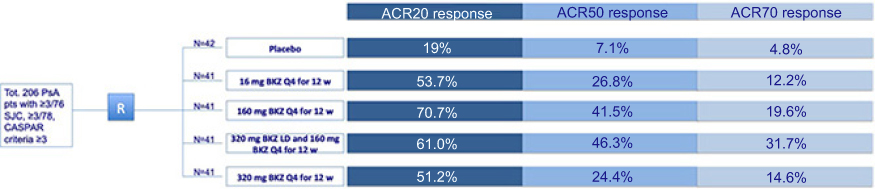 | Figure 3 Bimekizumab efficacy in treating psoriatic arthritis patients, assessed in a phase II trial. Clinical outcomes in psoriatic arthritis patients after 12-week therapy with placebo or different bimekizumab dosages. Abbreviations: ACR, American College of Rheumatology; BKZ, bimekizumab; CASPAR, Classification Criteria for Psoriatic Arthritis; pts: patients; Q4, every 4 weeks; R, randomization; SJC, swollen joint count; TJC, tender joint count; tot, total; w, weeks. |
These results were achieved in a mixed patient population constituted by both biologic naïve and biologic-exposed patients, with 19% of patients prior exposed to TNF inhibitors.21
Across bimekizumab groups, patients with skin involvement ≥3% body surface area, had PASI90 response in 20.7–53.8% compared to placebo (7.1%), while PASI75 was achieved by 44.8–76.9% of patients compared to placebo-treated patients (7.1%), at week 12.21,22 These response rates progressively increased at week 24 and week 48 achieving PASI90 response in a large proportion of patients across the different bimekizumab dose groups (45.5–78.6% at week 24, and 69.6–100% at week 48).
Safety data anaylsis revealed no apparent relationship between dose and TEAEs. Serious AEs were reported by 4.4% (9/206) of whole study population up to week 48 (1/206 [0.5%] receiving placebo and 8/206 [3.9%] receiving bimekizumab). The most common TEAE was nasopharyngitis (12.1%, 25/206), while oral candidiasis was observed in 4.9% (10/206) of all bimekizumab-treated cases at week 48. No deaths, cases of new-onset inflammatory bowel diseases, or major adverse cardiac events (MACE) were reported.21,22 Notwithstanding the limited number of patients is in line with phase II development, clinical outcomes were meaningful for a preliminary evaluation and interpretation, likely because of an extended observation period.
Preliminary data on clinical efficacy and safety of other agents neutralizing both IL-17A and IL-17FThe development of afasevikumab (NI-1401) is a human monoclonal IgG1κ antibody targeting IL-17A and IL-17F. One phase I trial in “autoimmune disorders” was performed, completing phase I, and phase II trial program was planned. Currently, no clinical data have been released. On the contrary clinical outcomes on ALX-0761 (M1095) have been recently published.23 ALX-0761 (M1095) was tested in one multicenter, double-blind, placebo-controlled dose-escalation phase Ib study that randomized 44 patients 4:1 to subcutaneous ALX-0761 (M1095) (30, 60, 120, or 240 mg) or placebo bi-weekly for 6 weeks, in 4four ascending dose cohorts.23 A favorable safety profile with no treatment-related serious AEsadverse events was reported in ALX-0761 (M1095)-–exposed patients. The most frequent TEtreatment-emergent AEs were pruritus (N=4) and headache (N=3); two2 patients withdrew due to AEs (injection site reaction, and elevated liver enzymes). Five of 10 M1095-treated patients (positive for anti-drug antibodies) showed treatment-emergent anti-drug antibody responses with no effect on ALX-0761 (M1095) exposure.23 Efficacy was assessed by both PASI score and static physician global assessment (sPGA) that showed dose-dependent improvements of skin lesions. Six weeks after the last dose of ALX-0761 (M1095), 100% and 56% of patients receiving the highest dose of ALX-0761 (M1095), 240 mg, achieved PASI90 and PASI 100 response, respectively. Improvements in static physician’s global assessment, and affected body surface area were also seen.23 Clinical amelioration was associated with dose-dependent reduction in dermal and epidermal CD3+ cell counts, epidermal thickness, and epidermal Ki67+ cell count. Moreover, histologic analysis revealed a decreased expression of pro-inflammatory and/or AMPantimicrobial peptide genes in lesional skin induced by IL-17A and IL-17F, whereas IL-17A- and IL-17F-induced genes in the blood were unaltered.23 Nevertheless, the interpretation of efficacy data is limited by the small sample size (8 patients each group, excepted the 240 mg- ALX-0761 (M1095) group having 9 subjects) and short-term treatment duration consisting of 6 weeks (total of administered injections: 3).
Conclusion
Preclinical studies have demonstrated a more potent suppression of inflammatory signals induced by dual blockade of IL-17A and IL-17F compared to the selective inhibition of a single cytokine, either IL-17A or IL-17F. This dual inhibition is able to suppress inflammation-linked gene expression, as well as disease-relevant immune cell migration.9–11 Bimekizumab represents a dual specific humanized monoclonal antibody showing a molecular structure designed to bind to both IL-17A and IL-17F at one site, that distinguishes bimekizumab from bispecific agents owing two different binding sites to IL-17A and IL-17F, such as afasevikumab and ALX-0761.18 These IL-17A/IL-17F bispecific biologic agents are being tested in phase I and II trials, while bimekizumab entered the phase III program. Preliminary data from the dose-ranging Phase II trial, BE ABLE, testing bimekizumab in plaque psoriasis patients are encouraging as bimekizumab was proven effective and rapid in clearing skin lesions and met the stringent study primary endpoint consisting ofachieving PASI90 response at week 12. Patients treated with highest bimekizumab doses obtained PASI90 response in 72.1–79.1%, while PASI100 response was achieved by 48.8–60% of patients. Mean absolute PASI score reduction confirmed the remarkable bimekizumab efficacy reaching a mean PASI score lower than five for all bimekizumab dose groups, particularly, the highest bimekizumab doses obtained residual mean PASI score close to one. Bimekizumab efficacy was also demonstrated in treating PsA, as revealed by the BE ACTIVE trial showing an elevated and rapid efficacy of bimekizumab in clearing skin and improving joint inflammation in psoriatic arthritis patients. Data are remarkable, especially because the study included ACR50 and PASI90, rigorous and meaningful efficacy endpoints, though data are limited to both short observational period and restricted patient cohort. Because neutropenia, worsening of inflammatory bowel diseases, and candida infections may be considered a potential class-effect of IL-17 and IL-17 receptor inhibitors, a clinically meaningful aspect that needs to be confirmed in the long-term period is the bimekizumab safety profile. Furthermore, long-term and comparative efficacy data related to the treatment of both plaque psoriasis and psoriatic arthritis will derived from the five trials constituting the current phase III program, in particular, data from the head-to-head studies testing bimekizumab vs ustekinumab (NCT03370133), adalimumab (NCT03412747), or secukinumab (NCT03536884) will be of great interest.24–26
Disclosure
Dr Andrea Chiricozzi has been an advisory board member and consultant, and has received fees and speaker’s honoraria or has participated in clinical trials for Abbvie, Biogen, Fresenius Kabi, Leo Pharma, Lilly, Novartis, UCB-Pharma, Sanofi, and Janssen. The other authors, Prof. Dr Clara De Simone, Dr Barbara Fossati, and Professor Ketty Peris report no conflicts of interest in this work.
References
1. Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64:ii14–ii17. doi:10.1136/ard.2004.032482
2. Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M. Scanning the immunopathogenesis of psoriasis. Int J Mol Sci. 2018;19:E179. doi:10.3390/ijms19010179
3. Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol. 2018;55(3):379–390. doi:10.1007/s12016-018-8702-3
4. Zhang X, Angkasekwinai P, Dong C, Tang H. Structure and function of interleukin-17 family cytokines. Protein Cell. 2011;2(1):26–40. doi:10.1007/s13238-011-1006-5
5. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. 2018;10:a028522. doi:10.1101/cshperspect.a028522
6. Wright JF, Bennett F, Li B, et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol. 2008;181:2799–2805.
7. Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190(5):2252–2262. doi:10.4049/jimmunol.1201505
8. Chiricozzi A, Suárez-Fariñas M, Fuentes-Duculan J, et al. Increased expression of interleukin-17 pathway genes in nonlesional skin of moderate-to-severe psoriasis vulgaris. Br J Dermatol. 2016;174:136–145. doi:10.1111/bjd.14034
9. Maroof A, Smallie T, Archer S, et al. Dual IL-17A and IL-17F inhibition with bimekizumab provides evidence for IL-17F contribution to immune-mediated inflammatory skin response. J Invest Dermatol. 2017;137:S120. doi:10.1016/j.jid.2017.02.722
10. Maroof A, Baeten D, Archer S, Griffiths M, Shaw S. IL-17F contributes to human chronic inflammation in synovial tissue: preclinical evidence with dual IL-17A and IL-17F inhibition with bimekizumab in psoriatic arthritis. Ann Rheum Dis. 2017;76:A13–A.
11. Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77:523–532. doi:10.1136/annrheumdis-2017-212127
12. van Baarsen LG, Lebre MC, van der Coelen D, et al. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther. 2014;16:426. doi:10.1186/s13075-014-0426-z
13. Norsgaard H, Hebsgaard J, Ewald D, et al. Multiple IL-17 cytokines, signalling through IL-17 receptor A, drive inflammatory pathways in psoriasis.
14. Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Investig Dermatol. 2011;131:677–687. doi:10.1038/jid.2010.340
15. Chiricozzi A, Nograles KE, Johnson-Huang LM, et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE. 2014;9:e90284. doi:10.1371/journal.pone.0090284
16. Guilloteau K, Paris I, Pedretti N, et al. Skin inflammation induced by the synergistic action of IL-17A, IL-22, Oncostatin M, IL-1β, and TNF-α recapitulates some features of psoriasis. J Immunol. 2010;184:5263–5270. doi:10.4049/jimmunol.0902464
17. Maroof A, Okoye R, Smallie T, et al. Bimekizumab dual inhibition of IL-17A and IL-17F provides evidence of IL-17F contribution to chronic inflammation in disease-relevant cells. Ann Rheum Dis. 2017;76(Suppl1):A1–A103.
18. Torres T, Romanelli M, Chiricozzi A. A revolutionary therapeutic approach for psoriasis: bispecific biological agents. Expert Opin Investig Drugs. 2016;25:751–754. doi:10.1080/13543784.2016.1187130
19. Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991–1001. doi:10.1111/bcp.13185
20. Papp KA, Merola JF, Gottlieb AB, et al. Dual neutralization of both interleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12-week randomized, double-blinded, placebo-controlled phase 2b trial. J Am Acad Dermatol. 2018;79:277–286.e10. doi:10.1016/j.jaad.2018.03.037
21. Ritchlin CT, Kavanaugh A, Merola JF, et al. Dual neutralization of IL-17A and IL-17F with bimekizumab in patients with active PsA: results from a 48-week phase 2b, randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheumatol. 2018;70(suppl):10.
22. Jancin B. Here comes bimekizumab, the newest IL-17 inhibitor. Available from:
23. Svecova D, Lubell MW, Casset-Semanaz F, Mackenzie H, Grenningloh R, Krueger JG. A randomized, double-blind, placebo-controlled phase 1 study of multiple ascending doses of subcutaneous M1095, an anti-interleukin-17A/F Nanobody®, in moderate-to-severe psoriasis. J Am Acad Dermatol. 2019;
24. Clinicaltrials.gov [homepage on the internet]. Phase 3 trial testing bimekizumab versus ustekinumab (BE VIVID). Available from: https://clinicaltrials.gov/ct2/show/NCT03370133?term=bimekizumab&phase=2&rank=5. Accessed May 17, 2019.
25. Clinicaltrials.gov [homepage on the internet]. Phase 3 trial testing bimekizumab versus adalimumab (BE SURE). Available from:
26. Clinicaltrials.gov [homepage on the internet]. Phase 3 trial testing bimekizumab versus secukinumab (BE RADIANT). Available from:
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.