")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Emerging treatment options for the management of pemphigus vulgaris
Authors Kridin K
Received 21 January 2018
Accepted for publication 4 March 2018
Published 27 April 2018 Volume 2018:14 Pages 757—778
DOI https://doi.org/10.2147/TCRM.S142471
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Khalaf Kridin
Department of Dermatology, Rambam Health Care Campus, Haifa, Israel
Abstract: Pemphigus vulgaris (PV) is a life-threatening disease belonging to the pemphigus group of autoimmune intra-epidermal bullous diseases of the skin and mucosae. The therapeutic management of PV remains challenging and, in some cases, conventional therapy is not adequate to induce clinical remission. The cornerstone of PV treatment remains systemic corticosteroids. Although very effective, long-term corticosteroid administration is characterized by substantial adverse effects. Corticosteroid-sparing adjuvant therapies have been employed in the treatment of PV, aiming to reduce the necessary cumulative dose of corticosteroids. Specifically, immunosuppressive agents such as azathioprine and mycophenolate mofetil are widely used in PV. More recently, high-dose intravenous immunoglobulins, immunoadsorption, and rituximab have been established as additional successful therapeutic options. This review covers both conventional and emerging therapies in PV. In addition, it sheds light on potential future treatment strategies for this disease.
Keywords: azathioprine, meycophenolate mofetil, rituximab, intravenous immunoglobulins, immunoadsporption, emerging
Introduction
Pemphigus vulgaris (PV) is a rare and life-threatening disease belonging to the pemphigus group of autoimmune intra-epidermal bullous diseases. Pathophysiologically, the underlying intraepithelial blister formation is caused by immunoglobulin G (IgG) autoantibodies against the desmosomal adhesion proteins (mainly desmoglein [Dsg]3, but also Dsg1) found on epidermal keratinocytes, thus resulting in acantholysis. PV is clinically characterized by widespread flaccid blisters and erosions of the skin and mucous membranes.1 The severity of PV refers to its progressive course, which is often accompanied by a high catabolic state with loss of body fluids and proteins as well as by secondary bacterial and viral infections that may lead to septicemia.2
Before the advent of systemic corticosteroids, the prognosis of pemphigus was almost fatal within the 2 years following initial presentation.2 The rapid tapering of corticosteroids after achieving control of the disease, as well as the introduction of novel immunosuppressants with less long-term adverse events, have decreased both morbidity and mortality of pemphigus patients in the past few decades.3 With proper treatment, the 1-year mortality rate of PV has been drastically reduced, but still stands at approximately 5%, with most deaths occurring due to secondary effects of immunosuppressive agents rather than as a result of the disease itself or disease sequelae.3–5
The aim of treatment in PV is to induce and maintain remission, which clinically corresponds to the cessation of new vesicle formation, healing of old erosions, and the completion of treatment tapering to maintenance doses.6 Subsequently, the real challenge is to prevent relapse in the long term and to avoid adverse events associated with the prolonged use of corticosteroids and immunosuppressive agents.2 There is a scarcity of high-quality randomized controlled trials (RCTs), partially due to the previous lack of standardized outcome measures. Given the rarity of the disease and, thus, the low number of patients available for studies, progress in RCTs has been slow. In many of these trials, sample sizes were small and occasionally deemed insufficient to yield conclusive results.7 Moreover, the durations of follow-up were variable across different RCTs. RCTs with larger sample sizes and longer follow-up periods, therefore, are highly necessary to conduct a long-term risk–benefit analysis of the therapeutic interventions reported in PV. Although an effort has been made to introduce common definitions and end points in PV treatment by the European Dermatology Forum (EDF)2 and the British Association of Dermatologists (BAD),8 there is no international consensus in regard to the treatment strategy for PV.
This article aims to assess and present an updated review of the literature on the efficacy and safety of the different interventions for the treatment of PV, as well as to discuss emerging therapies and concepts on the future management of the disease.
Corticosteroids
For decades, systemic corticosteroids have been used as the cornerstone of management for PV. The advent of systemic corticosteroids in the 1950s led to a dramatic reduction in mortality from 75% to 30% among patients with pemphigus.5 The efficacy of corticosteroids is well established. Control of disease activity is usually achieved within several weeks, whereas complete remission on minimal treatment (≤10 mg/day predniso(lo)ne) requires months; however, complete remission off treatment may require several months or even years of therapy.9
Recommendation in PV
The guidelines of the EDF and BAD define systemic corticosteroids as the first-line treatment of PV, recommending initial treatment with predniso(lo)ne at a dose of 0.5–1.5 mg/kg/day (EDF) and 1.0 mg/kg/day (BAD).2,8 If control of the disease is not achieved within 1–2 weeks, higher predniso(lo)ne doses (up to 2 mg/kg) may be administered. Progressive reduction of oral corticosteroid treatment should be commenced as soon as disease control is reached. Stepped tapering of predniso(lo)ne by a 25% reduction biweekly (or over a more extended time period if <20 mg/day) is recommended. If the reappearance of >3 lesions occurs during tapering of oral corticosteroid therapy, the recommendation is to readminister the last dose. At relapse, oral corticosteroid therapy should be increased until two steps back in the previous dose until control of the lesions is achieved, within 2 weeks.2 However, the optimal dose has not been thoroughly validated by RCTs.
Systemic corticosteroids can be combined with an immunosuppressive agent, particularly when complications due to prolonged use (>4 months), such as hypertension, diabetes mellitus, and osteoporosis, are expected.2 In cases where doses of predniso(lo)ne exceeding 100 mg/day are required, pulse treatment with either oral or intravenous steroids may be considered.10 The aim of high-dose pulse therapy is to reduce the duration, cumulative dose, and iatrogenic effects of corticosteroids.11 However, the benefit of oral corticosteroid pulses on top of conventional first-line treatment with oral predniso(lo)ne and immunosuppressive adjuvants is debatable.2,12,13
The main drawback of this treatment remains its substantial adverse effects due to the prolonged course and the high dosages required to induce remission (discussed in greater detail further). These multiple adverse effects may even contribute to poor prognostic outcomes and death.14 As these side effects correlate with the dose and duration of therapy, previous studies have attempted to identify the minimum dose required to induce and maintain remission.15
Mechanism of action
Corticosteroids function through interaction with the cytoplasmic corticosteroid receptor, resulting in upregulation of the expression of anti-inflammatory proteins and downregulation of the expression of pro-inflammatory proteins. The unbound corticosteroid enters cells and exerts its effects by binding to a cytoplasmic corticosteroid receptor, which leads to their translocation into the nucleus and the formation of a dimer that binds to corticosteroid response elements in the promoter region of certain genes. Approximately 10–100 genes in each cell are regulated directly by corticosteroids. Furthermore, the corticosteroid receptor inhibits transcription factors that have a dominant role in the inflammatory response as well as their co-activator molecules, such as cAMP response element binding protein, nuclear factor-κB (NF-κB), and activator protein 1 (AP-1).16–18
In B-cell-mediated diseases such as pemphigus, the therapeutic effect of corticosteroids may be mediated via downregulation of interleukin (IL)-2, which results in a reduction in both B-cell clone expansion and autoantibody synthesis. Moreover, the decrease in IL-2 suppresses cell-mediated immunity and reduces T-cell proliferation.19 The influence of corticosteroids on these multiple signal transduction pathways results in the anti-inflammatory, immunosuppressive, antiproliferative, and vasoconstrictive effects attributed to these compounds.20
Adverse events
Long-term systemic corticosteroid therapy at supraphysiologic doses is associated with an increased incidence of serious adverse events (summarized in Table 1).
 | Table 1 Adverse events of systemic corticosteroids divided by body systems |
Level of evidence
An RCT involving 22 patients with PV who were followed for 5 years did not detect any significant differences in terms of the duration of remissions and relapse rates in patients allocated to treatment with either low-dose oral prednisolone (45–60 mg/day) or high-dose oral prednisolone (120–180 mg/day) at the onset of PV.21
Two RCTs investigated the role of intravenous corticosteroid pulses in addition to oral predniso(lo)ne. In the first trial, one group received monotherapy with oral prednisone in an initial dose of 125 mg/day, whereas the other group was treated with three weekly pulses of intravenous betamethasone (20 mg/day for 4 days) in combination with oral prednisone (50 mg/day) during intervals. The pulse protocol was significantly superior in both, time to resolution of clinical manifestations, including oral lesions, and in safety profile.22 In the second trial, no statistical difference was observed between the addition of oral dexamethasone pulses (300 mg/day for three consecutive days) or placebo to the regimen of oral prednisolone (initially 80 mg/day, tapered over 19 weeks) in combination with azathioprine (3 mg/kg/day) in the following outcome measures: remission, death, relapse, and withdrawal rates. Moreover, the pulse treatment group experienced increased adverse events, with weight gain being the most commonly reported side effect.12 In summary, there is no evidence that pulsed corticosteroids are superior to conventional oral corticosteroids for maintenance of most cases of PV. However, short-term pulsed corticosteroids could be considered in recalcitrant PV to induce remission, especially if there has been no response to oral high doses.8
Adjuvant therapies
Immunosuppressive adjuvants can be administered in combination with systemic corticosteroids. According to the EDF guidelines, the combination of predniso(lo)ne with azathioprine or mycophenolate mofetil (MMF) is considered as second-line treatment, whereas the combination of predniso(lo)ne with rituximab, intravenous immunoglobulin (IVIg), immunoadsorption, cyclophosphamide, dapsone, or methotrexate is considered as third-line treatment in PV.2 The BAD guidelines advocate the use of azathioprine, MMF, and rituximab as first-line adjuvant therapies. If one of these agents fails to retain remission, switching to alternate first-line adjuvant agent is recommended as a second-line therapy. The administration of cyclophosphamide, IVIg, immunoadsorption, methotrexate, and plasmapheresis is recommended to be preserved as a third-line therapy.8
These adjuvant agents are commonly used in combination with corticosteroids, particularly in recalcitrant diseases, in cases of increased risk of corticosteroid therapy, when complications due to prolonged use of corticosteroid (>4 months) occur, or in dose dependency above the minimal therapy (>10 mg/day of predniso(lo)ne).2 However, there is only minimal evidence that the addition of adjuvants is superior to treatment with corticosteroids alone. Based on the current literature, adjuvants have only a steroid-sparing effect (allowing for reduced corticosteroid-related side effects) and may lead to steroid-free remission.2,7,23–27 A recent meta-analysis of clinical outcomes of RCTs investigating the efficacy of adjuvant therapy with azathioprine, MMF, cyclophosphamide, cyclosporine, IVIg, plasmapheresis, and infliximab in patients with PV concluded that adjuvants were not beneficial for achieving remission, but were found to collectively decrease the risk of relapse by 29% as compared to prednisolone monotherapy.28
In case of relapse in patients treated with oral corticosteroid monotherapy, the EDF guidelines recommend adding an adjuvant agent. If oral corticosteroids are already combined with an immunosuppressant during relapse, a change in first-line immunosuppressant or the use of a second-line immunosuppressant (eg, immunoadsorption, IVIg, or rituximab) may be discussed.2
Azathioprine
Mechanism of action
Azathioprine is a prodrug that converts non-enzymatically to 6-mercaptopurine after oral administration. This drug antagonizes purine metabolism and blocks the synthesis of DNA, RNA, and proteins. It may also impede cellular metabolism and inhibit mitosis. In addition to its effects on nucleic acid synthesis, azathioprine exerts its immunosuppressive effects in additional ways. It reversibly depletes the number of monocytes and Langerhans cells, reduces γ-globulin synthesis and T-cell lymphocyte function, affects the function of suppressor B cells, and may hinder the responses of B cells that depend on helper T cells.29–32
6-mercaptopurine can be inactivated to 6-methyl-mercaptopurine by the thiopurine methyltransferase (TPMT) enzyme. The activity of TPMT is determined genetically; approximately 10% of the population has a heterozygote genotype with intermediate enzymatic activity, and nearly 1 in 300 patients is either homozygous for mutant alleles or is a compound heterozygote with low enzymatic activity.33,34
Recommendations in PV
Azathioprine is one of the main adjuvants used in the treatment of PV and is recommended as a first-line adjuvant immunosuppressant, according to both the EDF and BAD guidelines.2,8
The dose of azathioprine is determined according to the activity of the TPMT enzyme involved in the metabolism of the drug. The measurement of TPMT activity before the initiation of azathioprine therapy is a promising approach for predicting both, possible toxicity and suboptimal doses of the drug.35 In patients with normal TPMT enzymatic activity, 2.0–2.5 mg/kg/day of azathioprine is usually administered, whereas 1 mg/kg/day is recommended for patients with intermediate or reduced TPMT activity. This drug should be avoided in patients without TPMT activity.36 A dosage of 50 mg/day should be initially started, and if no idiosyncratic reactions occur, the dose can be increased after 1 week to the optimal dose based on TPMT activity.
Adverse events
The safety profile of azathioprine is unfavorable, causing adverse reactions in 15%–30% of patients. Severe side effects include hematologic disturbances and myelosuppression (leukopenia, thrombocytopenia, anemia, and pancytopenia).31,37 Approximately 50% of myelosuppressive episodes occurring within the first 6 months of azathioprine therapy are attributable to low or intermediate TPMT enzymatic activity. Determining the TPMT enzymatic activity phenotype before commencing azathioprine has the potential to reduce myelosuppression by 25%–50%.31,34 However, some patients experience myelosuppression despite normal TPMT activity and, thus, all patients undergoing treatment with azathioprine need routine complete blood count monitoring throughout the treatment period.30,31 Besides genetic polymorphisms of TPMT, concurrent therapy with TPMT-inhibiting drugs, such as allopurinol or sulfasalazine, can also increase the risk of myelotoxicity.30 An abrupt increase in serum transaminase levels observed soon after administration of azathioprine is a clue for deficient TPMT activity.
Other severe adverse events are hepatotoxicity, pancreatitis, and arthralgia. Furthermore, long-term immunosuppression increases the risk of infections and neoplasia. Azathioprine is characterized by a mutagenic potential that leads to possible hematologic malignancies.38,39 Gastrointestinal symptoms (nausea, vomiting, anorexia, diarrhea, aphthous stomatitis), skin disorders (alopecia, maculopapular rash), and other rare adverse reactions may be observed.
Due to evidence of teratogenic risk, azathioprine use in pregnancy is not recommended; the same recommendation applies for breastfeeding because minimal concentrations of azathioprine and its metabolites have been found in breast milk.29,40
Level of evidence
Adjuvant azathioprine (2.5 mg/kg daily) with prednisolone was compared to prednisolone alone, adjuvant intravenous cyclophosphamide pulse therapy, and MMF in a four-regimen RCT involving 120 participants. This trial demonstrated that adjuvant azathioprine has a higher steroid-sparing effect (measured by cumulative corticosteroid dose) as compared to prednisolone alone and prednisolone with MMF.27
An RCT by Chams-Davatchi et al23 compared adjuvant azathioprine (2.5 mg/kg daily) to prednisolone alone in 56 participants. Over the study’s 1-year period of follow-up, azathioprine’s steroid-sparing effect was significant in the last trimester, but inconclusive in the first 9 months of treatment. Similarly, the activity of PV was comparable in the adjuvant-azathioprine and prednisolone-alone groups in the first 9 months after the initiation of therapy, but then, it was significantly lower in the azathioprine arm in months 10–12 following diagnosis.
A nonrandomized study compared high-dose oral prednisone daily (1.5 mg/kg/day) versus low-dose oral prednisone (40 mg/day) on alternate days plus azathioprine (100 mg/day) every day in 36 patients with PV. Both treatments resulted in high rates of clinical remission; however, the mean time to induction of remission was shorter in the monotherapy group and showed an increased rate of treatment-associated adverse events.41 In summary, despite the paucity of high-quality prospective RCTs, evidence exists to suggest that azathioprine has a superior corticosteroid-sparing effect relative to predniso(lo)ne monotherapy and MMF.8
Advantages and disadvantages
Azathioprine’s main advantages are its effective steroid-sparing effect and accessibility. Its main disadvantages are its adverse effects, which include increased risk of nausea, vomiting, myelosuppression, hepatitis, and pancreatitis, as well as the need for regular monitoring and its contraindication in pregnancy.
Mycophenolate mofetil
Mechanism of action
MMF is rapidly absorbed following oral administration and is then hydrolyzed to its active metabolite mycophenolic acid (MPA). The latter impairs the immune response via selectively inhibiting inosine monophosphate dehydrogenase, with resultant inhibition of the de novo pathway of purine synthesis in T and B cells. In contrast to most other cells, lymphocytes rely on the de novo pathway, more than the salvage pathway, for purine biosynthesis. As MPA specifically inhibits the de novo pathway, lymphocytes are the primary target of MPA action in vivo. This minimizes undesirable effects on other cell types and accounts for the favorable safety profile, as compared to other less selective adjuvant immunosuppressants, such as azathioprine. This mechanism results in the inhibition of lymphocyte proliferation and can cause suppression of both cellular and humoral immunity.35,42,43
Recommendation in PV
MMF is a safe steroid-sparing agent that is recommended as a first-line adjuvant immunosuppressant, based on both the EDF and BAD guidelines.2,8 It is typically prescribed in divided doses amounting to a total dose of 2–3 g/day, with reduced dosages in patients with renal impairment.44,45 Therapy should be started at a low dose of 500 mg daily and increased by 500 mg as tolerated until the desired final dose is reached. An eventual dose of 2 g/day is recommended for better gastrointestinal tolerance.2,35 Improvement is usually observed within 2 months of initiation.45 The relatively high cost and lack of availability as compared to other immunosuppressive agents limit the use of this agent.
Adverse events
MMF is generally well tolerated, and serious adverse events are rarely observed. The adverse events are predominantly gastrointestinal and include diarrhea, nausea, vomiting, and abdominal discomfort. These symptoms are dose dependent and can be attenuated by dose modification, gradual dosage increments, or ingesting the tablet along with food.45 Less frequent adverse effects include esophagitis, gastritis, genitourinary symptoms, hematologic abnormalities (<5% of patients),45 and opportunistic infections due to immunosuppression, including invasive cytomegalovirus infection.31,46 The intravenous administration of MMF is commonly complicated by superficial thrombophlebitis and thrombosis.31
Malignancies did not develop during the follow-up period among pemphigus patients who underwent MMF therapy;47 however, studies of transplant recipients on MMF have shown that it may increase the risk of some cancers, such as lymphomas and skin cancers.31,35 The use of MMF during pregnancy is associated with increased risk of spontaneous abortion and congenital malformations.40
Level of evidence
In an RCT, MMF (2 or 3 g/day) plus oral prednisone was compared with placebo plus prednisone in 96 patients with mild or moderate PV. Adjuvant MMF was not found to be superior to prednisone plus placebo in inducing disease control. However, the MMF group responded faster, reached a sustained response sooner, and showed a longer duration to relapse. The cumulative dose of prednisone was lower in the MMF arm, suggesting a significant steroid-sparing effect.25
In a prospective RCT including 40 patients with pemphigus (of whom 33 had PV), treatment with oral methylprednisolone plus adjuvant azathioprine was compared to oral methylprednisolone plus MMF. No significant differences in cumulative corticosteroid dose, efficacy, or adverse events were observed between the two immunosuppressive agents.24
The four-regimen RCT of Chams-Davatchi et al27 did not demonstrate any significant difference in efficacy or safety between treatment with prednisolone plus MMF (2 g/day), prednisolone as monotherapy, or prednisolone in combination with azathioprine and cyclophosphamide. In terms of its steroid-sparing effect, MMF was superior when compared to prednisolone alone, inferior when compared to azathioprine, and showed non-conclusive results when compared to cyclophosphamide.
In regard to the optimal dose in PV, Beissert et al’s25 study of 96 participants showed that the effect of a low to standard dose (2 g daily) versus a high dose (3 g daily) of MMF was inconclusive.
Advantages and disadvantages
MMF’s main advantages are its hepatic and renal safety as well as its effective steroid-sparing properties. Its main disadvantages include the need for ongoing monitoring and cost.
Rituximab
Mechanism of action
Rituximab is a monoclonal humanized antibody directed against the B-cell-specific cell surface antigen CD20. Rituximab binds to CD20-expressing B lymphocytes, which include the immature B cells in bone marrow, autoantigen-activated follicular B cells, autoantigen-activated marginal zone B cells, and memory B cells. B stem cells and plasma cells are not targeted. After the binding of rituximab to its cell-surface receptor, CD20-positive B cells are killed by a combination of antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, and apoptosis, and remain absent from the circulation for 6–12 months. The removal of mature CD20-positive B lymphocytes committed to differentiate into autoantibody-producing plasma cells is considered the major effect of rituximab and makes its use particularly attractive in autoimmune diseases wherein pathogenically relevant autoantibodies are present such as in pemphigus. Furthermore, rituximab achieves favorable effects in autoimmune diseases mediated by T cells, such as rheumatoid arthritis. This suggests additional mechanisms of action unrelated to plasma cell function, including autoantigen processing and presentation, help for autoreactive T cells, and production of T-cell-modulating cytokines.31 Because there is a close interaction between B and T cells, selective B-cell depletion indirectly results in a reduction of a subset of T lymphocytes.48,49
Though autoantibody titers are expected to decline with clinical improvement,48 a clinical response to treatment is not always closely reflected by changes in autoantibody titers;50 in some cases, patients who had entered a remission of their mucosal lesions continued to have high titers of anti-Dsg3 antibodies.50 Nonetheless, changes in anti-Dsg1 antibody levels are closely well correlated with the evolution of skin lesions, both in pemphigus foliaceus (PF) patients and in PV patients with mucosal and skin involvement.51
Recommendation in PV
Rituximab is indicated for patients who remain dependent on >10 mg prednisolone in combination with an immunosuppressive adjuvant, according to the EDF guidelines.2 More recently, the BAD guidelines advocated the employment of rituximab as a first-line therapy following the landmark study of Joly et al.8,52 Administration of rituximab based on the literature is either an intravenous 1,000 mg every 2 weeks or 375 mg/m2 each week for 4 consecutive weeks, following treatment regimens used in rheumatoid arthritis and lymphoma, respectively. The same dosage can be readministered in the case of clinical relapse.10 The majority of patients generally respond within the first 3 months following the initiation of rituximab; however, late responses (after almost 1 year) have also been anecdotally reported.31
Adverse events
Mild to moderate infusion-related reactions such as fever and chills/rigors may appear in most patients during the first rituximab infusion. Other frequent infusion-related symptoms include nausea, pruritus, angioedema, hypotension, bronchospasm, throat irritation, rhinitis, urticaria, vomiting, myalgia, headache, dizziness, and hypertension. In most cases, these reactions manifest 30–120 minutes subsequent to the beginning of the first infusion and usually subside with the slowing or interrupting of the rituximab infusion as well as with symptomatic treatment. A premedication with paracetamol and prednisone may reduce the probability of infusion-related adverse effects.51
Serious adverse events associated with rituximab have been reported, including those that may result in patient mortality. These include severe mucocutaneous reactions; hepatitis B reactivation with fulminant hepatitis; progressive, multifocal leukoencephalopathy; other viral and opportunistic infections; cardiac arrhythmias; renal toxicity; bowel obstruction and perforation; and hematologic disturbances, such as lymphopenia, neutropenia, and anemia. Although uncommon, some of these serious adverse reactions have also been described in patients with PV.30 Rituximab is contraindicated in pregnant or breastfeeding women and in individuals with hepatitis B or C, HIV, or sepsis.51,53,54 The incidence of serious infection was 3.9% in the weekly treatment protocol, but 15.21% in the biweekly protocol.55
Life-threatening adverse effects were noted as preferentially affecting individuals with underlying malignancy, in children or those older than 60 years, and in patients who had been on high-dose immunosuppressive regimens preceding (rather than concomitant) the treatment with rituximab.30,53 Overall, the prevalence of serious adverse events among patients with autoimmune bullous disease under rituximab seems to be higher than the prevalence observed in patients with systemic lupus erythematosus, dermatomyositis, and rheumatoid arthritis who were treated with the same agent.30,56
Level of evidence
When used as adjuvant therapy in non-randomized studies, rituximab led to complete remission in most of the patients with refractory PV and PF.57–60 The efficacy of rituximab in pemphigus was estimated only in one recent RCT. This prospective, multicenter, parallel-group, open-label RCT aimed to compare first-line rituximab combined with short-term prednisone (tapered over 3 or 6 months) versus prednisone alone (tapered over 12 or 18 months) in the treatment of pemphigus. The study demonstrated that adjuvant therapy with rituximab in combination with short-term prednisone (0.5–1.0 mg/kg/day) led to a greater proportion of patients achieving complete remission off-therapy, as compared with patients treated with prednisone (1.0–1.5 mg/kg/day) alone.52 In addition, more severe adverse events (grades 3–4) were reported in the prednisone-alone group than in the rituximab plus prednisone group, with diabetes and endocrine disorders being the most common of these events in both groups.52 Based on this landmark study, the United States Food and Drug Administration (FDA) granted a Breakthrough Therapy Designation to rituximab in PV. In 2015, the FDA granted Orphan Drug Designation to rituximab for the treatment of PV.
A notable phase III, randomized, double-blind, double-dummy, active-comparator, parallel-arm multicenter RCT (PEMPHIX study) is currently ongoing to evaluate the efficacy and safety of rituximab, compared with MMF, in patients with moderate to severe active PV requiring 60–120 mg/day oral prednisolone or equivalent.61 Another double-blind, parallel-group RCT is being conducted in India to compare the efficacy of rituximab versus rituximab plus intravenous cyclophosphamide pulse therapy.62
A meta-analysis on treatment with rituximab in severe pemphigus reviewed a total of 136 patients, including 103 patients with PV. This study found that approximately 95% of these patients experienced at least partial remission (defined as healing of at least 50% of lesions) following rituximab therapy, with 40% of them achieving complete remission (defined as clinical remission with no further therapy required). Clinical remission (defined as healing of all lesions with further immunosuppression required) occurred in 37% of the PV patients. Only 12% of the total patients experienced relapse, and 3% had worsening disease following rituximab.63
Advantages and disadvantages
As PV is a B-cell-mediated disease, rituximab’s main advantages are its high efficacy and specificity for B cells.64 Its main disadvantage is its potentially serious adverse events, mostly infection-related.
Cyclophosphamide
Mechanism of action
Cyclophosphamide is a synthetic alkylating agent with antineoplastic and immunosuppressive effects. In the liver, cyclophosphamide is converted to the active metabolites phosphoramide mustard and aldophosphamide, which bind to DNA and inhibit DNA replication and induce cell death, predominantly in rapidly dividing cells. This immunosuppressive agent is useful for the blockade of aberrant immune responses due to its inhibitory effects on proliferation, cytokine production, and lymphocyte-induced inflammation.29,65
Recommendation in PV
According to the EDF guidelines, cyclophosphamide is considered a second-line immunosuppressive adjuvant agent,2 whereas the BAD guidelines consider cyclophosphamide as a third-line therapy.8 The recommended oral dose is 2 mg/kg/day.35 Pulse intravenous cyclophosphamide has been tried as an alternative therapy, but failed to achieve therapeutic effect.66,67 Due to its unfavorable safety profile and associated serious adverse events, use of this agent is limited and is not recommended when patients respond to alternate adjuvants. Cyclophosphamide is rarely employed in most centers and is restricted to unresponsive or recalcitrant cases.51
Adverse events
Cyclophosphamide is characterized by a poor safety profile with frequent and substantial adverse effects, including chemotherapy-induced nausea and vomiting, diarrhea, lethargy, darkening of the skin/nails, alopecia, and changes in color and texture of the hair. Cyclophosphamide suppresses the production of blood cells from the bone marrow, causing leukopenia, anemia, and thrombocytopenia, and accounts for hemorrhagic cystitis, which can be prevented by adequate fluid intake and sodium 2-mercaptoethane sulfonate.
Cyclophosphamide may be carcinogenic, potentially leading to transitional cell carcinoma of the bladder as a long-term and dose-dependent complication.31 Moreover, it was causatively implicated in temporary or, rarely, permanent gonadal disturbances, including amenorrhea, azoospermia, and infertility. These are dose dependent, may be irreversible, and can occur after administration of as little as 6 g cyclophosphamide. Cyclophosphamide is a Pregnancy Category D drug that can lead to agenesis of the fingers and toes and an underdeveloped heart; thus, it must be avoided during pregnancy.40 Further, the drug is excreted in breast milk and could cause serious complications in the breastfed infant.30,31
Level of evidence
Adjuvant cyclophosphamide versus corticosteroid monotherapy was evaluated in three RCTs. A study involving 28 participants by Chrysomallis et al68 compared adjuvant oral cyclophosphamide (100 mg daily) with prednisolone alone as well as with adjuvant cyclosporine (5 mg/kg/day). Although they observed no difference in efficacy between treatments, the incidence of complications was higher with the combination treatment.
Adjuvant intravenous cyclophosphamide pulse therapy (15 mg/kg monthly) versus prednisolone alone was evaluated in an RCT involving 60 participants. No conclusive difference was noted between the two arms in remission and relapse rates, cumulative steroid doses, and adverse events.69 Of interest is the relatively acceptable safety profile of pulse therapy in this study.
Chams-Davatchi et al27 compared adjuvant cyclophosphamide pulse therapy (1 g monthly for 6 months, then 1 g every 2 months) with prednisolone alone as well as with adjuvant azathioprine and MMF. The study found the effect of cyclophosphamide pulse therapy to be inconclusive when compared with prednisolone alone as well as with adjuvant azathioprine and MMF in terms of the efficacy and steroid-sparing effect.
No significant difference was found between a pulse cyclophosphamide treatment protocol (500 mg intravenous cyclophosphamide in combination with 100 mg intravenous dexamethasone for three consecutive days) and the administration of oral methylprednisolone (initially 2 mg/kg/day) combined with oral azathioprine (2–2.5 mg/kg/day) in a multicenter prospective RCT with 22 patients with pemphigus (16 of which had PV).67
Dexamethasone–cyclophosphamide pulse therapy was evaluated in two RCTs from India. The first study allocated its 28 PV patients to two treatment arms. The first arm received intravenous dexamethasone 100 mg for three consecutive days each month, intravenous cyclophosphamide 500 mg monthly, and oral cyclophosphamide 50 mg daily. The second arm was treated with cyclophosphamide pulse therapy (15 mg/kg monthly) and prednisolone combination therapy. The first arm experienced a shorter time to reach a cutaneous, but not mucosal, response. The second arm achieved remission sooner, but experienced significantly more frequent steroid-related side effects.70 Another study of 19 PV patients compared two treatment regimens, with patients receiving either oral cyclophosphamide alone (50 mg daily) or a combination of intravenous dexamethasone 100 mg on three consecutive days monthly with intravenous cyclophosphamide 500 mg monthly as well as oral cyclophosphamide 50 mg daily on days between the pulses. No significant difference was seen between the two arms in regard to the relapse rate, Dsg1 and Dsg3 antibodies titers, and direct immunofluorescence grading.71
Overall, current studies support the use of cyclophosphamide for refractory pemphigus rather than as a first-line adjuvant.11
Advantages and disadvantages
The main advantage of cyclophosphamide is its potential steroid-sparing effect when azathioprine or MMF has failed. However, its main disadvantage is its severe adverse events profile, which includes infertility, cancer, sepsis, and lymphopenia.
Intravenous immunoglobulin
Mechanism of action
IVIg is an immunomodulating agent that has multiple activities. It is obtained from sterile, purified IgG products manufactured from pooled human plasma and typically contains more than 95% unmodified IgG, which has intact Fc-dependent effector functions and only trace amounts of IgA or IgM.51
Its mechanism of action is complex and targets various components of the innate and acquired immune system. The beneficial effect of IVIg may be achieved by one of the following pathways that are proposed to act synergistically to affect the disease pathogenesis: reduction of the titers of pathogenic autoantibodies; modulation of complement activation; saturation and blockade of Fc receptors (the Fc region of IgG facilitates interaction with and signaling through Fc receptors on B cells and phagocytes); suppression of various inflammatory mediators, including cytokines, chemokines, and metalloproteinases; suppression of activation, differentiation, and effector functions of dendritic, B, and Th-1 cells; and increasing the sensitivity to corticosteroids.72,73 In PV, it was found that IVIg selectively inhibits Dsg3 antibodies, decreases the number of circulating autoantibodies, and reduces blister formation in mice.73–76
Recommendation in PV
Since 1989, IVIg has been used as a component of adjuvant therapy for severe pemphigus.51 IVIg is usually administered in recalcitrant disease or in case of contraindications to immunosuppressive adjuvants, and is recommended as a second-line adjuvant by the EDF guidelines2 and as a third-line therapy by the BAD guidelines.8 Although diverse opinions exist in regard to the optimal dose, the conventional dose is 2 g/kg/cycle administered intravenously over two to five consecutive days per month.2 This therapy may need to be repeated monthly in recalcitrant cases.30,77 IVIg could be used as an adjuvant therapy to systemic corticosteroids and immunosuppressive adjuvants in recalcitrant cases.78
Following infusion, the half-life of this agent is 2–3 weeks. The average rate of clearance of pemphigus antibodies ranges between 30% and 70% at 1 week following administration.73,79,80
The rate of infusion is started slowly and gradually increased as tolerated, usually over 4–4.5 hours.30,74 During the infusion, vital signs should be monitored. Before administration, routine hematologic and biochemical investigations should be conducted. These include a complete blood count, renal and liver function tests, and levels of electrolytes, rheumatoid factor, serum cryoglobulin, and IgA.74
Adverse events
Patients treated with IVIg are not at high risk of infection or reactivation of chronic infections, as compared to those under conventional immunosuppressants. Conversely, it is being increasingly used for the treatment of certain bacterial or viral infectious diseases.81 The lack of immunosuppressive effect is considered a prominent advantage.35 Moreover, IVIg was recommended as the safest adjuvant treatment for pemphigus in pregnant women.40
Usually, IVIg-associated adverse reactions are mild and self-limiting. The incidence of adverse effects in patients treated for autoimmune diseases is usually <1%.30 Reactions such as headache, back pain, chills, flushing, fever, hypertension, myalgia, nausea, and vomiting seem to be related more to the infusion rate rather than to the dose. Erythema, pain, phlebitis, and eczematous plaques may occur at the infusion site. However, some rare serious adverse events were reported to coexist with IVIg in patients with PV, most prominently aseptic meningitis (particularly in patients who commonly experience episodes of migraine) and thrombotic complications (particularly in high infusion rates and high doses).82,83 Furthermore, other possible late adverse events were reported in association with IVIg, including acute renal failure, neutropenia, autoimmune hemolytic anemia, skin reactions, and arthritis.84 Anaphylactic reactions can arise in individuals with IgA deficiency.30
It is believed that the risk of complications, particularly in patients with underlying risk factors, can be alleviated via employment of thromboprophylaxis, slow infusion rates, hydration, and widening of the interval between IVIg cycles.35,85
Level of evidence
Two RCTs have been conducted to investigate the role of IVIg in the treatment of PV. The first study was a multicenter, randomized, placebo-controlled, double-blind trial originating from Japan in which one group of 20 patients received IVIg 200 mg/kg/day, a second group of 20 patients received IVIg 400 mg/kg/day, and a third group of 21 patients received placebo infusion. All interventions were administered over five consecutive days. The time until requirement of additional treatment (time to escape from the protocol) was significantly longer in the group treated with 400 mg/kg/day as compared with the placebo group. In addition, the activity scores of the disease were significantly decreased in both IVIg treatment groups.79
In addition, the beneficial effect of IVIg was shown in a randomized, placebo-controlled, crossover trial conducted in a single patient with severe PV, comprising two phases of six consecutive months of treatment with either IVIg or placebo infusion. When the patient was on IVIg, the mean daily prednisolone dose was significantly lower, as were the mean daily disease severity and monthly Dsg3 and Dsg1 levels (as measured by ELISA).78
Advantages and disadvantages
The main advantage of IVIg is its excellent safety profile, with adverse events being usually mild, such as headaches, flushing, or hypotension. Another advantage is its ease of delivery, requiring only a single cycle for a few days per month. Its main disadvantages include its high cost and poor availability.
Plasmapheresis
Mechanism of action
Plasmapheresis is an extracorporeal blood purification procedure in which plasma proteins are non-selectively removed from the circulation.35 In this procedure, the blood is continuously removed from the patient and separated into cellular components and plasma. The cellular compartments are then returned to the patient whereas the plasma is replaced with another fluid – often, albumin or fresh-frozen plasma.10
The benefit of removing plasma is the elimination of pathogenic autoantibodies from the circulation. The rationale behind plasmapheresis is based on the correlation observed between the titers of circulating anti-Dsg3 autoantibodies and disease activity.86 However, removal of the autoantibodies does not cease their production; conversely, pathogenic B lymphocytes increase the production of autoantibodies in the first 2 weeks following plasmapheresis to compensate for the initial reduction. Consequently, circulating levels of autoantibodies can be as high as or even higher than before the procedure.51 This rebound increase in antibody level can be inhibited by administering immunosuppressive agents prior to plasmapheresis.86,87 Despite this rebound effect, plasmapheresis results in lower long-term pemphigus antibody levels as compared to other therapies.88
Recommendations in PV
Plasmapheresis is a useful adjuvant therapy for quickly reducing the titers of circulating autoantibodies. This procedure should be considered for rapid control of severe pemphigus unresponsive to a combination of prednisone and immunosuppressive agents.77
No standardized protocol for the number and frequency of sessions of plasmapheresis exists; however, four or five plasma exchanges – each consisting of 1–1.5 plasma volumes over a period of 7–10 days – represent an adequate short-term treatment to remove 90% of the total initial body immunoglobulin burden.10,89 Other experts recommend 1–2 sessions per week.77 Plasmapheresis can be undertaken by using the conventional centrifugation device utilized in blood banks. Recently, the use of double-filtration plasmapheresis is increasing due to its higher safety profile.90 In double-filtration plasmapheresis, immunoglobulins are removed more selectively, and the loss of albumin is minimized.10
Concomitant immunosuppression with corticosteroids and cyclophosphamide prevents a post-plasmapheresis rebound increase in the production of autoantibodies.77
Adverse events
The safety profile of plasmapheresis is relatively high. The risk of associated infection is mainly attributed to the concomitant steroids and immunosuppressives.10 Other minor and transient adverse effects of plasma exchange that have been reported include thrombocytopenia, hypogammaglobulinemia, fluid overload leading to hypertension and pulmonary edema in patients with underlying congestive heart failure, hypoproteinemia, anemia, leukopenia, and hypocalcemia.91 The rapid fluid shift that occurs due to the removal of proteins (and in which osmotic pressure is maintained) can lead to disturbance of homeostasis and severe problems in patients with compromised cardiac function.10,51
Level of evidence
Small trials have demonstrated the effectiveness of plasmapheresis in inducing partial or complete remission;86,88,92 however, a multicenter RCT with 40 participants suggested that plasma exchange with low-dose steroids is ineffective as a therapy for pemphigus.93 In this study, the clinical outcomes of 18 patients treated with prednisolone (0.5 mg/kg/day) alone were compared with those of 22 patients who underwent 10 plasmapheresis procedures within 4 weeks in combination with the same prednisolone protocol. No difference was seen between the two groups in regard to the control rate, serum titers of autoantibodies, or cumulative prednisolone doses. Moreover, four deaths due to sepsis were recorded in the plasmapheresis arm.93
Immunoadsorption
Mechanism of actions
Immunoadsorption – also known as immunoapheresis – is another extracorporeal blood purification procedure that leads to the rapid removal of circulating autoantibodies against Dsg1 and Dsg3. Whereas plasmapheresis inadvertently removes clotting factors, albumin, and hormones that need to be replaced, immunoadsorption is more specific – only IgGs and immune complexes are attracted to the adsorber and removed from the circulation.35,51 Thus, immunoadsorption is more efficient and safe, relative to plasmapheresis.94 It was claimed that IgG concentration against Dsg1 and Dsg3 could be reduced by 75%, following a single immunoadsorption, and by 95%, following three immunoadsorptions on three consecutive days.95
Recommendations in PV
Immunoadsorption is recommended as a second-line adjuvant agent by the EDF guidelines2 and as a third-line therapy by the BAD guidelines.8 It is indicated in patients with recalcitrant PV who have not sufficiently responded to first-line treatment – that is, corticosteroids in combination with azathioprine or mycophenolate.2 The recommended schedule is four treatments of immunoadsorption on four consecutive days (2.5-fold plasma volume per day), repeated after 4 weeks, if needed.2 Treatment could be undertaken in combination with immunosuppressive agents, such as rituximab and cyclophosphamide.96,97 The efficacy of immunoadsorption is increased substantially in combination with systemic immunosuppressive drugs.2
Immunoadsorption is superior to plasmapheresis in terms of efficacy and safety; however, the high cost of the adsorbers is the chief limiting factor. Unlike plasmapheresis, substitution of plasma components such as fresh-frozen plasma or human albumin is not required.
Adverse events
Usually, immunoadsorption is highly tolerable. However, limited adverse effects, including hypotension, anaphylaxis, bradycardia, infarction, deep venous thrombosis, and herpes zoster infection, were reported.35 Contraindications include severe systemic infections, cardiovascular diseases, treatment with angiotensin-converting enzyme inhibitors, hypersensitivity against components of the immunoadsorption column, and hemorrhagic diathesis.2,10
Level of evidence
Immunoadsorption has been successfully used, leading to noticeable improvement within weeks in individual patients with refractory PV.98,99 However, the exact efficacy of this procedure is difficult to estimate because this treatment is most often used with concomitant immunosuppressants, IVIg, or rituximab in recalcitrant cases.51,100,101 RCTs to evaluate this intervention in PV are yet to be conducted.
A review by Schmidt and Zillikens102 evaluated 40 cases of PV treated by immunoadsorption. The review revealed that immunoadsorption induced complete remission and clinical remission in approximately 20% and 50% of patients, respectively. Due to the limited amount of evidence, the effect of immunoadsorption in PV remains inconclusive. An RCT on immunoadsorption for pemphigus has been listed in the US National Institutes of Health ongoing trials register.103
Advantages and disadvantages
The main advantage of immunoadsorption is its presumed rapid efficacy and favorable safety profile. Its main disadvantage is its poor availability and high cost.
Dapsone
Mechanism of action
Dapsone is a drug that has both antimicrobial/antiprotozoal properties and anti-inflammatory effects. Additionally, dapsone seems to be capable of suppressing tumor necrosis factor (TNF)-α and IL-8 at the level of mRNA, and at higher concentrations, may inhibit B-cell proliferation.37
Recommendation in PV
Dapsone is recommended in combination with systemic corticosteroids at a dose of 100 mg/day (or ≤1.5 mg/kg/day) as a second-line steroid-sparing agent.2 Prior to the initiation of treatment, serum glucose-6-phosphate dehydrogenase (G6PD) activity should be tested.
Dapsone appears to benefit some PV patients in the maintenance phase of their disease; however, as there is a lack of evidence in regard to the use of dapsone in PV, it is difficult to evaluate its utility.51
Level of evidence
The efficacy of dapsone as a corticosteroid-sparing agent in the maintenance phase of PV has been evaluated in 1 RCT. In this study, 19 patients with pemphigus in whom tapering of the corticosteroid dose to <15 mg/day had failed at least twice when using a standard regimen were randomized to either dapsone or placebo. The ability of these patients to adjust to tapering of corticosteroids to ≤7.5 mg/day was used as the primary endpoint. This outcome measure favored the dapsone-treated arm, but was not statistically significant.104 A meta-analysis study that compiled clinical data from 55 pemphigus patients found that 34 PV and 12 PF patients responded to dapsone. In this study, hemolysis was the most common adverse reaction.105
Advantages and disadvantages
The main advantage of dapsone is its relatively superior safety profile. Its disadvantages include the lack of evidence in regard to its efficacy and the increased risk of hematologic side effects, such as hemolytic anemia. This requires ongoing monitoring, especially in elderly patients.
Methotrexate
Mechanism of action
Methotrexate is an antimetabolite drug originally synthesized for the treatment of leukemia. It inhibits dihydrofolate reductase, which results in decreased methionine, purine, and thymidylate synthesis and, thus, decreased DNA synthesis.35 Because of its ability to suppress cell-mediated and humoral immune responses, its utilization has been extended to several autoimmune diseases, including rheumatoid arthritis.106
Recommendation in PV
Methotrexate at a dosage of 10–20 mg/week is currently under consideration as a second-line steroid-sparing option in PV according to the EDF guidelines.2
Adverse events
Despite concerns about the adverse effects of methotrexate, doses up to 25 mg/week are relatively safe, whereas higher doses may be associated with unwanted effects. The main adverse events reported among pemphigus patients undergoing treatment with methotrexate are nausea and infection, including pneumonia and reactivation of tuberculosis.51
Level of evidence
There is a scarcity of literature assessing the efficacy of methotrexate in PV treatment. Prospective RCTs evaluating the role of methotrexate in PV have not been conducted yet. A recent retrospective single-center study reported that 21 out of 25 patients were able to taper corticosteroids following 6 months of adjuvant treatment with 15 mg methotrexate per week.107 Another retrospective single-center study showed that 16 of 23 PV patients on methotrexate were eventually weaned completely off prednisone, with a mean time to discontinuation of 18 months.108
Disadvantages
There is a very poor level of evidence suggesting its use in PV.
Cyclosporine
Mechanism of action
Cyclosporine is a potent immunosuppressant that can effectively suppress immune responses through inhibition of the phosphatase activity of calcineurin. This leads to the downregulation of several transcription factors, especially the nuclear factor of activated T lymphocytes. Moreover, it reduces matrix metalloproteinase-9 expression and blocks both c-Jun N-terminal kinase (JNK) and the p38 signaling pathways.109,110 Therefore, cyclosporine could reversibly suppress both humoral and cellular immunity.
Recommendation in PV
Cyclosporine treatment of 3–5 mg/kg/day is infrequently added to systemic corticosteroids as second-line adjuvant therapy in PV.31 Current data are insufficient to support cyclosporine use in PV, and it is not recommended by the EDF or BAD guidelines.30
Adverse events
The main adverse reactions to cyclosporine therapy are renal dysfunction, hypertension, tremor, hirsutism, and gingival hyperplasia. Low serum magnesium has been reported in some, but not all, patients exhibiting convulsions under cyclosporine therapy. It is probable that adverse events are more frequent in the setting of combined corticosteroid and cyclosporine therapy.51 However, it is considered the relatively safest immunosuppressive drug in pregnancy (FDA Pregnancy Category C) for the treatment of PV.30,31
Level of evidence
The evaluation of cyclosporine in two RCTs failed to demonstrate any advantage of cyclosporine in PV. Moreover, greater adverse events and toxicity were detected in the cyclosporine treatment arm.68,111 In their study on 28 patients with PV, Chrysomallis et al compared treatments between three groups: one group received corticosteroid monotherapy as 40 mg/day of prednisone equivalent, whereas the other two groups received 40 mg/day corticosteroids combined with either 100 mg oral cyclophosphamide administered daily or 5 mg/kg/day cyclosporine. This study reported an inconclusive effect of cyclosporine in all outcome measures, and that the toxicities were encountered more frequently in combination treatment.68 Ioannides et al conducted another RCT involving 33 pemphigus patients (29 with PV) in which prednisolone (1 mg/kg/day) alone was compared with a combined regimen of prednisolone (1 mg/kg/day) and oral cyclosporine (5 mg/kg/day). This study showed no advantage of this adjuvant drug over treatment with corticosteroids alone.111
Disadvantages
There is a very poor level of evidence suggesting its use in PV.
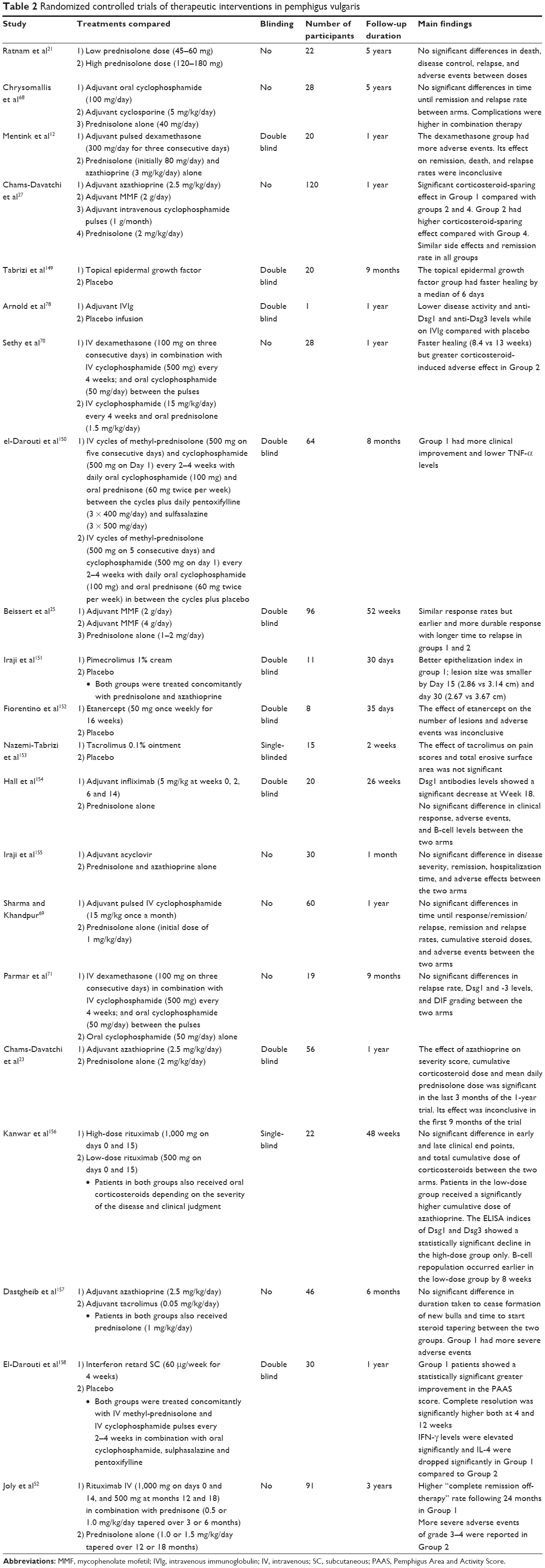
Table 2 summarizes RCTs evaluating different interventions in patients with pemphigus.
 | Table 2 Randomized controlled trials of therapeutic interventions in pemphigus vulgaris |
Potential future treatments
Chimeric antigen receptor therapy
Dsg-specific immune suppression by specifically targeting B and T cells involved in the production of pathogenic pemphigus autoantibodies is under development. Recently, for example, the possibility of using modified chimeric antigen receptor (CAR) therapy to target Dsg3-specific B cells was proposed; human T cells were engineered to express a chimeric autoantibody receptor (CAAR) consisting of Dsg3. In the murine model, Dsg3 CAAR-T cells exhibit specific cytotoxicity against B cells bearing anti-Dsg3 B-cell receptors in vitro and specifically eliminate Dsg3-specific B cells in vivo. This strategy would directly eliminate anti-Dsg3 memory B cells and indirectly eliminate Dsg3-specific short-lived plasma cells that produce disease-causing antibodies. CAAR-T cells may provide an effective strategy for specific targeting of autoreactive B cells in PV without inducing general immunosuppression.112 This approach can be considered a therapeutic strategy in patients carrying autoantibodies directed against Dsg3 and other well-defined target antigens such as Dsg1. However, its usefulness needs to be assessed in human disease where additional target antigens may be present.
This innovative approach would represent an ideal therapeutic strategy because the target antigens and pathophysiological mechanisms of pemphigus have been well characterized.
T-cell immunotherapy
It is known that T-cell activation is necessary for the initiation and coordination of the autoantibody response in PV.113,114 It is conceivable to attenuate T-cell function at several points by utilizing monoclonal antibodies to block specific accessory co-receptors, co-stimulatory molecules, or cytokines.
Antigen-specific T and B cells must interact via the molecules CD154 and CD40, respectively, in order to produce autoantibodies. Blocking CD154 with the use of an anti-CD154 monoclonal antibody in a mouse model prevented the production of anti-Dsg3 IgG and the subsequent development of the PV phenotype.115 In other mouse models, the transfer of Dsg3-specific CD4+ T cells can induce a PV phenotype, with IL-4 production playing a critical role in this process.116
Altered peptide ligands (APLs) may be utilized in an immune-based treatment strategy targeting the T-cell level. APLs are peptide analogs with one or more amino acid substitutions at major TCR contact residues. APLs could be engineered to prevent the interaction between autoreactive T cells and autoantigen peptides that present on disease-associated HLA class II molecules and, thus, prevent the start of the autoimmune process. Instead, APLs could lead to the induction of an incomplete response by antigen-specific T cells, resulting in T-cell functional tolerance. A phase I clinical trial in PV patients studied the effects of systemic PI-0824 – a Dsg3 peptide found to be immunodominant in a small subset of patients. The aim of this therapeutic strategy is to induce T-cell tolerance and abrogate the source of T-cell help required for the production of autoantibodies directed against Dsg3. Neither a significant change in anti-Dsg3 antibodies nor a clinical response following the intravenous administration of Dsg3 peptides was observed in this study.117
BAFF and APRIL inhibitors
B-cell activating factor (BAFF) is a member of the TNF superfamily of cytokines and is a critical activator of B-cell development and differentiation.118 A proliferation-inducing ligand (APRIL) – another TNF superfamily ligand – is also involved in B-cell development.119 It was demonstrated that BAFF and APRIL lead to the promotion of Ig switching to the IgG, IgE, and IgA subclasses.120,121 Contrasting with the significant correlation between BAFF levels and the activity of some autoimmune diseases, namely systemic lupus erythematosus and rheumatoid arthritis,122 serum levels of BAFF and APRIL were not found to be elevated in patients with PV.123 Furthermore, rituximab use in PV patients was found to lead to a significant elevation of serum BAFF levels but to a decrease in anti-Dsg1 and anti-Dsg3 autoantibody titers.124 In the same study, an inverse relationship between BAFF levels and peripheral CD19+ levels in PV patients treated with rituximab was observed. Although BAFF and APRIL were not established as dominant players in the immunopathogenesis of PV, further studies are warranted to clarify their role in this disease. Clearly, an improved mechanistic understanding of the role of BAFF in PV pathogenesis is necessary before inhibitors of this factor, such as belimumab, can be considered for the treatment of PV.125
There may be promise in studying the therapeutic efficacy of targeting other B-cell surface molecules besides CD20, such as CD19 and CD22; however, research in this area remains limited.125
P38MAPK signaling pathway inhibitor
IgG-induced phosphorylation has been demonstrated to activate p38 mitogen-activated protein kinase (p38MAPK) and heat shock protein (HSP) 27, which causes downstream remodeling of the actin cytoskeleton and retraction of keratin and contributes to the loss of cell–cell adhesion. This signaling pathway is initiated following the binding of IgG autoantibody to keratinocytes and may be involved in the induction of acantholysis.126 Experiments utilizing human cultured keratinocytes have suggested that inhibition of p38MAPK prevents the phosphorylation of HSP27 and, thus, also prevents early cytoskeletal changes.126 This hypothesis was strengthened when SB202190 – an inhibitor of p38MAPK – was shown to prevent blister formation by inhibiting IgG-activated signaling in a mouse model of PV.127 Moreover, enhancement of anti-Dsg1 IgG antibodies and blister formation in PF in a p38MAPK-dependent mechanism could lend weight to the role of p38MAP in blister formation in pemphigus.128 However, although inhibition of the p38MAPK signaling pathway protects against blistering in PV, Mao et al129 showed that blistering can also occur in mice lacking the major p38MAPK isoform. Therefore, it was concluded that p38MAPK is not essential for the loss of intercellular adhesion in PV, but may function downstream to augment blistering.
Studies examining p38MAPK inhibitors in animal models revealed severe adverse effects (mainly hepatotoxicity, undefined gastrointestinal toxicity, and an unusual inflammatory response in the central nervous system).130 To date, there has been only one clinical trial conducted to assess the use of oral p38MAPK inhibitors in the prevention and healing of blisters in PV patients. The trial was terminated early (due to high hepatotoxicity) without providing any conclusions on the efficacy of this agent.125 Although this specific inhibitor was not associated with favorable clinical outcomes, P38MAPK remains a viable target for potential treatment options.
Other signaling pathways inhibitors
Elevation of cAMP levels has been shown experimentally to interfere with signaling pathways and prevent blister formation in both in vitro and in vivo mouse models.131 Elevated cAMP is thought to interfere with the previously described p38MAPK activation pathway and, thus, results in blockade of the loss of intercellular adhesion, depletion of cellular Dsg3, and induction of morphologic changes by antibodies of PV patients in cultured keratinocytes.131 It is well-established that β-adrenergic receptor agonists increase cAMP levels.131,132 Because keratinocytes predominantly express β2 receptors, the use of specific β2 agonists could theoretically be utilized therapeutically to increase cAMP levels;125 moreover, c-Myc and epidermal growth factor receptor signaling were shown as other pathways with possible involvement in the pathogenesis of PV.133,134 These may be attractive therapeutic targets in the future.35
Bruton’s tyrosine kinase inhibitor
Bruton’s tyrosine kinase (BTK) inhibition is supposed to target several pathways and cell types implicated in inflammation and autoimmunity. These include modulation of B-cell receptor-mediated B-cell pathways, as well as inhibition of FcR-induced cytokine release from monocytes and macrophages and FcεR-induced mast cell degranulation and granulocyte migration as well as mediator release.135 Mutations in the gene encoding BTK result in X-linked agammaglobulinemia type 1, which represents an immunodeficiency associated with failure to produce mature B lymphocytes, and is characterized by a failure of Ig heavy chain rearrangement.136
Preliminary results of a phase II open-label cohort study examining reversible covalent BTK inhibitor PRN1008 in adult patients with PV showed promising efficacy and steroid-sparing effect.137 It is noteworthy that the reversible, covalent, oral, small-molecule BTK inhibitor PRN473 demonstrated good response in canine PF.135 Recently, PRN1008 has been granted Orphan Drug Designation by the United States FDA for the treatment of patients with PV.
Disclosure
The author reports no conflicts of interest in this work.
References
Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366(9479):61–73. | ||
Hertl M, Jedlickova H, Karpati S, et al. Pemphigus. S2 Guideline for diagnosis and treatment – guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29(3):405–414. | ||
Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus. An update. Arch Dermatol. 1996;132(2):203–212. | ||
Kridin K, Zelber-Sagi S, Bergman R. Pemphigus vulgaris and Pemphigus foliaceus: differences in epidemiology and mortality. Acta Derm Venereol. 2017;97(9):1095–1099. | ||
Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in patients with pemphigus. Acta Derm Venereol. 2017;97(5):607–611. | ||
Murrell DF, Dick S, Ahmed AR, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58(6):1043–1046. | ||
Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903–908. | ||
Harman KE, Brown D, Exton LS, et al. British Association of Dermatologists’ guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177(5):1170–1201. | ||
Almugairen N, Hospital V, Bedane C, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol. 2013;69(4):583–588. | ||
Gregoriou S, Efthymiou O, Stefanaki C, Rigopoulos D. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521–527. | ||
Dick SE, Werth VP. Pemphigus: a treatment update. Autoimmunity. 2006;39(7):591–599. | ||
Mentink LF, Mackenzie MW, Tóth GG, et al. Randomized controlled trial of adjuvant oral dexamethasone pulse therapy in pemphigus vulgaris: PEMPULS trial. Arch Dermatol. 2006;142(5):570–576. | ||
Tóth GG, Westerlaken BO, Eilders M, Laseur M, Jonkman MF, Uges DR. Dexamethasone pharmacokinetics after high-dose oral therapy for pemphigus. Ann Pharmacother. 2002;36(6):1108–1109. | ||
Ahmed AR, Moy R. Death in pemphigus. J Am Acad Dermatol. 1982;7(2):221–228. | ||
Fernandes NC, Perez M. Treatment of pemphigus vulgaris and pemphigus foliaceus: experience with 71 patients over a 20 year period. Rev Inst Med Trop Sao Paulo. 2001;43(1):33–36. | ||
Fauci AS, Dale DC, Balow JE. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med. 1976;84(3):304–315. | ||
Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med. 1993;119(12):1198–1208. | ||
Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J. 2006;27(2):413–426. | ||
Nelson HS, Leung DYM, Bloom JW. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2003;111(1):3–22. | ||
Liu D, Ahmet A, Ward L, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013;9(1):30. | ||
Ratnam KV, Phay KL, Tan CK. Pemphigus therapy with oral prednisolone regimens A 5-year study. Int J Dermatol. 1990;29(5):363–367. | ||
Femiano F, Gombos F, Scully C. Pemphigus vulgaris with oral involvement: evaluation of two different systemic corticosteroid therapeutic protocols. J Eur Acad Dermatol Venereol. 2002;16(4):353–356. | ||
Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, et al. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27(10):1285–1292. | ||
Beissert S, Werfel T, Frieling U, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of pemphigus. Arch Dermatol. 2006;142(11):1447–1454. | ||
Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol. 2010;130(8):2041–2048. | ||
Ioannides D, Apalla Z, Lazaridou E, Rigopoulos D. Evaluation of mycophenolate mofetil as a steroid-sparing agent in pemphigus: a randomized, prospective study. J Eur Acad Dermatol Venereol. 2012;26(7):855–860. | ||
Chams-Davatchi C, Esmaili N, Daneshpazhooh M, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol. 2007;57(4):622–628. | ||
Atzmony L, Hodak E, Leshem YA, et al. The role of adjuvant therapy in pemphigus: a systematic review and meta-analysis. J Am Acad Dermatol. 2015;73(2):264–271. | ||
Sandborn WJ. State-of-the-art: immunosuppression and biologic therapy. Dig Dis. 2010;28(3):536–542. | ||
Schiavo AL, Puca RV, Ruocco V, Ruocco E. Adjuvant drugs in autoimmune bullous diseases, efficacy versus safety: facts and controversies. Clin Dermatol. 2010;28(3):337–343. | ||
Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30(1):78–83. | ||
Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111(8):1122–1124. | ||
Aberer W, Wolff-Schreiner EC, Stingl G, Wolff K. Azathioprine in the treatment of pemphigus vulgaris: a long-term follow-up. J Am Acad Dermatol. 1987;16(3 Pt 1):527–533. | ||
Anstey AV, Wakelin S, Reynolds NJ. Azathioprine: guidelines for prescribing azathioprine in dermatology. In: Cox NH, English JS, editors. British Association of Dermatologists’ Management Guidelines. Chichester: Wiley-Blackwell; 2011:219–228. | ||
Tavakolpour S. Current and future treatment options for pemphigus: is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133–142. | ||
Meggitt SJ, Anstey AV, Mohd Mustapa MF, Reynolds NJ, Wakelin S. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165(4):711–734. | ||
Kasperkiewicz M, Schmidt E, Zillikens D. Current therapy of the pemphigus group. Clin Dermatol. 2012;30(1):84–94. | ||
Mabrouk D, Gürcan HM, Keskin DB, Christen WG, Ahmed AR. Association between cancer and immunosuppressive therapy – analysis of selected studies in pemphigus and pemphigoid. Ann Pharmacother. 2010;44(11):1770–1776. | ||
Kwong YL, Au WY, Liang RH. Acute myeloid leukemia after azathioprine treatment for autoimmune diseases: association with -7/7q-. Cancer Genet Cytogenet. 1998;104(2):94–97. | ||
Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77(1). | ||
Chaidemenos G, Apalla Z, Koussidou T, Papagarifallou I, Ioannides D. High dose oral prednisone vs. prednisone plus azathioprine for the treatment of oral pemphigus: a retrospective, bi-centre, comparative study. J Eur Acad Dermatol Venereol. 2011;25(2):206–210. | ||
Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14 (Suppl 1):s2–s8. | ||
Olejarz W, Bryk D, Zapolska-Downar D. Mycophenolate mofetil – a new atheropreventive drug? Acta Pol Pharm. 2014;71(3):353–361. | ||
Nousari HC, Sragovich A, Kimyai-Asadi A, Orlinsky D, Anhalt GJ. Mycophenolate mofetil in autoimmune and inflammatory skin disorders. J Am Acad Dermatol. 1999;40(2 Pt I):265–268. | ||
Orvis AK, Wesson SK, Breza TS, Church AA, Mitchell CL, Watkins SW. Mycophenolate mofetil in dermatology. J Am Acad Dermatol. 2009;60(2):183–199. | ||
Ensley RD, Bristow MR, Olsen SL, et al. The use of mycophenolate mofetil (RS-61443) in human heart transplant recipients. Transplantation. 1993;56(1):75–82. | ||
Mimouni D, Anhalt GJ, Cummins DL, Kouba DJ, Thorne JE, Nousari HC. Treatment of pemphigus vulgaris and pemphigus foliaceus with mycophenolate mofetil. Arch Dermatol. 2003;139(6):739–742. | ||
Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128(12):2850–2858. | ||
Zambruno G, Borradori L. Rituximab immunotherapy in pemphigus: therapeutic effects beyond B-cell depletion. J Invest Dermatol. 2008;128(12):2745–2747. | ||
Mouquet H, Musette P, Gougeon ML, et al. B-cell depletion immunotherapy in pemphigus: effects on cellular and humoral immune responses. J Invest Dermatol. 2008;128(12):2859–2869. | ||
Kim J, Daniel BS, Werth VP, Joly P, Murrell DF. The treatment of pemphigus vulgaris and pemphigus foliaceus. In: Murrell DF, editor. Blistering Diseases: Clinical Features, Pathogenesis, Treatment. Berlin: Springer; 2015:523–531. | ||
Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al; French study group on autoimmune bullous skin diseases. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389(10083):2031–2040. | ||
Schmidt E, Bröcker EB, Goebeler M. Rituximab in treatment-resistant autoimmune blistering skin disorders. Clin Rev Allergy Immunol. 2008;34(1):56–64. | ||
Hertl M, Zillikens D, Borradori L, et al. Recommendations for the use of rituximab (anti-CD20 antibody) in the treatment of autoimmune bullous skin diseases. J Dtsch Dermatol Ges. 2008;6(5):366–373. German [with English abstract]. | ||
Zakka LR, Shetty SS, Ahmed AR. Rituximab in the treatment of pemphigus vulgaris. Dermatol Ther (Heidelb). 2012;2(1):17. | ||
Lapeyre H, Joly P. [Rituximab: mabthera]. Ann Dermatol Venereol. 2007;134(12):968–971. | ||
Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355(17):1772–1779. | ||
Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545–552. | ||
Colliou N, Picard D, Caillot F, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. 2013;5(175):175ra30. | ||
Cianchini G, Lupi F, Masini C, Corona R, Puddu P, De Pità O. Therapy with rituximab for autoimmune pemphigus: results from a single-center observational study on 42 cases with long-term follow-up. J Am Acad Dermatol. 2012;67(4):617–622. | ||
Hoffmann-La Roche. A study to evaluate the efficacy and safety of rituximab versus mycophenolate mofetil (MMF) in participants with pemphigus vulgaris (PV). Available from: https://clinicaltrials.gov/ct2/show/study/NCT02383589?cond=pemphigus&draw=2&rank=11&view=results. NLM identifier: NCT02383589. Accessed February 23, 2018. | ||
Uprety Shraddha. Comparative study of rituximab versus combination of rituximab and intravenous cyclophosphamide in severe pemphigus. Available from: https://clinicaltrials.gov/ct2/show/NCT01974518?cond=pemphigus&draw=5&rank=3. NLM identifier: NCT01974518. Accessed February 24, 2018. | ||
Schmidt E, Goebeler M, Zillikens D. Rituximab in severe pemphigus. Ann N Y Acad Sci. 2009;1173:683–691. | ||
Rosenbach M, Murrell DF, Bystryn JC, et al. Reliability and convergent validity of two outcome instruments for pemphigus. J Invest Dermatol. 2009;129(10):2404–2410. | ||
Awad A, Stüve O. Cyclophosphamide in multiple sclerosis: scientific rationale, history and novel treatment paradigms. Ther Adv Neurol Disord. 2009;2(6):357–368. | ||
Akhtar SJ, Hasan MU. Treatment of pemphigus: a local experience. J Pak Med Assoc. 1998;48(10):300–304. | ||
Rose E, Wever S, Zilliken D, Linse R, Haustein UF, Bröcker EB. Intravenous dexamethasone-cyclophosphamide pulse therapy in comparison with oral methylprednisolone-azathioprine therapy in patients with pemphigus: results of a multicenter prospectively randomized study. J Dtsch Dermatol Ges. 2005;3(3):200–206. | ||
Chrysomallis F, Ioannides D, Teknetzis A, Panagiotidou D, Minas A. Treatment of oral pemphigus vulgaris. Int J Dermatol. 1994;33(11):803–807. | ||
Sharma VK, Khandpur S. Evaluation of cyclophosphamide pulse therapy as an adjuvant to oral corticosteroid in the management of pemphigus vulgaris. Clin Exp Dermatol. 2013;38(6):659–664. | ||
Sethy PK, Khandpur S, Sharma VK. Randomized open comparative trial of dexamethasone-cyclophosphamide pulse and daily oral cyclophosphamide versus cyclophosphamide pulse and daily oral prednisolone in pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 2009;75(5):476–482. | ||
Parmar NV, Kanwar AJ, Minz RW, et al. Assessment of the therapeutic benefit of dexamethasone cyclophosphamide pulse versus only oral cyclophosphamide in phase II of the dexamethasone cyclophosphamide pulse therapy: a preliminary prospective randomized controlled study. Indian J Dermatol Venereol Leprol. 2013;79(1):70–76. | ||
Hartung HP. Advances in the understanding of the mechanism of action of IVIg. J Neurol. 2008;255 (Suppl 3):3–6. | ||
Aoyama Y. What’s new in i.v. immunoglobulin therapy and pemphigus: high-dose i.v. immunoglobulin therapy and its mode of action for treatment of pemphigus. J Dermatol. 2010;37(3):239–245. | ||
Prins C, Gelfand EW, French LE. Intravenous immunoglobulin: properties, mode of action and practical use in dermatology. Acta Derm Venereol. 2007;87(3):206–218. | ||
Green MG, Bystryn JC. Effect of intravenous immunoglobulin therapy on serum levels of IgG1 and IgG4 antidesmoglein 1 and antidesmoglein 3 antibodies in pemphigus vulgaris. Arch Dermatol. 2008;144(12):1621–1624. | ||
Mimouni D, Blank M, Ashkenazi L, et al. Protective effect of intravenous immunoglobulin (IVIG) in an experimental model of pemphigus vulgaris. Clin Exp Immunol. 2005;142(3):426–432. | ||
Amagai M. Pemphigus. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Madrid: Elsevier; 2012:461–474. | ||
Arnold DF, Burton J, Shine B, Wojnarowska F, Misbah SA. An “n-of-1” placebo-controlled crossover trial of intravenous immunoglobulin as adjuvant therapy in refractory pemphigus vulgaris. Br J Dermatol. 2009;160(5):1098–1102. | ||
Amagai M, Ikeda S, Shimizu H, et al; Pemphigus Study Group. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595–603. | ||
Bystryn JC, Jiao D, Natow S. Treatment of pemphigus with intravenous immunoglobulin. J Am Acad Dermatol. 2002;47(3):358–363. | ||
Ferrara G, Zumla A, Maeurer M. Intravenous immunoglobulin (IVIg) for refractory and difficult-to-treat infections. Am J Med. 2012;125(10):1036.e1–e8. | ||
Sheehan DJ, Lesher JL Jr. Deep venous thrombosis after high-dose intravenous immunoglobulin in the treatment of pemphigus vulgaris. Cutis. 2004;73(6):403–406. | ||
Ventura F, Rocha J, Fernandes JC, Machado A, Brito C. Recalcitrant pemphigus vulgaris: aseptic meningitis associated with intravenous immunoglobulin therapy and successful treatment with rituximab. Int J Dermatol. 2013;52(4):501–502. | ||
Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. 2005;29(3):173–184. | ||
Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol. 2007;33(1–2):67–77. | ||
Turner MS, Sutton D, Sauder DN. The use of plasmapheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2000;43(6):1058–1064. | ||
Roujeau JC, Andre C, Joneau Fabre M, et al. Plasma exchange in pemphigus: uncontrolled study of ten patients. Arch Dermatol. 1983;119(3):215–221. | ||
Tan-Lim R, Bystryn JC. Effect of plasmapheresis therapy on circulating levels of pemphigus antibodies. J Am Acad Dermatol. 1990;22(1):35–40. | ||
Ranugha PS, Kumari R, Kartha LB, Parameswaran S, Thappa DM. Therapeutic plasma exchange as a crisis option in severe pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 2012;78(4):508–510. | ||
Kasuya A, Moriki M, Tatsuno K, Hirakawa S, Tokura Y. Clearance efficacy of autoantibodies in double filtration plasmapheresis for pemphigus foliaceus. Acta Derm Venereol. 2013;93(2):181–182. | ||
Yeh SW, Sami N, Ahmed RA. Treatment of pemphigus vulgaris: current and emerging options. Am J Clin Dermatol. 2005;6(5):327–342. | ||
Sagi L, Baum S, Gendelman V, Trau H, Barzilai A. The role of therapeutic plasma exchange in pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2011;25(1):82–86. | ||
Guillaume JC, Roujeau JC, Morel P, et al. Controlled study of plasma exchange in pemphigus. Arch Dermatol. 1988;124(11):1659–1663. | ||
Braun N, Kadar JG, Risler T. Therapeutic immunoadsorption – its role in clinical practice. Transfus Sci. 1998;19 Suppl:65–69. | ||
Meyersburg D, Schmidt E, Kasperkiewicz M, Zillikens D. Immunoadsorption in dermatology. Ther Apher Dial. 2012;16(4):311–320. | ||
Kasperkiewicz M, Shimanovich I, Meier M, et al. Treatment of severe pemphigus with a combination of immunoadsorption, rituximab, pulsed dexamethasone and azathioprine/mycophenolate mofetil: a pilot study of 23 patients. Br J Dermatol. 2012;166(1):154–160. | ||
Zillikens D, Derfler K, Eming R, et al. Recommendations for the use of immunoapheresis in the treatment of autoimmune bullous diseases. J Dtsch Dermatol Ges. 2007;5(10):881–887. | ||
Lüftl M, Stauber A, Mainka A, Klingel R, Schuler G, Hertl M. Successful removal of pathogenic autoantibodies in pemphigus by immunoadsorption with a tryptophan-linked polyvinylalcohol adsorber. Br J Dermatol. 2003;149(3):598–605. | ||
Frost N, Messer G, Fierlbeck G, Risler T, Lytton SD. Treatment of pemphigus vulgaris with protein A immunoadsorption: case report of long-term history showing favorable outcome. Ann N Y Acad Sci. 2005;1051:591–596. | ||
Eming R, Hertl M. Immunoadsorption in pemphigus. Autoimmunity. 2006;39(7):609–616. | ||
Schneider KM. Plasmapheresis and immunoadsorption: different techniques and their current role in medical therapy. Kidney Int Suppl. 1998;64:S61–S65. | ||
Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res. 2010;302(4):241–253. | ||
Zhao CY, Murrell DF. Pemphigus vulgaris: an evidence-based treatment update. Drugs. 2015;75(3):271–284. | ||
Werth VP, Fivenson D, Pandya AG, et al. Multicenter randomized, double-blind, placebo-controlled, clinical trial of dapsone as a glucocorticoid-sparing agent in maintenance-phase pemphigus vulgaris. Arch Dermatol. 2008;144(1):25–32. | ||
Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol. 2009;10(6):383–396. | ||
Cipriani P, Ruscitti P, Carubbi F, Liakouli V, Giacomelli R. Methotrexate: an old new drug in autoimmune disease. Expert Rev Clin Immunol. 2014;10(11):1519–1530. | ||
Baum S, Greenberger S, Samuelov L, et al. Methotrexate is an effective and safe adjuvant therapy for pemphigus vulgaris. Eur J Dermatol. 2012;22(1):83–87. | ||
Tran KD, Wolverton JE, Soter NA. Methotrexate in the treatment of pemphigus vulgaris: experience in 23 patients. Br J Dermatol. 2013;169(4):916–921. | ||
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47(2–3):119–125. | ||
Doller A, Akool el-S, Müller R, et al. Molecular mechanisms of cyclosporin A inhibition of the cytokine-induced matrix metalloproteinase-9 in glomerular mesangial cells. J Am Soc Nephrol. 2007;18(2):581–592. | ||
Ioannides D, Chrysomallis F, Bystryn JC. Ineffectiveness of cyclosporine as an adjuvant to corticosteroids in the treatment of pemphigus. Arch Dermatol. 2000;136(7):868–872. | ||
Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179–184. | ||
Hertl M, Amagai M, Sundaram H, Stanley J, Ishii K, Katz SI. Recognition of desmoglein 3 by autoreactive T cells in pemphigus vulgaris patients and normals. J Invest Dermatol. 1998;110(1):62–66. | ||
Chow S, Rizzo C, Ravitskiy L, Sinha AA. The role of T cells in cutaneous autoimmune disease. Autoimmunity. 2005;38(4):303–317. | ||
Aoki-Ota M, Kinoshita M, Ota T, et al. Tolerance induction by the blockade of CD40/CD154 interaction in pemphigus vulgaris mouse model. J Invest Dermatol. 2006;126(1):105–113. | ||
Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, Kuwana M. Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine pemphigus ulgaris. J Immunol. 2008;181(2):1526–1535. | ||
Anhalt G, Werth V, Strober B, et al. An open-label phase I clinical study to assess the safety of PI-0824 in patients with pemphigus vulgaris. J Invest Dermatol. 2005;125(5):1088. | ||
Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189(11):1747–1756. | ||
Hahne M, Kataoka T, Schröter M, et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J Exp Med. 1998;188(6):1185–1190. | ||
Litinskiy MB, Nardelli B, Hilbert DM, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3(9):822–829. | ||
Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18(5):263–275. | ||
Tangye SG, Bryant VL, Cuss AK, Good KL. BAFF, APRIL and human B cell disorders. Semin Immunol. 2006;18(5):305–317. | ||
Asashima N, Fujimoto M, Watanabe R, et al. Serum levels of BAFF are increased in bullous pemphigoid but not in pemphigus vulgaris. Br J Dermatol. 2006;155(2):330–336. | ||
Nagel A, Podstawa E, Eickmann M, Müller HH, Hertl M, Eming R. Rituximab mediates a strong elevation of B-cell-activating factor associated with increased pathogen-specific IgG but not autoantibodies in pemphigus vulgaris. J Invest Dermatol. 2009;129(9):2202–2210. | ||
Sinha AA, Hoffman MB, Janicke EC. Pemphigus vulgaris: approach to treatment. Eur J Dermatol. 2015;25(2):103–113. | ||
Berkowitz P, Hu P, Liu Z, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280(25):23778–23784. | ||
Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103(34):12855–12860. | ||
Yoshida K, Ishii K, Shimizu A, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci. 2017;85(3):197–207. | ||
Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. 2011;286(2):1283–1291. | ||
Sweeney SE, Firestein GS. Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann Rheum Dis. 2006;65 (Suppl 3):iii83–iii88. | ||
Spindler V, Vielmuth F, Schmidt E, Rubenstein DS, Waschke J. Protective endogenous cyclic adenosine 5′-monophosphate signaling triggered by pemphigus autoantibodies. J Immunol. 2010;185(11):6831–6838. | ||
Grando SA, Pittelkow MR, Schallreuter KU. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J Invest Dermatol. 2006;126(9):1948–1965. | ||
Williamson L, Raess NA, Caldelari R, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25(14):3298–3309. | ||
Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem. 2013;288(13):9447–9456. | ||
Outerbridge C, Davis D, White S, et al. A new treatment for autoimmune blistering diseases – the efficacy of the Bruton’s tyrosine kinase inhibitor PRN473 in canine pemphigus foliaceus. J Am Acad Dermatol. 2016;74(5):AB141. | ||
Smith CE, Berglöf A. X-linked agammaglobulinemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. SourceGeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301626. Accessed February 23, 2018. | ||
Principia Biopharma, Inc. A study of PRN1008 in adult patients with pemphigus vulgaris. Available from: https://clinicaltrials.gov/ct2/show/NCT02704429?cond=pemphigus&rank=10. NLM identifier: NCT02704429. Accessed February 23, 2018. | ||
Iqbal MM. Osteoporosis: epidemiology, diagnosis, and treatment. South Med J. 2000;93(1):2–18. | ||
Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken). 2010;62(11):1515–1526. | ||
Schadt C, Jackson S. Glucocorticoids. In: Bolognia JL, Schaffer JV, Jorizzo JL, editors. Dermatology. 4th ed. Philadelphia: Elsevier; 2017:2186–2199. | ||
Desforges JF, Mankin HJ. Nontraumatic necrosis of bone (osteonecrosis). N Engl J Med. 1992;326(22):1473–1479. | ||
Gupta Y, Gupta A. Glucocorticoid-induced myopathy: pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17(5):913–916. | ||
Allen DB. Growth suppression by glucocorticoid therapy. Endocrinol Metab Clin North Am. 1996;25(3):699–717. | ||
Renfro L, Snow JS. Ocular effects of topical and systemic steroids. Dermatol Clin. 1992;10(3):505–512. | ||
Whitworth JA. Mechanisms of glucocorticoid-induced hypertension. Kidney Int. 1987;31(5):1213–1224. | ||
Piper JM, Ray WA, Daugherty JR, Griffin MR. Corticosteroid use and peptic ulcer disease: role of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;114(9):735–740. | ||
Amber KT, Lamberts A, Solimani F, et al. Determining the incidence of Pneumocystis pneumonia in patients with autoimmune blistering diseases not receiving routine prophylaxis. JAMA Dermatol. 2017;153(11):1137–1141. | ||
Fardet L, Flahault A, Kettaneh A, et al. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br J Dermatol. 2007;157(1):142–148. | ||
Tabrizi MN, Chams-Davatchi C, Esmaeeli N, et al. Accelerating effects of epidermal growth factor on skin lesions of pemphigus vulgaris: a double-blind, randomized, controlled trial. J Eur Acad Dermatol Venereol. 2007;21(1):79–84. | ||
el-Darouti M, Marzouk S, Abdel Hay R, et al. The use of sulfasalazine and pentoxifylline (low-cost antitumour necrosis factor drugs) as adjuvant therapy for the treatment of pemphigus vulgaris: a comparative study. Br J Dermatol. 2009;161(2):313–319. | ||
Iraji F, Asilian A, Siadat AH. Pimecrolimus 1% cream in the treatment of cutaneous lesions of pemphigus vulgaris: a double-blind, placebo-controlled clinical trial. J Drugs Dermatol. 2010;9(6):684–686. | ||
Fiorentino DF, Garcia MS, Rehmus W, Kimball AB. A pilot study of etanercept treatment for pemphigus vulgaris. Arch Dermatol. 2011;147(1):117–118. | ||
Nazemi-Tabrizi MJ, Hatami P, Ghiasi M, Daneshpazhooh M, Chams-Davatchi C. Randomized trial of tacrolimus 0.1% ointment versus triamcinolone acetonide 0.1% paste in the treatment of oral pemphigus vulgaris. Iran J Dermatol. 2012;15(60):42–46. | ||
Hall RP 3rd, Fairley J, Woodley D, et al. A multicentre randomized trial of the treatment of patients with pemphigus vulgaris with infliximab and prednisone compared with prednisone alone. Br J Dermatol. 2015;172(3):760–768. | ||
Iraji F, Faghihi G, Siadat AH. The efficacy of acyclovir in treatment of the pemphigus vulgaris. J Res Med Sci. 2013;18(11):976–978. | ||
Kanwar AJ, Vinay K, Sawatkar GU, et al. Clinical and immunological outcomes of high- and low-dose rituximab treatments in patients with pemphigus: a randomized, comparative, observer-blinded study. Br J Dermatol. 2014;170(6):1341–1349. | ||
Dastgheib L, Sadati MS, Baghernejhad M. Assessment of the adjuvant effect of tacrolimus in the management of pemphigus vulgaris: a randomized controlled trial. J Dermatolog Treat. 2015;26(1):90–93. | ||
El-Darouti MA, Hegazy RA, Abdel Hay RM, et al. Study of T helper 1 and T helper 2 responses in pemphigus vulgaris patients receiving interferon alpha 2a injections in addition to a standard protocol therapy: a randomized controlled trial. Arch Dermatol Res. 2015;307(4):299–307. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.