")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 1
Emerging role of the peroxisome proliferator-activated receptor-gamma in hepatocellular carcinoma
Received 8 May 2014
Accepted for publication 19 June 2014
Published 26 August 2014 Volume 2014:1 Pages 127—135
DOI https://doi.org/10.2147/JHC.S48512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Hui-Tzu Hsu
Views: 3186
Hui-Tzu Hsu,1 Chin-Wen Chi1,2
1Department and Institute of Pharmacology, School of Medicine, National Yang-Ming University, Taipei, Taiwan; 2Department of Medical Research, Taipei Veterans General Hospital, Taipei, Taiwan
Abstract: Hepatocellular carcinoma (HCC) is the major leading cause of cancer death worldwide. Hepatitis B virus, hepatitis C virus, alcohol consumption, non-alcoholic fatty liver disease, and diabetes are the major risks for developing HCC. Until now, recurrence and metastasis are the major cause of death in HCC patients. Therefore, identification of new effective molecular targets is an urgent need for treatment of HCC. Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear receptor which could be activated by PPARγ agonists such as thiazolidinediones, and natural PPARγ ligand (such as 15-deoxy-∆12,14-prostaglandin J2, 15d-PGJ2). Increasing in vitro and in vivo evidence has demonstrated that PPARγ agonists exhibit an inhibitory role on tumor cell growth, migration, and invasion, suggesting that PPARγ activation may play an important role in the regulation of growth of HCC. It has been reported that PPARγ activation by thiazolidinediones or overexpression of PPARγ by virus-mediated gene transfer has shown growth inhibitory effects in hepatoma cells, but the expression level of PPARγ in HCC tissues still remains conflicting. Notably, a novel PPARγ agonist, honokiol, has recently been found to activate the PPARγ /RXR heterodimer, and has also exhibited significant anti-cancer effects in hepatoma cells. In the present review, we summarized studies on the role and the molecular regulation of PPARγ in HCC development in vitro and in vivo. PPARγ has the potential to be a therapeutic target for future treatment of HCC.
Keywords: hepatocellular carcinoma, peroxisome proliferator-activated receptor γ, thiazolidinediones, honokiol, microRNA
Hepatocellular carcinoma
Liver cancer is the second leading cause of cancer death in males worldwide.1 Hepatocellular carcinoma (HCC) is the major subtype among liver cancers. Eighty percent of HCC occurred in Asia and Africa, where the high prevalence of hepatitis B virus (HBV) and hepatitis C virus (HCV) infections is highly correlated to inflammation of the liver, leading to the subsequent development of HCC.2 Additionally, patients with diabetes not only have an increased risk of HCC3 but also have a poorer survival rate after curative therapy for HCC.4 On the other hand, obesity, dietary aflatoxin B1 exposure, and excessive alcohol consumption are also major risk factors for the development of HCC.5,6 Moreover, liver cirrhosis is a well-known risk factor for HCC (Figure 1). Inflammatory cytokine-induced signaling plays an important role in liver cirrhosis7 and carcinogenesis of HCC.8 In western countries, 30%–40% of HCC cases are attributable to non-alcoholic fatty liver disease (NAFLD) or metabolic syndrome.9,10 Until now, surgical resection has been the best treatment for HCC, especially for patients with a tumor ≤5 cm in diameter. In addition, liver transplantation provides another option for curing HCC but is limited by the number of available donors.11 Recurrence and metastasis are the major causes of increased mortality after treatments such as transcatheter arterial chemoembolization, chemotherapy, and radiotherapy.12,13 Therefore, understanding the pathogenesis and development of HCC can help us to identify new therapeutic targets and to develop new therapeutic agents or strategies to increase the effectiveness of treatment for patients with HCC.
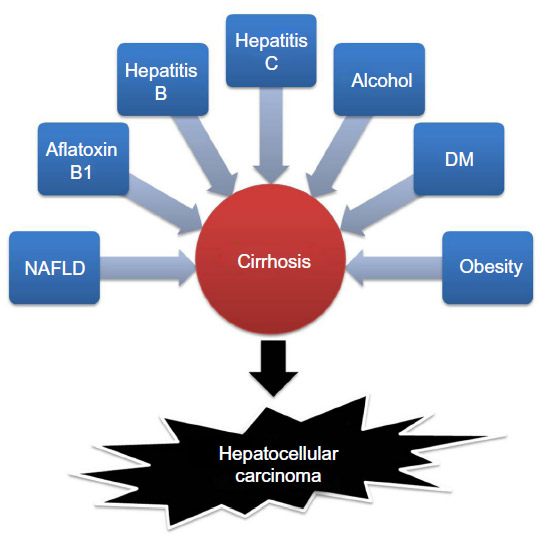 | Figure 1 Risk factors for hepatocellular carcinoma development. |
Targeted therapy has significantly advanced treatment of HCC as chemotherapy for HCC has been met with limited success for many years as a result of high expression of multidrug resistance in HCC.14,15 In the past few years, a small multikinase (vascular endothelial growth factor [VEGF] receptor, platelet-derived growth factor receptor, and Raf kinases) inhibitor, sorafenib (Nexavar®, Bayer and Onyx Pharmaceuticals, South San Francisco, California, USA), was the first targeted therapeutic agent that showed a significant increase in the survival of HCC patients.16–18 It has long been known that HCC tissue has VEGF expression.19 Most recently, it has been reported that decreased plasma VEGF, a major mediator of angiogenesis in HCC, with a level >5% at 8 weeks after sorafenib treatment was highly associated with favorable overall survival in advanced HCC patients.20 It is interesting that the VEGF level in these patients showed an increase at 4 weeks after sorafenib treatment and this may have resulted from sorafenib-induced hypoxia in tumor cells leading to increased secretion of VEGF. However, only those patients who had a >5% decreased VEGF level at 8 weeks after starting sorafenib treatment had favorable overall survival. The mechanism for this observation is not clear but it is likely that decreased secretion of VEGF from tumor cells may be important. This report demonstrated that sequential measurement of VEGF is important for the prediction of sorafenib treatment’s efficacy in advanced HCC patients.20
In addition to targeted therapy, there are many anti-inflammatory herbal medicines such as baicalein,21 curcumin,22 quercetin,23 resveratrol,24 and honokiol25–27 which also exhibited an inhibitory role on cell growth, migration, and invasion of HCC.28 Recently, a natural biphenolic compound honokiol was found to function as a novel non-adipogenic peroxisome proliferator-activated receptor γ (PPARγ) agonist of adipocyte.29 Notably, PPARγ is not only involved in anti-inflammatory pathways, but also exerted cell growth inhibition and anti-metastasis in HCC.30 Moreover, synthetic PPARγ agonists, thiazolidinediones (TZDs), also showed anti-tumor effects on HCC.30 Consequently, the function, molecular pathways, and herbal medicine involved in PPARγ signaling pathways will be novel targets for potential treatment of HCC.
Molecular structure of PPARγ
PPARγ is a ligand-activated nuclear hormone receptor. Once ligand binding has occurred, PPARγ heterodimerizes with retinoid X receptor (RXR) and this complex subsequently recruits coactivators or corepressors, to regulate the target genes related to lipid and glucose metabolisms as well as inflammation.31 Human PPARγ protein is composed of four functional domains including an N-terminal A/B domain (containing activation function 1, AF1), a DNA-binding domain containing two zinc fingers, a hinge domain, and a C-terminal ligand binding domain (LBD) (containing AF2) (Figure 2).32 The A/B domain is associated with ligand-independent transactivation through phosphorylation.33 This phosphorylation decreased transcriptional activity of PPARγ by mitogen-activated protein kinase34 and 5′-AMP-activated protein kinase to repress ligand-dependent effects including lipid and glucose metabolisms.35 The LBD is responsible for ligand-dependent transactivation and is the crucial regulatory domain for the heterodimerization which is capable of recruiting coactivators and corepressors necessary for the subsequent transcriptional regulation (Figure 3).36 PPARγ can be activated by the natural ligands such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2),37 lysophosphatidic acid,38 and nitrolinoleic acid.39 PPARγ can also be activated by synthetic ligands including the TZDs such as rosiglitazone, pioglitazone, ciglitazone, and troglitazone.40,41 There are two PPARγ isoforms expressed in mouse42 and human,43 γ1 and γ2, with different length of N-terminus. PPARγ2 has an additional 28 amino acids at its N-terminus resulting from different promoter use and alternative RNA splicing.42 PPARγ1 is expressed in various tissues including adipose tissue, heart, skeletal muscle, liver, kidney, intestines, colon, kidney, pancreas, and spleen.44 Whereas PPARγ2 is expressed mainly in adipose tissue.45,46 A previous study reported that PPARγ1 and PPARγ2 have distinctive activation capacities of insulin, the authors isolated PPARγ1 and PPARγ2 N termini with activation domains, and the activation capacities of PPARγ2 was 5–6 fold greater than that of PPARγ1.33 A better understanding of the molecular structure of PPARγ will provide more opportunities to design ligands to regulate the receptor-mediated biological activities.
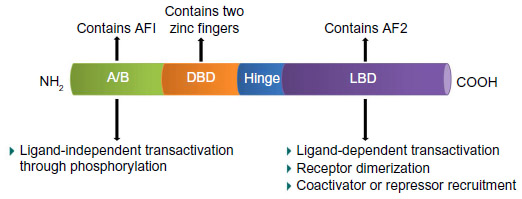 | Figure 2 Schematic diagram of the functional domain of PPARγ. |
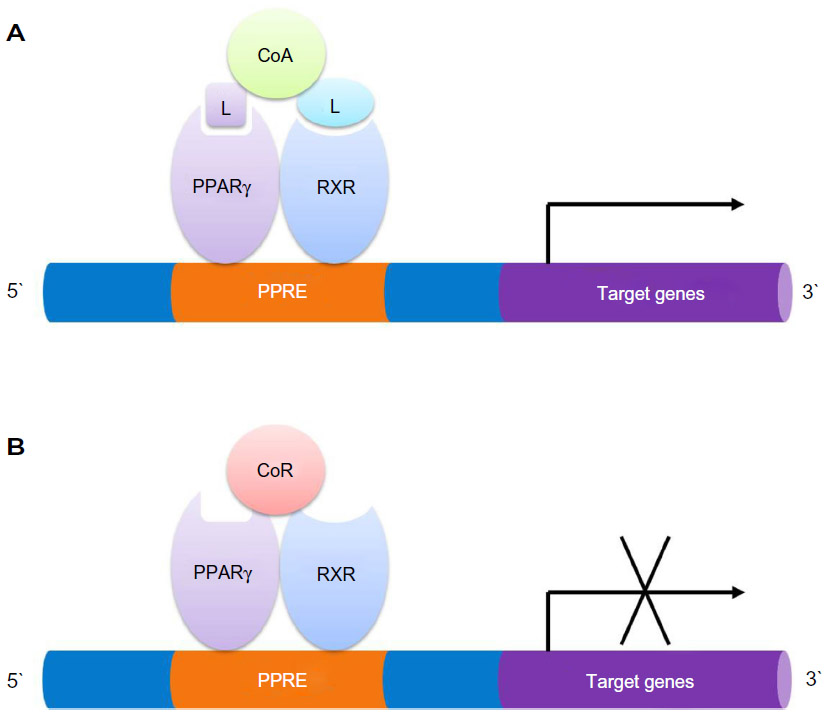 | Figure 3 Binding of PPARγ:RXR heterodimers to the PPAR response element. |
Expression of PPARγ in HCC
The expression of PPARγ in human HCC tissues showed conflicting results from previous studies. Koga et al found there was no significant difference of PPARγ protein expression between HCC tissues and adjacent non-tumorous liver tissues from five patients with HCV-associated HCC.47 Interestingly, Schaefer et al found increased expression of PPARγ protein in tumors from 20 HCC patients by immunohistochemistry but no expression in non-tumorous liver distal to the tumor.48 In contrast, Yu et al found a significantly reduced PPARγ expression in HCC tissues compared with adjacent non-tumorous liver in 20 HCC patients, as assessed by Western blot.49 More recently, Lin et al found a significant increase in mRNA expression of PPARγ by reverse transcription polymerase chain reaction (RT-PCR) in tumor tissues compared with normal liver in 16 HCC patients. In addition, they also examined the expression of PPARγ in HepG2, Hep3B, Huh-7, and HA22T hepatoma cell lines by RT-PCR and Western blot analyses. The protein level (Hep3B>Huh-7=HA22T>HepG2) of PPARγ did not show exactly the same patterns as compared with mRNA level (Hep3B>HepG2>Huh-7>HA22T).50 Therefore, studies using a larger sample size are required in order to elucidate the functional importance and the clinical significance of PPARγ in HCC. A better understanding of the expression of PPARγ in HCC tissues may help us to elucidate its role in the pathogenesis of HCC.
Functions of PPARγ and PPARγ ligands in HCC
Deficiency and overexpression of PPARγ in the progression of HCC
The role of PPARγ in hepatocarcinogenesis was examined by Yu et al using PPARγ-deficient (PPARγ+/−) and wild-type (PPARγ+/+) mice in a diethylnitrosamine-induced HCC model. They found increased hepatocellular carcinogenesis in PPARγ+/− mice in the diethylnitrosamine-induced HCC model as compared to PPARγ+/+ mice, and also found that rosiglitazone decreased the incidence of HCC in PPARγ+/+ mice compared to PPARγ+/− mice, indicating that PPARγ acted as a tumor suppressor in hepatocarcinogenesis.51 Furthermore, adenovirus-mediated mouse PPARγ overexpression in Hep3B hepatoma cells showed significant inhibition of cell growth, increased cell apoptosis through intrinsic (caspase-3, 7, 9, Bax, and poly[ADP-ribose] polymerase) and extrinsic (Fas, tumor necrosis factor-α, and caspase-8) pathways, and cell cycle arrest in G2/M phase through phosphorylation of G2/M phase inhibitors cell division cycle 25 c (cdc25c) and cdc2. In addition, PPARγ overexpression decreased cell viability after rosiglitazone treatment in Hep3B cells.51 Recently, overexpression of mouse PPARγ in MHCC97L and BEL-7404 hepatoma cells treated with rosiglitazone showed a significant inhibition of migration and invasion through upregulation of E-cadherin and tissue inhibitor of metalloproteinase (TIMP) 3 and downregulation of heparanase (HPSE), matrix metalloproteinase (MMP) 9, and MMP13.52 Most recently, adenovirus-mediated PPARγ gene transfer in Hep3B cells exhibited a dramatic increase of Glu/Asp-rich carboxyl-terminal domain, 2 (CITED2) expression in mRNA and protein levels, resulting in G1-S phase arrest and cell growth inhibition.53 However, it will be more interesting if the direct effect of PPARγ ligands on the expression of CITED2 is known in the PPARγ transfected cells. Apparently, PPARγ appears to play a tumor suppressor role in HCC (Figure 4).
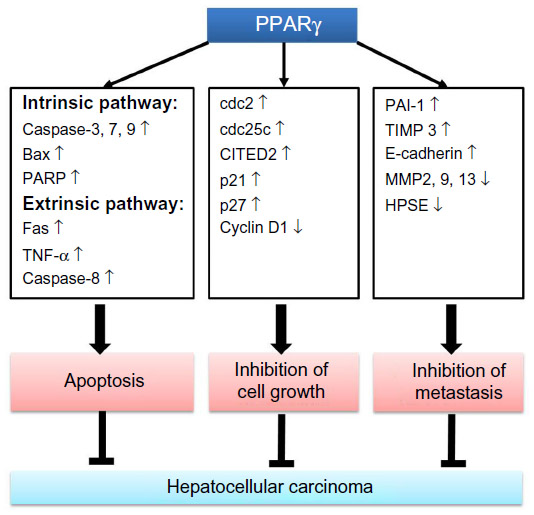 | Figure 4 PPARγ signaling in hepatocellular carcinoma. |
PPARγ agonists inhibit cell growth and induce apoptosis in HCC
Both natural and synthetic PPARγ agonists exhibited growth inhibitory effects in hepatoma cells. TZDs are synthetic PPARγ agonists, used as insulin sensitizers for the treatment of patients with type II diabetes mellitus (DM). PPARγ agonists exert their functions through PPARγ-dependent and PPARγ-independent pathways.54,55 Many studies have reported that TZDs had growth inhibitory effects, such as troglitazone that not only inhibited cell proliferation through increasing p21 and p2747 but also induced apoptosis in hepatoma cells mediated through PI3K-Akt pathway.56 Additionally, troglitazone suppressed COX-2 expression and increased p27 expression to inhibit cell growth in Hep3B and Huh7 hepatoma cells.49 Moreover, rosiglitazone upregulated phosphatase and tensin homolog (PTEN) and downregulated COX-2 expression via PPARγ signaling pathway, leading to the growth inhibition in BEL-7402 and Huh7 cells.57 A natural ligand of PPARγ, 15d-PGJ2, induced apoptosis via activation of NFκB in HepG2 and SK-Hep1 cells.58 Furthermore, 15d-PGJ2 and pioglitazone induced apoptosis possibly through caspase-dependent pathways in HBV-associated cell lines.59 It is interesting that HepG2 and Hep3B cells are less invasive as compared to other hepatoma cells such as QGY7703 cells,60 further investigation using overexpression of PPARγ in low endogenous PPARγ expression hepatoma cells may provide more valuable information. These results together suggest that activation of PPARγ can induce cell growth inhibition and apoptosis in hepatoma cells.
PPARγ ligands inhibit migration, invasion, and metastasis in HCC
PPARγ agonists not only have cell growth inhibitory effects but can also inhibit migration, invasion, and metastasis of hepatoma cells. Rosiglitazone inhibited migration through upregulation of PTEN accompanied with decreased phosphorylation of Akt and focal adhesion kinase in hepatoma cell line BEL-7404.61 Rosiglitazone and troglitazone inhibited cell migration via upregulation of E-cadherin expression in HepG2 cells.54 A recent study demonstrated that activation of PPARγ by rosiglitazone decreased lung metastasis in an orthotopic HCC mouse model.52 PPARγ agonist GW1929 decreased invasion ability via upregulation of plasminogen activator inhibitor-1 (PAI-1) in HepG2 and Hepa1-6 cells. However, the invasion ability was restored after pretreatment of PPARγ antagonist GW9662 in Hepa1-6 cells.62 Interestingly, PPARγ antagonists GW9662 and T0070907 also inhibited cell migration and invasion through induction of vimentin degradation and inactivation of focal adhesion kinase in poorly differentiated hepatoma cell lines SH-J1 and HLE with high expression of PPARγ.63 At present, the detailed mechanism of PPARγ antagonist-induced inhibition of migration in HCC cells is not clear. It is possible that an off target effect exists for PPARγ antagonist in HCC cells, but further experiments using PPARγ knockdown HCC cells will be able to clarify whether this effect is dependent on PPARγ or not. Together, these results clearly showed that both PPARγ agonists and antagonists exhibited an inhibitory role on migration and invasion in hepatoma cells, the detailed molecular mechanism of PPARγ upregulated genes in relation to migration and invasion still needs further investigation.
Honokiol serves as a novel non-adipogenic PPARγ agonist
Honokiol, a natural biphenolic compound derived from the stem and bark of the plant Magnolia officinalis, exhibits anti-angiogenic and pro-apoptotic activity in cancer cells.64 Recent studies found that honokiol, a natural rexinoid, can serve as an RXR agonist in the activation of RXR heterodimers.65,66 In addition, honokiol potentiated the PPARγ agonist rosiglitazone induced activation of PPARγ/RXR heterodimers.67 Most recently, one study has found that honokiol directly bound to purified PPARγ LBD and served as a novel non-adipogenic PPARγ agonist to stimulate glucose uptake in 3T3-L1 adipocytes.29 Additionally, it has been reported that honokiol possesses a wide variety of pharmacological actions with low toxicity such as anti-inflammatory (in lipopolysaccharide-stimulated human monocyte-derived dendritic cells through inhibition of NF-κB signaling),68 antioxidant,69 anti-thrombosis,70 and neuroprotective.71 Moreover, its anti-tumor effects have been found in different types of cancers such as colorectal cancer,72 gastric cancer,73 and HCC.25–27 Honokiol also exhibited an inhibitory role on hepatoma cells by activating the p38 mitogen-activated protein kinase and caspase 3 pathways to induce apoptosis in HepG2 cells,25 upregulated Ras GTPase-activating-like protein expression to inhibit HepG2 cell migration.26 Recently, it has been reported that honokiol inhibited STAT3 to reduce cell proliferation in HCC cells.27 Collectively, results from these studies show that honokiol has the potential to become a novel agent to treat HCC.
Crosstalk between PPARγ and TGF-β
Transforming growth factor-β (TGF-β) induces cell growth, cell migration, and epithelial to mesenchymal transition as a key driver to promote HCC progression.74 There are two mechanisms: intrinsic and extrinsic activities involved in TGF-β activation pathway. Once the cell polarity has been lost, acquisition of motile ability, and epithelial to mesenchymal transition are considered as intrinsic changes of the tumor cells.75 Extrinsic factors are involved in the changes of tumor microenvironment including angiogenesis, inflammation, and fibroblast activation.74 Therefore, TGF-β can be considered as a therapeutic target in HCC. Interestingly, PPARγ activation plays an inhibitory role to repress the expression of TGF-β via dephosphorylation of zinc finger transcription factor-9, and this dephosphorylation was induced by PTEN-mediated p70 ribosomal S6 kinase-1 inhibition in mouse fibroblast cells.76 However, there is no available information to present on the regulation between PPARγ and TGF-β in HCC. Therefore, further investigations are needed to address this issue in HCC. In the pharmacological manipulation, the crosstalk between PPARγ and TGF-β can be applied for treating TGF-β mediated fibrosis or cancer metastasis.
Interaction between microRNAs and PPARγ in HCC
The function of microRNAs (miRNAs) in cancers has been studied extensively in recent years. miRNAs are a group of small (20–22 nucleotides) molecule, endogenous, and non-coding RNAs that play an important role in regulating gene expression by targeting mRNAs 3′-untranslated region for cleavage or translational repression to silence gene expression in eukaryotes.77 miRNAs may serve as tumor suppressors or oncogenes which are involved in many biological functions including development, cell proliferation, differentiation, metabolism, and apoptosis.78 Many studies have demonstrated that miRNAs are important regulators of gene expression in HCC cells. Recently, it has been reported that PPARγ and RXRα were regulated by miR-27a in HCC cells to reduce lipid synthesis.79 Additionally, miR-122 is liver-specific miRNA which is significantly reduced in HCC, and is correlated with poor prognosis and metastasis.80 Most recently, it has been reported that restoration of miR-122 expression in HCC cells reduced AKT3 levels, inhibited cell migration and proliferation, and induced apoptosis, suggesting that miR-122 functions as a tumor suppressor.81 It has been reported that some miRNAs could target PPARγ and mediate signal transduction of cellular functions in HCC cells. Interestingly, a recent study clearly showed that miR-122 can be upregulated in hepatoma cells via overexpression of PPARγ by PPARγ/RXRα complex.82 These studies together suggest that the reciprocal regulation between miR-122 and PPARγ is worthy of further investigation.
Use of TZDs and the risk of HCC in patients with type II DM
Several cohort studies have reported that TZDs used in DM patients may influence the risk for HCC. The association between HCC and DM has been reviewed using epidemiological data. A previous study has shown that DM patients have a 2–3 fold increased risk of HCC from a population based case control study in the USA, despite the presence of other major HCC risk factors including HBV or HCV infections or alcoholic liver disease.3 Similarly, results from a population-based cohort study conducted using the Taiwan National Health Insurance Research Database showed an increased incidence of HCC in DM patients. Moreover, this study also showed that the use of TZDs or metformin reduced the risk of HCC development in DM patients.83 A similar study also showed a significant decrease of liver cancer incidence in type II DM patients treated with rosiglitazone or pioglitazone.84 In contrast, a systematic review and meta-analysis using data from Medline, EMBASE, and Web of Science up to August 2012 showed that TZDs did not change the risk of HCC in patients with type II DM.85 Whether this reduced risk is due to control of diabetes which reduces stress on the liver leading to reduced risk of cancer, or due to PPAR related effects is not clear at present and worthy of further investigation. These results together suggest that the underlying functional role of TZDs in HCC development in type II DM patients needs further investigation.
Conclusion
Current in vitro and in vivo evidence supports the fact that PPARγ activation exerted an inhibitory role on cell growth, migration, and invasion in HCC cells. Most of the studies focused on the effects of TZDs (PPARγ agonist) in HCC cells, while TZDs are currently only used for treatment of type II DM patients. TZDs have known adverse effects such as heart failure.86 However, a combination of TZDs with chemotherapeutic drugs for local delivery via transcatheter arterial chemoembolization method may provide an alternative approach for HCC treatment. On the other hand, in view of the recent finding that honokiol has anti-cancer effects, the molecular mechanisms of honokiol-induced cell growth, apoptosis, migration, and invasion needs further investigation in hepatoma cells using in vitro and in vivo models. More importantly, studies focusing on the differential miRNAs expression in HCC, especially miR122 which was regulated by PPARγ in hepatoma cells, may be a potential target for herbal drugs and PPARγ agonists/antagonists. A better understanding of the molecular regulation on anti-cancer effects in PPARγ-mediated signaling may contribute to the development of novel combination therapy for future treatment of HCC.
Disclosure
The authors have no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | |
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264–1273. | |
Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case control study. Gut. 2005;54(4):533–539. | |
Shau WY, Shao YY, Yeh YC, et al. Diabetes mellitus is associated with increased mortality in patients receiving curative therapy for hepatocellular carcinoma. Oncologist. 2012;17(6):856–862. | |
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. | |
Wu HC, Santella R. The role of aflatoxins in hepatocellular carcinoma. Hepat Mon. 2012;12(10 HCC):e7238. | |
Yang YY. Can serum cytokines predict hepatic cytokine expression in liver cirrhosis? J Chin Med Assoc. 2011;74(11):485–486. | |
Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. | |
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med. 2003;348(17):1625–1638. | |
El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4(3):369–380. | |
Maggs JR, Suddle AR, Aluvihare V, Heneghan MA. Systematic review: the role of liver transplantation in the management of hepatocellular carcinoma. Aliment Pharmacol Ther. 2012;35(10):1113–1134. | |
Tang ZY. Hepatocellular carcinoma – cause, treatment and metastasis. World J Gastroenterol. 2001;7(4):445–454. | |
Flores A, Marrero JA. Emerging Trends in Hepatocellular Carcinoma: Focus on Diagnosis and Therapeutics. Clin Med Insights Oncol. 2014;8:71–76. | |
Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686–695. | |
Shen DW, Lu YG, Chin KV, Pastan I, Gottesman MM. Human hepatocellular carcinoma cell lines exhibit multidrug resistance unrelated to MRD1 gene expression. J Cell Sci. 1991;98(Pt 3):317–322. | |
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | |
Worns MA, Koch S, Niederle IM, et al. The impact of patient and tumour baseline characteristics on the overall survival of patients with advanced hepatocellular carcinoma treated with sorafenib. Dig Liver Dis. 2013;45(5):408–413. | |
Yamaguchi R, Yano H, Iemura A, Oqasawara S, Haramaki M, Kojiro M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology. 1998;28(1):68–77. | |
Tsuchiya K, Asahina Y, Matsuda S, et al. Changes in plasma vascular endothelial growth factor at 8 weeks after sorafenib administration as predictors of survival for advanced hepatocellular carcinoma. Cancer. 2014;120(2):229–237. | |
Chen K, Zhang S, Ji Y, et al. Baicalein inhibits the invasion and metastatic capabilities of hepatocellular carcinoma cells via down-regulation of the ERK pathway. PLoS One. 2013;8(9):e72927. | |
Xu MX, Zhao L, Deng C, et al. Curcumin suppresses proliferation and induces apoptosis of human hepatocellular carcinoma cells via the wnt signaling pathway. Int J Oncol. 2013;43(6):1951–1959. | |
Zhao JL, Zhao J, Jiao HJ. Synergistic growth-suppressive effects of quercetin and cisplatin on HepG2 human hepatocellular carcinoma cells. Appl Biochem Biotechnol. 2014;172(2):784–791. | |
Yeh CB, Hsieh MJ, Lin CW, et al. The antimetastatic effects of resveratrol on hepatocellular carcinoma through the downregulation of a metastasis-associated protease by SP-1 modulation. PLoS One. 2013;8(2):e56661. | |
Deng J, Qian Y, Geng L, et al. Involvement of p38 mitogen-activated protein kinase pathway in honokiol-induced apoptosis in a human hepatoma cell line (hepG2). Liver Int. 2008;28(10):1458–1464. | |
Liang S, Fu A, Zhang Q, et al. Honokiol inhibits HepG2 migration via down-regulation of IQGAP1 expression discovered by a quantitative pharmaceutical proteomic analysis. Proteomics. 2010;10(7):1474–1483. | |
Rajendran P, Li F, Shanmugam MK, et al. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J Cell Physiol. 2012;227(5):2184–2195. | |
Hu Y, Wang S, Wu X, et al. Chinese herbal medicine-derived compounds for cancer therapy: a focus on hepatocellular carcinoma. J Ethnopharmacol. 2013;149(3):601–612. | |
Atanasov AG, Wang JN, Gu SP, et al. Honokiol: a non-adipogenic PPARgamma agonist from nature. Biochim Biophys Acta. 2013; 1830(10):4813–4819. | |
Wu CW, Farrell GC, Yu J. Functional role of peroxisome-proliferator-activated receptor gamma in hepatocellular carcinoma. J Gastroenterol Hepatol. 2012;27(11):1665–1669. | |
Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20(5):649–688. | |
Zieleniak A, Wojcik M, Wozniak LA. Structure and physiological functions of the human peroxisome proliferator-activated receptor gamma. Arch Immunol Ther Exp (Warsz). 2008;56(5):331–345. | |
Werman A, Hollenberg A, Solanes G, et al. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor gamma (PPARgamma). Differential activity of PPARgamma1 and -2 isoforms and influence of insulin. J Biol Chem. 1997;272(32):20230–20235. | |
Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272(8):5128–5132. | |
Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans. 2003;31(Pt 1):224–227. | |
Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol. 1997;9(2):222–232. | |
Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83(5):803–812. | |
McIntyre TM, Pontsler AV, Silva AR, et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci U S A. 2003;100(1):131–136. | |
Schopfer FJ, Lin Y, Baker PR, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A. 2005;102(7):2340–2345. | |
Murphy GJ, Holder JC. PPAR-gamma agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol Sci. 2000;21(12):469–474. | |
Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep. 2013;13(3):329–341. | |
Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A. 1995;92(17):7921–7925. | |
Elbrecht A, Chen Y, Cullinan CA, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun. 1996;224(2):431–437. | |
Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications – a review. Nutr J. 2014;13:17. | |
Fajas L, Auboeuf D, Raspe E, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272(30):18779–18789. | |
Saraf N, Sharma PK, Mondal SC, Garg VK, Singh AK. Role of PPARg2 transcription factor in thiazolidinedione-induced insulin sensitization. J Pharm Pharmacol. 2012;64(2):161–171. | |
Koga H, Sakisaka S, Harada M, et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology. 2001;33(5):1087–1097. | |
Schaefer KL, Wada K, Takahashi H, et al. Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005;65(6):2251–2259. | |
Yu J, Qiao L, Zimmermann L, et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology. 2006;43(1):134–143. | |
Lin YM, Velmurugan BK, Yeh YL, et al. Activation of estrogen receptors with E2 downregulates peroxisome proliferator-activated receptor gamma in hepatocellular carcinoma. Oncol Rep. 2013;30(6):3027–3031. | |
Yu J, Shen B, Chu ES, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51(6):2008–2019. | |
Shen B, Chu ES, Zhao G, et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br J Cancer. 2012;106(9):1486–1494. | |
Cheung KF, Zhao J, Hao Y, et al. CITED2 is a novel direct effector of peroxisome proliferator-activated receptor gamma in suppressing hepatocellular carcinoma cell growth. Cancer. 2013;119(6):1217–1226. | |
Lee HJ, Su Y, Yin PH, Lee HC, Chi CW. PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer Res. 2009;29(12):5057–5063. | |
Uray IP, Rodenberg JM, Bissonnette RP, Brown PH, Mancini MA. Cancer-preventive rexinoid modulates neutral lipid contents of mammary epithelial cells through a peroxisome proliferator-activated receptor gamma-dependent mechanism. Mol Pharmacol. 2012;81(2):228–238. | |
Mishra P, Paramasivam SK, Thylur RP, Rana A, Rana B. Peroxisome proliferator-activated receptor gamma ligand-mediated apoptosis of hepatocellular carcinoma cells depends upon modulation of PI3Kinase pathway independent of Akt. J Mol Signal. 2010;5:1–17. | |
Cao LQ, Wang XL, Wang Q, et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARgamma signaling pathway. Acta Pharmacol Sin. 2009;30(9):1316–1322. | |
Okano H, Shiraki K, Inoue H, et al. 15-deoxy-delta-12-14-PGJ2 regulates apoptosis induction and nuclear factor-kappaB activation via a peroxisome proliferator-activated receptor-gamma-independent mechanism in hepatocellular carcinoma. Lab Invest. 2003;83(10):1529–1539. | |
Shim J, Kim BH, Kim YI, et al. The peroxisome proliferator-activated receptor gamma ligands, pioglitazone and 15-deoxy-Delta(12,14)-prostaglandin J(2), have antineoplastic effects against hepatitis B virus-associated hepatocellular carcinoma cells. Int J Oncol. 2010;36(1):223–231. | |
Zhang NN, Sun QS, Chen Z, Liu F, Jiang YY. Homeostatic regulatory role of Pokemon in NF-kappaB signaling: stimulating both p65 and IkappaBalpha expression in human hepatocellular carcinoma cells. Mol Cell Biochem. 2013;372(1–2):57–64. | |
Zhang W, Wu N, Li Z, Wang L, Jin J, Zha XL. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol Ther. 2006;5(8):1008–1014. | |
Pang X, Wei Y, Zhang Y, Zhang M, Lu Y, Shen P. Peroxisome proliferator-activated receptor-gamma activation inhibits hepatocellular carcinoma cell invasion by upregulating plasminogen activator inhibitor-1. Cancer Sci. 2013;104(6):672–680. | |
Kim KR, Choi HN, Lee HJ, et al. A peroxisome proliferator-activated receptor gamma antagonist induces vimentin cleavage and inhibits invasion in high-grade hepatocellular carcinoma. Oncol Rep. 2007;18(4):825–832. | |
Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11(5):1139–1148. | |
Kotani H, Tanabe H, Mizukami H, Makishima M, Inoue M. Identification of a naturally occurring rexinoid, honokiol, that activates the retinoid X receptor. J Nat Prod. 2010;73(8):1332–1336. | |
Perez E, Bourguet W, Gronemeyer H, de Lera AR. Modulation of RXR function through ligand design. Biochim Biophys Acta. 2012;1821(1):57–69. | |
Kotani H, Tanabe H, Mizukami H, Amagaya S, Inoue M. A naturally occurring rexinoid, honokiol, can serve as a regulator of various retinoid x receptor heterodimers. Biol Pharm Bull. 2012;35(1):1–9. | |
Li CY, Chao LK, Wang SC, et al. Honokiol inhibits LPS-induced maturation and inflammatory response of human monocyte-derived dendritic cells. J Cell Physiol. 2011;226(9):2338–2349. | |
Zhao C, Liu ZQ. Comparison of antioxidant abilities of magnolol and honokiol to scavenge radicals and to protect DNA. Biochimie. 2011;93(10):1755–1760. | |
Teng CM, Chen CC, Ko FN, et al. Two antiplatelet agents from Magnolia officinalis. Thromb Res. 1988;50(6):757–765. | |
Chen C, Lin JK, Liu SH, et al. Characterization of neurotoxic effects of NMDA and the novel neuroprotection by phytopolyphenols in mice. Behav Neurosci. 2010;124(4):541–553. | |
Lai YJ, Lin CI, Wang CL, et al. Expression of survivin and p53 modulates honokiol-induced apoptosis in colorectal cancer cells. J Cell Biochem. Epub June 6, 2014. | |
Liu SH, Shen CC, Yi YC, et al. Honokiol inhibits gastric tumourigenesis by activation of 15-lipoxygenase-1 and consequent inhibition of peroxisome proliferator-activated receptor-gamma and COX-2-dependent signals. Br J Pharmacol. 2010;160(8):1963–1972. | |
Giannelli G, Villa E, Lahn M. Transforming growth factor-beta as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014;74(7):1890–1894. | |
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | |
Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70(1):415–425. | |
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. | |
Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11(4):441–450. | |
Shirasaki T, Honda M, Shimakami T, et al. MicroRNA-27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J Virol. 2013;87(9):5270–5286. | |
Tsai WC, Hsu PW, Lai TC, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49(5):1571–1582. | |
Nassirpour R, Mehta PP, Yin MJ. miR-122 regulates tumorigenesis in hepatocellular carcinoma by targeting AKT3. PLoS One. 2013;8(11):e79655. | |
Song K, Han C, Zhang J, et al. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology. 2013;58(5):1681–1692. | |
Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol. 2012;107(1):46–52. | |
Chang CH, Lin JW, Wu LC, Lai MS, Chuang LM, Chan KA. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology. 2012;55(5):1462–1472. | |
Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108(6):881–891. | |
Marks DH. Drug utilization, safety and clinical use of Actos and Avandia. Int J Risk Saf Med. 2013;25(1):39–51. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.