")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 2
Emerging role of Hpo signaling and YAP in hepatocellular carcinoma
Authors Valero III V, Pawlik T, Anders R
Received 7 April 2014
Accepted for publication 29 May 2014
Published 18 June 2015 Volume 2015:2 Pages 69—78
DOI https://doi.org/10.2147/JHC.S48505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Vicente Valero III,1 Timothy M Pawlik,1 Robert A Anders2
1Department of Surgery, Division of Surgical Oncology, 2Department of Pathology, Sidney Kimmel Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Abstract: Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third most common cause of cancer-related mortality worldwide. Due to the poor prognosis and limited therapeutic options, there is great interest in further understanding better the molecular underpinnings and potential molecular targets associated with HCC. The Hippo (Hpo) signaling pathway and YAP, its principal downstream effector, represent an innovative area of research in HCC. Pioneered in Drosophila melanogaster, the Hpo cascade controls tissue homeostasis including organ size, cell proliferation, apoptosis, as well as cell-cycle regulation and differentiation. This conserved kinase cascade in mammals depends on central control by the tumor suppressor mammalian sterile 20-like kinase 1/2 (Mst1/2). The Mst1/2 commences the downstream kinase cascade, ultimately activating the oncoprotein YAP and allowing its physical association with downstream targets to enhance the gene expression signatures that are involved in proliferation and survival. Alterations in YAP expression and defective regulation of other key Hpo pathway members, such as Mst1/2, Salvador, neurofibromatosis and Mer (Nf2/mer), large tumor suppressor homolog 1/2 (Lats1/2), and Mps one binder kinase activator-like 1A and 1B (Mob1) drive carcinogenesis in animal models. The dysregulation of the Hpo pathway – resulting in an unchecked activation of YAP – culminates in the development of a broad range of human tumor types, including HCC. The abrogation of Mst1/2-mediated YAP phosphorylation permits YAP entry into the nucleus in murine models and functions similarly in human HCCs. Chemoresistance mechanisms displayed by HCC tumors occur in a YAP-dependent manner. The HCC specimens exhibit YAP overexpression, and YAP serves as an independent prognostic marker for disease-free survival and overall survival in patients with HCC. Recently, the small molecule inhibitor, verteporfin has been shown to attenuate YAP activity in murine models, perhaps offering a novel therapeutic approach for patients with advanced HCC.
Keywords: hepatocellular carcinoma, yes-associated-protein, Hippo signaling, liver cancer, hepatic malignancy
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer worldwide and the third most common cause of cancer-related mortality, with 746,000 deaths each year; the estimated survival of patients with advanced HCC is <10%.1,2 Nearly 80% of HCC cases occur in the developing world; however, the incidence of HCC in the USA has increased over the last several decades with the incidence rising from 3.1 per 100,000 persons in the 1990s to 5.1 per 100,000 in the 2000s.3
The increased incidence of HCC in the Western hemisphere is most directly attributable to the hepatitis C virus (HCV) infection, which accounts for approximately one-half of HCC cases.4 While some data suggest that the incidence of HCC will continue to rise, based on the increased prevalence of HCV in the USA, other data imply that the peak of the HCV epidemic in the West has been reached.5,6
The metabolic risk factors for HCC, such as diabetes and obesity, have also resulted in an increased incidence of nonalcoholic fatty liver disease and, in turn, NASH has been associated with the increased incidence of primary liver cancer in the USA.6 In the Eastern hemisphere, hepatitis B virus (HBV) continues to be a main cause of HCC, due to the endemicity observed in adults of this geographic region and the paucity of vaccination programs before the 1980s.4,7 However, the institution of vaccination campaigns has begun to decrease the incidence of HBV infection seen in children.7 Similarly, the ubiquitous spread of HBV and HCV continues to drive the incidence of HCC in eastern Asia and sub-Saharan Africa.4
While resection, ablation, or transplantation is indicated for patients with early-stage disease, the treatment options for patients with advanced HCC are limited. Patients with early-stage disease who are treated with resection or transplantation can expect a 5-year survival of 40%–75%; in contrast, survival among patients with locally advanced disease is dismal, with a 5-year survival ranging from 5%–13%.3,8 Chemotherapeutic options are available; however, they provide limited treatment response.9 For example, studies using cytotoxic agents, such as doxorubicin or gemcitabine plus oxaliplatin, show a response rate as low as 18% and combination therapy with cisplatin, interferon-alpha-2b, doxorubicin, and 5-fluorouracil (PIAF) report response rates of 26%.9
Targeting signaling pathways for therapeutic use in oncology has begun to take root over the last decade. Biologic agents, such as sorafenib, bevacizumab, and erlotinib, add more to the clinicians’ armamentarium; however, more options are needed for patients.9 In a recent Phase III study, the biologic agent sorafenib was shown to increase median survival and the time to radiologic progression by nearly 3 months for patients treated with sorafenib than for those given a placebo.10 Given these data, a better and more fundamental understanding of the genetic and epigenetic mechanisms of hepatocarcinogenesis is needed. This, in turn, may help lead to the discovery of novel treatment regimens for patients with unresectable disease.
Several groups have reported that the dysregulation of the Hippo (Hpo) signaling pathway can lead to HCC formation.11–17 YAP, the downstream effector of the Hpo pathway, controls a myriad of protein targets that influence gene expression.16,18 The emergence of the Hpo-signaling pathway in HCC development will hopefully further delineate the molecular drivers of HCC while providing the clinician avenues for risk stratification and novel therapeutic strategies.
Hippo signaling pathway
The Hpo signaling cascade regulates the expression of genes favorable to cell cycle progression, proliferation, differentiation, and survival.18 The canonical pathway members Hpo, Warts (Wrts), and Salvador (Sav) were discovered through genetic screens in Drosophila melanogaster, while searching for suppressors of tissue growth.16 The origin of the pathway names arises from the hippopotamus-like phenotype observed in D. melanogaster when the members of the pathway are mutated. The pathway plays an integral role in determining organ size and the growth control regulation.16,19,20
Core components of the pathway are conserved in mammals and act in a kinase cascade that exhibits a complex network of crosstalk with other important signaling pathways, including the TGFβ/SMAD, WNT/β-catenin, PI3-kinase/AKT, Jnk, Hedgehog, Jak/Stat, Notch, and apoptotic pathways.18 Ultimately, YAP is the effector of the Hpo pathway and coordinates interactions with these signaling pathways by the induction of gene expression.16,18 The Hpo pathway also mediates intercellular signaling responding to external cellular signals, leading to cell contact-driven proliferation and growth.16,18
The individual components of Hpo signaling were linked to pivotal intracellular processes prior to the delineation of the pathway. For example, YAP was identified as the first WW domain containing protein and was reported to enhance the transcription of genes, serving a constellation of cellular functions while the transcriptional coactivator with PDZ-binding motif (TAZ) was linked to cell differentiation.21–26 Mammalian sterile 20-like kinase 1/2 (Mst1/2) was identified as a promoter of apoptosis via the histone modification of H2B and via FOXO-mediated apoptosis following oxidative stress.27,28 Concurrently, there are reports that implicate the scaffolding protein Sav or WW45 as having a role in cell cycle entry and exit.29 Finally, the large tumor suppressor homolog 1/2 (Lats1/2) was observed to regulate mitosis and cytokinesis by modulating CDC2, LIMK1, and zyxin.30–32
The Hpo core signal cascade commences with the Mst1/2-mediated phosphorylation of the Lats1/2 kinases (Figure 1). The adaptor protein WW45 couples the Mst1/2–Lats1/2 interaction, increasing the efficiency of the reaction, while the Mps one binder kinase activator-like 1A and 1B (collectively, Mob1) enhances the kinase activity of Lats1/2. These series of events culminate in YAP phosphorylation, inactivating the oncoprotein’s ability to induce target gene expression. The protein phosphorylation is a central tenant of transcriptional regulation acting to sequester proteins inside the cytoplasm via coupling reactions by the 14-3-3 proteins.33 Phosphorylated YAP is competent to bind these 14-3-3 proteins, which prevents its intranuclear transport, thereby rendering it powerless over its transcriptional targets.25,34 YAP acts as a transcriptional coactivator, which partners with transcription factor TEAD to initiate the transcription on a multitude of genes involved in cell proliferation, cell contact, and apoptosis, including c-Myc, survivin, SOX4, cyclin D, and CTGF.18,35 Alternatively, YAP phosphorylation results in proteasomal degradation, eliminating its ability to bind protein targets and hindering its transcriptional coactivator role.36 Neurofibromatosis 2/merlin (Nf2/mer) and kidney- and brain-expressed protein are upstream components of the Hpo pathway and act by translating signals from the cell membrane to Mst1/2.16,37 The Nf2/mer inactivation results in carcinogenesis, behaving antagonistically to YAP as YAP activation leads to the development of cancer.37 Importantly, YAP appears to be an effector of Nf2/Mer as deletion of YAP in Nf2-deficient mice abrogates the uncontrolled cell growth typically observed in the Nf2-deficient phenotype.37
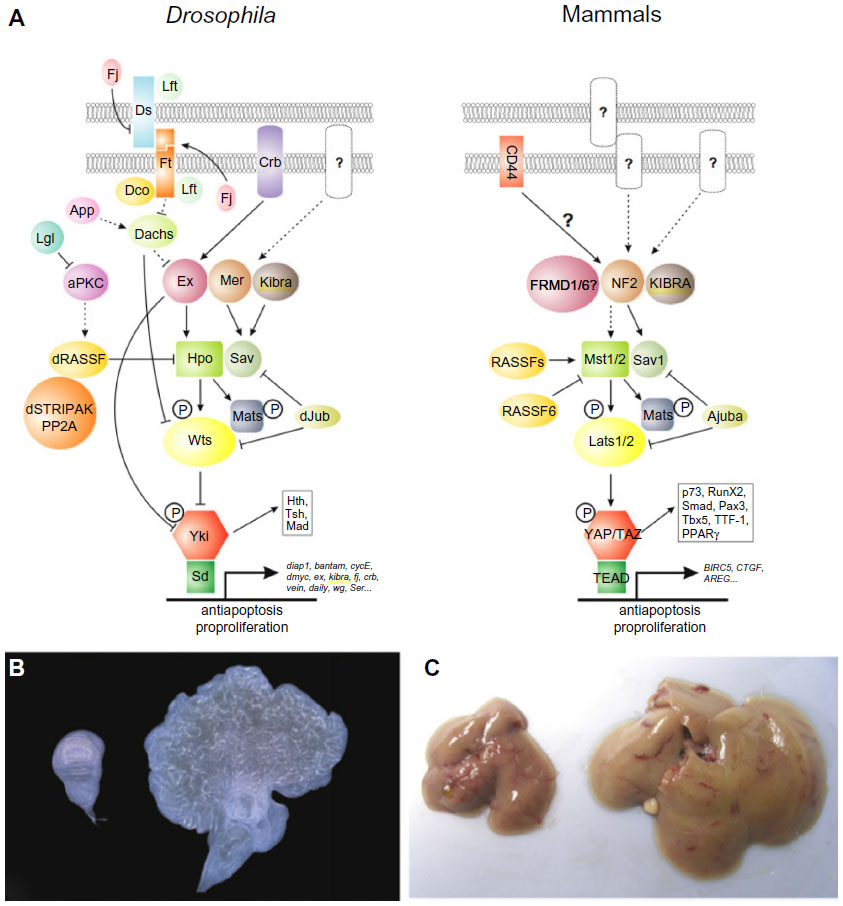 | Figure 1 Hippo pathway signaling in Drosophila and mammals. |
Hpo pathway members in cancer
The genetic alterations and epigenetic events in the Hpo kinase cassette drive tumor progression in cancer cell lines, animal models, and human cancers. While the genetics of the tumor suppressor members of Hpo signaling play a role, the epigenetic changes through the CpG island methylation and microRNA (miR) also prove to be the prevalent mechanisms of tumorigenesis.16,38,39
The Hpo pathway is pivotal in the control of cell growth and dysregulation of the signaling cascade in mammals, leading to carcinogenesis. The mammalian Hpo orthologs – Mst1 and Mst2 – serve as the critical regulators of the Hpo pathway. Epigenetic silencing with the hypermethylation of the CpG island promoter of Mst1/2 is observed in human soft tissue sarcomas, resulting in the alteration of the Sav–RASSF1–Hpo tumor suppressor pathway.40
In colorectal cancer, the cytoplasmic loss of Mst1/2 is predictive of more aggressive tumor biology and is associated with a higher T stage and N stage, as well as vascular invasion and worse overall survival (P<0.05).41 The mechanism of this phenomena in HCC remains unknown as downregulation of Mst1/2 was not found to be related to promoter methylation.41 Interestingly, in prostate cancer, the attenuation of the Mst1/2 expression as manifested through the activation of the PI3K/AKT/mTOR pathway has been associated with the more advanced stages of disease.42
WW45, the human Sav ortholog, helps stabilize Hpo, promoting the phosphorylation of Lats1/2, thereby regulating YAP/TAZ and promoting cell cycle exit and apoptosis.16 WW45 maps to the chromosomal region 14q13–14q23, a locus that is frequently deleted in human renal cell carcinoma, ovarian cancers, and mesothelioma, thereby substantiating its importance in regulating carcinogenesis.43 Along with the large multigenic deletions of WW45, various human cancer cell lines, including those of colorectal, renal, and ovarian lineage harbor single base substitutions in the WW45 gene.29 Tumorigenesis is driven by the mutations in Sav, leading to elevated cyclin E and Drosophila inhibitor of apoptosis 1 levels, resulting in delayed cell cycle exit and impaired apoptosis in D. melanogaster.29
Lats1/2, the mammalian ortholog of Wrts, operates as the molecular off switch to YAP, as its phosphorylation by Lats1/2 downregulates gene expression signatures associated with proliferation and survival.16 Two distinct Lats1/2-mediated mechanisms result in YAP inactivation via Ser-127 phosphorylation-mediated spatial regulation and the Ser-381 phosphorylation-mediated temporal regulation.36 The first mechanism precipitates YAP phosphorylation at the Ser-127 residue, leading to the 14-3-3-protein binding, sequestration from the nucleus, and the inhibition of gene expression. Moreover, YAP function is alternatively mitigated via Ser-381 phosphorylation by casein kinase 1 that ushers the polyubiquitination of YAP, followed by the E3 ubiquitin ligase degradation.36 The epigenetic modifications are implicated in the dysregulation of Lats1/2 activity. For example, the hypermethylation of the Lats1/2 promoter induces carcinogenesis and is identified in human astrocytomas and breast cancers.44,45
In astrocytomas, the promoter hypermethylation of Lats1/2 occurred in 63.6% and 71.5% of CpG islands, respectively, with a corresponding decrease in the messenger RNA (mRNA) expression, demonstrating the critical role that Lats1/2 plays in cell cycle modulation.44 Breast cancers also exhibit this hypermethylation pattern of the Lats1/2 promoter regions (56.7%; 50.0%) and correlate to a biologically aggressive phenotype.45 Recent reports indicate the novel microRNA-mediated silencing of Lats1/2 by miR-372 and miR-373 leads to the development of human testicular germ cell tumors by regulating the p53 pathway.46
The Hpo pathway member Mob1 functions to strengthen the kinase activity of Lats1/2 following phosphorylation by Mst1/2. This increased kinase activity serves as another decelerating force in YAP/TAZ target gene expression thereby inducing growth inhibition and tumor supression.47 Genetic variants in Mob1 were identified in mammary- and melanocyte-derived carcinomas resulting in increased cell proliferation and defective apoptosis.47 Similarly, evaluation of the colorectal cancer patients revealed significantly lower mRNA expression of Mob1 and clinicopathologic variables, such as tumor size, lymphatic invasion, and metastasis were more frequent as the Mob1 expression declined (P<0.05).48 This same decrement in the mRNA expression is reported in non-small-cell lung cancer; however, the decrease was more pronounced in early stage tumors, indicating an early phase phenomenon in lung carcinoma.49
Protein phosphatase-1, a recently identified activator of YAP, acts to dephosphorylate the oncoprotein, allowing its entry into the nucleus and transcriptional acceleration of key proliferative genes.50 Investigators noted that protein phosphatase-1 inhibition occurred following the administration of okadaic acid, thereby increasing the percentage of phosphorylated YAP and abrogating its transcriptional coactivator potential; this may serve as a possible target for modulating Hpo signaling.50
Regulation and function of YAP/TAZ in Hpo pathway
The regulation of YAP/TAZ activation is pivotal to cellular homeostasis as they are the final downstream effectors of the Hpo kinase pathway. In their active form, YAP/TAZ possess a strong transcriptional coactivation domain and interact with the DNA-binding proteins that influence gene transcription of merlin, SMAD7, CTNNB1, ERBB4, SRC, ABL, CDH1, PPARγ, RUNXs, p73, PAX3, PAX8, TTF-1, and many more (Figure 2).18,22,25 In healthy tissue, YAP/TAZ phosphorylation acts a molecular brake triggered by cell–cell contact. In the functional cells, this cell density-dependent activation of the Hpo kinase cascade is required for contact inhibition in mammalian cell lines, corroborating its role in controlling proliferation and, ultimately, organ size.51 Important to invasive cancers, the TAZ induction was reported to promote the epithelial–mesenchymal transition by forming spindle-shaped cells in the culture and upregulation of vimentin, N-cadherin, and fibronectin; the epithelial–mesenchymal transition was mitigated by the Hpo pathway tumor suppressor Lats1/2.34
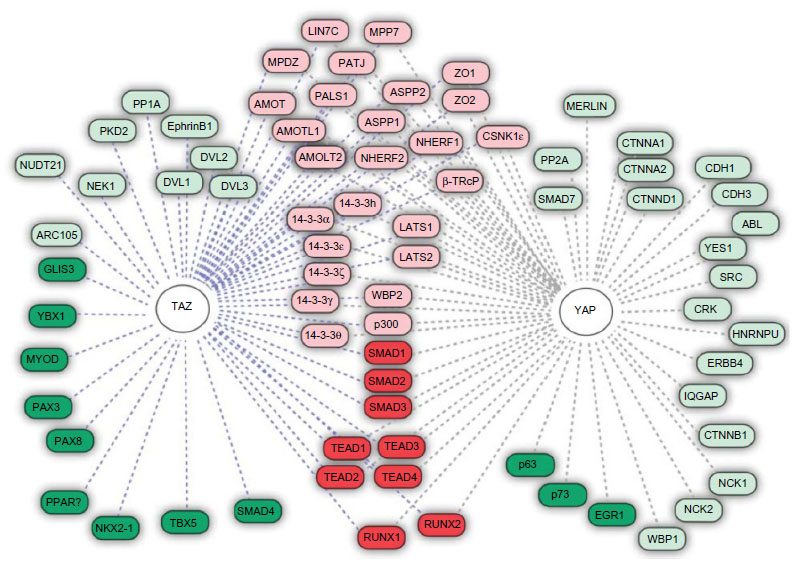 | Figure 2 YAP target proteins. |
The activation of the YAP/TAZ-TEAD transcription factor complex initiates carcinogenesis; in addition, amplification of the YAP gene has been reported in a plethora of human and murine tumors.16 For example, the chromosomal amplification of the 11q22 region has been implicated in various tumor types and is home to the YAP gene locus; examination of YAP expression in resected colonic adenocarcinoma, lung adenocarcinoma, and ovarian serous exhibited intense and diffuse signals in both the nuclear and cytoplasmic compartments.21,52 In the HCC cell lines, there is a marked increase in YAP expression as compared to the human hepatocyte lines and the short hairpin ribonucleic acid-mediated knockdown of YAP resulted in a 50% decrease in cell viability.53 In addition, YAP has been reported to be linked to initiation and recurrence in medulloblastomas, with expression upregulated in cells following irradiation especially in the perivascular niche.54 Furthermore, YAP is known to be overexpressed in the areas of the brain responsible for harboring progenitor cells; in contrast, little immunolabeling of YAP has been observed in the cerebral cortex, thereby suggesting that YAP may be important in brain development.55 YAP expression is enhanced in brain cancer and loss of function results in decreased cell growth in vitro.55
The association between elevated bile acids and carcinogenesis is well-known, and recent reports indicate that this process is YAP mediated.56 Bile acids act as upstream regulators of YAP via the induction of the scaffold protein IQGAP1, and patients with known biliary stasis exhibit increased expression of YAP and IQGAP1.56 Interestingly, Sonic hedgehog induces YAP expression and promotes its nuclear transport in neuronal stem cells, representing a novel effector for Sonic hedgehog signaling.54 Investigators have also observed that coamplification of the BIRC3 gene, an inhibitor of apoptosis and a mammalian homolog of the Drosophila inhibitor of apoptosis 1 located adjacent to YAP, confers a selective advantage to primary lung carcinomas, suggesting synergistic effect on tumorigenesis as seen in D. melanogaster.57
Hpo pathway members in HCC
Consonant with the fundamental roles of Hpo signaling, alterations of Mst1/2, Sav, Lats1/2, and Nf2/mer lead to excess proliferation and – in some cases – carcinogenesis in the mammalian liver. Across the globe, HBV is the predominant etiology precipitating HCC, and YAP activation may play a role. YAP expression is markedly elevated in samples of HBV-induced HCC, HBV-infected cell lines, and HBV X protein (HBx) transgenic mice.58 HBx upregulates YAP by the CRE-dependent HBx-binding of the YAP promoter with subsequent activation of YAP target genes. These data further confirm the importance of the Hpo pathway in cancer and, possibly, offer novel methods of treating patients with known HBV infection to prevent progression to HCC.
The modulation of the Hpo pathway members alters YAP activity. For example, the transgenic Mst1/2 double-knockout mice exhibit abrogation of YAP phosphorylation and a profound nuclear accumulation of YAP.59 Other studies have shown that the specific deletion of Mst1/2 leads to the development of multiple foci of liver tumors in mice by 5–6 months.13 The importance of deleting both Mst1/2 has been demonstrated, as deleting either Mst1 or Mst2 alone did not potentiate liver size or the number of tumor foci in the liver.13 These data strongly suggest a pivotal role of Mst1/2 in YAP activity. In fact, nearly 30% of human HCCs have a loss of YAP phosphorylation, due to the defective Mst1/2 activity culminating in carcinogenesis.59
Introducing the active form of Mst1/2 or using upstream effectors of Mst1/2 activation may decrease the gene expression of the target genes that can lead to uncontrolled cell growth, culminating in HCC formation. The attenuation of the Hpo pathway also results after the liver-specific knockout of WW45. This leads to progressive liver enlargement secondary to the aberrant expansion of hepatic progenitor cells resulting in the development of HCC.13,60 These data indicate the importance that Sav plays critical role in differentiation of progenitor cells. Delayed proliferation arrest is mechanism, not accelerated proliferation.61 There is, however, a requirement for Mst1/2 deletion concurrent with Sav1 deletion to see the downstream attenuation of the Hpo pathway.
The levels of phosphorylated YAP and Lats1/2 are not affected in the Sav1 mutants alone, suggesting that the Mst1/2 kinase activity may be a more prominent driver of carcinogenesis.13 The Sav1-deficient mice do develop tumors; however, the course of tumor development is more indolent with a mean tumor onset of 14 months.13 The liver-specific inactivation of NF2/mer results in a dramatic expansion of hepatic stem cells without changing differentiated hepatocytes.62 The analysis of Lats1/2 and YAP/TAZ phosphorylation in Mst1/2- or Sav-livers has exhibited mixed results concerning the need for Mst1/2 or Sav in Lats1/2 phosphorylation.59,61 The pathway also appears to be specific to hepatic tissues, as Mst1/2 are not required for YAP phosphorylation in mouse fibroblasts.59 This tissue specific divergence from the defined pathway points to the presence of noncanonical members of the cascade that remain to be defined.
The biologic importance of Hpo signaling in hepatocellular carcinogenesis is reinforced by studies of YAP in transgenic murine models. The harmonious orchestration of cell proliferation and apoptosis is crucial in attaining proper organ size and maintaining cellular homeostasis. In a transgenic murine model, YAP induction resulted in uniform liver expansion and after 8 weeks of YAP induction, numerous nodules appeared throughout the hepatic parenchyma (Figure 3).12 The nodules were composed of discrete areas of proliferative hepatocytes that compressed the adjacent parenchyma and displayed the pathognomonic features of HCC. Similarly, the liver-specific deletion of both Mst1/2 in hepatocytes results in significant liver growth due to uncontrolled cell proliferation.13 After 6 months, Mst1/2 conditional mutant livers exhibited numerous liver tumors and an increase in the fraction of progenitor cells, demonstrating that the combined activities of Mst1/2 act as redundant tumor suppressors in hepatocytes.13,14 These data prove that the transgenic overexpression of YAP leads to the dysregulation of organ size and eventual hepatocellular carcinoma.12
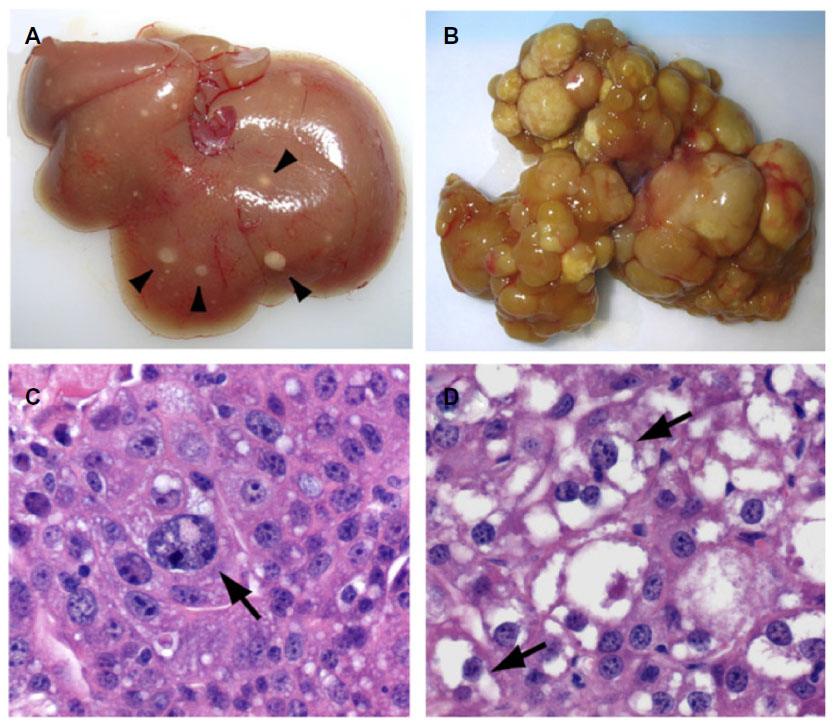 | Figure 3 Hepatocellular carcinoma development following YAP induction in a transgenic murine model. |
Clinical implication of aberrant Hpo signaling
The clinical significance of YAP/TAZ overexpression was investigated in 177 patients with HCC.63 Yap was expressed in 62.1% of the HCC tumor specimens compared with 9.0% in matched normal specimens (P<0.0001). Most accumulation was noted in the nucleus of tumor cells and overexpression of YAP was significantly associated with a higher serum α-fetoprotein level (P<0.001) and worsening tumor grade (P=0.021).63 Nuclear YAP was also an independent predictor of HCC-specific disease-free survival (hazard ratio, 1.653; 95% confidence interval 1.081–2.528; P=0.02), implicating YAP expression in early disease recurrence.63 The increased YAP induction was also predictive of the overall survival (hazard ratio, 2.148; 95% confidence interval 1.255–3.677; P=0.005) with YAP-negative tumors demonstrating a 5-year survival of 58% versus 36% for patients with YAP-positive tumors.63
Survivin is a member of the antiapoptotic protein family and also has been shown to be correlated with a poor prognosis among patients with HCC.64 Recently, YAP expression has been shown to be increased along with survivin in patients with HCC. It appears that YAP actually potentiates oncogenic effects on the cell via survivin upregulation resulting in carcinogenesis.64
Many patients with HCC present with advanced disease. Unfortunately, HCC is highly resistant to most systemic cytotoxic chemotherapy agents, leaving the oncologist with limited treatment options.65 Resistance to doxorubicin is observed in many patients with HCC, and the YAP connection to drug resistance has recently been elucidated.66 YAP induction in HCC cell lines leads to increased resistance to doxorubicin-induced apoptosis; whereas, suppression of YAP expression via RNA interference has been noted to reverse YAP-augmented chemoresistance.66 These effects are mediated through the MAP kinase pathway activation rather than an AKT-driven mechanism of chemoresistance.66 This mechanism of chemoresistance is supported after treatment with the MEK1/2 inhibitor U0126 prior to doxorubicin administration, which significantly decreases cell viability in YAP overexpressing HCC lines.66 Identifying patients with YAP-induced chemoresistance may alter and enhance the efficacy of current drug regimens, leading to improved clinical outcomes in HCC.
The pharmacologic targeting of aberrant YAP induction may further elucidate the Hpo-signaling network and lead to the development of novel drugs against YAP-driven human cancers.67 Previous studies demonstrate that YAP’s oncogenic capacity is mediated by TEAD factors, rendering it pharmacologically targetable.35 Transgenic murine models that overexpress YAP do not develop HCC when crossed with mice featuring a truncated form of TEAD2 that lacks its DNA-binding domain.67 The abrogation of YAP hyperactivity also resulted in mice with Nf2/merlin-deficient livers after crossing with mice harboring the defective TEAD2 gene, further strengthening the therapeutic role of targeting the TEAD/YAP complex.67 Using a drug-screening library, the small molecule inhibitor verteporfin was discovered to prevent the physical association between YAP/TEAD by selectively binding YAP and changing its conformational structure to preclude any interaction with TEAD.67 Verteporfin administration has been shown to decrease liver size in YAP transgenic mice and has no effect on the tissue homeostasis of wild-type mice, highlighting the compound’s potential for therapeutic use in patients with HCC.67 Verteporfin exhibits several key features that make its clinical implementation for HCC promising. Although it was principally designed for use in phototherapy, it does not require photoactivation to bind and to inhibit YAP activity. Moreover, verteporfin use is safe as it has been successful in mouse models, and the human Phase I/Phase II clinical trials reported that verteporfin use is feasible and safe with minimal phototoxicity noted.68,69 These results stress the need for future clinical trials in patients with HCC.
Conclusion
HCC remains a disease with a poor prognosis, due to the ineffective therapies for advanced disease. Recent studies have further elucidated the inner workings of the Hpo-signaling cascade and unmasked this critical pathway’s role in developmental and cancer biology. The Hpo pathway drives hepatocarcinogenesis, and YAP serves as a prognostic marker in HCC. The seminal discoveries of the Hpo pathway and YAP have opened up a new field of study focused on implementation of biologically active molecules that can alter the aberrant Hpo cascade and, ultimately, restore YAP target genes to wild-type expression levels. The modulation of the Hpo pathway offers hope for future therapies to improve the survival of this devastating disease. Further study into the upstream regulators of the pathway is needed. YAP plays a fundamental role in cancer initiation. In the future, YAP or one of its targets may serve as a diagnostic marker for early detection in high-risk individuals, as well as a target for improved therapeutics.
Disclosure
The authors report no conflicts of interest in this work. The study was supported by NIH grant T32DK007713 (V.V.).
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. | |
Nathan H, Schulick RD, Choti MA, Pawlik TM. Predictors of survival after resection of early hepatocellular carcinoma. Ann Surg. 2009;249(5):799–805. | |
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. | |
Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl:S2–S6. | |
Mizokami M, Tanaka Y. Molecular evolutionary analysis predicts the incidence of hepatocellular carcinoma in the United States and Japan. Cancer Chemother Pharmacol. 2004;54 Suppl 1:S83–S86. | |
McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011;15(2):223–243, vii–x. | |
André F. Hepatitis B epidemiology in Asia, the Middle East and Africa. Vaccine. 2000;18 Suppl 1:S20–S22. | |
Duffy JP, Hiatt JR, Busuttil RW. Surgical resection of hepatocellular carcinoma. Cancer J. 2008;14(2):100–110. | |
Shindoh J, Kaseb A, Vauthey JN. Surgical strategy for liver cancers in the era of effective chemotherapy. Liver Cancer. 2013;2(1):47–54. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Zender L, Spector MS, Xue W, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125(7):1253–1267. | |
Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. | |
Lu L, Li Y, Kim SM, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107(4):1437–1442. | |
Song H, Mak KK, Topol L, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A. 2010;107(4):1431–1436. | |
Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20(6):638–646. | |
Pan D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010;19(4):491–505. | |
Kowalik MA, Saliba C, Pibiri M, et al. Yes-associated protein regulation of adaptive liver enlargement and hepatocellular carcinoma development in mice. Hepatology. 2011;53(6):2086–2096. | |
Mauviel A, Nallet-Staub F, Varelas X. Integrating developmental signals: a Hippo in the (path)way. Oncogene. 2012;31(14):1743–1756. | |
Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9(5):534–546. | |
Karpowicz P, Perez J, Perrimon N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development. 2010;137(24):4135–4145. | |
Overholtzer M, Zhang J, Smolen GA, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103(33):12405–12410. | |
Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18(9):2551–2562. | |
Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9(8):2145–2152. | |
Hong JH, Hwang ES, McManus MT, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005; 309(5737):1074–1078. | |
Kanai F, Marignani PA, Sarbassova D, et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19(24):6778–6791. | |
Komuro A, Nagai M, Navin NE, Sudol M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem. 2003;278(35):33334–33341. | |
Cheung WL, Ajiro K, Samejima K, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113(4):507–517. | |
Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125(5):987–1001. | |
Tapon N, Harvey KF, Bell DW, et al. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110(4):467–478. | |
Tao W, Zhang S, Turenchalk GS, et al. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet. 1999;21(2):177–181. | |
Yang X, Yu K, Hao Y, et al. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6(7):609–617. | |
Hirota T, Morisaki T, Nishiyama Y, et al. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J Cell Biol. 2000;149(5):1073–1086. | |
Muslin AJ, Xing H. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal. 2000;12(11–12):703–709. | |
Lei QY, Zhang H, Zhao B, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28(7):2426–2436. | |
Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. | |
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24(1):72–85. | |
Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19(1):27–38. | |
Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16(19):1895–1904. | |
Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126(4):767–774. | |
Seidel C, Schagdarsurengin U, Blümke K, et al. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog. 2007;46(10):865–871. | |
Minoo P, Baker K, Baumhoer D, Terracciano L, Lugli A, Zlobec I. Urokinase-type plasminogen activator is a marker of aggressive phenotype and an independent prognostic factor in mismatch repair-proficient colorectal cancer. Hum Pathol. 2010;41(1):70–78. | |
Cinar B, Fang PK, Lutchman M, et al. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007;26(21):4523–4534. | |
Valverde P. Cloning, expression, and mapping of hWW45, a novel human WW domain-containing gene. Biochem Biophys Res Commun. 2000;276(3):990–998. | |
Jiang Z, Li X, Hu J, et al. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res. 2006;56(4):450–458. | |
Takahashi Y, Miyoshi Y, Takahata C, et al. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res. 2005;11(4):1380–1385. | |
Voorhoeve PM, le Sage C, Schrier M, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124(6):1169–1181. | |
Lai ZC, Wei X, Shimizu T, et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120(5):675–685. | |
Kosaka Y, Mimori K, Tanaka F, Inoue H, Watanabe M, Mori M. Clinical significance of the loss of MATS1 mRNA expression in colorectal cancer. Int J Oncol. 2007;31(2):333–338. | |
Sasaki H, Kawano O, Endo K, et al. Human MOB1 expression in non-small-cell lung cancer. Clin Lung Cancer. 2007;8(4):273–276. | |
Wang P, Bai Y, Song B, et al. PP1A-mediated dephosphorylation positively regulates YAP2 activity. PLoS One. 2011;6(9):e24288. | |
Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. | |
Steinhardt AA, Gayyed MF, Klein AP, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39(11):1582–1589. | |
Li H, Wolfe A, Septer S, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32(1):38–47. | |
Fernandez-L A, Northcott PA, Dalton J, et al. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009;23(23):2729–2741. | |
Orr BA, Bai H, Odia Y, Jain D, Anders RA, Eberhart CG. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J Neuropathol Exp Neurol. 2011;70(7):568–577. | |
Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013;5(4):1060–1069. | |
Dai Z, Zhu WG, Morrison CD, et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum Mol Genet. 2003;12(7):791–801. | |
Zhang T, Zhang J, You X, et al. Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells. Hepatology. 2012;56(6):2051–2059. | |
Zhou D, Conrad C, Xia F, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16(5):425–438. | |
Lee KP, Lee JH, Kim TS, et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(18):8248–8253. | |
Lee JH, Kim TS, Yang TH, et al. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008;27(8):1231–1242. | |
Benhamouche S, Curto M, Saotome I, et al. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 2010;24(16):1718–1730. | |
Xu MZ, Yao TJ, Lee NP, et al. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115(19):4576–4585. | |
Bai H, Gayyed MF, Lam-Himlin DM, et al. Expression of Yes-associated protein modulates Survivin expression in primary liver malignancies. Hum Pathol. 2012;43(9):1376–1385. | |
Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686–695. | |
Huo X, Zhang Q, Liu AM, et al. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol Rep. 2013;29(2):840–846. | |
Liu-Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300–1305. | |
Ayaru L, Wittmann J, Macrobert AJ, Novelli M, Bown SG, Pereira SP. Photodynamic therapy using verteporfin photosensitization in the pancreas and surrounding tissues in the Syrian golden hamster. Pancreatology. 2007;7(1):20–27. | |
Huggett MT, Jermyn M, Gillams A, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110(7):1698–1704. | |
Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell. Cell. 2005;122(3):421–434. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.