")
Back to Journals » ImmunoTargets and Therapy » Volume 9
Emerging Role of Fractalkine in the Treatment of Rheumatic Diseases
Authors Tanaka Y , Hoshino-Negishi K, Kuboi Y, Tago F, Yasuda N, Imai T
Received 20 August 2020
Accepted for publication 9 October 2020
Published 4 November 2020 Volume 2020:9 Pages 241—253
DOI https://doi.org/10.2147/ITT.S277991
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Yoshiya Tanaka,1 Kana Hoshino-Negishi,2 Yoshikazu Kuboi,2 Fumitoshi Tago,3 Nobuyuki Yasuda,2 Toshio Imai2
1First Department of Internal Medicine, University of Occupational and Environmental Health, Fukuoka, Japan; 2 KAN Research Institute Inc, Hyogo, Japan; 3Eisai Co. Ltd., Tokyo, Japan
Correspondence: Yoshiya Tanaka
First Department of Internal Medicine, University of Occupational and Environmental Health, 1-1 Iseigaoka, Yahata-Nishi, Kitakyushu, Fukuoka 807-8555, Japan
Tel +81-93-603-1611
Fax +81-93-691-9334
Email [email protected]
Abstract: Rheumatoid arthritis (RA) is an autoimmune disorder that affects joints and is characterized by synovial hyperplasia and bone erosion associated with neovascularization and infiltration of proinflammatory cells. The introduction of biological disease-modifying anti-rheumatic drugs has dramatically changed the treatment of RA over the last 20 years. However, fewer than 50% of RA patients enter remission, and 10– 15% are treatment refractory. There is currently no cure for RA. Fractalkine (FKN, also known as CX3CL1) is a cell membrane-bound chemokine that can be induced on activated vascular endothelial cells. FKN has dual functions as a cell adhesion molecule and a chemoattractant. FKN binds specifically to the chemokine receptor CX3CR1, which is selectively expressed on subsets of immune cells such as patrolling monocytes and killer lymphocytes. The FKN–CX3CR1 axis is thought to play important roles in the initiation of the inflammatory cascade and can contribute to exacerbation of tissue injury in inflammatory diseases. Accordingly, studies in animal models have shown that inhibition of the FKN–CX3CR1 axis not only improves rheumatic diseases but also reduces associated complications, such as pulmonary fibrosis and cardiovascular disease. Recently, a humanized anti-FKN monoclonal antibody, E6011, showed promising efficacy with a dose-dependent clinical response and favorable safety profile in a Phase 2 clinical trial in patients with RA (NCT02960438). Taken together, the preclinical and clinical results suggest that E6011 may represent a new therapeutic approach for rheumatic diseases by suppressing a major contributor to inflammation and mitigating concomitant cardiovascular and fibrotic diseases. In this review, we describe the role of the FKN–CX3CR1 axis in rheumatic diseases and the therapeutic potential of anti-FKN monoclonal antibodies to fulfill unmet clinical needs.
Keywords: fractalkine, CX3CR1, humanized anti-fractalkine monoclonal antibody (E6011), CD16+ monocyte, rheumatic diseases
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder that primarily affects joints. The disease is characterized by synovial hyperplasia and bone erosion associated with neovascularization, infiltration of proinflammatory cells, and increased cytokine production. These pathological inflammatory features are generated locally by the selective invasion and accumulation of immune cells in the lesion.1 The step-wise process by which immune cells are recruited from the blood, extravasate through interactions with vascular endothelial cells, and migrate into tissue is a tightly regulated process involving a number of chemotactic factors and cell adhesion molecules.2
Chemokines are a family of molecules that play important roles in the migration of leukocytes through binding to specific cell-surface receptors.3 The approximately 50 members of the chemokine family are classified into CC, CXC, CX3C, and C subfamilies,3 whereas the 19 known chemokine receptors are all members of the G protein-coupled 7-transmembrane superfamily of receptors.3,4 The first step of leukocyte migration to sites of inflammation involves transient and weak selectin-mediated interactions between rolling leukocytes and vascular endothelial cells (Figure 1A). Next, integrins expressed by leukocytes are activated by chemokines presented on glycosaminoglycans. This is followed by firm adhesion of the leukocytes to the endothelium, extravasation, and transmigration into the tissue, where the cells move along a chemoattractant gradient towards the site of inflammation (Figure 1A).5–8 The soluble form of macrophage inflammatory protein-1β (MIP-1β) is one example of a chemokine that induces firm adhesion of T cells to endothelial cells. We reported that MIP-1β immobilized by binding to cell-surface proteoglycans induces integrin-mediated adhesion of T cells much more efficiently than does soluble MIP-1β. At one time, chemokines were thought to be exclusively secreted as soluble molecules that were indispensable to forming a local chemokine gradient via binding to proteoglycans;9 however, it is now known that chemokines are also synthesized as membrane-bound molecules that do not interact with proteoglycans.4
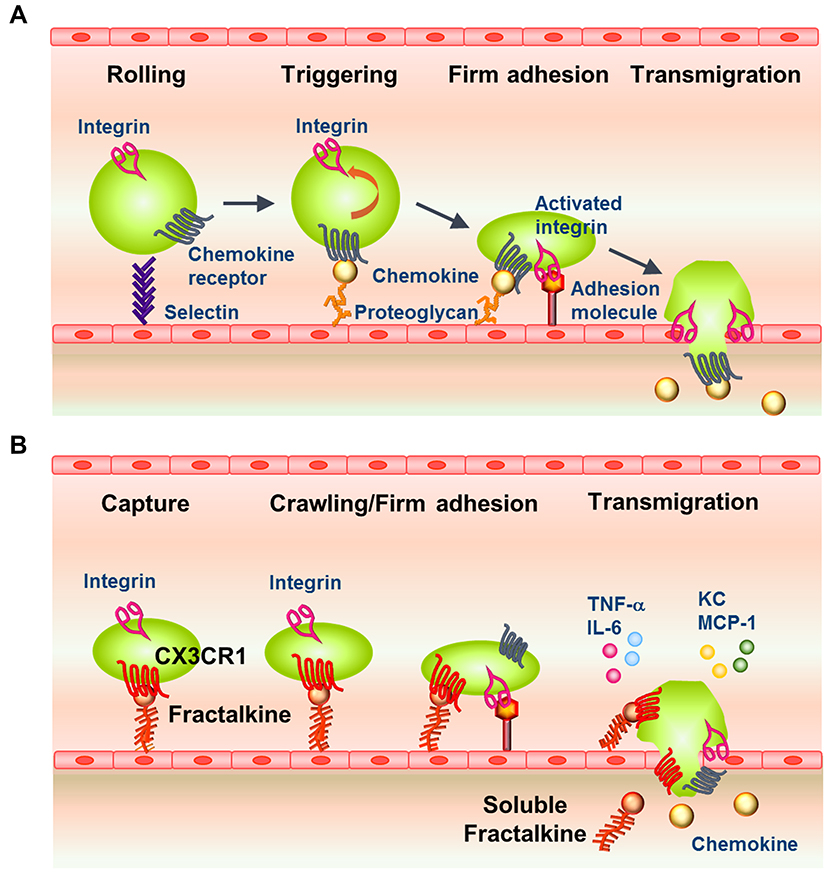 |
Figure 1 Classical and Fractalkine–CX3CR1-Mediated Pathways of Leukocyte Recruitment to Inflamed Tissue. (A) Model of the classical pathway for leukocyte extravasation into sites of inflammation via an adhesion and transmigration cascade. Leukocytes adhere to the endothelial layer through selectins (tethering and rolling), which is followed by engagement of chemokine receptors and integrin activation (firm adhesion), and transmigration into the underlying tissue. (B) Model of the involvement of fractalkine-mediated pathways in the adhesion and transmigration of CX3CR1high leukocytes from the circulation into inflamed tissue. Fractalkine–CX3CR1 engagement enhances the transient capture and attachment of leukocytes to endothelial cells, which is followed by crawling/firm adhesion (activation of integrins by chemokines), production of inflammatory cytokines, and transmigration through the endothelial layer to the sites of inflammation. |
Fractalkine (FKN, also known as CX3CL1) is the only known member of the CX3C chemokine family and is a functionally unique membrane-bound chemokine possessing multiple biological functions.2 Auffray et al showed that FKN–CX3CR1 interactions enable a subset of monocytes, known as patrolling cells, to crawl along the resting endothelium in a manner dependent on both FKN and the integrin LFA-1.10 Under inflammatory conditions, binding of FKN to CX3CR1 plays multiple roles in maintaining immune homeostasis by supporting the movement of patrolling monocytes on vascular endothelial cells, facilitating the rapid migration of circulating leukocytes into inflamed tissues (Figure 1B), and contributing to the survival of leukocyte subsets. Considering these roles, it is not surprising that the FKN–CX3CR1 axis is involved in the pathogenesis of many inflammatory diseases.
In this review, we focus on the physiological and pathological roles of FKN in various rheumatic diseases. We also discuss the therapeutic potential of the anti-FKN monoclonal antibody (mAb) E6011 (Eisai Co. Ltd.),11 which has a distinct mode of action compared with cytokine inhibitors and holds promise as a strategy to meet the unmet medical needs of patients with rheumatic diseases.
Physiological Functions of the FKN–CX3CR1 Axis
FKN is the sole member of the CX3C-type chemokine family and consists of a chemokine domain, a mucin domain, and a transmembrane domain with a short cytoplasmic tail.12 FKN is also unique compared with other classical secreted chemokines in that its membrane-bound form is fully functional as an adhesion molecule,4 thereby obviating the need for an association with proteoglycans. Indeed, cells expressing CX3CR1, the FKN receptor, bind rapidly and with high affinity to plate-immobilized FKN as well as to FKN-expressing cells.4,13,14 The soluble form of FKN is generated by proteolytic cleavage mediated by ADAM 10 (a disintegrin-like metalloproteinase 10) or ADAM17 (also known as tumor necrosis factor-α converting enzyme).15,16
Membrane-bound FKN is expressed on endothelial cells, fibroblast-like synoviocytes (FLSs), intestinal epithelial cells, osteoblasts, neurons, and astrocytes.17 FKN is upregulated on several of these cell types, especially endothelial cells, FLSs, and intestinal epithelium, upon exposure to inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1α (IL-1α), and interferon-γ (IFN-γ).18
The FKN receptor CX3CR1 is expressed on subsets of cytotoxic lymphocytes, including natural killer cells, effector memory T cells, and γδ+ T cells, all of which express the lytic molecules perforin and granzyme B and exhibit marked cytotoxicity.4 CX3CR1 is also expressed on monocytes/macrophages, dendritic cells, and osteoclast precursors (OCPs).19,20 Circulating peripheral blood monocytes can be classified into three subsets: CD14highCD16− (classical), CD14highCD16int (intermediate), and CD14intCD16high (non-classical monocytes).20–22 Among these three subsets, intermediate and non-classical monocytes are known to express high levels of CX3CR1.23,24 Consequently, FKN preferentially mediates the migration of the two CX3CR1high subsets (intermediate and non-classical monocytes; hereafter referred to as CD16+ monocytes).24
In mice, CX3CR1high monocytes have been shown to play a role in monitoring vascular abnormalities by acting as patrolling cells, as described above. Haskell et al reported that CX3CR1-expressing cells adhere more rapidly to immobilized FKN than they do to vascular cell adhesion molecule-1 on endothelial cells,25 suggesting that the FKN–CX3CR1 interaction may be dominant in vivo.
FKN–CX3CR1 Signaling Cascades
FKN–CX3CR1 binding also triggers G protein-mediated signaling that enhances the avidity of integrin–ligand binding.13,26,27 Activation of CX3CR1 by FKN initiates a signaling cascade through Gαi/o that involves activation of extracellular signal-regulated kinase (a mitogen-activated protein kinase), phosphoinositide 3-kinase, Akt/protein kinase B, and mobilization of intracellular Ca2+. These signals promote the survival of CX3CR1high macrophages.28–31 These events result in firm attachment of CX3CR1high monocytes to the vascular endothelium, and the cells then produce chemokines and cytokines that initiate local inflammation and recruit neutrophils and CX3CR1low monocytes to the lesion (Figure 1B).32 Through these mechanisms, FKN facilitates the rapid recruitment and extravasation of circulating leukocytes,26 thereby amplifying the inflammatory reaction.
White et al reported that FKN contributes to the survival of macrophages in both mice and humans.29 In CX3CR1-deficient mice, macrophage survival is impaired under normal physiological conditions as well as during liver inflammation and atherosclerosis.
FKN–CX3CR1 Axis Involvement in the Pathogenesis of Rheumatic Diseases
RA
The synovium is the primary target of immune cells in RA, and infiltrated activated macrophages and lymphocytes are abundant in the affected synovial tissue. These cells secrete a variety of inflammatory cytokines that further activate joint-resident cells such as FLSs, chondrocytes, and osteoclasts. In turn, the locally amplified tissue inflammation results in hyperproliferation of FLSs and joint destruction through activation of osteoclasts and copious production of matrix metalloproteinases.33
Joint-infiltrated CX3CR1high T cells strongly adhere to FLSs in the synovium in an FKN-dependent manner, and they also produce IFN-γ and exhibit cytotoxic activity.34 The number of circulating CX3CR1high T cell is also elevated in the circulation of RA patients.35 A recent global transcriptomics study suggested that peripheral T helper (Tph) cells express CX3CR1. Tph cells can activate B cells and induce antibody production, which may contribute to tissue inflammation in RA.36
In mice with type II collagen-induced arthritis, a model of RA, administration of an anti-FKN mAb efficiently suppresses arthritis, as reflected by a decrease in arthritis score and a reduction in cartilage oligomeric matrix protein and matrix metalloproteinase-3 levels in plasma.37,38 In contrast, anti-FKN treatment did not affect plasma levels of serum amyloid A, anti-type II collagen antibody, TNF-α, or IL-6, but it significantly suppressed TNF-α and IL-6 mRNA expression in the affected joints.38 Moreover, an increase in cell death in the inflamed synovium could be detected immediately after the administration of an anti-FKN mAb to mice with type II collagen-induced arthritis.39 In these studies, histological analysis showed suppression of cartilage and bone destruction accompanied by a marked decrease in the number of osteoclasts.37,38 These results establish that an anti-FKN mAb can suppress local joint destruction in models of RA.
A study of human TNF transgenic mice, which express human TNF-α but are mouse Ccr2-deficient and lack classical monocytes, showed that joint destruction is predominantly mediated by osteoclasts differentiated from OCPs, which are themselves derived from CD115+CX3CR1highLy6clowCCR2low non-classical monocytes.40 The interaction between FKN and CX3CR1 is important for normal osteoclast differentiation and efficient bone resorption in normal mice.41–43 FKN is highly expressed on osteoblasts located on the bone surface in conditions associated with inflamed joints, such as RA. In addition, immobilized FKN is involved in firm adhesion of CX3CR1-expressing OCPs to the plate.41,43,44
FKN blockade has been shown to inhibit migration of macrophages and OCPs into the inflamed synovium.37,38 Interestingly, drugs currently used for RA treatment, such as etanercept (TNF-α inhibitor) and tofacitinib (Janus kinase [JAK] inhibitor), do not directly inhibit OCP migration.45 These results suggest that anti-FKN mAbs may act through multiple modes of action; namely, an anti-inflammatory effect via inhibition of the accumulation of inflammatory cells; induction of FLS cell death; and a bone-preserving effect via a reduction of osteoclasts in the affected joints. These observations further support the possible utility of anti-FKN mAbs as an alternative therapy for RA.
Interstitial lung disease (ILD) is a common extraarticular complication of RA that worsens the prognosis and increases mortality in refractory cases.46 The role of FKN in the pathogenesis of ILD is currently unknown; however, the FKN–CX3CR1 axis has been implicated in lung involvement in patients with systemic sclerosis (SSc)47,48 and amyopathic dermatomyositis (ADM),49,50 suggesting that this mechanism might also be involved in RA-associated ILD. In a bleomycin-induced model of pulmonary fibrosis, mice lacking Cx3cr1 had reduced levels of pulmonary fibrosis, suggesting that FKN contributes to an inflammatory state that exacerbates ILD.51 Thus, further studies should investigate anti-FKN therapy as a potential treatment for RA-associated ILD.
SSc
SSc is characterized by fibrosis and vascular alterations that affect various organs, including the skin, lungs, esophagus, intestines, and heart. Hasegawa et al reported that patients with SSc have large numbers of CX3CR1-expressing macrophages in the lung and skin tissues compared with healthy subjects. SSc patients show strong FKN expression on endothelial cells in the skin and on endothelial cells, type II pneumocytes, and airway epithelial cells in the lungs.47,48 Several studies, including a large cohort study of 292 SSc patients, have shown that serum FKN concentrations are higher in SSc patients than in healthy subjects, and high FKN is associated with a higher titer of anti-topoisomerase-I-antibody, the presence of digital ischemia, and more severe pulmonary fibrosis.47,48,52 Serum FKN is decreased by glucocorticoid treatment with or without cyclophosphamide.47 Polymorphisms at positions 249I and 280M of the CX3CR1 sequence are thought to be associated with pulmonary arterial hypertension, which is a life-threatening complication in SSc patients.53 These findings suggest that FKN is a biomarker of SSc-related ILD and may contribute to the pathogenesis of SSc. Cultured normal human lung fibroblasts produce FKN after incubation with IL-1β and IFN-γ,54 which are stimulators of pulmonary fibrosis and inflammation.55 Thus, pulmonary fibrosis might be exacerbated by FKN induced in inflammatory conditions.
Studies in mice have demonstrated that the FKN–CX3CR1 axis is an important contributor to the accumulation of macrophages and fibroblasts at wound sites.56 In the bleomycin-induced skin fibrosis model of SSc, serum FKN and lesional FKN expression are increased. Administration of an anti-FKN mAb or Cx3cr1 deficiency significantly suppresses the dermal thickness, collagen content, and capillary loss caused by bleomycin.57 In a murine model of SSc induced by transforming growth factor-β and connective tissue growth factor, skin fibrosis and macrophage infiltration are attenuated by anti-FKN mAb treatment or Cx3cr1 deficiency.58 Anti-FKN mAb administration to bleomycin-treated mice suppresses skin fibrosis and skin infiltration of CX3CR1high cells, monocytes/macrophages, and CD3+ T cells.57 In addition, FKN is reportedly involved in liver and kidney fibrosis in mice, both of which are attenuated by Cx3cr1 deficiency.59–62 Although the role of FKN in the pathogenesis of pulmonary fibrosis is not fully understood, increased expression of CX3CR1 on fibroblasts and M2 type macrophages, which play a pivotal role in fibrosis, is observed in the bleomycin-induced pulmonary fibrosis mouse model. Notably, this effect is attenuated in Cx3cr1-deficient mice via a reduction in fibrocyte and M2 macrophage infiltration.51
Polymyositis and Dermatomyositis (PM and DM)
PM and DM are inflammatory diseases involving infiltration of T cells and macrophages into the muscles, and both diseases are often complicated by pulmonary fibrosis. The affected muscle tissue of patients with PM or DM and the lungs of those with ILD express FKN on infiltrated mononuclear cells and endothelial cells, and infiltrated T cells and macrophages in these organs express CX3CR1.63 Levels of ADAM17, which cleaves FKN to generate a soluble form, is significantly higher in the serum of patients with inflammatory myositis than in healthy subjects,64 suggesting that FKN is secreted into the lung tissue. Similarly, serum FKN is higher in PM or DM patients than in healthy subjects63,65 and its level correlates with disease activity, as reflected by serum creatine kinase, manual muscle tests, and alveolar–arterial oxygen pressure difference.63 Notably, in ADM patients who have antibodies against CADM-140/MDA5 (clinically amyopathic dermatomyositis-140/melanoma differentiation-associated gene 5), serum FKN levels and the anti-CADM-1/MDA5 antibody titer not only correlated with each other but also both correlated with disease activity.49,50 Takada et al also reported that the anti-CADM-140/MDA5 antibody titer can predict the course of rapidly progressing ADM-related ILD.50
Anti-FKN mAb administration ameliorates myositis in mice with experimental autoimmune myositis, a model of human PM.66 In these mice, FKN is expressed on infiltrated mononuclear cells and endothelial cells in the affected muscle, and CX3CR1 is expressed on CD4+ and CD8+ T cells and macrophages.66
Systemic Lupus Erythematosus (SLE)
SLE is an autoimmune disease that can affect many organs, including the skin, joints, central nervous system, and kidneys.67 The serum concentration of FKN is higher in patients with SLE than in healthy subjects or patients with RA or primary Sjogren’s syndrome.68,69 In addition, serum FKN correlates significantly with the disease activity index of SLE patients, as well as with biomarkers such as anti-double stranded DNA antibodies, anti-Sm antibodies, immune complexes, and complement hemolytic activity (CH50).69
SLE-related lupus nephritis (LN) is a significant cause of morbidity and mortality.70 The LN classification proposed by the International Society of Nephrology/Renal Pathological Society (ISN/RPS) is used to provide information on disease activity and/or chronicity and to guide treatment.71 Glomerular expression of FKN and kidney infiltration by CX3CR1highCD16+ monocytes are both elevated in patients with ISN/RPS class III or IV LN who present with proliferative glomerulonephritis.72 Reflecting the local pathology, FKN levels in serum and urine are also higher in patients with class III or IV LN than in patients with other classes.73 Similarly, in MRL/lpr mice, which spontaneously develop a form of glomerulonephritis that resembles class IV LN, FKN expression and CD16+ monocyte infiltration in glomeruli both increase in parallel with LN progression.74 Moreover, the mononuclear cell infiltration and glomerular damage in these mice are reduced by administration of an FKN antagonist.74,75 These findings therefore support the involvement of FKN in the pathogenesis of LN via recruitment of monocytes into the kidney.
FKN levels are significantly higher in cerebrospinal fluid samples from patients with SLE-associated involvement of the central nervous system (termed neuropsychiatric SLE) than from healthy subjects, and the FKN levels correlate with treatment effects.76 Similarly, both cerebrospinal fluid and serum levels of FKN are significantly higher in patients with diffuse neuropsychiatric SLE than in patients with other SLE subtypes or in healthy subjects.76 These results further support a role for FKN in the pathogenesis of SLE, at least neuropsychiatric SLE, and suggest that FKN could serve as a therapeutic target and/or a biomarker for SLE disease activity.
IgG4-Related Disease (IgG4-RD)
IgG4-RD is a relatively recently recognized immunological disease characterized by an elevated serum IgG4 concentration and immune-mediated fibroinflammatory processes. Infiltration of IgG4-positive plasma cells is observed in various organs, including the lacrimal glands, salivary glands, pancreas, kidneys, lungs, and retroperitoneum.77 The main histopathological findings of the involved organs are storiform fibrosis formed by spindle cells that resemble fibroblasts, obliterative phlebitis, and ectopic lymphoid structures.
A recent study indicated that CX3CR1-expressing Tph-like cells, which can recruit B cells and T follicular helper cells, contribute to the typical pathological findings of tissue injury and ectopic lymphoid structure formation in IgG4-RD. Patients with this disorder have elevated levels of CX3CR1high Tph-like cells in the blood, and the percentage of these cells correlates positively with the number of involved organs and the IgG4-RD Responder Index score. CX3CR1high Tph-like cells express abundant levels of cytotoxic mediators such as granzyme A and perforin, leading to pathological tissue damage in IgG4-RD lesions. Thus, CX3CR1high Tph-like cells could be a potential clinical biomarker and/or a therapeutic target for inhibiting the progression of IgG4-RD.78
Cardiovascular Disease (CVD)
Epidemiological studies have shown that RA is associated with a significantly increased risk of CVD-related morbidity and mortality.79 Numerous reports have documented the involvement of the FKN–CX3CR1 axis in atherosclerosis and cardiovascular events.80,81 The FKN–CX3CR1 axis participates in the atherosclerotic pathological process by mediating the recruitment of leukocytes and their interaction with vascular cells.82 Interestingly, polymorphisms in the human CX3CR1 gene are genetic risk factors for coronary artery disease and atherosclerosis. These polymorphisms are associated with a significant decrease in the number of FKN-binding sites per cell.83
Clinical research has shown that elevated CD16+ monocyte counts are associated with an increased risk of cardiovascular events.84 CD14lowCD16high monocytes are associated with more advanced vascular dysfunction, as measured by nitric oxide bioavailability and vascular production of reactive oxygen species.85 Plasma FKN levels are significantly increased in patients with unstable angina pectoris and plaque rupture compared with healthy subjects.86 These studies, supported by a growing body of evidence demonstrating the significant role of CD16+ monocytes in atherosclerosis development, suggest that CD16+ monocytes are a potential target for the development of new therapeutic strategies in atherosclerosis.84
Cx3cr1 deficiency has been shown to prevent the development of arteriosclerosis in apolipoprotein E-deficient (Apoe−/−) mice.87,88 CX3CR1 is expressed on intimal dendritic cells, and these cells are less abundant in the aortas of Cx3cr1−/−/Apoe−/− mice compared with Apoe−/− mice.89 In this study, Cx3cr1 deficiency was found to impair the accumulation of dendritic cells in the aortic wall and markedly reduce the atherosclerotic burden.89
Clinical Development of E6011, a Humanized Anti-FKN mAb, in RA
Insufficiently treated RA leads to severe joint damage, disability, decreased quality of life, and other comorbidities. At present, the predominant treatments are disease-modifying anti-rheumatic drugs (DMARDs). They include conventional synthetic DMARDs, of which methotrexate is the anchor drug, as well as biological and synthetic DMARDs that target TNF-α, the IL-6 receptor, T cell costimulatory molecules, CD20 on B cells, and intracellular signaling molecules such as JAKs. Recent guidelines for the management of RA recommended that sustained remission or low disease activity should be rapidly attained in every patient.90–92 However, about 50–70% of patients fail to achieve remission or to maintain low disease activity, even if they initially respond well to current therapies.93,94
We recently conducted a phase 2, multicenter, randomized, double-blind, placebo-controlled study of the humanized anti-FKN mAb E6011 to evaluate its safety and efficacy in Japanese RA patients inadequately responding to methotrexate (NCT02960438).95 Patients were randomly assigned to receive placebo or E6011 at 100 mg, 200 mg, or 400/200 mg in a 2:1:2:2 ratio. Subjects in the 100 mg, 200 mg, and placebo groups were dosed at Weeks 0, 1, and 2, and every 2 weeks subsequently; subjects in the 400/200 mg group received 400 mg at Weeks 0, 1, 2, 4, 6, 8, and 10 and then received 200 mg every 2 weeks subsequently. During the 24-week double-blind period, patients received the study drug subcutaneously at Weeks 0, 1, and 2, and then every 2 weeks thereafter. The primary endpoint was the American College of Rheumatology 20 (ACR20) response rate at Week 12. Using a non-responder imputation (NRI) method, the response rates were 37.0%, 39.3%, 48.1%, and 46.3% in the placebo, 100 mg, 200 mg, and 400/200 mg groups, respectively (not statistically significant). However, the secondary endpoint (ACR20 response rate at Week 24) was significantly different for the 200 mg group (53.7%, P < 0.025) and 400/200 mg group (57.4%, P < 0.025) compared with the placebo group (35.2%). In the biomarker analysis, we focused on CD16+ monocytes due to their importance in RA pathophysiology and their high expression of the FKN receptor CX3CR1. The full patient population was dichotomized into “high” and “low” CD16+ monocyte subgroups using a cutoff value of 10.35%, the median percentage of CD16+ monocytes at baseline. Subjects in the high group (>10.35% CD16+ monocytes at baseline) showed a greater dose-dependent ACR20 response compared with subjects in the low group at Week 24: the response rates were 30.0% vs 43.3%, 46.7% vs 20.0%, 57.7% vs 54.5%, and 69.6% vs 45.5% for the placebo, 100, 200, and 400/20 mg groups, respectively [NRI method]; Figure 2). These results indicated that the baseline percentage of CD16+ monocytes could predict the response to E6011, and additionally suggest the possibility of a precision medicine approach to E6011 therapy, although further research in this area will be needed.
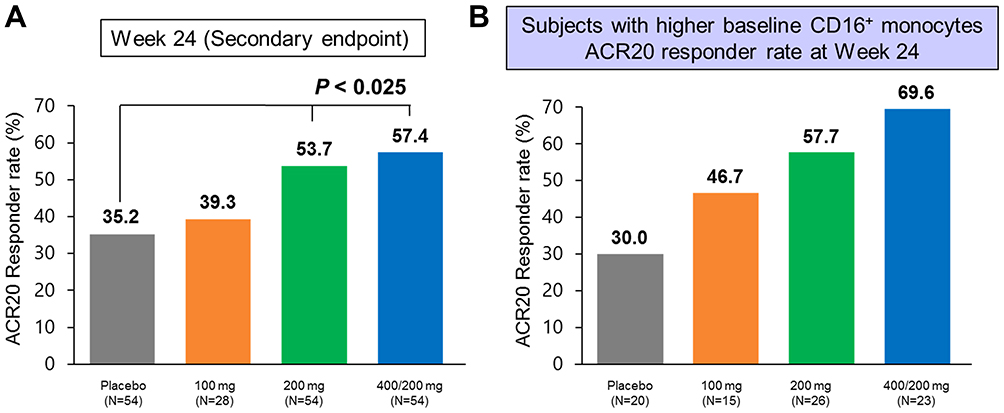 |
Figure 2 Results of a Phase 2 Clinical Trial of E6011, a Humanized Anti- FKN mAb, in Subjects with RA (NCT02960438). (A) ACR20 response rate of the full cohort at Week 24 (NRI). (B) ACR20 response rate at Week 24 in the patient subset with a high percentage of CD16+ monocytes at baseline. Subjects were divided into high and low groups using the median percentage of CD16+ monocytes at baseline (10.35%). Reproduced from ACR/ARP Annu Meet, A Phase 2 Study of E6011, an Anti-Fractalkine Monoclonal Antibody, in Patients with Rheumatoid Arthritis Inadequately Responding to Biologics, Tanaka T, Takeuchi T, Yamanaka H, et al. 9(Supplement 70):1-3553, copyright 2018, with permission from BMJ Publishing Group Ltd.45 |
In this clinical study, adverse events that occurred in ≥5% of subjects in any E6011 group were nasopharyngitis, upper respiratory tract infection, stomatitis, bronchitis, back pain, pharyngitis, and dental caries. Thus, E6011 was well tolerated with no notable safety concerns at doses of 100, 200, and 400/200 mg when administered subcutaneously for 24 weeks.
In a second study of E6011 in RA patients with an inadequate response to biologics, subcutaneous administration of E6011 at 400 mg was well tolerated but did not show significant efficacy compared with placebo at Week 12 (NCT02960490).96 However, an exploratory pharmacokinetic exposure analysis indicated that subjects with higher serum trough concentrations of E6011 showed a trend towards efficacy, albeit not significant. Based on these results, further investigation of E6011 is warranted to determine the optimal clinical dose and evaluation period in RA.
Future Perspectives for the Treatment of RA with E6011
Despite considerable advances in RA treatments and strategies, some patients fail to achieve and/or sustain remission. Those patients require new treatment options with novel mechanisms of action. Several therapeutic antibodies are in clinical development for RA, including modulators of inflammatory cytokines (IL-6, IL-10), inflammatory growth factors (granulocyte-macrophage colony-stimulating factor), and adhesion molecules (cadherin-11) (Table 1).97 Among these investigational drugs, inhibition of cadherin-11 is a particularly novel approach that targets synovial fibroblasts in RA.98 Although it will be interesting to evaluate the efficacy of adding anti-cadherin-11 mAb (RG6125) on top of anti-TNF therapy in RA patients with an inadequate response to anti-TNF alone, unfortunately, no discernable therapeutic effect of RG6125 in combination with TNF blockers has been demonstrated to date.98
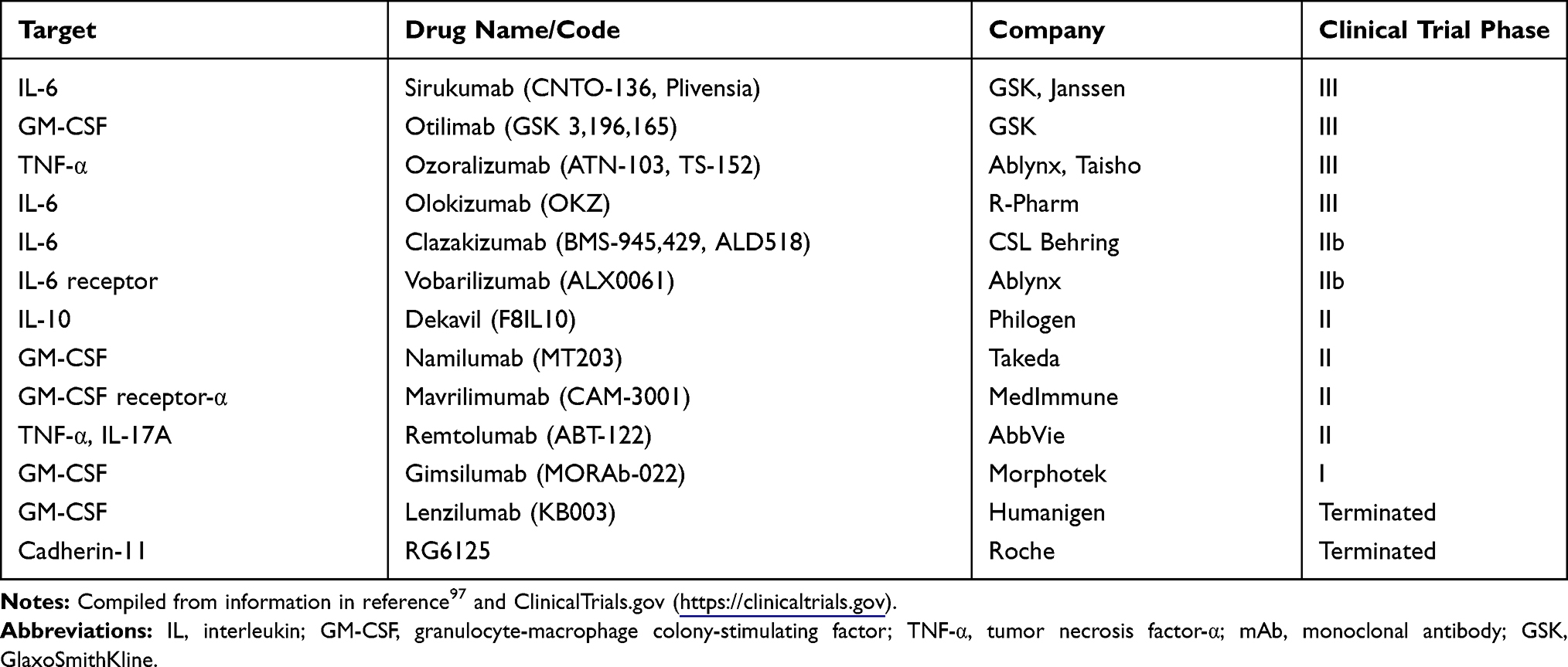 |
Table 1 Cytokine-Targeting Therapies in Development for RA |
In addition to monotherapy with new drugs, there is a pressing need to investigate novel combinations or sequential treatments with targeted therapies for patients refractory to currently available therapies,97,99 who are arguably the patient population with the most urgent unmet medical needs.100 It should be emphasized that patients with a history of treatment with multiple biologics and/or small molecules should not be excluded from clinical trials. Of particular interest is the testing of combination therapies in refractory patients, which should also be studied alongside novel targeted therapies. E6011 is a biologic classified as a cell trafficking inhibitor but it is not a direct cytokine inhibitor. Theoretically, adding E6011 on top of anti-cytokine therapy may be a feasible option for treatment-refractory RA patients.
Presentations at the 2019 Advances in Targeted Therapies meeting emphasized the need to better define “refractory” states both phenotypically and molecularly. Recently, Tasaki et al reported a longitudinal study that monitored the drug response of RA patients using multi-omics analysis of peripheral blood constituents.101 Even RA patients who achieved clinical remission by treatment with tocilizumab or infliximab may not reach molecular remission, which is defined as a molecular profile similar to that of healthy individuals. Interestingly, that study found that the transcriptional residual molecular signature (RMS) of CD16+ monocytes is upregulated in RA patients compared with healthy individuals,101 suggesting the possibility that the CD16+ monocyte RMS may be indicative of incomplete response to treatment. Moreover, the CD16+ monocyte RMS observed in RA patients was also found in patients with inflammatory bowel disease and obesity.101 Thus, these results suggest that the clinical importance of the transcriptional RMS may not be limited to RA, and that CD16+ monocytes could be a biomarker to identify molecular remission in patients with diseases other than RA. Based on these observations, we speculate that E6011 monotherapy or combination therapy with other biologics or JAK inhibitors could be an option to achieve molecular remission in refractory RA patients.
Conclusion
The preceding discussion highlighted the need to study novel targeted therapies, novel combinations, and new sequential treatment strategies with existing therapies for the treatment of refractory RA.100 By blocking cell adhesion and signaling, the anti-FKN mAb E6011 has a distinct mode of action from other drugs currently under investigation for RA, which include cytokine/cytokine receptor inhibitors (eg, infliximab, tocilizumab), modulators of T cell costimulation (eg, abatacept), and JAK inhibitors (eg, tofacitinib, baricitinib). This unique feature may allow E6011 to be used not only for monotherapy but also for combination therapy with other drugs for patients with refractory RA. E6011 may also reduce the risk of CVD in RA patients by decreasing the abundance of CD16+ monocytes, thereby acting as a cardioprotective drug. As noted above, recent studies have shown that the CD16+ monocytes transcriptional RMS may be useful as a hallmark for molecular remission in RA. The results of clinical and preclinical studies indicate that E6011 also has the potential to promote “total health care” in patients with rheumatic and other diseases involving CD16+ monocytes, and could present a new therapeutic strategy for the treatment of patients with refractory RA.
Abbreviations
ACR20, American College of Rheumatology 20; ADAM 10, a disintegrin-like metalloproteinase 10; ADM, amyopathic dermatomyositis; Apoe−/−, apolipoprotein E-deficient; CADM-140/MDA5, clinically amyopathic dermatomyositis-140/melanoma differentiation-associated gene 5; CH50, complement hemolytic activity; CVD, cardiovascular disease; DM, dermatomyositis; DMARDs, disease-modifying anti-rheumatic drugs; FKN, fractalkine; FLS, fibroblast-like synoviocyte; IFN-γ, interferon-γ; IL-1α, interleukin-1α; ILD, interstitial lung disease; ISN/RPS, International Society of Nephrology/Renal Pathological Society; JAK, Janus kinase; LN, lupus nephritis; mAb, monoclonal antibody; MIP-1β, macrophage inflammatory protein-1β; NRI, non-responder imputation; OCP, osteoclast precursor; PM, polymyositis; RA, rheumatoid arthritis; RMS, residual molecular signature; SLE, systemic lupus erythematosus; SSc, systemic sclerosis; TNF-α, tumor necrosis factor-α; Tph, peripheral T helper cell.
Acknowledgments
We thank Anne M. O’Rourke, PhD from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript. We also thank our colleagues at KAN Research Institute, Inc. and Eisai Co., Ltd. for helpful discussions and advice.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Y. Tanaka received consulting fees, speaking fees, and/or honoraria from Abbvie, Chugai, Daiichi-Sankyo, Bristol Myers Squibb, Mitsubishi-Tanabe, Astellas, Takeda, Pfizer, Teijin, Asahi-kasei, YL Biologics, Sanofi, Janssen, Eli Lilly, and GlaxoSmithKline, and received research grants from Mitsubishi-Tanabe, Takeda, Daiichi-Sankyo, Chugai, Bristol Myers Squibb, MSD, Astellas, Abbvie, and Eisai. K.
Hoshino-Negishi reports personal fees from KAN Research Institute, Inc., during the conduct of the study, is a shareholder of Eisai Co., Ltd. and an employee of KAN Research Co., Ltd. (a subsidiary of Eisai Co., Ltd.).
Y. Kuboi reports personal fees from Eisai. Co. Ltd, during the conduct of the study; In addition, Mr Yoshikazu Kuboi has a patent COMPOSITIONS AND METHODS FOR TREATING INFLAMMATORY DISORDERS issued, is a shareholder of Eisai Co., Ltd. and an employee of KAN Research Co., Ltd.
F. Tago is an employee of Eisai Co., Ltd.
N. Yasuda reports personal fees from Eisai. Co., Ltd., during the conduct of the study, is a shareholder of Eisai Co., Ltd. and an employee of KAN Research Co., Ltd.
T. Imai reports personal fees from KAN Research Institute, Inc., during the conduct of the study; In addition, Dr Toshio Imai has a patent COMPOSITIONS AND METHODS FOR TREATING INFLAMMATORY DISORDERS issued; is a shareholder of Eisai Co., Ltd. and an employee of KAN Research Co., Ltd. The authors report no other potential conflicts of interest in this work.
References
1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. doi:10.1056/NEJMra1004965
2. Imai T, Yasuda N. Therapeutic intervention of inflammatory/immune diseases by inhibition of the fractalkine (CX3CL1)-CX3CR1 pathway. Inflamm Regen. 2016;36(1):9. doi:10.1186/s41232-016-0017-2
3. Yoshie O, Imai T, Nomiyama H. Chemokines in immunity. Adv Immunol. 2001;78:57–110.
4. Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91(4):521–530. doi:10.1016/S0092-8674(00)80438-9
5. Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91(10):3527–3561.
6. Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100(12):3853–3860. doi:10.1182/blood.V100.12.3853
7. Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2(2):123–128.
8. Worthylake RA, Burridge K. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr Opin Cell Biol. 2001;13(5):569–577. doi:10.1016/S0955-0674(00)00253-2
9. Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature. 1993;361(6407):79–82. doi:10.1038/361079a0
10. Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–670. doi:10.1126/science.1142883
11. Tanaka Y, Takeuchi T, Umehara H, et al. Safety, pharmacokinetics, and efficacy of E6011, an antifractalkine monoclonal antibody, in a first-in-patient Phase 1/2 study on rheumatoid arthritis. Mod Rheumatol. 2018;28(1):58–65. doi:10.1080/14397595.20-17.1337056
12. Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640–644. doi:10.1038/385640a0
13. Goda S, Imai T, Yoshie O, et al. CX3C-chemokine, fractalkine-enhanced adhesion of THP-1 cells to endothelial cells through integrin-dependent and -independent mechanisms. J Immunol. 2000;164(8):4313–4320. doi:10.4049/jimmunol.164.8.4313
14. Fong AM, Robinson LA, Steeber DA, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188(8):1413–1419. doi:10.1084/jem.188.8.1413
15. Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J Biol Chem. 2001;276(48):44622–44626. doi:10.1074/jbc.M107327200
16. Garton KJ, Gough PJ, Blobel CP, et al. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem. 2001;276(41):37993–38001.
17. Kim KW, Vallon-Eberhard A, Zigmond E, et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood. 2011;118(22):e156–167.
18. Silverman MD, Zamora DO, Pan Y, et al. Constitutive and inflammatory mediator-regulated fractalkine expression in human ocular tissues and cultured cells. Invest Ophthalmol Vis Sci. 2003;44(4):1608–1615. doi:10.1167/iovs.02-0233
19. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5:514.
20. Nishimura M, Umehara H, Nakayama T, et al. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme B+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J Immunol. 2002;168(12):6173–6180. doi:10.4049/jimmunol.168.12.6173
21. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi:10.1016/S1074-7613(03)00174-2
22. Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–80. doi:10.1182/blood-2010-02-258558
23. Wong KL, Tai JJ, Wong WC, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):e16–31. doi:10.1182/blood-2010-12-326355
24. Ancuta P, Rao R, Moses A, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197(12):1701–1707. doi:10.1084/jem.20022156
25. Haskell CA, Cleary MD, Charo IF. Molecular uncoupling of fractalkine-mediated cell adhesion and signal transduction. Rapid flow arrest of CX3CR1-expressing cells is independent of G-protein activation. J Biol Chem. 1999;274(15):10053–10058. doi:10.1074/jbc.274.15.10053
26. Umehara H, Bloom E, Okazaki T, Domae N, Imai T. Fractalkine and vascular injury. Trends Immunol. 2001;22(11):602–607. doi:10.1016/S1471-4906(01)02051-8
27. Umehara H, Goda S, Imai T, et al. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol Cell Biol. 2001;79(3):298–302. doi:10.1046/j.1440-1711.2001.01004.x
28. Korbecki J, Simińska D, Kojder K, et al. Fractalkine/CX3CL1 in neoplastic processes. Int J Mol Sci. 2020;21(10):3723. doi:10.3390/ijms21103723
29. White GE, Greaves DR. Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler Thromb Vasc Biol. 2012;32(3):589–594. doi:10.1161/ATVBAHA.111.237412
30. Lionakis MS, Swamydas M, Fischer BG, et al. CX3CR1-dependent renal macrophage survival promotes candida control and host survival. J Clin Invest. 2013;123(12):5035–5051. doi:10.1172/JCI71307
31. Lee M, Lee Y, Song J, Lee J, Chang SY. Tissue-specific role of CX(3)CR1 expressing immune cells and their relationships with human disease. Immune Netw. 2018;18(1):e5. doi:10.4110/in.2018.18.e5
32. Carlin LM, Stamatiades EG, Auffray C, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153(2):362–375. doi:10.1016/j.cell.2013.03.010
33. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–3545. doi:10.1172/JCI36389
34. Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52(5):1392–1401. doi:10.1002/art.21140
35. Nanki T, Imai T, Nagasaka K, et al. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46(11):2878–2883. doi:10.1002/art.10622
36. Rao DA, Gurish MF, Marshall JL, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542(7639):110–114. doi:10.1038/nature20810
37. Nanki T, Urasaki Y, Imai T, et al. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004;173(11):7010–7016. doi:10.4049/jimmunol.173.11.7010
38. Hoshino-Negishi K, Ohkuro M, Nakatani T, et al. Role of anti-fractalkine antibody in suppression of joint destruction by inhibiting migration of osteoclast precursors to the synovium in experimental arthritis. Arthritis Rheumatol. 2019;71(2):222–231. doi:10.1002/art.40688
39. Hoshino-Negishi K, Ohkuro M, Nakatani T, et al. Anti-fractalkine monoclonal antibody ameliorates joint destruction in collagen-induced arthritis model by inhibiting migration and survival of osteoclast precursor cells. ANZBMS Annu Sci Meet. 2018.
40. Puchner A, Saferding V, Bonelli M, et al. Non-classical monocytes as mediators of tissue destruction in arthritis. Ann Rheum Dis. 2018;77(10):1490–1497. doi:10.1136/annrheumdis-2018-213250
41. Koizumi K, Saitoh Y, Minami T, et al. Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J Immunol. 2009;183(12):7825–7831. doi:10.4049/jimmunol.0803627
42. Hoshino A, Ueha S, Hanada S, et al. Roles of chemokine receptor CX3CR1 in maintaining murine bone homeostasis through the regulation of both osteoblasts and osteoclasts. J Cell Sci. 2013;126(Pt 4):1032–1045. doi:10.1242/jcs.113910
43. Matsuura T, Ichinose S, Akiyama M, Kasahara Y, Tachikawa N, Nakahama KI. Involvement of CX3CL1 in the migration of osteoclast precursors across osteoblast layer stimulated by interleukin-1ss. J Cell Physiol. 2017;232(7):1739–1745. doi:10.1002/jcp.25577
44. Isozaki T, Kasama T, Takahashi R, et al. Synergistic induction of CX3CL1 by TNF alpha and IFN gamma in osteoblasts from rheumatoid arthritis: involvement of NF-kappa B and STAT-1 signaling pathways. J Inflamm Res. 2008;1:19–28.
45. Tanaka T, Takeuchi T, Yamanaka H, et al. A Phase 2 Study of E6011, an Anti-Fractalkine Monoclonal Antibody, in Patients with Rheumatoid Arthritis Inadequately Responding to Biologics. ACR/ARP Annu Meet. 2018;9(Supplement 70):1–3553.
46. Hyldgaard C, Hilberg O, Pedersen AB, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis. 2017;76(10):1700–1706. doi:10.1136/annrheumdis-2017-211138
47. Hasegawa M, Sato S, Echigo T, Hamaguchi Y, Yasui M, Takehara K. Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann Rheum Dis. 2005;64(1):21–28. doi:10.1136/ard.2003.018705
48. Hoffmann-Vold AM, Weigt SS, Palchevskiy V, et al. Augmented concentrations of CX3CL1 are associated with interstitial lung disease in systemic sclerosis. PLoS One. 2018;13(11):e0206545. doi:10.1371/journal.pone.0206545
49. Hayashi M, Aoki A, Asakawa K, Sakagami T, Kikuchi T, Takada T. Cytokine profiles of amyopathic dermatomyositis with interstitial lung diseases treated with mycophenolate. Respirol Case Rep. 2017;5(4):e00235.
50. Takada T, Aoki A, Asakawa K, et al. Serum cytokine profiles of patients with interstitial lung disease associated with anti-CADM-140/MDA5 antibody positive amyopathic dermatomyositis. Respir Med. 2015;109(9):1174–1180. doi:10.1016/j.rmed.2015.07.004
51. Ishida Y, Kimura A, Nosaka M, et al. Essential involvement of the CX3CL1-CX3CR1 axis in bleomycin-induced pulmonary fibrosis via regulation of fibrocyte and M2 macrophage migration. Sci Rep. 2017;7(1):16833. doi:10.1038/s41598-017-17007-8
52. Benyamine A, Magalon J, Cointe S, et al. Increased serum levels of fractalkine and mobilisation of CD34(+)CD45(-) endothelial progenitor cells in systemic sclerosis. Arthritis Res Ther. 2017;19(1):60. doi:10.1186/s13075-017-1271-7
53. Marasini B, Cossutta R, Selmi C, et al. Polymorphism of the fractalkine receptor CX3CR1 and systemic sclerosis-associated pulmonary arterial hypertension. Clin Dev Immunol. 2005;12(4):275–279. doi:10.1080/17402520500303297
54. Isozaki T, Otsuka K, Sato M, et al. Synergistic induction of CX3CL1 by interleukin-1beta and interferon-gamma in human lung fibroblasts: involvement of signal transducer and activator of transcription 1 signaling pathways. Transl Res. 2011;157(2):64–70. doi:10.1016/j.trsl.2010.11.007
55. Elias JA, Freundlich B, Kern JA, Rosenbloom J. Cytokine networks in the regulation of inflammation and fibrosis in the lung. Chest. 1990;97(6):1439–1445. doi:10.1378/chest.97.6.1439
56. Ishida Y, Gao JL, Murphy PM. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J Immunol. 2008;180(1):569–579. doi:10.4049/jimmunol.180.1.569
57. Luong VH, Utsunomiya A, Chino T, et al. Inhibition of the progression of skin inflammation, fibrosis, and vascular injury by blockade of the CX(3) CL1/CX(3) CR1 pathway in experimental mouse models of systemic sclerosis. Arthritis Rheumatol. 2019;71(11):1923–1934. doi:10.1002/art.41009
58. Arai M, Ikawa Y, Chujo S, et al. Chemokine receptors CCR2 and CX3CR1 regulate skin fibrosis in the mouse model of cytokine-induced systemic sclerosis. J Dermatol Sci. 2013;69(3):250–258. doi:10.1016/j.jdermsci.2012.10.010
59. Wasmuth HE, Zaldivar MM, Berres ML, et al. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48(2):208–215.
60. Shimizu K, Furuichi K, Sakai N, et al. Fractalkine and its receptor, CX3CR1, promote hypertensive interstitial fibrosis in the kidney. Hypertens Res. 2011;34(6):747–752. doi:10.1038/hr.2011.23
61. Engel DR, Krause TA, Snelgrove SL, et al. CX3CR1 reduces kidney fibrosis by inhibiting local proliferation of profibrotic macrophages. J Immunol. 2015;194(4):1628–1638. doi:10.4049/jimmunol.1402149
62. Peng X, Zhang J, Xiao Z, Dong Y, Du J. CX3CL1-CX3CR1 interaction increases the population of Ly6C(-)CX3CR1(hi) macrophages contributing to unilateral ureteral obstruction-induced fibrosis. J Immunol. 2015;195(6):2797–2805. doi:10.4049/jimmunol.1403209
63. Suzuki F, Kubota T, Miyazaki Y, et al. Serum level of soluble CX3CL1/fractalkine is elevated in patients with polymyositis and dermatomyositis, which is correlated with disease activity. Arthritis Res Ther. 2012;14(2):R48. doi:10.1186/ar3761
64. Nishimi A, Isozaki T, Nishimi S, et al. ADAM-17 is expressed in the inflammatory myopathy and is involved with interstitial lung disease. Clin Rheumatol. 2018;37(4):1017–1024. doi:10.1007/s10067-018-4014-5
65. Yajima N, Wakabayashi K, Odai T, et al. Clinical features of hemophagocytic syndrome in patients with dermatomyositis. J Rheumatol. 2008;35(9):1838–1841.
66. Suzuki F, Nanki T, Imai T, et al. Inhibition of CX3CL1 (fractalkine) improves experimental autoimmune myositis in SJL/J mice. J Immunol. 2005;175(10):6987–6996. doi:10.4049/jimmunol.175.10.6987
67. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039.
68. Sato E, Iikuni N, Yoshio T, Minota S, Kamatani N, Okamoto H. Soluble fractalkine in the cerebrospinal fluid of patients with neuropsychiatric lupus. Ann Rheum Dis. 2006;65(9):1257–1259. doi:10.1136/ard.2005.051276
69. Yajima N, Kasama T, Isozaki T, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52(6):1670–1675. doi:10.1002/art.21042
70. Danila MI, Pons-Estel GJ, Zhang J, Vila LM, Reveille JD, Alarcon GS. Renal damage is the most important predictor of mortality within the damage index: data from LUMINA LXIV, a multiethnic US cohort. Rheumatology (Oxford). 2009;48(5).
71. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5).
72. Yoshimoto S, Nakatani K, Iwano M, et al. Elevated levels of fractalkine expression and accumulation of CD16+ monocytes in glomeruli of active lupus nephritis. Am J Kidney Dis. 2007;50(1):47–58. doi:10.1053/j.ajkd.2007.04.012
73. Lan L, Han F, Lang X, Chen J, Jia Z. Monocyte chemotactic protein-1, fractalkine, and receptor for advanced glycation end products in different pathological types of lupus nephritis and their value in different treatment prognoses. PLoS One. 2016;11(7):e0159964. doi:10.1371/journal.pone.0159964
74. Nakatani K, Yoshimoto S, Iwano M, et al. Fractalkine expression and CD16+ monocyte accumulation in glomerular lesions: association with their severity and diversity in lupus models. Am J Physiol Renal Physiol. 2010;299(1):F207–216.
75. Inoue A, Hasegawa H, Kohno M, et al. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2005;52(5):1522–1533. doi:10.1002/art.21007
76. Guo L, Lu X, Wang Y, Bao C, Chen S. Elevated levels of soluble fractalkine and increased expression of CX3CR1 in neuropsychiatric systemic lupus erythematosus. Exp Ther Med. 2017;14(4):3153–3158. doi:10.3892/etm.2017.4862
77. Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1–14. doi:10.3109/s10165-011-0508-6
78. Yabe H, Kamekura R, Yamamoto M, et al. Cytotoxic Tph-like cells are involved in persistent tissue damage in IgG4-related disease. Mod Rheumatol. 2020:1–12. doi:10.1080/14397595.2020.1719576
79. Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52(2):402–411. doi:10.1002/art.20853
80. Ghattas A, Griffiths HR, Devitt A, Lip GYH, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. 2013;62(17):1541–1551. doi:10.1016/j.jacc.2013.07.043
81. Skoda M, Stangret A, Szukiewicz D. Fractalkine and placental growth factor: a duet of inflammation and angiogenesis in cardiovascular disorders. Cytokine Growth Factor Rev. 2018;39:116–123. doi:10.1016/j.cytogfr.2017.12.001
82. Liu H, Jiang D. Fractalkine/ CX3CR1 and atherosclerosis. Clin Chim Acta. 2011;412(13–14):1180–1186. doi:10.1016/j.cca.2011.03.036
83. Moatti D, Faure S, Fumeron F, et al. Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood. 2001;97(7):1925–1928. doi:10.1182/blood.V97.7.1925
84. Idzkowska E, Eljaszewicz A, Miklasz P, Musial WJ, Tycinska AM, Moniuszko M. The role of different monocyte subsets in the pathogenesis of atherosclerosis and acute coronary syndromes. Scand J Immunol. 2015;82(3):163–173. doi:10.1111/sji.12314
85. Urbanski K, Ludew D, Filip G, et al. CD14(+)CD16(++) “nonclassical” monocytes are associated with endothelial dysfunction in patients with coronary artery disease. Thromb Haemost. 2017;117(5):971–980. doi:10.1160/TH16-08-0614
86. Li RJ, Yang M, Li JF, Xue L, Chen YG, Chen WQ. Circulating CD36 and fractalkine levels are associated with vulnerable plaque progression in patients with unstable angina pectoris. Clin Exp Pharmacol Physiol. 2014;41(11):863–869. doi:10.1111/1440-1681.12302
87. Combadière C, Potteaux S, Gao JL, et al. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107(7):1009–1016. doi:10.1161/01.CIR.0000057548.68243.42
88. Landsman L, Bar-On L, Zernecke A, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113(4):963–972. doi:10.1182/blood-2008-07-170787
89. Liu P, Yu YR, Spencer JA, et al. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28(2):243–250. doi:10.1161/ATVBAHA.107.158675
90. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977.
91. Smolen JS, Breedveld FC, Burmester GR, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis. 2016;75(1):3–15. doi:10.1136/annrheumdis-2015-207524
92. Tanaka Y. Current concepts in the management of rheumatoid arthritis. Korean J Intern Med. 2016;31(2):210–218. doi:10.3904/kjim.2015.137
93. Iannone F, Sinigaglia L, Favalli EG, et al. Drug survival of adalimumab in patients with rheumatoid arthritis over 10 years in the real-world settings: high rate remission together with normal function ability. Clin Rheumatol. 2016;35(11):2649–2656. doi:10.1007/s10067-016-3349-z
94. Iannone F, Ferraccioli G, Sinigaglia L, et al. Real-world experience of tocilizumab in rheumatoid arthritis: sub-analysis of data from the Italian biologics’ register GISEA. Clin Rheumatol. 2018;37(2):315–321. doi:10.1007/s10067-017-3846-8
95. Tanaka Y, Takeuchi T, Yamanaka H, et al. OP0223 efficacy and safety of e6011, an anti-fractalkine monoclonal antibody, in mtx-ir patients with rheumatoid arthritis. Ann Rheum Dis. 2019;78(Suppl 2):188.
96. Tanaka Y, Takeuchi T, Yamanaka H, et al. SAT0126 a phase 2 study of e6011, an anti-fractalkine monoclonal antibody, in patients with rheumatoid arthritis inadequately responding to biologics. Ann Rheum Dis. 2019;78(Suppl 2):1131–1132.
97. Senolt L. Emerging therapies in rheumatoid arthritis: focus on monoclonal antibodies. F1000Res. 2019;8.
98. Finch R, Sostelly A, Sue-Ling K, et al. Op0224 Results of a phase 2 study of Rg6125, an anti-cadherin-11 monoclonal antibody, in rheumatoid arthritis patients with an inadequate response to anti-TNF-alpha therapy. Oral Presentations. 2019.
99. Winthrop KL, Weinblatt ME, Bathon J, et al. Unmet need in rheumatology: reports from the targeted therapies meeting 2019. Ann Rheum Dis. 2020;79(1):88–93. doi:10.1136/annrheumdis-2019-216151
100. Smolen JS, Landewe RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–699.
101. Tasaki S, Suzuki K, Kassai Y, et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun. 2018;9(1):2755. doi:10.1038/s41467-018-05044-4
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.