")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 6
Emerging role of cell polarity proteins in breast cancer progression and metastasis
Authors Chatterjee S, McCaffrey L
Received 17 October 2013
Accepted for publication 18 November 2013
Published 9 January 2014 Volume 2014:6 Pages 15—27
DOI https://doi.org/10.2147/BCTT.S43764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Sudipa June Chatterjee, Luke McCaffrey
Rosalind and Morris Goodman Cancer Centre, Department of Oncology, McGill University, Montreal, QC, Canada
Abstract: Breast cancer is a heterogeneous group of diseases that frequently exhibits loss of growth control, and disrupted tissue organization and differentiation. Several recent studies indicate that apical–basal polarity provides a tumor-suppressive function, and that disrupting polarity proteins affects many stages of breast cancer progression from initiation through metastasis. In this review we highlight some of the recent advances in our understanding of the molecular mechanisms by which loss of apical–basal polarity deregulates apoptosis, proliferation, and promotes invasion and metastasis in breast cancer.
Keywords: apical, basal, oncogene, tumor suppressor, proliferation, apoptosis
Pathophysiology of breast cancer
The normal mammary gland architecture consists of a bilayer of epithelial cells; the inner layer is made from polarized luminal cells that are surrounded by an outer myoepithelial cell layer, which contacts the basement membrane. Breast cancer develops through a step-wise progression from benign epithelial atypia and atypical ductal hyperplasia (ADH), to malignant ductal carcinoma in situ (DCIS), or more rarely lobular carcinoma in situ (LCIS), and finally to invasive ductal carcinoma (IDC).1 Epithelial atypia and hyperplasia are precursor lesions that are clonal and are characterized by multi-layering of ducts (Figure 1).2–4 DCIS lesions consist of malignant cell masses that lack ductal organization, express E-cadherin, can have the presence of small disordered luminal spaces, and are enclosed by an intact basement membrane (Figure 1).3,4
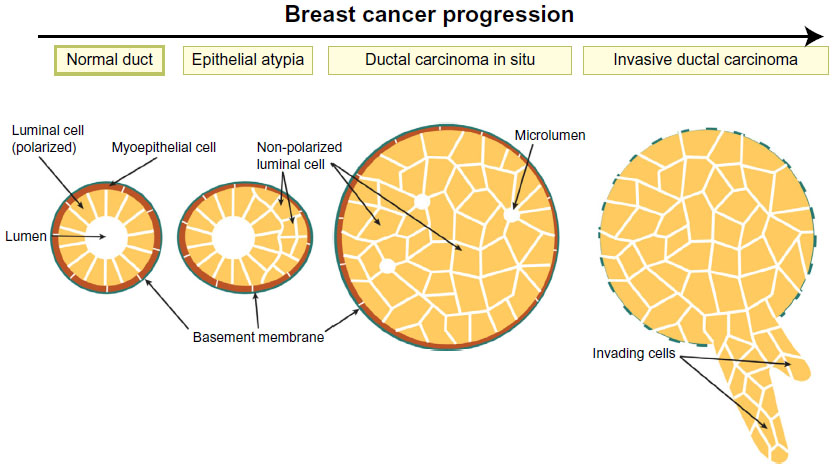 | Figure 1 Breast tumor progression. |
Invasive ductal carcinomas breach the basement membrane (Figure 1),5,6 and can be divided into subtypes that have distinct histological features, molecular expression signatures, and clinical outcomes.7 Luminal A breast cancers express markers of luminal ductal cells such as cytokeratin 8, as well as estrogen and progesterone hormone receptors and have a good prognosis. Luminal B cancers also express luminal cell markers, but have higher proliferation and a worse prognosis than Luminal A. Human epidermal growth factor receptor 2 (HER2) positive tumors have amplification of the HER2/ErbB2 growth factor receptor and also have poor prognosis. Finally, triple negative cancers lack expression of estrogen, progesterone, and HER2 receptors and have the poorest prognosis. Triple negative cancers can be further subdivided into basal-like, which express markers of basal myoepithelial and mesenchymal markers; and claudin-low cancers, which are enriched in stem cell markers and have low levels of differentiation markers and genes associated with adherens junctions (AJs) and tight junctions (TJs).8,9
The normal mammary gland is hierarchically organized with a multipotent stem cell population that can give rise to more committed progenitors, lineage restricted myoepithelial and luminal cell progenitors, which produce differentiated myoepithelial and luminal epithelial cells respectively. Remarkably, gene expression signatures of different breast cancer subtypes resemble different populations along the differentiation hierarchy, and therefore it has been postulated that different breast cancer subtypes may be derived from specific stem cell (claudin-low), progenitor (basal-like, HER2), and differentiated populations (luminal A/B).10,11
Most breast cancer-related deaths occur because of metastasis to secondary sites. Metastasis has classically been considered a late event in cancer progression. However, dissemination of cells from a primary breast lesion may occur early in tumor progression, indicating that tumor progression and tumor dissemination may occur through distinct mechanisms.12,13 Remarkably, even non-metastatic or non-transformed breast cells are capable of disseminating and surviving in the lung microenvironment.13,14
Epithelial cell polarity
In the breast, luminal epithelial cells exhibit apical–basal polarity, which is the asymmetric distribution of proteins and lipids to distinct membrane domains. The apical domain faces the central lumen of the duct, into which milk is secreted during pregnancy. The basolateral domain makes contact with neighboring luminal cells as well as myoepithelial cells and the basement membrane (Figure 2). Cell–cell adhesions between luminal cells are mediated by AJs, which maintain intercellular adhesion through E-cadherin by linking to the actin cytoskeleton through α- and β-catenin adaptors. Desmosomes also mediate cell–cell adhesions, which link to the cytokeratin cytoskeleton through transmembrane desmocollin and desmoglein proteins. Finally, apical TJs associate with adjacent cells through transmembrane proteins including claudins, occludins, and junction adhesion molecule A (JAM-A), and are anchored to the actin cytoskeleton through the adaptor zonula occludens 1 (ZO-1). TJs create a selective permeability barrier between cells, and also separate the apical and basolateral membrane domains.15
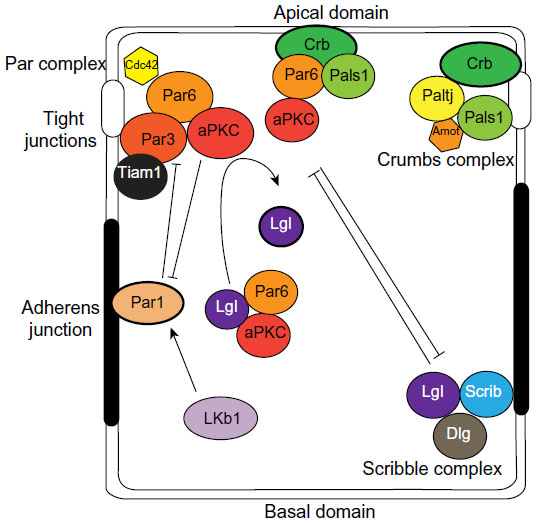 | Figure 2 Polarity complexes in epithelial cells. |
Apical–basal polarity is established and maintained by an evolutionarily conserved group of proteins that assemble into dynamic protein complexes (Figure 2).16–18 The Par complex consists of the multi-domain scaffolding protein, Par3, the adaptor Par6, atypical protein kinase C (aPKC), and the small GTPase cell division control protein 42 (Cdc42). Par3 binds directly with phospholipids at the plasma membrane and with the tight junction protein JAM-A19 and recruits Par6 and aPKC to the plasma membrane where Cdc42 induces a conformation change in Par6 that enables aPKC activation. The membrane localization of Par3, and subsequently Par6 and aPKC, is partly restricted by another Par protein, Par1b, which localizes to the basolateral domain. Par1b phosphorylates Par3, which creates a binding site for 14-3-3 proteins (also called Par5), and causes Par3 to dissociate from the cell cortex.20,21 In this way, basolateral Par1b excludes the Par complex from the basolateral domain and restricts it apically. Conversely, aPKC phosphorylates Par1b to exclude it from the apical membrane.22
Another Par protein is liver kinase b1 (Lkb1; also known as Par4), which phosphorylates and activates a family of 13 AMP-activated protein kinase (AMPK)-related kinases to regulate cell growth, metabolism and cell polarity.23 One of the substrates is Par1b, which provides a mechanism by which Lkb1 regulates apical–basal polarity.24 In addition, AMPK, which is phosphorylated and activated by Lkb1, is also implicated in modulating cell polarity by regulating tight junction formation.25 Importantly, expression of a constitutively active AMPK rescues polarity defects in Lkb1-mutants, indicating that AMPK is a direct downstream effector of Lkb1 that regulates polarity.26
In epithelial cells, the Crumbs complex is comprised of the transmembrane protein Crumbs 3 (Crb), which binds the multi-domain proteins Pals1 (also known as MPP5), Pals1-associated tight junction protein (Patj; also known as INADL), and angiomotin (Amot), which are required for TJ formation and to specify the apical membrane.27
The Scribble complex is positioned at the basolateral membrane and consists of the scaffolding proteins Scribble (Scrib), Discs-large (Dlg), and the adaptor lethal giant larvae (Lgl2; also known as Hugl2). Scrib localization at the basolateral membrane is dependent on cell–cell adhesion mediated by E-cadherin.28 In return, the Scrib complex is necessary to maintain E-cadherin-mediated adhesions, and specifies the basolateral membrane.29 Furthermore the Scrib complex opposes apical membrane identity and in Drosophila, expression of Scrib mutants cause the delocalization of apical proteins to all cell surfaces.30
The three polarity complexes are complementary and act together to establish and maintain polarity. However, the complexes are dynamic and can interact with each other to regulate apical–basal polarity (Figure 2). For example, Lgl2 associates with Par6 and aPKC, which is important for trafficking of Par6/aPKC proteins. Par3 competes with Lgl2 for binding, and activated aPKC phosphorylates and inhibits Lgl2. Moreover, Par6 can interact with Pals1 and Crb of the Crumbs complex, and Pals1 is required for normal localization of the Par complex.31,32 Crb competes with Par3 for binding Par6, which localizes Par6/aPKC to the apical membrane in polarized cells.
The role of polarity proteins in cancer signaling
At a fundamental level, breast and other epithelial cancers are characterized by a loss of growth control. This can occur at both qualitative and quantitative levels, whereby both the organization or differentiation state of cells (qualitative growth) and number of cells (quantitative growth) are deregulated. Emerging evidence indicates that cell polarity proteins have key roles in regulating both quantitative and qualitative aspects of epithelial growth control. Maintaining apical–basal polarity is important because it controls the localization of key mediators of signaling pathways involved in regulating proliferation, apoptosis, differentiation, and tissue organization. In this way, apical–basal polarity is a crucial mechanism to ensure that the communication between a cell and its microenvironment is correctly interpreted.
A number of mechanisms can account for altered expression of apical–basal polarity proteins in breast and other cancers. For example, amplification of the aPKCι gene (PRKCI) has been observed in high-grade serous ovarian cancers, non-small-cell lung cancer and esophageal squamous cell carcinoma.33–35 Furthermore, the Par6β gene, PARD6B, is frequently amplified in breast cancer.36 Conversely, gene deletions for Par6γ (PARD6G), Par3 (PARD3), or Dlg1 (DLG1) have been documented in multiple cancer types.37 In addition, epigenetic changes can alter expression in tumors, and hypermethylation of the DLG3 promoter results in inactivation in colorectal cancer.38 The expression of polarity proteins can also be affected by aberrant transcriptional regulation, and several polarity proteins are suppressed by transcription factors that promote tumorigenesis, including Zeb1/2 and Snail.39,40 Mutations can also affect polarity signaling. Mutations have been identified for aPKC, which affect its kinase activity or substrate specificity.41,42 Furthermore, expression of Scrib with a mutation that disrupts its localization has many of the same tumorigenic effects as loss of Scrib.43 Moreover, numerous intragenic deletions have been identified in Par3 that remove domains that are necessary for localization and interactions with downstream effectors.37 In this paper we will discuss evidence that polarity proteins regulate diverse signaling pathways and that disrupted polarity signaling contributes to multiple stages of cancer progression, from initiation through invasion.
Tissue organization and cell polarity
The earliest stages of breast tumorigenesis involve loss of bilayer organization, with ducts becoming multilayered (Figure 1). Cells that no longer contact the lumen lose apical–basal polarity and eventually form solid masses that may contain micro-lumina (Figure 1). Interestingly, this process resembles normal events during mammary branching morphogenesis, when ducts become multilayered at the leading tips of growing ducts. The inner cells within the multilayered epithelium transiently lose apical–basal polarity, with aPKC colocalizing with basolateral proteins β-catenin and Scrib, and Par3 becoming delocalized to the cytoplasm.44
Loss of an apical–basal protein is sufficient to induce the multilayered phenotype observed during early stages of breast cancer progression. For example, depletion of Par3 from the normal mammary epithelium in mice results in dilated, multilayered ducts that resemble epithelial atypia.45 Although Par3-depleted ducts still contain a central lumen and retain tight junctions, some aspects of apical–basal polarity are lost since they no longer have aPKC restricted to the apical membrane.45 This mirrors human breast cancer, in which Par3 is frequently downregulated and aPKC is mislocalized from the apical membrane.46–48 Furthermore, depletion of the basolateral protein Scrib from mammary epithelial cells leads to solid ducts with no luminal space, which is closer to DCIS, and appears more advanced than the loss of Par3 phenotype.43 Loss of Lkb1 also leads to loss of apical–basal polarity in mammary ducts, which fail to form a lumen when cultured in 3D extracellular matrix.49 Therefore, polarity proteins are essential regulators of mammary epithelial integrity, and disruption of individual polarity proteins is sufficient to induce lesions that are characteristic of early stages of breast cancer.
How might disruption of cell polarity affect tissue organization? A number of different mechanisms could account for generating a multilayered epithelium: 1) cells could switch to a more mesenchymal phenotype and migrate out of the plane of the epithelium; 2) cells could remain epithelial and rearrange to become multilayered; or 3) cells could misorient the plane of cell division from parallel to perpendicular to the epithelial layer (Figure 3). While some epithelial cells undergo an epithelial–mesenchymal transition (EMT) to become more migratory and leave the epithelial layer during embryonic development,50 there is little evidence that this occurs during early stages of breast cancer when cells predominantly retain epithelial markers, including E-cadherin.3 During mammary gland development, the epithelia can transiently lose polarity. For example, during branching morphogenesis, multi-layering occurs in the absence of EMT, and instead individual cells lose polarity and rearrange within the epithelium.44,51 Furthermore, during lactation, polarity is partially lost, with apical proteins Ezrin and Na/H Exchange Regulatory Factor 1 (NHERF1) becoming basolateral and interacting with β-catenin and E-cadherin. This reorganization is required to correctly localize the prolactin receptor,52 indicating that there is crosstalk between apical–basal polarity and hormone signaling.
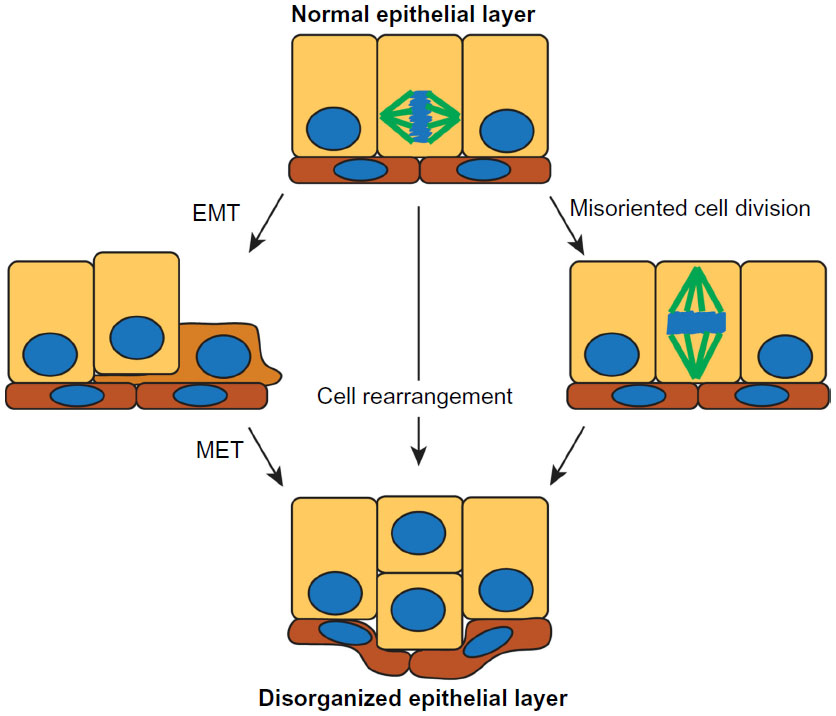 | Figure 3 Potential mechanisms by which normal epithelial organization can be lost early in the progression of breast cancer. |
In several epithelial systems, the polarity machinery orients cell division, and depleting Par3, Par6, aPKC, Cdc42, or Dlg misorients cell divisions, causing defects in tissue organization (Figure 4).53–59 The main link between polarity and spindle orientation is the adaptor protein Leu-Gly-Asn repeat-enriched (LGN; also known as Partner of Inscuteable [Pins]) protein, which binds to the heterotrimeric G-protein, Gαi, a myristoylated protein anchored to the plasma membrane.60 LGN also binds to Dlg, which positions LGN at the lateral membrane.61,62 In order to segregate the mitotic chromosomes, mitotic spindle poles need to anchor to the lateral membranes, which is achieved through the binding of LGN to nuclear mitotic apparatus (NuMA; also known as Mud) protein, which can bind astral microtubules as well as the force generating microtubule motor protein dynein.63 A function for the Par complex is to prevent LGN from localizing to the apical membrane to reinforce correct spindle orientation. Apically restricted aPKC phosphorylates LGN, which then binds 14-3-3 to induce a conformation change that releases it from the membrane, thereby excluding LGN from the apical membrane.54
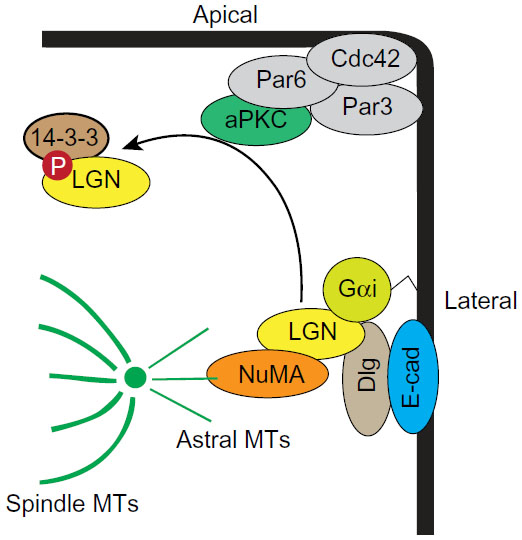 | Figure 4 Model for oriented cell division. |
Our understanding of the mechanisms of how spindles are oriented is incomplete and additional mechanisms may be involved. For example, deletion of β1-integrin from the basal layer of the mammary epithelium causes spindle orientation defects, with mixing of the basal and luminal layers.64 Furthermore, deletion of β1-integrin from the mammary gland was recently shown to block apical–basal polarity, by deregulating trafficking of apical proteins.65 Therefore, a potential mechanism by which loss of β1-integrin misorients cell divisions is by mislocalizing apical–basal polarity proteins. Support for the idea that spindle orientation defects can induce early tumor-like events comes from Drosophila, where disruption of Dlg or Scrib misorients the cell division plane, and cells that divide out of the plane undergo apoptosis,59 indicating that normal tissues can eliminate delaminated cells. Remarkably, however, when apoptosis is blocked, the delaminated cells develop into disorganized tumor-like masses.59
Polarity proteins balance proliferation and apoptosis
The ability to balance proliferation and apoptosis is a fundamental requirement for epithelial homeostasis, and altering this balance promotes tumor proliferation. For example, apoptosis and proliferation are interconnected through a process of compensatory proliferation.66 When epithelial cells are damaged and cannot be repaired they initiate an apoptotic program that involves secreting morphogens to stimulate the surrounding cells to proliferate and replace the damaged cells. One cue that triggers apoptosis in damaged cells may be the integrity of apical–basal polarity itself, since loss of components of the Par or Scrib complexes can induce apoptosis.45,53,67 Interestingly, blocking apoptosis induced by disruption of the Par complex leads to compensatory proliferation and epithelial overgrowth in Drosophila.66 Remarkably, although depleting Dlg or Crb induces apoptosis, unlike depleting the Par complex, it does not induce compensatory proliferation, indicating that the three complexes induce apoptosis through different mechanisms.66 A compensatory-like mechanism may be at play in mammary epithelial cells, because loss of Par3 induces both apoptosis and proliferation in the developing mouse mammary gland and enhances mammary tumor growth.45,46
The Par complex may also regulate proliferation of cancer cells through additional mechanisms. Phosphatidylinositide 3-kinase (PI3K) is a prominent player in the survival pathway, promoting growth and proliferation, and has an antagonistic relationship with the tumor suppressor phosphatase and tensin homolog (PTEN). Par3 binds PTEN, which targets it to tight junctions and is necessary to maintain apical–basal polarity.68 Par3 also binds to the catalytic p110 subunit of PI3K, which is necessary for polarity in migrating cells.69 Interactions between PI3K/PTEN and the Par complex are likely important in both physiological and cancer contexts. Most breast cancers have at least one member of the PI3K pathway altered, and activation of this pathway is significant in basal-like breast cancers.70 In breast cancer and glioblastoma cell lines expressing oncogenic PI3K or loss of PTEN, aPKC expression and activity are upregulated, which blocks oncogene-induced senescence, thereby promoting proliferation (Figure 5).71 Furthermore, in estrogen receptor positive breast cancer cells, estradiol promotes cell proliferation by stimulating an interaction between the p85 regulatory subunit of PI3K, aPKC, Src, and ERα.72 Therefore, balanced PI3K/PTEN signaling likely has an important function in linking cell polarity and growth control, and additional work should further clarify this relationship in cancer.
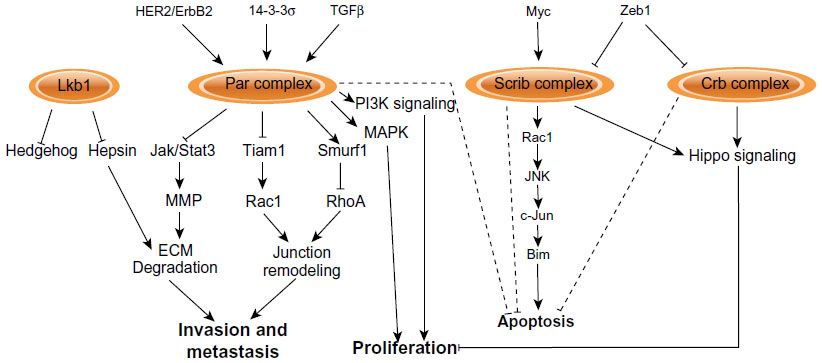 | Figure 5 Signaling pathways regulated by apical–basal polarity complexes. |
Epithelial polarity and growth control through Hippo signaling
Another mechanism that can control epithelial growth is through the transcription co-factor Yes-associated protein 1 (Yap1) or the related Taz (Figure 6). Yap1/Taz nuclear localization and activity is associated with oncogenic transformation in mammary epithelial cells, and also with invasion of lobular breast cancers and metastasis.73–76 Furthermore, activation of Taz sustains breast cancer stem cell self-renewal.77 However, Yap1 may also be a tumor suppressor in some breast cancers since it is in a region of frequent loss-of-heterozygosity (LOH) and depletion of Yap1 from breast cancer cell lines increased their survival, invasiveness, and tumor growth in xenografts.78 These apparent differences may be due to the ability of Yap1/Taz to interact with multiple transcription factors. For example, binding to the TEAD family of transcription factors regulates expression of genes involved in proliferation,79 whereas binding p73 regulates apoptotic genes in response to DNA damage.80 There are a number of mechanisms that can regulate Yap1/Taz nuclear localization and activation, including the Hippo pathway, cell–cell adhesion, cell tension through the actin cytoskeleton, G protein-coupled receptor (GPCR) signaling, tyrosine kinases, and cell polarity.81,82 The Hippo signaling pathway is a negative regulator of Yap1/Taz and consists of a kinase cascade in which Mst1/2 phosphorylates Lats1/2, which subsequently phosphorylates and inactivates Yap1 by creating a 14-3-3 binding site required to export Yap1/Taz from the nucleus.81 Hippo signaling regulates contact-inhibited growth through Yap1/Taz by binding to cell polarity complexes, which sequesters Yap1/Taz out of the nucleus (Figure 6). Yap1 interacts with multiple components of the Crumbs complex and tight junctions, including Pals1, Patj, and Amot, and depleting Crb or Pals1 causes Yap1/Taz to accumulate in the nucleus in mammary epithelial cells.83 Regulation of the Yap1/Taz by the Crumbs complex is dependent on Mst1/2 and Lats1/2; however, the mechanism by which the complexes function is incompletely understood. One possibility is that the apical cortex acts as a scaffold to regulate Hippo-mediated inhibition of Yap1/Taz. Indeed, Lats1/2 is recruited to the plasma membrane by neurofibromin 2 (NF2), which is apically localized in epithelial cells,84 and kidney and brain expressed protein (Kibra), which interacts apically with the Par complex, can regulate Lats1/2 activity.85–87 Therefore, the Crumbs complex links cell density with proliferation control. The ability of Yap1/Taz to interact with multiple members of the Crumbs complex may mean that Yap1/Taz most efficiently binds to a complete Crumbs complex, ensuring that it is sequestered out of the nucleus in cells only when apical–basal polarity is fully established.
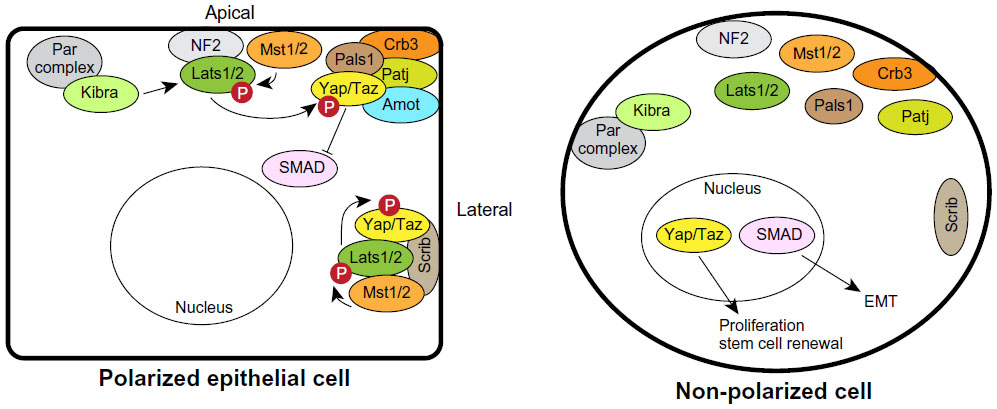 | Figure 6 Regulation of Hippo-Yap1/Taz signaling by polarity complexes. The apical Crumbs and lateral Scrib complexes can sequester Yap1/Taz and the Hippo pathway kinases to prevent proliferation in polarized epithelial cells (left). Loss of polarity inactivates the Hippo pathway and releases Yap1/Taz to enter the nucleus to regulate proliferation (right). The Crumbs complex can also regulate SMADs to sensitize cells to epithelial-mesenchymal transition. |
Basolateral polarity complexes can also regulate Hippo signaling. For example, in breast cancer, Scrib acts as a scaffold to assemble Mst1/2, Lats1/2 and Taz.77 Either loss or mislocalization of Scrib blocks the ability of Mst1/2 to activate Lats1/2, resulting in sustained Taz activation.77 Therefore, the Hippo pathway may have multiple interactions with cell polarity complexes as a mechanism to sense epithelial integrity. An advantage of this would be that the cell would be sensitive to relatively minor disruptions in apical–basal polarity, which could enable a graded activation of Yap1. Further studies should enhance our understanding of the complexity by which polarity controls Hippo signaling in development and cancer.
Cell polarity and the EMT
A widely accepted paradigm for cancer progression is that epithelial cells undergo a mesenchymal transition, during which they lose apical–basal polarity and intercellular adhesions, and express mesenchymal genes, such as N-cadherin and vimentin.50 Single mesenchymal cells escape from the epithelial tumor mass and enter the lymphatic system or bloodstream through which they disseminate. At ectopic sites in the body, the tumor cells extravasate, revert to an epithelial phenotype, and colonize surrounding tissue to form metastases. However, EMT is not essential for tumor invasion, as epithelial cells can collectively invade.6,88 Furthermore, circulating tumor cells (CTCs) isolated from cancer patients show expression of markers for both mesenchymal and epithelial cells,89 indicating that multiple mechanisms may be involved by which cells can disseminate. This may depend on the breast cancer subtype, since CTCs from patients with triple negative breast cancers have a mesenchymal phenotype, whereas CTCs from patients with lobular cancer express epithelial markers, similar to the primary tumor.89
EMT is regulated by multiple transcription factors including Snail, Slug, Twist, Zeb1/2, FOXC2, KLF8, and E47, which are able to suppress epithelial genes and activate a mesenchymal expression program.50 In particular, Zeb1 and Snail repress the expression of an array of epithelial polarity genes, including E-cadherin, Crumbs, Patj and Lgl.39,90,91 The suppression of apical–basal polarity is considered an invariable feature of EMT, and re-expression of Lgl2 blocks Zeb1 and Snail-induced EMT.90,91 Importantly, a feedback cycle involving members of the miR-200 family maintains epithelial characteristics by suppressing expression of transforming growth factor β (TGFβ) and Zeb1.92,93 In the mammary gland, the micro ribonucleic acid (miRNA) miR-200a maintains apical–basal polarity and its knockdown suppresses claudin-3 and Par6β expression, which compromises apical–basal polarity and lumen formation.94
Although loss of apical–basal polarity may be necessary for EMT, loss of polarity proteins is not sufficient to induce EMT. For example, although the simultaneous depletion of two polarity proteins (Dlg, AF-6, Scrib) induces invasion of breast epithelial cells, it does not induce EMT.95 Furthermore, loss of Par3 in vivo induces invasion and metastasis, however this occurs in the absence of overt EMT.46,47 Taken together, these data argue that tumors can use multiple mechanisms for dissemination, including but not limited to EMT, and that loss of apical–basal polarity is not sufficient to induce EMT. Interestingly, Par6 is actually necessary to dissolve TJs during TGF-β-induced EMT, and blocking Par6 activity prevents EMT.96 In this model, the TGF-β receptor binds and phosphorylates Par6, which then recruits the E3 ubiquitin ligase Smurf1 to degrade RhoA (Ras homolog gene family member A) and destabilize TJs.96
An EMT phenotype has also been associated with mammary stem cell characteristics, which have been linked to increased breast tumor-initiating capacity, resistance to radiation and chemotherapy, and metastasis.97,98 Interestingly, EMT is able to stimulate self-renewal of breast cancer stem cells by activating Taz through delocalizing Scrib.77 This highlights the complexity of signaling regulated by polarity proteins, which can lead to crosstalk between pathways in the progression of breast cancer. The Crumbs complex also mediates crosstalk between EMT and the Hippo signaling pathway. TGF-β induces EMT through SMAD signaling in NMuMG (normal murine mammary gland) mammary epithelial cells. Surprisingly, in polarized cells, Yap1/Taz sequesters SMADs in the cytoplasm to inhibit their activity, which is controlled by binding of Yap1/Taz to the Crumbs complex.83 Therefore disruption of the Crumbs complex sensitizes cells to EMT by delocalizing Yap1/Taz and SMADs.
Specific evidence of the importance of polarity proteins in breast cancer
Cooperation between cell polarity proteins, oncogenes, and tumor suppressors
In this section we will examine evidence for a direct role of polarity proteins in breast cancer progression and metastasis, with a focus primarily on studies utilizing mouse models.
HER2/ErbB2 is amplified in approximately 20%–30% of breast cancers and is associated with increased proliferation, reduced apoptosis, and disrupted epithelial organization and apical–basal polarity. Loss of apical–basal polarity is dependent on Par6/aPKC, and activated ErbB2 is able to bind Par6/aPKC and displace Par3; therefore, activation of ErbB2 affects apical–basal polarity by disrupting the Par complex (Figure 5).42,99 Although Par6 is over-expressed in human breast cancers and is able to induce proliferation in mammary epithelial cells through the mitogen activated protein kinase (MAPK) pathway in vitro,100 Par6 and aPKC are not necessary for ErbB2-dependent proliferation.99
The contribution of the Par polarity complex in HER2/ErbB2-positive breast cancers may occur at multiple levels. The 14-3-3σ tumor suppressor gene is frequently lost in ErbB2 amplified tumors and genetic deletion of the 14-3-3σ locus in mice disrupts cell–cell junctions and apical–basal polarity.101 14-3-3σ binds Par3, and loss of 14-3-3σ mislocalizes Par3 from the plasma membrane to induce polarity defects and accelerate ErbB2-dependent tumor onset.102 Therefore, the Par complex is targeted and disrupted by oncogenes and tumor suppressors to perturb apical–basal polarity during breast cancer progression (Figure 5).
Loss of Par3 has also been shown to promote breast cancer progression by collaborating with oncogenes. Depletion of Par3 from the normal mammary gland increases both apoptosis and proliferation, which offset each other and palpable tumors never form.46,47 However, in the presence of either oncogenic Notch1 intracellular domain (NICD) or activated RasV12, loss of Par3 robustly promotes tumor growth.46 This effect may depend on the oncogene, because loss of Par3 has no effect on primary tumor size in an ErbB2 model.47
Loss of polarity proteins can also cooperate with the Myc oncogene to promote tumor growth. Loss of Scrib in Myc-driven mammary tumors enhances primary tumor size, in this case by blocking Myc-induced apoptosis.43 In this model, Scrib acts as a scaffold to coordinate a Rac/JNK/c-Jun pathway that regulates expression of the pro-apoptotic protein Bim (Figure 5).43 Loss of Lkb1 is also able to promote primary tumor growth in Myc-induced mammary tumors.49 In this case, loss of Lkb1 is associated with disrupted localization of apical–basal polarity proteins, but does not affect Myc-induced apoptosis.49 Instead, a mechanism by which Lkb1 may regulate mammary tumor size is through Hedgehog signaling (Figure 5); depletion of Lkb1 causes upregulation of Hedgehog pathway genes Sonic hedgehog (Shh), Smoothened (Smo), Gli1, and Patched (Ptch).103 Importantly, Lkb1 and the expression of Hh pathway components are negatively correlated in human breast cancer samples, and inhibition of Hh signaling enhances apoptosis in Lkb1-deficient tumor cells.103 Lkb1 is also an important regulator of AMPK, a central regulator of metabolic function, which may represent another mechanism by which Lkb1 could regulate breast cancer growth.23
A role for polarity proteins in breast cancer invasion and metastasis
The transition to invasive breast cancers involves degradation of the basement membrane, providing an opportunity for cells to escape the primary tumor mass and invade surrounding tissue. The basement membrane can also exert a polarizing cue to epithelial cells to establish and maintain apical–basal polarity in the mammary epithelium.65,104 However, signaling between apical–basal polarity proteins and the extracellular is bidirectional and disrupted expression of polarity proteins also impinges on extracellular matrix (ECM) integrity. For example, loss of Lkb1 in the mammary gland causes fragmentation of the basement membrane through mislocalization of the serine protease hepsin (Figure 5).49 Furthermore, loss of Par3 in NICD-expressing mammary tumors alters the expression of numerous ECM-related genes, and leads to degradation of the ECM and defects in cell-matrix adhesion.46 Therefore, the disruption of some polarity proteins may contribute to breast cancer progression by degrading the basement membrane and extracellular matrix, which may be important for the switch from carcinoma in situ to invasive carcinoma. Indeed, this appears to be the case; loss of Par3 in both NICD and RasV12 breast cancer models promotes local invasion and lung metastasis. Loss of Par3 causes activation of Janus kinase/signal transducer and activator of transcription (Jak/Stat) signaling, which through matrix metalloproteinases (MMPs) and likely other targets, remodels the ECM and enables Par3-deficient cells to invade and metastasize (Figure 5).46 This is dependent on aPKC, which is mislocalized and activated in Par3-depleted cells.46 Concomitant depletion of Par3 and inhibition of Jak/Stat3 or MMP9 partially inhibits invasion and metastasis, indicating that additional mechanisms may be at play.46 Interestingly, another study identified a complementary pathway for loss of Par3 to induce invasion and metastasis in ErbB2 breast cancers.47 In this case, invasion and metastasis caused by loss of Par3 involves the loosening of adherens junctions, but without inducing EMT.47 In this model, loss of Par3 activates the GTPase Rac1 by delocalizing the Rac activator Tiam1,47 and Tiam1 itself is necessary for ErbB2-mediated mammary tumor growth and metastasis (Figure 5).105 The activated Rac then induces uncontrolled cortical actin dynamics, which decreases E-cadherin stability and cell–cell cohesion to prime cells to become more invasive and metastatic.47
Par6 has also been implicated in breast cancer metastasis, although the mechanism is distinct than for Par3. In this case, Par6 expression is required for TGF-β-induced metastasis by destabilizing tight junctions (Figure 5).96,106 Expression of an inactive Par6 mutant that uncouples it from the TGF-β receptor, restores tight junction formation, and blocks metastasis to the lungs.106
Clinical perspectives
A challenge with breast cancer is that it is a heterogeneous disease, with diverse molecular subtypes driven by distinct oncogenes and tumor suppressors. Therefore, polarity proteins may have distinct functions in different breast cancer subtypes. It is interesting to note that The Cancer Genome Atlas (TCGA) breast data set reveals that expression of at least one core polarity protein is deregulated in 65% of Luminal A/B, 79% of HER2, and 95% of basal-like breast cancers.107 Furthermore, since polarity proteins function to regulate signaling moieties by spatially restricting them, other factors such as mutations that affect protein–protein interactions, or even localization could be important. For example, Scrib is mislocalized in DCIS, and mislocalization of Scrib can mimic some, but not all, cancer-related functions observed when Scrib is depleted.43,108
A current clinical challenge is assessing early breast lesions and predicting which are benign and which may progress to invasive disease.109 It is recognized that like more advanced tumors, DCIS also has multiple distinct subtypes, and up to 50% of DCIS foci will progress to invasive carcinoma.4 One marker for this transition is E-cadherin, which is expressed in early DCIS, but is absent from IDC.109 Analysis of additional polarity markers and their relationship with breast cancer progression may identify changes that enhance diagnostic or prognostic decisions at early stages.
An intriguing question is whether polarity proteins could represent therapeutic targets. Since many polarity proteins act as scaffolding or adaptor proteins without any enzymatic activity, it may be possible to target specific protein–protein interaction domains using available high-through put screening methods for small molecule inhibitors of phosphorylation-dependent protein–protein interactions.110 For example, this may have utility in Par6 over-expressing breast tumors to uncouple Par6 from TGF-β receptor in the treatment of metastatic breast cancer.106 A more feasible approach may be to inhibit the polarity kinases. Several small molecule aPKC inhibitors have been developed,111–113 and inhibiting aPKC in Par3 over-expressing breast cancer cells sensitizes them to chemotherapy.114 Alternatively, a deeper understanding of the signaling pathways that are hyper-activated in cells with altered polarity may also identify pathways with established therapeutic targets.
Finally, although still somewhat controversial, breast cancer stem cells have been identified that have increased tumor-initiating capacity, resistance to chemotherapy and radiation, and metastatic potential.115–117 Moreover, the claudin-low subtype displays an enhanced stem cell signature.118 Mammary stem cells divide asymmetrically and p53-null and ErbB2 tumors in mice have an expanded stem cell population due to a switch to symmetric, self-renewing divisions.119 In numerous stem cell systems, the Par complex is involved in regulating asymmetric cell divisions,120 and an important future goal will be to determine how disruption of polarity proteins affects stem cell properties during breast cancer progression and treatment. A deeper understanding of the mechanisms of stem cell renewal may hold promise for identifying new ways to manage breast cancer patients clinically.
Conclusion
Our understanding of the roles of polarity proteins in breast cancer progression is still in its infancy. However, our current state of knowledge indicates that the core polarity proteins interact with a wide array of signaling pathways, and function in context-specific ways that may be important to many, if not all, stages of cancer progression. A more thorough understanding of the mechanisms that lead to altered apical–basal polarity, and the consequences of these changes in different breast subtypes, as well as in the context of diverse oncogenes and tumor suppressors will lead to a further understanding of the mechanisms that regulate breast cancer progression and metastasis.
Acknowledgments
Research by LM is supported by grants from the Terry Fox Research Institute (1009) and Canadian Institutes of Health Research (MOP-119482). LM is a FRQS Research Scholar. SJC is supported by the McGill Integrated Cancer Research Training Program.
Disclosure
The authors declare no conflicts of interest in this work.
References
Bombonati A, Sgroi DC. The molecular pathology of breast cancer progression. J Pathol. 2011;223(2):307–317. | |
Schnitt SJ. The diagnosis and management of pre-invasive breast disease: flat epithelial atypia – classification, pathologic features and clinical significance. Breast Cancer Res. 2003;5(5):263–268. | |
Zagouri F, Sergentanis TN, Zografos GC. Precursors and preinvasive lesions of the breast: the role of molecular prognostic markers in the diagnostic and therapeutic dilemma. World J Surg Oncol. 2007; 5:57. | |
Ellis IO. Intraductal proliferative lesions of the breast: morphology, associated risk and molecular biology. Mod Pathol. 2010;23 Suppl 2: S1–S7. | |
Schedin P, Elias A. Multistep tumorigenesis and the microenvironment. Breast Cancer Res. 2004;6(2):93–101. | |
Nguyen-Ngoc KV, Cheung KJ, Brenot A, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A. 2012; 109(39):E2595–E2604. | |
Malhotra GK, Zhao X, Band H, Band V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther. 2010;10(10):955–960. | |
Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8(5):R76. | |
Metzger-Filho O, Sun Z, Viale G, et al. Patterns of Recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: results from international breast cancer study group trials VIII and IX. J Clin Oncol. 2013;31(25):3083–3090. | |
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–4124. | |
Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009;23(22):2563–2577. | |
Dumont N, Liu B, Defilippis RA, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia. 2013;15(3):249–262. | |
Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321(5897):1841–1844. | |
Goodison S, Kawai K, Hihara J, et al. Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin Cancer Res. 2003;9(10 Pt 1):3808–3814. | |
Bazzoun D, Lelièvre S, Talhouk R. Polarity proteins as regulators of cell junction complexes: implications for breast cancer. Pharmacol Ther. 2013;138(3):418–427. | |
Tepass U. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol. 2012;28:655–685. | |
Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000; 289(5476):113–116. | |
Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52(3):311–320. | |
Ebnet K, Suzuki A, Horikoshi Y, et al. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J. 2001;20(14):3738–3748. | |
Yang Z, Xue B, Umitsu M, Ikura M, Muthuswamy SK, Neel BG. The signaling adaptor GAB1 regulates cell polarity by acting as a PAR protein scaffold. Mol Cell. 2012;47(3):469–483. | |
Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119(Pt 6):979–987. | |
Suzuki A, Hirata M, Kamimura K, et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr Biol. 2004;14(16):1425–1435. | |
Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. | |
Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23(4):833–843. | |
Zhang L, Li J, Young LH, Caplan MJ. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc Natl Acad Sci U S A. 2006;103(46):17272–17277. | |
Lee JH, Koh H, Kim M, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447(7147):1017–1020. | |
Fogg VC, Liu CJ, Margolis B. Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells. J Cell Sci. 2005;118(Pt 13):2859–2869. | |
Navarro C, Nola S, Audebert S, et al. Junctional recruitment of mammalian Scribble relies on E-cadherin engagement. Oncogene. 2005;24(27):4330–4339. | |
Qin Y, Capaldo C, Gumbiner BM, Macara IG. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol. 2005;171(6):1061–1071. | |
Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403(6770):676–680. | |
Wang Q, Hurd TW, Margolis B. Tight junction protein Par6 interacts with an evolutionarily conserved region in the amino terminus of PALS1/stardust. J Biol Chem. 2004;279(29):30715–30721. | |
Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol. 2003;5(2):137–142. | |
Eder AM, Sui X, Rosen DG, et al. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A. 2005; 102(35):12519–12524. | |
Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280(35):31109–31115. | |
Yang YL, Chu JY, Luo ML, et al. Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer. 2008;47(2):127–136. | |
Cunliffe HE, Jiang Y, Fornace KM, Yang F, Meltzer PS. PAR6B is required for tight junction formation and activated PKC¶ localization in breast cancer. Am J Cancer Res. 2012;2(5):478–491. | |
Rothenberg SM, Mohapatra G, Rivera MN, et al. A genome-wide screen for microdeletions reveals disruption of polarity complex genes in diverse human cancers. Cancer Res. 2010;70(6):2158–2164. | |
Feng X, Chen K, Ye S, et al. MPP3 inactivation by promoter CpG islands hypermethylation in colorectal carcinogenesis. Cancer Biomark. 2012;11(2–3):99–106. | |
Aigner K, Dampier B, Descovich L, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26(49):6979–6988. | |
Russ A, Louderbough JM, Zarnescu D, Schroeder JA. Hugl1 and Hugl2 in mammary epithelial cells: polarity, proliferation, and differentiation. PLoS One. 2012;7(10):e47734. | |
Galvez AS, Duran A, Linares JF, et al. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29(1):104–115. | |
Linch M, Sanz-Garcia M, Rosse C, et al. Regulation of polarized morphogenesis by protein kinase C iota in oncogenic epithelial spheroids. Carcinogenesis. Epub 2013 Nov 30. | |
Zhan L, Rosenberg A, Bergami KC, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135(5):865–878. | |
Ewald AJ, Huebner RJ, Palsdottir H, et al. Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J Cell Sci. 2012;125(Pt 11):2638–2654. | |
McCaffrey LM, Macara IG. The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 2009;23(12):1450–1460. | |
McCaffrey LM, Montalbano J, Mihai C, Macara IG. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell. 2012;22(5):601–614. | |
Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell–cell cohesion. Nat Cell Biol. 2013;15(2):189–200. | |
Kojima Y, Akimoto K, Nagashima Y, et al. The overexpression and altered localization of the atypical protein kinase C lambda/iota in breast cancer correlates with the pathologic type of these tumors. Hum Pathol. 2008;39(6):824–831. | |
Partanen JI, Tervonen TA, Myllynen M, et al. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc Natl Acad Sci U S A. 2012;109(7):E388–E397. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14(4):570–581. | |
Morales FC, Hayashi Y, van Pelt CS, Georgescu MM. NHERF1/EBP50 controls lactation by establishing basal membrane polarity complexes with prolactin receptor. Cell Death Dis. 2012;3:e391. | |
Durgan J, Kaji N, Jin D, Hall A. Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J Biol Chem. 2011;286(14):12461–12474. | |
Hao Y, Du Q, Chen X, et al. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr Biol. 2010;20(20):1809–1818. | |
Jaffe AB, Kaji N, Durgan J, Hall A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J Cell Biol. 2008;183(4):625–633. | |
Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437(7056):275–280. | |
Zheng Z, Zhu H, Wan Q, et al. LGN regulates mitotic spindle orientation during epithelial morphogenesis. J Cell Biol. 2010;189(2):275–288. | |
Žigman M, Trinh le A, Fraser SE, Moens CB. Zebrafish neural tube morphogenesis requires Scribble-dependent oriented cell divisions. Curr Biol. 2011;21(1):79–86. | |
Nakajima Y, Meyer EJ, Kroesen A, McKinney SA, Gibson MC. Epithelial junctions maintain tissue architecture by directing planar spindle orientation. Nature. 2013;500(7462):359–362. | |
Du Q, Macara IG. Mammalian Pins is a conformational switch that links NuMA to heterotrimeric G proteins. Cell. 2004;119(4):503–516. | |
Bergstralh DT, Lovegrove HE, St Johnston D. Discs large links spindle orientation to apical-basal polarity in Drosophila epithelia. Curr Biol. 2013;23(17):1707–1712. | |
Johnston CA, Hirono K, Prehoda KE, Doe CQ. Identification of an Aurora-A/PinsLINKER/Dlg spindle orientation pathway using induced cell polarity in S2 cells. Cell. 2009;138(6):1150–1163. | |
Kotak S, Gönczy P. Mechanisms of spindle positioning: cortical force generators in the limelight. Curr Opin Cell Biol. 2013;25:741–748. | |
Taddei I, Deugnier MA, Faraldo MM, et al. Beta1 integrin deletion from the basal compartment of the mammary epithelium affects stem cells. Nat Cell Biol. 2008;10(6):716–722. | |
Akhtar N, Streuli CH. An integrin-ILK-microtubule network orients cell polarity and lumen formation in glandular epithelium. Nat Cell Biol. 2013;15(1):17–27. | |
Warner SJ, Longmore GD. Context-dependent compensatory proliferation in epithelial homeostasis and tumorigenesis. Cell Cycle. 2010;9(20):4037–4038. | |
Warner SJ, Yashiro H, Longmore GD. The Cdc42/Par6/aPKC polarity complex regulates apoptosis-induced compensatory proliferation in epithelia. Curr Biol. 2010;20(8):677–686. | |
Feng W, Wu H, Chan LN, Zhang M. Par-3-mediated junctional localization of the lipid phosphatase PTEN is required for cell polarity establishment. J Biol Chem. 2008;283(34):23440–23449. | |
Itoh N, Nakayama M, Nishimura T, et al. Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton (Hoboken). 2010;67(5):297–308. | |
López-Knowles E, O’Toole SA, McNeil CM, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126(5):1121–1131. | |
Paget JA, Restall IJ, Daneshmand M, et al. Repression of cancer cell senescence by PKCι. Oncogene. 2012;31(31):3584–3596. | |
Castoria G, Migliaccio A, Di Domenico M, et al. Role of atypical protein kinase C in estradiol-triggered G1/S progression of MCF-7 cells. Mol Cell Biol. 2004;24(17):7643–7653. | |
Chan SW, Lim CJ, Loo LS, Chong YF, Huang C, Hong W. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J Biol Chem. 2009;284(21):14347–14358. | |
Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109(37):E2441–E2450. | |
Vlug EJ, van de Ven RA, Vermeulen JF, Bult P, van Diest PJ, Derksen PW. Nuclear localization of the transcriptional coactivator YAP is associated with invasive lobular breast cancer. Cell Oncol (Dordr). 2013;36(5):375–384. | |
Steinhardt AA, Gayyed MF, Klein AP, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39(11):1582–1589. | |
Cordenonsi M, Zanconato F, Azzolin L, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147(4):759–772. | |
Yuan M, Tomlinson V, Lara R, et al. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008;15(11):1752–1759. | |
Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. | |
Lapi E, Di Agostino S, Donzelli S, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell. 2008;32(6):803–814. | |
Lin JI, Poon CL, Harvey KF. The Hippo size control pathway – ever expanding. Sci Signal. 2013;6(259):pe4. | |
Boggiano JC, Fehon RG. Growth control by committee: intercellular junctions, cell polarity, and the cytoskeleton regulate Hippo signaling. Dev Cell. 2012;22(4):695–702. | |
Varelas X, Samavarchi-Tehrani P, Narimatsu M, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev Cell. 2010;19(6):831–844. | |
Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154(6):1342–1355. | |
Moleirinho S, Chang N, Sims AH, et al. KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene. 2013;32(14):1821–1830. | |
Yoshihama Y, Sasaki K, Horikoshi Y, et al. KIBRA suppresses apical exocytosis through inhibition of aPKC kinase activity in epithelial cells. Curr Biol. 2011;21(8):705–711. | |
Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell. 2010;18(2):288–299. | |
Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188(1):11–19. | |
Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–584. | |
Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68(2):537–544. | |
Kashyap A, Zimmerman T, Ergül N, et al. The human Lgl polarity gene, Hugl-2, induces MET and suppresses Snail tumorigenesis. Oncogene. 2013;32(11):1396–1407. | |
Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop – a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11(9):670–677. | |
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. | |
Nagaoka K, Zhang H, Watanabe G, Taya K. Epithelial cell differentiation regulated by MicroRNA-200a in mammary glands. PLoS One. 2013;8(6):e65127. | |
Chatterjee S, Seifried L, Feigin ME, et al. Dysregulation of cell polarity proteins synergize with oncogenes or the microenvironment to induce invasive behavior in epithelial cells. PLoS One. 2012;7(4):e34343. | |
Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307(5715):1603–1609. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Jain P, Alahari SK. Breast cancer stem cells: a new challenge for breast cancer treatment. Front Biosci (Landmark Ed). 2011;16:1824–1832. | |
Aranda V, Haire T, Nolan ME, et al. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol. 2006;8(11):1235–1245. | |
Nolan ME, Aranda V, Lee S, et al. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008;68(20):8201–8209. | |
Ling C, Zuo D, Xue B, Muthuswamy S, Muller WJ. A novel role for 14-3-3 sigma in regulating epithelial cell polarity. Genes Dev. 2010;24(9):947–956. | |
Ling C, Su VM, Zuo D, Muller WJ. Loss of the 14-3-3σ tumor suppressor is a critical event in ErbB2-mediated tumor progression. Cancer Discov. 2012;2(1):68–81. | |
Zhuang Z, Wang K, Cheng X, et al. LKB1 inhibits breast cancer partially through repressing the Hedgehog signaling pathway. PLoS One. 2013;8(7):e67431. | |
Weaver VM, Lelièvre S, Lakins JN, et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2(3):205–216. | |
Strumane K, Rygiel T, van der Valk M, Collard JG. Tiam1-deficiency impairs mammary tumor formation in MMTV-c-neu but not in MMTV-c-myc mice. J Cancer Res Clin Oncol. 2009;135(1):69–80. | |
Viloria-Petit AM, David L, Jia JY, et al. A role for the TGFbeta-Par6 polarity pathway in breast cancer progression. Proc Natl Acad Sci U S A. 2009;106(33):14028–14033. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Elsum IA, Humbert PO. Localization, not important in all tumor-suppressing properties: a lesson learnt from scribble. Cells Tissues Organs. 2013;198(1):1–11. | |
van de Vijver MJ. Biological variables and prognosis of DCIS. Breast. 2005;14(6):509–519. | |
Watanabe N, Osada H. Phosphorylation-dependent protein-protein interaction modules as potential molecular targets for cancer therapy. Curr Drug Targets. 2012;13(13):1654–1658. | |
Kjær S, Linch M, Purkiss A, et al. Adenosine-binding motif mimicry and cellular effects of a thieno[2,3-d]pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem J. 2013;451(2):329–342. | |
Erdogan E, Lamark T, Stallings-Mann M, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281(38):28450–28459. | |
Pillai P, Desai S, Patel R, et al. A novel PKC-ι inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int J Biochem Cell Biol. 2011;43(5):784–794. | |
Jin YT, Ying XX, Hu YH, Zou Q, Wang HY, Xu YH. aPKC inhibitors might be the sensitizer of chemotherapy and adoptive immunotherapy in the treatment of hASIPa-overexpressed breast cancer. Oncol Res. 2008;17(2):59–68. | |
Herschkowitz JI, Zhao W, Zhang M, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A. 2012;109(8):2778–2783. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | |
Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. | |
Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138(6):1083–1095. | |
Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11(12):849–860. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.