")
Back to Journals » The Application of Clinical Genetics » Volume 8
Emerging pharmacologic treatment options for fragile X syndrome
Authors Schaefer T, Davenport M, Erickson C
Received 17 May 2014
Accepted for publication 11 December 2014
Published 7 April 2015 Volume 2015:8 Pages 75—93
DOI https://doi.org/10.2147/TACG.S35673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
Tori L Schaefer, Matthew H Davenport, Craig A Erickson
Division of Child and Adolescent Psychiatry, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA
Abstract: Fragile X syndrome (FXS) is the most common single gene cause of intellectual disability and autism spectrum disorder. Caused by a silenced fragile X mental retardation 1 gene and the subsequent deficiency in fragile X mental retardation protein, patients with FXS experience a range of physical, behavioral, and intellectual debilitations. The FXS field, as a whole, has recently met with some challenges, as several targeted clinical trials with high expectations of success have failed to elucidate significant improvements in a variety of symptom domains. As new clinical trials in FXS are planned, there has been much discussion about the use of the commonly used clinical outcome measures, as well as study design considerations, patient stratification, and optimal age range for treatment. The evidence that modification of these drug targets and use of these failed compounds would prove to be efficacious in human clinical study were rooted in years of basic and translational research. There are questions arising as to the use of the mouse models for studying FXS treatment development. This issue is twofold: many of the symptom domains and molecular and biochemical changes assessed and indicative of efficacy in mouse model study are not easily amenable to clinical trials in people with FXS because of the intolerability of the testing paradigm or a lack of noninvasive techniques (prepulse inhibition, sensory hypersensitivity, startle reactivity, or electrophysiologic, biochemical, or structural changes in the brain); and capturing subtle yet meaningful changes in symptom domains such as sociability, anxiety, and hyperactivity in human FXS clinical trials is challenging with the currently used measures (typically parent/caregiver rating scales). Clinicians, researchers, and the pharmaceutical industry have all had to take a step back and critically evaluate the way we think about how to best optimize future investigations into pharmacologic FXS treatments. As new clinical trials are coming down the drug discovery pipeline, it is clear that the field is moving in a direction that values the development of molecular biomarkers, less subjective quantitative measures of symptom improvement, and rating scales developed specifically for use in FXS in conjunction with drug safety. While summarizing preclinical evidence, where applicable, and discussing challenges in FXS treatment development, this review details both completed clinical trials for the targeted and symptomatic treatment of FXS and introduces novel projects on the cusp of clinical trial investigation.
Keywords: anxiety, intellectual disability, autism, clinical trial, fragile X syndrome
Introduction
Pharmacologic treatment development in fragile X syndrome (FXS) is complicated by a number of factors. These range from genetic/epigenetic and sex influences and include variability in the severity and presence of behavioral endophenotypes such as autism-like behavior and hyperactivity. In addition, the interaction between the causal relationship between these biologic factors and behavioral presentation is unclear, making patient stratification in clinical trials difficult. Sensitive methods capable of ascertaining reliable treatment response in behavioral domains such as sociability and anxiety (two major symptoms of FXS) are not readily available or have yet to be validated for specific use in FXS populations. The goal of this review is to summarize preclinical rationale and results for recently completed clinical studies while discussing challenges and future treatment options in FXS human study. This review organizes clinical pharmacologic FXS treatment interventions into four sections: restoration of excitatory/inhibitory balance, modification of fragile X mental retardation protein (FMRP) targets, symptom-based treatments, and novel treatments in early clinical development. Completed pharmacologic clinical trials in the first two sections have been summarized in Table 1, and the trials in the third section are summarized in Table 2.
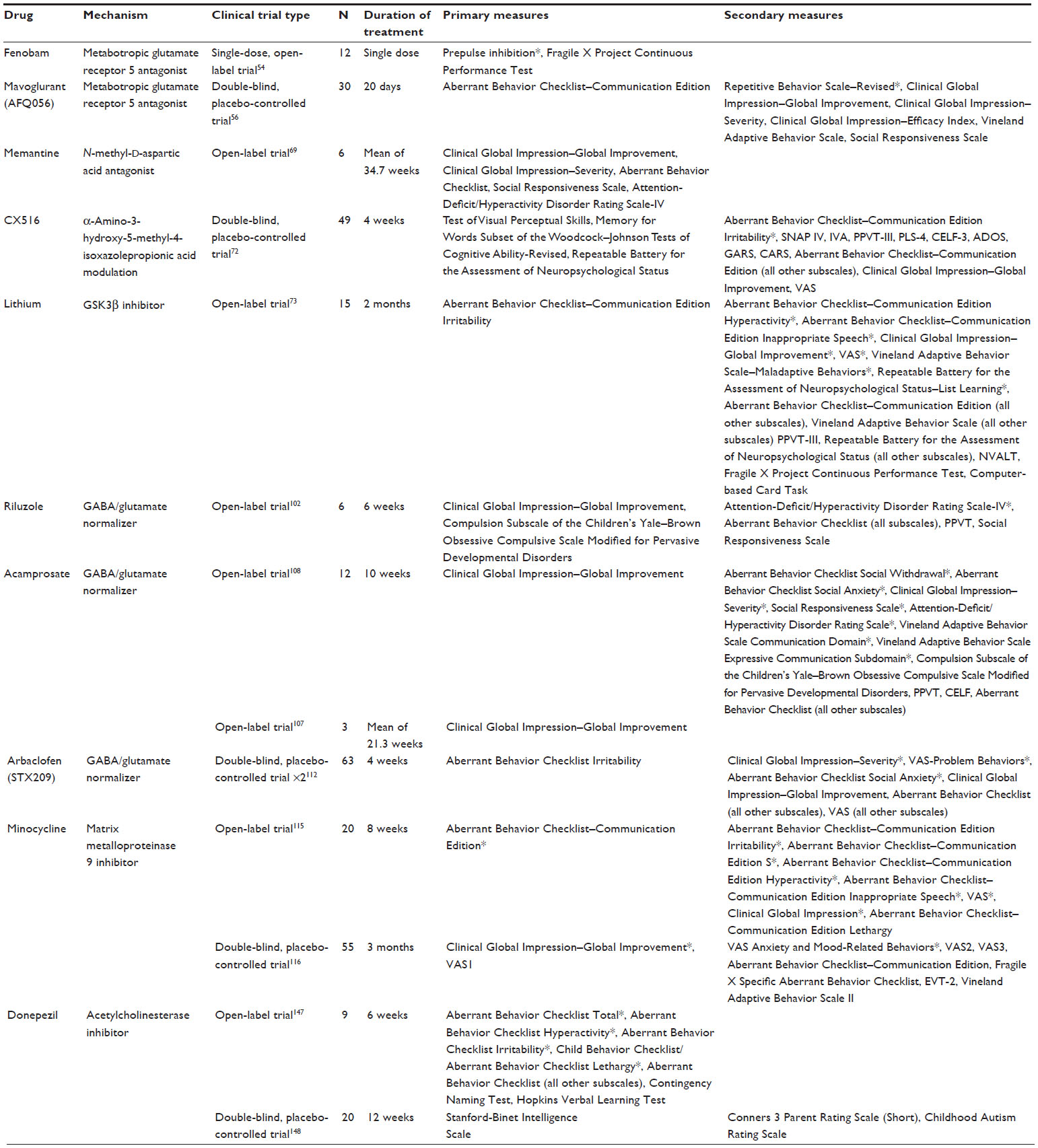 | Table 1 Clinical trials: restoration of excitatory/inhibitory balance and modification of fragile X mental retardation protein targets |
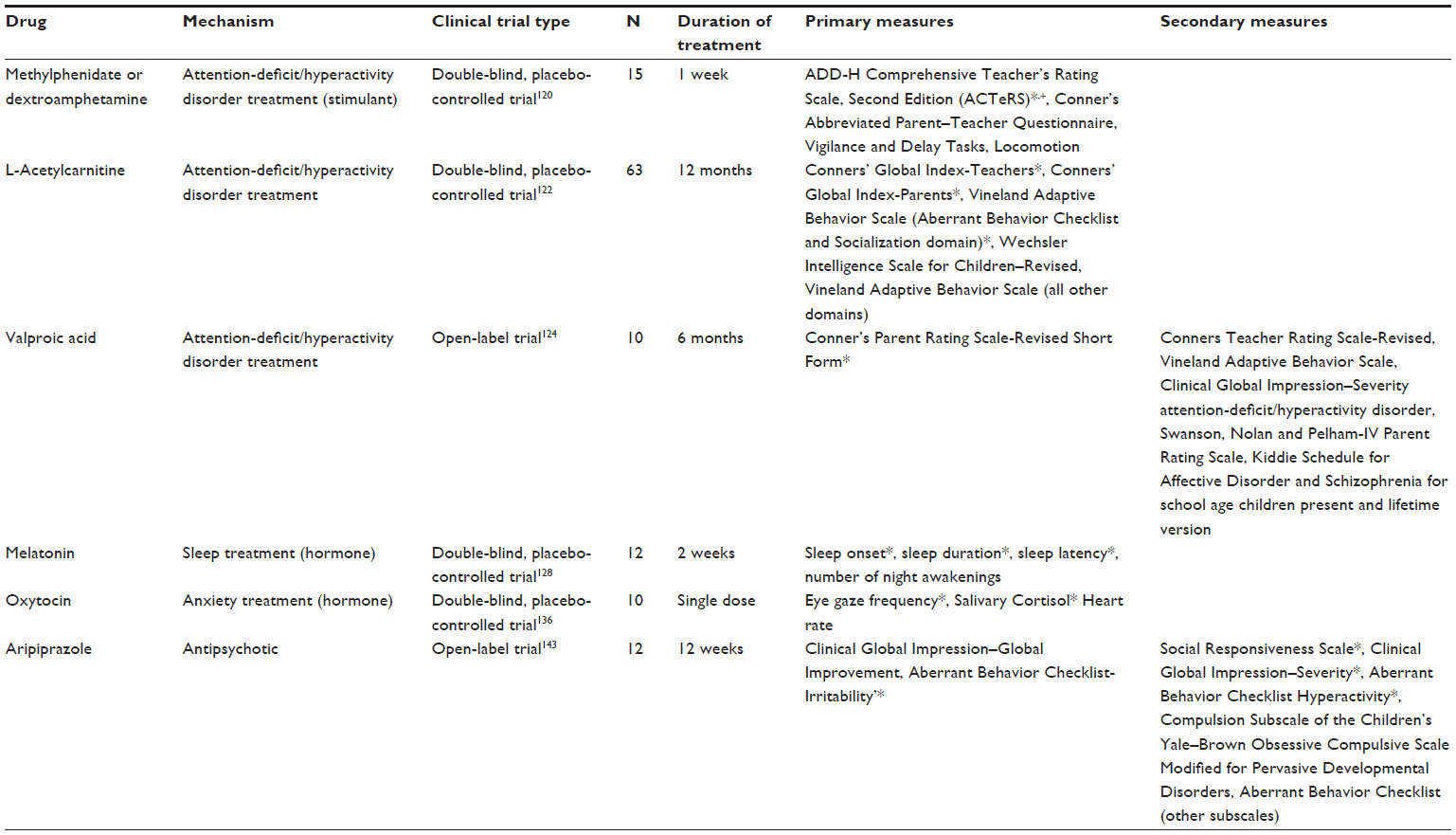 | Table 2 Clinical trials: symptom-based treatments |
Since the 1980s, few behavioral intervention studies in FXS have been published; however, there have been a wealth of pharmacologic studies reported.1 Some drugs have only been tested in a small number of people in an open-label format, whereas others have been the focus of large, multinational placebo-controlled trials. The biggest concern when conducting a clinical trial is ensuring the safety of all participants. For this reason, many initial investigations into a drug’s effectiveness in human study begin in a small, unblinded, non-placebo-controlled environment. These types of open-label trials cannot necessarily indicate that a drug will be or is effective in a population, but rather, positive results can serve as an indication that further blinded, placebo-controlled study in a larger population may be warranted. In addition, results from small controlled trials must also be interpreted with caution, as they may not readily extrapolate to a larger population.
FXS is the most common single-gene cause of inherited developmental disability, occurring in 1:4,000 males and 1:4,000–6,000 females.2,3 Physically, FXS is characterized by macrocephaly, a narrow, elongated face and prominent ears; high arched palate; flat feet; hyperextensible joints; and macroorchidism, although not every individual will have these characteristics or a similar degree of presentation.4 Individuals with FXS often have increased anxiety, obsessive compulsive disorder-like behavior, attention-deficit/hyperactivity disorder (ADHD) symptoms, increased risk for seizures, self-injurious behavior, hyperarousal to sensory stimuli, perseverative language, sleep issues, aggression, and impaired cognition, but again this can vary widely.4–6 Furthermore, there is a significant clinical overlap between FXS and Autism Spectrum Disorder (ASD), with approximately two in every three males diagnosed with FXS exhibiting features consistent with the broad autism phenotype, although there are likely qualitative differences in this behavior domain between these two disorders.7
FXS is typically the result of an unstable cytosine guanine guanine (CGG) trinucleotide repeat expansion (200+ repeats) within the promoter of the fragile X mental retardation 1 gene (FMR1), which resides on the X chromosome.8,9 The trinucleotide repeat expansion is usually inherited from a maternal carrier (55–200 CGG repeats) whose gene undergoes a further expansion. Subsequent methylation (fully versus partially methylated) and near or full silencing of the FMR1 gene and messenger RNA (mRNA) leads to the production of little or no FMRP and results in FXS.10 In vivo study has shown that the initiation of epigenetic silencing of the FMR1 gene is thought to be caused by a type of RNA-directed gene silencing in which the 5′ untranslated region that contains the transcribed CGG repeat sequences directly binds to the CGG repeat expansion in the DNA adjacent to the FMR1 promoter, but the process of subsequent methylation or maintenance of gene silencing is still unknown.11 FXS can also be caused by intragenetic point mutations in the FMR1 gene, although this is much less common.12,13
The influence of changes in DNA (genetic/epigenetic), mRNA, and level of protein production on the severity of behavior or domains affected in an individual with FXS is not entirely clear, making patient stratification a challenge. Traditionally, FXS clinical trial inclusion criteria require confirmation of full mutation, or greater than 200 CGG repeats, by southern blot analysis. More recent clinical trials have started adding additional stipulations based on methylation status or blood FMRP expression as a way to identify patients most severely affected or likely to respond the drug treatment, although this method has been largely unsuccessful. Inclusion criteria for psychological characteristics typically include stipulations on the severity of symptoms and are often related to the primary endpoint of the trial.
Sex considerations have not been a factor in past FXS clinical trials and may also add to the challenges of obtaining significant improvement with treatment. Males typically suffer more severe symptoms than females, as a result of X inactivation patterns leading to significant variation in the female phenotype. However, even within a single sex, there is tremendous heterogeneity in the behavioral presentation of FXS.14,15 Mosaicism in FXS in both males and females (methylation mosaicism, somatic mosaicism) further complicates the prediction of behavioral outcomes, can also result in variable phenotypes, and has often been overlooked when identifying individuals for clinical trial participation in the past.16
Success in FXS clinical trials is complicated further by the role FMRP plays in brain function. FMRP is an mRNA binding protein and has been shown to bind to and regulate the expression of hundreds of mRNAs in the central nervous system.17,18 The lack of FMRP in FXS leads to long, spindly, immature dendritic spines and negatively affects synaptic transmission, synaptic plasticity, learning and memory, and other behavior.19–27 Although the function of FMRP is complicated, much of FMRP’s synaptic effect is thought to be related to its role as a translational repressor of the group 1 metabotropic glutamate receptor (mGluR1/5) pathway.28 Loss of FMRP within this signaling pathway leads to excessive protein production and increased internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR).29 In addition to increased glutamatergic signaling, inhibitory signaling is reduced in FXS.30–33 The excitatory/inhibitory imbalance has been the major focus of targeted treatment trials in both model systems and humans with FXS.
The Fmr1 knockout (KO) mice mirror many of the morphologic, behavioral, electrophysiologic, and physical phenotypes that are present in humans with FXS, but they can be similarly variable in penetrance and severity across background strains and study design. It is important to note that the Fmr1 KO mice that are most often used to model human full-mutation FXS do not have an expanded CGG repeat domain. Instead, the Fmr1 gene has been artificially manipulated at the genetic level, such that no FMRP is produced and little or no Fmr1 mRNA is expressed.34,35 Fmr1 KO mice exhibit similar changes in dendritic morphology, with immature, long, spindly dendritic spines being typical.21 When placed in an open field, Fmr1 KO mice tend to make more photocell interruptions, indicative of hyperactivity, which can be observed in a subset of individuals with FXS.34,36 Although subject to high levels of variability, mild cognitive deficits have been reported, including deficits in spatial memory, conditioned fear, and novel object recognition.37–39 Fmr1 KO mice and humans with FXS are more prone to sensory hypersensitivity,25 and in mice, this can result in increased susceptibility to audiogenic seizures and increased acoustic startle response (reviewed by Rotschafer and Razak40). Fmr1 KO mice also show increased burying in the marble burying test, indicating that they are prone to repetitive behaviors, which are common in FXS patients.41 Physically, the mice exhibit macroorchidism that becomes more severe over time, which is also observed in people with FXS.37 The similarities between FXS humans and Fmr1 KO mice have led to the discovery of putative drug targets and the preclinical study of pharmacologic agents to effectively modulate behavioral, morphologic, electrophysiologic, and molecular endpoints. Recent clinical trials have been initiated after positive results in rodent studies, although translation of positive effects has been a challenge for a number of reasons and will be discussed later.
Restoration of excitatory/inhibitory balance
Metabotropic glutamate receptor 5 antagonists
In the past 10 years, FXS research has seen a tremendous growth in the study of new targeted pharmacologic treatments. A significant effort has been made to identify agents that restore excitatory/inhibitory balance in the FXS brain. Within this focus, modification of mGluR5 signaling has received both preclinical and clinical attention. The mGluR theory of fragile X postulates that the loss of FMRP leads to an excessive activation of group I mGluRs, specifically mGLuR5, and that this increase in mGluR activation is primarily responsible for the FXS phenotype.28 This theory was proposed on the basis of four key observations: FMRP acts as a translational repressor at the synapse,42 protein synthesis at the synapse is potentially triggered by the activation of group I mGluRs,43 the absence of FMRP increases the expression of several downstream consequences of mGluR activation,24,44 and many of the results of mGluR activation depend on the synaptic translation of mRNA.45–48 It has also been shown that the introduction of the mGluR group I agonist (S)-3,5-dihydroxyphenylglycine to hippocampal slices causes an increase in long-term depression (LTD) in CA1 cells of the hippocampus, likely because of the increased internalization of AMPA downstream of excessive mGluR5-dependent protein synthesis.49 LTD is important in the maintenance of neural plasticity. Excessive mGluR activation leading to an increase in LTD or synaptic weakening could be one of the main causes behind the loss of neural plasticity in FXS. In confirmation of the mGluR theory, it has been shown in animal models that mGluR5 antagonists have the ability to rescue many of the prominent phenotypes seen in the Fmr1 KO mice, including restoration of AMPAR expression, improved structure of dendritic spines, and correction of several behavioral deficits.50,51 In addition, the genetic reduction of mGluR5 levels in Fmr1 KO mice (thus lowering the mGluR5-dependent synaptic protein synthesis) has been shown to normalize protein levels and rescue the dendritic spine phenotype, as well as behavioral phenotypes such as inhibitory avoidance extinction, although a more recent and thorough study showed very little behavioral improvement with this method.52,53
The first report describing the use of selective mGluR5 antagonists to treat FXS in humans was an open-label, single-dose (50–150 mg) trial for the drug fenobam.54 Fenobam is a highly potent mGluR5 antagonist, similar in function to 2-methyl-6-(phenylethynyl)pyridine (MPEP), the mGluR5 antagonist used in many FXS mouse trials. Fenobam had previously been the subject of a Phase II trial in the 1980s, examining its potential as an anxiolytic. These trials were inconclusive but turned up no major health concerns aside from a small number of subjects reporting odd central nervous system-related perception issues such as vertigo or paraesthesias after continual use. To avoid any potential central nervous system-related adverse effects, the study was a single-dose trial focusing on sensory gating, attention, and inhibition.
Twelve subjects took part in the fenobam trial, six males and six females, all of whom had FXS. The prepulse inhibition (PPI) test was used as the primary measure of sensorimotor gating, as well as measuring inhibitory control. To measure attention, impulsivity, and inhibition, the California Fragile X Project Continuous Performance Test (FXCPT) was used, with the commission score as the focus of analysis. During the trial, there were no adverse effects reported, with all participants finishing the trial. The PPI test showed improvement in six of the twelve patients (four of six males and two of six females). Improvement was defined as an increase of at least 20% compared with baseline. The proportion of subjects demonstrating improvement was significant (P=0.01) when compared with untreated individuals from a previous study with a similar cohort. There was no significant change in the FXCPT, possibly as a result of ceiling effects, with almost all subjects having high baseline scores. However, the most impaired subject on the FXCPT did show significant improvement.54 Cognition was improved in a mouse study assessing fenobam in the Fmr1 KO and demonstrated improvement in associative learning and avoidance behavior in mutant mice while being tested in an Erasmus Ladder paradigm.55
The only published placebo-controlled trial of an mGluR5 antagonist in FXS was a double-blind crossover controlled study of the drug mavoglurant (AFQ056).56 Mavoglurant was initially tested for treatment of smoking cessation and anxiety but did not yield a positive outcome.57 In addition, it has been shown to alleviate complications caused by chronic levodopa use.58 The FXS trial involved 30 subjects, all of whom completed their active drug phase, with one participant who dropped out of the study while receiving placebo because of a drug-unrelated adverse event. Of the 30 FXS adult males enrolled and treated for 28 days each with placebo and mavoglurant, 24 experienced some form of adverse event, most of which were mild or moderate in severity. The primary measure used to assess efficacy was the Aberrant Behavior Checklist–Communication Edition (ABC-C), with the Clinical Global Impression (CGI) scale, Vineland Adaptive Behavior Scale (VABS), Repetitive Behavior Scale–Revised, Visual Analogue Scale (VAS), and Social Responsiveness Scale (SRS) used as secondary measures. No significant improvement was found using the primary measure, but there was a significant improvement in the Repetitive Behavior Scale–Revised (P=0.046). The investigator was able to identify a seven-subject subset with full methylation of the FMR1 promoter that did show significant improvement on the ABC total score during mavoglurant use. It was proposed that mGluR5 antagonists may improve behavior solely within that subset of individuals who have full methylation of the FMR1 promoter.56 Mavoglurant was recently the subject of a multinational Phase III study in individuals with FXS. Patients were stratified (only fully methylated individuals were allowed in the trial) in an attempt to reduce variability in the population and increase response to the drug; however, after negative results in the Phase III study, Novartis announced the discontinuation of the mavoglurant FXS development program (fraxa.org). The novel mGluR5 negative modulator, RO4917523, is still in Phase III study (clinicaltrials.gov, NCT01750957); the likelihood of using mGluR5 modulators as a successful single pharmacologic treatment in FXS is dwindling, but combined pharmacotherapy with additional targeted treatments may be an option.
N-methyl-D-aspartic acid modulators
Memantine, a drug commonly used to treat Alzheimer’s disease, is another potential FXS therapy aimed at modifying the function of FMRP targets and rectifying the gamma-aminobutyric acid (GABA)/glutamate imbalance by modifying ionotropic glutamate receptor activity. Memantine is an uncompetitive antagonist of N-methyl-D-aspartic acid receptors,59 but it has also been shown to act as an antagonist at nicotinic acetylcholine receptors and can affect dopamine (DA) and 5-HT signaling.60–62 FMRP has been shown to associate with glutamate receptor subunits NR1 and NR2B in the neocortex and/or hippocampus of Fmr1 KO mice.63 The degree and direction of N-methyl-D-aspartic acid dysfunction in Fmr1 KO mice is unclear and appears to be dependent on age and brain region;24,64–66 however, in vitro study of memantine in Fmr1 KO-cultured cerebellar granule cells demonstrated stimulatory effects of dendritic spine maturation and excitatory synapse formation in treated versus nontreated cells.67 In human open-label study of memantine in children with autism or pervasive developmental disorder (PDD), 70% of subjects treated with memantine showed significant improvement.68 To investigate memantine’s potential as a treatment for FXS, an open-label, six-subject trial was performed.69 The FXS memantine study evaluated subjects using the CGI-Global Improvement (CGI-I) as a primary measure with a secondary battery consisting of the CGI-Severity (CGI-S), ABC, SRS, and ADHD Rating Scale-IV scales. Four of the six subjects were considered treatment responders according to their CGI-I score, whereas the remaining two patients began to suffer from increased irritability during the trial, leading to drug discontinuation. There were no statistically significant changes found in any of the secondary measures. Improvement in the primary endpoint suggests that memantine may be beneficial to people with FXS; however, the small sample size makes interpretation of efficacy difficult, and the increase in significant irritability observed in two of the six patients suggests caution should be used before expanding its use in this population. Furthermore, the nonselective nature of memantine, and specifically its effects on 5-HT2A and D2 receptors, may contribute to the increased irritability reported in clinical study in both PDD and FXS.62,69,70
AMPA modulation
Along with glutamate receptor antagonists, ampakine compounds are potential treatments acting within the glutamate signaling pathway. Acting downstream of the mGluRs,28 AMPARs are excessively internalized as a result of increased mGlur5 signaling after loss of FMRP.29 This hypersensitive internalization and altered trafficking of AMPARs is thought to underlie FXS-associated deficits in long-term potentiation (LTP) and LTD. In addition, reduced levels of GluR1 and GluR2 AMPAR subunits have been reported in the cortex of Fmr1 KO mice.71 The goal of positive modulation of AMPAR is potentiation of AMPA signaling and subsequent normalization of LTD, LTP, and improved cognition.
To date, there has been one clinical trial assessing the efficacy of an ampakine compound in FXS. This trial was a randomized, double-blind, placebo-controlled trial of the drug CX516, an AMPAR-positive allosteric modulator.72 Of the 49 subjects who began the trial, two did not finish: one because of loss of interest and the other because of severe rash. The study found no significant change in memory, the primary outcome being evaluated, or in any secondary measure for language, attention, behavior, or overall function when compared with placebo. Because of the low potency of CX516, in addition to the short treatment period (4 weeks), it is unclear whether AMPA-mediated neurotransmission is a viable target for FXS treatment.
GSK3 modulation
Although lithium has been used to treat mood instability and aggression in FXS for some time, anecdotal evidence has been the only indication of its efficacy. The first and only trial for lithium in FXS was a 15 subject, open-label, add-on trial.73 There were a large number of adverse events during the 2-month trial (79 recorded). Events were typically moderate to mild, and none warranted removal from the trial. The primary outcome measure was the ABC-C Irritability subscale, with secondary measures comprised of the other ABC-C subscales, VAS, VABS, CGI-I, FXCPT, and several behavior-specific tasks. The cohort showed significant improvement based on the total ABC-C score after lithium treatment, with the Hyperactivity and Inappropriate Speech subscores showing significance on their own. Significance was not reached by the Irritability subscale, which was the primary measure. The CGI and VAS also showed significant improvements. The physiologic rationale for studying lithium in FXS involves manipulation of glycogen synthase kinase 3 (GSK3). It has been reported that adult FXS mice have normal levels of the two GSK3 isoforms, but regulation of these proteins by inhibitory phosphorylation is inadequate.74 In several mouse studies and in Drosophila, lithium, and other more selective GSK3 inhibitors were reported to increase phosphorylation of GSK3, improve behavior, and correct neuronal signaling and dendritic spine morphology.74–82 Recently, more specific inhibitors of GSK3 were also reported to have beneficial effects in the Fmr1 KO mouse, including improvements in long-term potentiation and cognitive tasks involving the hippocampus.83,84 These studies suggest that pharmacologic manipulation/inhibition of GSK3 may be beneficial in FXS (reviewed by Portis et al85), although the use of lithium to target this pathway may not be ideal. The high number of adverse events and worry about its use in younger children may deter its widespread use, but nonetheless the available data suggest the development of safe and specific modulators of the GSK3 pathway may be valuable for future study in humans with FXS.
GABA modulators
Loss of FMRP is not only associated with elevated levels of excitatory neurotransmission but also results in deficits in inhibitory neurotransmission involving the major inhibitory neurotransmitter GABA.28,86 In the knockout mouse model, impaired GABAergic neurotransmission has been demonstrated in many areas of the brain, including the striatum, amygdala, somatosensory cortex, and hippocampus.86–90 Deficits have been demonstrated in GABA(A) receptors, and GABA(B) modulators show some beneficial effects, although direct evidence of GABA(B) dysfunction has not been demonstrated at this time. In the Drosophila and mouse models of FXS, GABAergic compounds have been shown to reverse many of the related behavioral and morphologic phenotypes associated with FXS in these systems,91,92 suggesting GABA(A) receptors are sensitive to GABAergic drugs and that stimulation of GABA receptors could be a viable treatment for FXS. Treatment with GABA agonists could work directly by rectifying the inhibitory/excitatory imbalance through increased GABA signaling, or indirectly with the GABA agonists working as negative modulators of glutamate signaling.93
Preclinical data implicating GABA(A) dysregulation in FXS includes evidence that FMRP transcriptionally regulates GABA(A) receptor subunit mRNA expression with reductions in GABA(A) receptor mRNA noted in Fmr1 KO mice lacking FMRP.94 In addition, GABA(A) receptor expression has been shown to be significantly down-regulated in a number of brain regions in Fmr1 KO mice that are important for behavior, including the hippocampus and amygdala.87,89,90,95,96 In animal models of FXS, GABA(A) agonism has shown significant promise as a pharmacotherapy target. Regarding preclinical treatment, alphaxalone, a neuroactive steroid with multiple potential pharmacodynamics effect, including modulation of nicotinic acetylcholine receptors, activation of chloride channels, and nonselective GABA(A) agonism, was associated with reductions in anxiety and rescue of audiogenic seizures in Fmr1 KO mice.91 Also in Fmr1 KO mice, the GABA(A) extrasynaptic δ-subunit agonist gaboxadol attenuated neuron excitability deficits in the basolateral amygdala86 and reduced hyperactivity and mildly attenuated PPI, but did not correct deficits in cued fear or startle response.39 Taurine, another GABA(A) receptor agonist, improved memory acquisition and retention in a passive avoidance task, although baseline deficits were not observed in the Fmr1 KO mice compared with wild-type (WT).97 Direct application of taurine to hippocampal slices did not alter pair-pulse depression deficits, potentially indicating a reduction in binding sites on GABA(A) receptors.98
The first clinical study in FXS of a drug using GABA agonism was an open-label trial of the drug riluzole. Riluzole is approved by the US Food and Drug Administration (FDA) for treating amyotrophic lateral sclerosis. Although the precise mechanism of the drug is unknown, it is thought that its action is related to the attenuation of glutamate-induced excitotoxicity.99 Preclinical studies have also implied that riluzole works to potentiate postsynaptic GABA(A) receptors,100 which have been shown to be significantly down-regulated in FXS, in addition to blocking GABA uptake.101 The preliminary clinical study of riluzole involved six patients, all of whom completed the study.102 The primary measure used was the CGI-I, with secondary measures including all ABC subscales, the SRS, and the CGI-S. Throughout the trial, there were no severe or serious adverse events, with the most common adverse event being mild tiredness. Of the six participants, only one met the criteria for positive treatment response. There were no significant improvements found in the primary or secondary measures.
Acamprosate, a drug approved by the FDA for the maintenance of abstinence from alcohol, is another potential treatment targeting glutamate/GABA imbalance. Despite having been shown to not directly interact with several glutamate or GABA receptor subtypes at a significant level in a Xenopus oocyte model,103 acamprosate is believed to have pleiotropic effects on both GABA and glutamate neurotransmission.104 As with riluzole, the exact mechanism of acamprosate is unknown, but in animal models, it has been shown to exhibit GABA(A) agonism105 and has been implicated to have an effect as an mGluR5 antagonist.106 The first trial of acamprosate in FXS was a three-subject, open-label trial.107 All three subjects showed significant improvement, as measured by the CGI-I, especially in social impairment and communication deficits.108 After the initial trial, acamprosate was the subject of an open-label study in youth with FXS. Of the 13 subjects enrolled, one did not complete the trial after failing to meet eligibility criteria. Throughout the trial, there were no severe or serious adverse events. The primary measure was the CGI-I with secondary measures, including all subscales of the ABC, SRS, CGI-S, and VABS. Of the twelve subjects, nine met the criteria for positive treatment response based on the CGI-I scores, with significant improvement in the Social Withdrawal, Hyperactivity, and Social Avoidance subscales of the ABC, the CGI-S, the SRS, and the Expressive Communication Subdomain of the VABS. Acamprosate is currently the subject of a Phase II, double-blind, placebo-controlled trial in FXS (clinicaltrials.gov, NCT01911455).
Ganaxolone is a synthetic neurosteroid modulator of GABA(A) receptors that selectively binds to the neurosteroid binding site of GABA(A) receptors, as opposed to the benzodiazepine binding site.109 In preclinical study, acute in vivo dosing prevented audiogenic seizures in the Fmr1 KO mice.91 Ganaxolone is the focus of an ongoing proof-of-concept clinical phase 2, double-blind, placebo-controlled trial in FXS (clinicaltrials.gov, NCT01725152). The main focus of symptom improvement is in anxiety and attention domains.
Arbaclofen (STX209) is another potential targeted treatment aimed at rectifying the GABA/glutamate imbalance in FXS. Unlike riluzole and acamprosate, arbaclofen acts as a GABA(B) agonist, as opposed to a GABA(A) agonist. Activation of presynaptic GABA(B) receptors has been shown to reduce the amount of glutamate released into the synapse, as well as increase levels of GABA release in mouse WT hippocampal neurons.93 In the Fmr1 KO mouse model, acute arbaclofen treatment has been shown to reduce susceptibility to audiogenic seizures.110 In addition, arbaclofen normalized dendritic spine density in the visual cortex and attenuated basal protein synthesis in hippocampal synaptoneurosomes, but no behavioral endophenotypes with baseline deficits between WT and Fmr1 KO mice were assessed in this study.111 In a clinical study of arbaclofen, 63 subjects participated in the double-blind, placebo-controlled trial.112 Of these 63 individuals, three subjects dropped out of the study because of increased irritability. Hospitalization for behavioral management was required for one subject who had two similar hospitalizations in the past. Within the whole 63 subject cohort, significant improvement was only found in the VAS-problem behaviors and the ABC-Social Avoidance. When looking at a more impaired subset (ABC-LSW≤8; n=27), significant improvement was seen in the ABC-Social Avoidance, CGI-I, CGI-S, and Vineland Socialization raw score. Arbaclofen was further studied in a Phase III study in youth and adults with FXS. After negative Phase III study results, the arbaclofen development program in FXS was ended by Seaside Therapeutics.
Targets of FMRP
One potential method of FXS treatment is the inhibition of proteins whose synthesis would typically be regulated by FMRP. Matrix metalloproteinase 9 (MMP9) is one such downstream target. MMPs have been shown to be instrumental in modulating synaptic physiology and plasticity in hippocampal neurons.113 In the Fmr1 KO mouse model, levels of MMP9 are shown to be elevated in the hippocampus, possibly contributing to the phenotypes expressed in FXS.114 Early treatment with minocycline, a MMP9 inhibitor, has been shown to promote the maturation of the typically immature dendritic spines seen in the FXS mice and reduced locomotor behavior that influenced marble burying and elevated plus maze behavior.114 These observations may not necessarily have had a direct effect on anxiety or repetitive behavior, as these tests are sensitive to basal changes in locomotor activity.
Two clinical trials investigating minocycline in FXS have been executed. The first, an open-label add-on trial, was conducted in 20 subjects aged 13–35 years, with one dropping out as a result of adverse effects. This preliminary study did show significant improvement in the ABC-Irritability subscale, with VAS and CGI warranting further study.115 The second trial was a double-blind, placebo-controlled trial in children and adolescents.116 The study involved 66 subjects aged 3.5–16 years, with 55 completing the first round of treatment and 48 completing the entire 3-month trial. There were 144 adverse events during the trial, the majority of which were mild and with no significant difference between placebo and treatment groups. Five subjects developed brown and yellow tooth discolorations, a known adverse effect of minocycline, most of which resolved with time. This is abnormal, as typically the discoloration is permanent. The study used the CGI-I and VAS1 (severity of target behavior 1) as the primary measures. The secondary battery included ABC-C, VABS-II, VAS2 (target behavior 2), and VAS3 (target behavior 3). Minocycline treatment showed significant improvement in CGI-I only, and although significant, this improvement was modest. Secondary measures were not significantly altered, although ad hoc analysis of the VAS for anxiety- and mood-related behavior showed improvement. Several potential biomarkers for brain function and treatment response were assessed throughout this trial. MMP9 was decreased as intended,117 and event-related potential amplitude in a passive auditory oddball paradigm was significantly altered in individuals receiving minocycline treatment, although a much larger sample size is needed to validate these results.118
Symptom-based treatments
ADHD treatments
ADHD is highly prevalent among boys with FXS. In a 37 subject cohort, 73% had ADD ratings of 15 or higher on the Conner’s abbreviated scale.119 Even patients who do not demonstrate clear hyperactivity will often suffer from inattentiveness, especially in the classroom. Because of the prevalence of ADHD symptoms in people with FXS, a number of studies have aimed to alleviate hyperactivity and inattentiveness.
The earliest published clinical trial targeting ADHD symptoms was a double-blind, placebo-controlled trial of the stimulants methylphenidate and dextroamphetamine. In the double-blind crossover study of stimulants (randomized 1 week of treatment with placebo, methylphenidate, and dextroamphetamine each), individuals with FXS were assessed using the Conner’s Abbreviated Parent–Teacher Questionnaire and the ADD-H: Comprehensive Teacher Rating Scale.120 Observation for specific ADHD behaviors, locomotion quantification using an actometer, and impulsivity in the Delay and attentiveness in the Vigilance tasks were also performed. Significant improvement was only seen on the ADD-H: Comprehensive Teacher Rating Scale, with methylphenidate showing improvement in the social skills factor and attention. Significant changes were not found on any measure for dextroamphetamine. Ten of the 15 patients were judged to be clinical responders, according to parent and teacher reports at the end of the study.
A recent study showed that L-carnitine has the potential to reduce hyperactive and impulsive behaviors in boys (non-FXS) with ADHD without the adverse effects often associated with stimulant medication.121 A double-blind, placebo-controlled study was done to examine the effects of L-acetylcarnitine (LAC) on the ADHD symptoms of boys with FXS.122 The trial initially had 63 subjects, with 56 completing 1 year of treatment and 51 being included in statistical analysis. The study was justified by previous findings that LAC acts to inhibit the in vitro cytogenetic expression of the fragile site associated with FXS.123 The measures used were the CGI-Teachers, CGI-Parents, VABS, and Wechsler Intelligence Scale for Children–Revised. Of the patients who did not complete treatment, none were a result of adverse effects of LAC. Significant improvement was found in the CGI-Parents, CGI-Teachers, VABS Socialization domain, and VABS ABC domain.
The most recent clinical trial to treat ADHD symptoms in FXS was an open-label trial of valproic acid (VPA).124 VPA is a known antiepileptic drug that has been used in number of seizure disorders. VPA has been prescribed off-label for FXS and could potentially act as a demethylating agent by inhibiting histone deacetylase.125,126 The primary measure in the VPA FXS study was the Conner’s Parent Rating Scale–Revised Short Form with the Conner’s Teacher Rating Scale–Revised, VABS–survey form, and CGI-S as secondary measures after 6 months of treatment. Of the ten patients enrolled, eight finished the trial, with two (twins) withdrawing because of worsening behavior. There were no severe adverse events during the trial. The only significant improvement was for hyperactivity symptoms according to the CGI-S, with a variety of other parameters showing improvement, but not reaching significance. Overall, clinical treatment of ADHD symptoms in the context of FXS continues to focus on stimulant usage. Published data supporting use of other agents approved by the FDA, including atomoxetine, clonidine, and guanfacine, specifically in persons with FXS, remains limited. Use of novel agents such as LAC or VPA remains limited in the clinical setting for symptoms and behaviors of ADHD in this population.
Sleep treatments
Sleep problems are common in children suffering with FXS. In a parent/caregiver report, 32% of youth with FXS were noted to have some form of sleep issue, with problems falling asleep and night awakenings being the most common issues.127 Melatonin was systematically studied in FXS as the subject of a 4-week, placebo-controlled, double-blind, cross-over design trial in children with developmental delay (12 total, six with FXS).128 The study measured sleep onset time, total sleep duration, sleep onset latency, and the number of night awakenings (2 weeks receiving active drug; 2 weeks receiving placebo). Melatonin treatment leads to significantly longer sleep duration, shorter sleep latency, and earlier onset of sleep. There was also a reduction in number of night awakenings, but it did not reach significance. Treatment with melatonin was studied in the Fmr1 KO mouse model of FXS, with a focus on its effects at preventing oxidative stress. This study suggests that chronic melatonin treatment can improve glutathione levels, lipid peroxidation, and several types of behavior in treated KO mice, although these types of endpoints have not been studied clinically.129
Anxiety treatments
Heightened anxiety is one of the more socially debilitating facets of the FXS phenotype. Subjects, and males in particular, often exhibit severe eye gaze avoidance and hyperarousal.130 Oxytocin (OT) is a hormone produced in the hypothalamus that could diminish stress-induced behavior. OT receptors are found in a variety of socially relevant and stress-sensitive areas of the brain. It has recently been shown that OT treatment can be effective in improving social decision making, repetitive behaviors, and visual scanning of faces in patients with high-functioning autism and Asperger’s syndrome.131,132 Intranasal dosing has been shown to attenuate stress in monkeys and rodents, as well as alter emotional regulation in humans.133–135 On this basis, a double-blind, placebo-controlled, single-dose trial to study the effects of intranasal OT in males with FXS was completed. The eight study participants who finished the study experienced no adverse events over the course of the trial.136 The study measured eye gaze frequency, heart rate, respiratory sinus arrhythmia, heart rate variability, and concentration of salivary cortisol during a social proximity and social interaction test. There was significant improvement in eye gaze frequency and a reduction in salivary cortisol concentrations. This study suggests the need for future controlled trials of OT, possibly in combination with behavioral treatment programs.
Although prospective treatment trials assessing the efficacy of selective serotonin reuptake inhibitors (SSRIs) has not been completed in FXS, prospective studies in children with ASD and a recent retrospective chart review and survey in FXS suggest these agents may be beneficial to individuals with FXS. The SSRI sertraline is approved by the FDA as a treatment for obsessive compulsive disorder in children (aged 6–17 years). A case series of nine children with ASD demonstrated that low-dose sertraline improved anxiety, irritability, and transition-induced behavioral deterioration in 89% of the children.137 A retrospective chart review of 45 children with FXS, 11 of whom were treated with sertraline and 34 who were not treated with sertraline, suggests sertraline treatment may improve both expressive and receptive language development.138 Importantly, a survey found that the SSRI fluoxetine reportedly caused the most behavioral activation in FXS compared with other drugs in its class; however, it may be beneficial for people with autism, social anxiety, or selective mutism.6,139 Despite limited published data, SSRI use targeting anxiety in persons with FXS is widespread. Clinical consensus on FXS appears to hold that SSRIs may be better tolerated in persons with FXS compared with youth with idiopathic ASD, where SSRI controlled trials have noted significant adverse effect rates and a lack of efficacy targeting repetitive behavior.
Irritability treatments
Atypical or newer-generation antipsychotics are often used by clinicians to treat the irritability associated with FXS.140 Despite their high rate of clinical use, there has been little systematic study of the efficacy of these antipsychotics within the FXS population. Only one newer-generation antipsychotic has been the subject of clinical trial in FXS. Aripiprazole is a partial D2 and 5-HT1A agonist and a 5-HT2A antagonist.141,142 It has been approved by the FDA in a number of disorders, including autism, mainly as a treatment for irritability. The clinical trial in FXS was a 12-week open-label trial comprising twelve subjects.143 The CGI-I and ABC-I were used to determine treatment responders with the SRS, additional ABC subscales, CGI-S, and Compulsion Subscale of the Children’s Yale–Brown Obsessive Compulsive Scale Modified for PDDs used as secondary measures. Two patients dropped out of the study because of adverse events. Ten of the twelve subjects were considered treatment responders, with significant improvement found on the CGI-S, Children’s Yale–Brown Obsessive Compulsive Scale Modified for PDDs, and SRS.
Novel treatments in early development
The cholinergic system is a potential target for treatment in FXS. FMR1 is highly expressed in cholinergic neurons during early development,144 and the cholinergic pathways in the forebrain and hippocampus are known to be involved in several cognitive functions impaired in FXS.145,146 Diminished choline levels were reported in a small 1H magnetic resonance spectroscopy study involving nine individuals with FXS.147 The KO mouse model also shows aberrant cholinergic function in the subiculum.33 These studies suggest that inhibiting the breakdown of acetylcholine may benefit individuals with FXS. Donepezil is an FDA-approved drug to treat dementia in Alzheimer’s disease and was studied in an open-label trial in FXS because it is an acetylcholinesterase inhibitor. The 6-week trial involved eight individuals with FXS, all of whom completed the trial. No severe adverse effects were reported. The assessment battery included the Contingency Naming Test to look at working memory and mental flexibility, the Hopkins Verbal Learning Test to look at auditory learning, the ABC, and the Achenbach Child and Adult Behavior Checklists. Donepezil treatment was shown to produce significant improvements in the Contingency Naming Test, ABC-Total Score, ABC-Hyperactivity, ABC-Irritability, and Achenbach Child and Adult Behavior Checklists.147 An additional placebo-controlled study was completed in fragile X to evaluate the effectiveness and safety of donepezil in 20 subjects over the course of 12 weeks.148 Donepezil was well tolerated with only mild adverse effects, all of which resolved spontaneously. This trial did not find a significant effect of treatment on any of its outcome measures. These measures included change from baseline on the Stanford–Binet Intelligence Scale, Conners 3 Parent Rating Scale (short), and Childhood Autism Rating Scale. A placebo-controlled trial of donepezil in FXS is ongoing (clinicaltrials.gov, NCT01120626).
Several new projects are being initiated to determine their efficacy as potential treatments for FXS. Metadoxine extended release (Alcobra) has been studied as a treatment for ADHD and is also being studied as a potential treatment in FXS in a Phase IIb clinical study (clinicaltrials.gov, NCT02126995). In addition, NNZ-2566 (Neuren), an insulin-like growth factor 1 agonist with anti-inflammatory and neuroprotective effects,149,150 is also being studied in FXS (clinicaltrials.gov, NCT01894958). Peer-reviewed manuscripts reporting the efficacy of these agents in Fmr1 KO mice have not been published, although respective company websites report significant improvement in multiple facets of FXS-associated deficits in FXS mouse models.
Discussion
The FXS field is moving forward after the failure of several large targeted treatment trials; however, significant efforts are being made to complete more rigorous preclinical testing and development of clinical tools and biomarkers that aid in capturing clinical improvement. Before a recent surge in targeted treatments for FXS, symptom management received the most attention, focusing on the use of FDA-approved treatments for ADHD symptoms, anxiety, irritability, and sleep issues. Largely because of preclinical work done in the Fmr1 KO mouse model, more recent treatment discovery has concentrated on targeting underlying molecular mechanisms thought to be contributing to the wide range of endophenotypes associated with FXS. These treatments aim to restore not only the behavioral phenotypes but also those at a cellular level, including restoration of synaptic plasticity and aberrant cellular signaling, although it is unclear whether these cellular changes will result in improved function in clinical study. Although largely unsuccessful clinically, targeted treatments receiving the most attention have been focused on rescuing the excitatory/inhibitory signaling imbalance via mGlur5 antagonism, which is believed to underlie much of the disorder. Additional pharmacologic agents under current clinical investigation include several that aim to restore FRMP targets to normal levels.
In mouse model study, it is important to recognize the limitations of the data and of the model system. In Fmr1 KO mouse studies, critical interpretation, replication, or further building on initial results needs to receive more attention. In addition, negative molecular, behavioral, and electrophysiologic data are not always reported, and this can skew the perception of a treatment’s efficacy. Specifically pertaining to behavioral analyses, it is critical that authors be aware of potential confounding results. Many tests in rodent study can be influenced by the basal locomotor activity of the mice.151 For example, if a treatment reduces locomotor behavior in the Fmr1 KO mice and it reduces marble burying and time spent in the open of the elevated zero or plus maze, disentangling the effects on anxiety and repetitive behavior becomes a challenge. It is also important to note whether there was a baseline deficit between the vehicle-treated WT and Fmr1 mutant mice when interpreting the effects of pharmacologic treatment. Without rigorous assessment, interpretation, and disclosure of all tests in preclinical models, it is difficult to get a clear picture of the robustness of a treatment, and hence it appears that everything “cures” the mouse model, when in fact it does not. This is not to say that an improvement in a single behavior domain (observed in several different tasks) in a FXS mouse study is not an important finding. As many behavior domains are affected in individuals with FXS, even a drug that only improves one of these domains would be incredibly beneficial.
Even with behavioral (limited in many cases) and molecular and electrophysiologic improvement/normalization (although direct correlation to functional improvement is unknown) in the mouse model and improvements in small clinical trials (not unlike improvements in the mouse model), expansion into larger populations has met with very little success in human study to date. It is important to keep in mind that the human equivalent of many of the outcome measures or symptom domains assessed in mouse model study are not typically assessed in human clinical trials (ie, audiogenic seizure susceptibility, object memory, PPI), or the human symptom domain is not robustly affected or easily replicated in the mouse (social preference, inhibitory control, and sustained attention in the five-choice serial reaction time test152), and therefore efficacy in that domain cannot be adequately assessed in a preclinical mouse study. For example, one of the most commonly used FXS preclinical mouse behavior tests is the audiogenic seizure paradigm. Several mGlur5 modulators and many other agents that target excitatory/inhibitory imbalance were successful at preventing seizures produced by a tone of approximately 120 dB in Fmr1 KO mice.50,51 This type of test may be an indication that the drug is effectively attenuating hyperexcitability in the mouse brain, but this is not something that is assessed in human clinical trials, and the direct effect of this attenuation is not a clear correlation to other symptom domains, such as anxiety or inattentiveness. In addition, mavoglurant was shown to improve deficits in inhibition of the startle response after a prepulse in the mouse model, but PPI was not assessed in the clinical trial (FXS individuals do not easily tolerate this procedure).153 Extrapolation of positive mouse results in one behavior test, or molecular/electrophysiologic improvement in a single brain region, should not be expected to have a global improvement in human clinical study, nor positive effects in other symptom domains. These findings can serve as a positive indication that a drug may be efficacious, and further study should be initiated. There are tests that can be performed on both mouse models and humans, such as PPI; however, these tests are difficult for people with FXS to tolerate, and when they are used in clinical study, the cohort assessed tend to be skewed to include only higher-functioning individuals.54 Great benefit could be gained by developing testing paradigms that are amenable to mouse models as well as individuals with FXS.
Genetic rescue in the Fmr1 KO mouse by creating double mutants that harbor a mutated/deficient allele such as mGlur5,53 muscarinic M4,41 p70 ribosomal S6 kinase,154 or amyloid β-protein precursor (APP)155 can be powerful experiments. For example, if the double mutant has a less severe phenotype than the Fmr1 KO alone, it is a good indication that the second gene is able to modulate the effects of the disease-causing mutation. One must keep in mind, however, that this type of experiment is very different than administering a receptor-modifying drug to an adolescent, adult, or even a child. The double-mutant mouse will have been “treated” with this second genetic manipulation from conception, every day, all day, rather than the typically short-duration period of treatment in a FXS clinical trial, in which the optimal dose is unknown and the highest tolerable dose is often used. Genetic rescue in Fmr1 KO mice with a single deficient mGlur5 allele was very limited and did not improve audiogenic seizures, anxiety- and perseverative-related behavior, sensorimotor gating, memory, or motor responses, suggesting that these receptors, at this level of inhibition, may not be dominant modifiers of Fmr1 KO endophenotypes.53 Both genetic studies and pharmacologic treatment of potential mechanisms using the compound that will move into clinical study will give greater insight into the likelihood of success in people with FXS.
Moving forward in FXS clinical study, efforts are being made to improve the ability to detect treatment-induced improvement. It is apparent that the clinical measures used to track improvement in FXS clinical study (parent reports and rating scales, clinical impression) are not adequate, and significant efforts are being made to rectify this issue.156 Efforts to validate the Fragile X Symptom Rating Scale, a scale specific to FXS, are underway in several ongoing trials, including NNZ-2566 (clinicaltrials.gov, NCT01894958). Recent work has focused on the use of the Pediatric Anxiety Rating Scale specifically revised for FXS.157 In addition, patient stratification will likely aid in pairing pharmacologic agents with patients most likely to respond positively, although the best methods used to accomplish this are currently not known. After post hoc analyses, several studies used symptom severity (social withdrawal; arbaclofen) or methylation status (mavoglurant) to stratify patients. In both instances, these attempts have not resulted in positive treatment response when repeated in large trials. Although somewhat controversial, the expression level of FMRP is positively correlated and methylation status of the FMR1 gene have been negatively correlated with cognitive ability, whereas little correlation between CGG repeat number and cognition is thought to exist.158,159 Importantly, it has been shown that repeat number can vary in different tissue types from the same individual carrying permutation alleles, suggesting that numbers obtained from blood mononuclear cells (the cell type used for FXS diagnostic purposes) may not always directly translate to the brain.160 Again, this disconnect can add to the variability of the syndrome and complicate efforts to use blood results for clinical trial inclusion/exclusion criteria.
The development and improvement of target-based therapies in FXS is expanding, as is our understanding of the molecular and cellular role FMRP plays in signaling processes, synaptic plasticity, and behavior. Clinical, translational, and basic research discoveries are creating waves of novel target and treatment development possibilities for individuals with FXS. A major goal in future FXS study, and in neuroscience in general, is determining whether “reversal” of lifelong aberrant synaptic functioning can or will translate into improved behavioral functioning. The FXS field is moving toward pharmacologic treatment trials at the youngest possible age in an effort to correct synaptic plasticity deficits early on in development. Drug tolerability/toxicity will be a critical factor in determining which treatment mechanisms, drugs, and targets are appropriate for early intervention. In addition, progress in clinical trial endpoint and biomarker development are becoming priorities in the field.156,161 Molecular biomarkers including, but not limited to, cyclic adenosine 3′, 5′-monophosphate,162 ERK,102,163 brain-derived neurotrophic factor,108 amyloid beta-protein precursor and cleavage proteins,164 MMP9,117 and event-related potential118 (the latter may only be applicable to higher-functioning individuals, as many with FXS cannot tolerate the procedure) are being explored as indicators of treatment response and signaling proficiency, and may be used to determine likelihood of treatment response on an individualized basis in the future.
Disclosure
Dr Erickson is a consultant to and holds equity in Confluence Pharmaceuticals; is the inventor on intellectual property describing methods to treat Fragile X Syndrome held by Indiana University School of Medicine and Cincinnati Children’s Hospital Medical Center; and has been a consultant to Alcobra Pharmaceuticals. Dr Schaefer is the inventor in intellectual property describing methods to treat Fragile X Syndrome held by Cincinnati Children’s Hospital Medical Center. The authors report no further conflicts of interest in this work.
References
Weiskop S, Richdale A, Matthews J. Behavioural treatment to reduce sleep problems in children with autism or fragile X syndrome. Dev Med Child Neurol. 2005;47(2):94–104. | |
Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64(1):196–197. | |
Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol Assess. 2003;7(16):1–106. | |
Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666–672. | |
Tsiouris JA, Brown WT. Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs. 2004;18(11):687–703. | |
Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378–390. | |
Kaufmann WE, Cortell R, Kau AS, et al. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A(3):225–234. | |
Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–1058. | |
Pieretti M, Zhang FP, Fu YH, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–822. | |
Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–340. | |
Colak D, Zaninovic N, Cohen MS, et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343(6174):1002–1005. | |
Grønskov K, Brøndum-Nielsen K, Dedic A, Hjalgrim H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur J Hum Genet. 2011;19(4):489–491. | |
Myrick LK, Nakamoto-Kinoshita M, Lindor NM, Kirmani S, Cheng X, Warren ST. Fragile X syndrome due to a missense mutation. Eur J Hum Genet. 2014;22(10):1185–1189. | |
Loesch DZ, Hay DA, Sutherland GR, et al. Phenotypic variation in male-transmitted fragile X: genetic inferences. Am J Med Genet. 1987;27(2):401–417. | |
Bennetto L, Pennington BF, Porter D, Taylor AK, Hagerman RJ. Profile of cognitive functioning in women with the fragile X mutation. Neuropsychology. 2001;15(2):290–299. | |
Stöger R, Genereux DP, Hagerman RJ, Hagerman PJ, Tassone F, Laird CD. Testing the FMR1 promoter for mosaicism in DNA methylation among CpG sites, strands, and cells in FMR1-expressing males with fragile X syndrome. PLoS ONE. 2011;6(8):e23648. | |
Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. | |
Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. | |
Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107(41):17768–17773. | |
Desai NS, Casimiro TM, Gruber SM, Vanderklish PW. Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol. 2006;96(4):1734–1745. | |
Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21(14):5139–5146. | |
Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci U S A. 2001;98(13):7101–7106. | |
Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25(32):7385–7392. | |
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–7750. | |
Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103(4):1043–1050. | |
Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav Pharmacol. 2004;15(5–6):433–442. | |
Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus. 2002;12(1):39–46. | |
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. | |
Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104(39):15537–15542. | |
Martin BS, Corbin JG, Huntsman MM. Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J Neurophysiol. 2014;112(4):890–902. | |
Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. 2007;412(3):227–232. | |
Gantois I, Vandesompele J, Speleman F, et al. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21(2):346–357. | |
D’Antuono M, Merlo D, Avoli M. Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience. 2003;119(1):9–13. | |
The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78(1):23–33. | |
Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21(3):549–555. | |
Backes M, Genç B, Schreck J, Doerfler W, Lehmkuhl G, von Gontard A. Cognitive and behavioral profile of fragile X boys: correlations to molecular data. Am J Med Genet. 2000;95(2):150–156. | |
Kooy RF, D’Hooge R, Reyniers E, et al. Transgenic mouse model for the fragile X syndrome. Am J Med Genet. 1996;64(2):241–245. | |
King MK, Jope RS. Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav. 2013;12(7):723–731. | |
Olmos-Serrano JL, Corbin JG, Burns MP. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci. 2011;33(5):395–403. | |
Rotschafer SE, Razak KA. Auditory processing in fragile x syndrome. Front Cell Neurosci. 2014;8:19. | |
Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav Brain Res. 2012;228(1):1–8. | |
Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–487. | |
Weiler IJ, Irwin SA, Klintsova AY, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94(10):5395–5400. | |
Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25(35):8048–8055. | |
Karachot L, Shirai Y, Vigot R, Yamamori T, Ito M. Induction of long-term depression in cerebellar Purkinje cells requires a rapidly turned over protein. J Neurophysiol. 2001;86(1):280–289. | |
Zho WM, You JL, Huang CC, Hsu KS. The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci. 2002;22(20):8838–8849. | |
Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–1257. | |
Raymond CR, Thompson VL, Tate WP, Abraham WC. Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci. 2000;20(3):969–976. | |
Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36(11–12):1517–1532. | |
Michalon A, Sidorov M, Ballard TM, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74(1):49–56. | |
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49(7):1053–1066. | |
Dölen G, Osterweil E, Rao BS, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56(6):955–962. | |
Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res. 2011;223(2):310–321. | |
Berry-Kravis E, Hessl D, Coffey S, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–271. | |
Vinueza Veloz MF, Buijsen RA, Willemsen R, et al. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012;11(3):325–331. | |
Jacquemont S, Curie A, des Portes V, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3(64):64ra1. | |
Spooren W, Gasparini F. mGlu5 receptor antagonists: a novel class of anxiolytics? Drug News Perspect. 2004;17(4):251–257. | |
Berg D, Godau J, Trenkwalder C, et al. AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov Disord. 2011;26(7):1243–1250. | |
Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ; Memantine Study Group. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. | |
Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312(3):1195–1205. | |
Buisson B, Bertrand D. Open-channel blockers at the human alpha4beta2 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53(3):555–563. | |
Nakaya K, Nakagawasai O, Arai Y, et al. Pharmacological characterizations of memantine-induced disruption of prepulse inhibition of the acoustic startle response in mice: involvement of dopamine D2 and 5-HT2A receptors. Behav Brain Res. 2011;218(1):165–173. | |
Schütt J, Falley K, Richter D, Kreienkamp HJ, Kindler S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem. 2009;284(38):25479–25487. | |
Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28(31):7847–7862. | |
Pilpel Y, Kolleker A, Berberich S, et al. Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J Physiol. 2009;587(Pt 4):787–804. | |
Eadie BD, Cushman J, Kannangara TS, Fanselow MS, Christie BR. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus. 2012;22(2):241–254. | |
Wei H, Dobkin C, Sheikh AM, Malik M, Brown WT, Li X. The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS ONE. 2012;7(5):e36981. | |
Chez MG, Burton Q, Dowling T, Chang M, Khanna P, Kramer C. Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J Child Neurol. 2007;22(5):574–579. | |
Erickson CA, Mullett JE, McDougle CJ. Open-label memantine in fragile X syndrome. J Autism Dev Disord. 2009;39(12):1629–1635. | |
Erickson CA, Chambers JE. Memantine for disruptive behavior in autistic disorder. J Clin Psychiatry. 2006;67(6):1000. | |
Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19(2):138–151. | |
Berry-Kravis E, Krause SE, Block SS, et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol. 2006;16(5):525–540. | |
Berry-Kravis E, Sumis A, Hervey C, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29(4):293–302. | |
Min WW, Yuskaitis CJ, Yan Q, et al. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. 2009;56(2):463–472. | |
Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol. 2010;79(4):632–646. | |
Liu ZH, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis. 2012;45(3):1145–1152. | |
Yu F, Wang Z, Tchantchou F, Chiu CT, Zhang Y, Chuang DM. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma. 2012;29(2):362–374. | |
Choi CH, Schoenfeld BP, Bell AJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380: 106–119. | |
Choi CH, McBride SM, Schoenfeld BP, et al. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology. 2010;11(3):347–362. | |
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE. 2010;5(3):e9706. | |
Chen X, Sun W, Pan Y, et al. Lithium ameliorates open-field and elevated plus maze behaviors, and brain phospho-glycogen synthase kinase 3-beta expression in fragile X syndrome model mice. Neurosciences (Riyadh). 2013;18(4):356–362. | |
McBride SM, Choi CH, Wang Y, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45(5):753–764. | |
Chen T, Lu JS, Song Q, et al. Pharmacological rescue of cortical synaptic and network potentiation in a mouse model for fragile X syndrome. Neuropsychopharmacology. 2014;39(8):1955–1967. | |
Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry. 2014;75(3):198–206. | |
Portis S, Giunta B, Obregon D, Tan J. The role of glycogen synthase kinase-3 signaling in neurodevelopment and fragile X syndrome. Int J Physiol Pathophysiol Pharmacol. 2014;4(3):140–148. | |
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30(29):9929–9938. | |
El Idrissi A, Yan X, L’Amoreaux W, Brown WT, Dobkin C. Neuroendocrine alterations in the fragile X mouse. Results Probl Cell Differ. 2012;54:201–221. | |
D’Hulst C, Heulens I, Brouwer JR, et al. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009;1253: 176–183. | |
D’Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30(8):425–431. | |
D’Hulst C, De Geest N, Reeve SP, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–245. | |
Heulens I, D’Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229(1):244–249. | |
Chang S, Bray SM, Li Z, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4(4):256–263. | |
Isaacson JS, Hille B. GABA(B)-mediated presynaptic inhibition of excitatory transmission and synaptic vesicle dynamics in cultured hippocampal neurons. Neuron. 1997;18(1):143–152. | |
Hong A, Zhang A, Ke Y, El Idrissi A, Shen CH. Downregulation of GABA(A) β subunits is transcriptionally controlled by Fmr1p. J Mol Neurosci. 2012;46(2):272–275. | |
D’Hulst C, Heulens I, Brouwer JR, et al. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009;1253: 176–183. | |
El Idrissi A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005;377(3):141–146. | |
El Idrissi A, Boukarrou L, Dokin C, Brown WT. Taurine improves congestive functions in a mouse model of fragile X syndrome. Adv Exp Med Biol. 2009;643:191–198. | |
El Idrissi A, Neuwirth LS, L’Amoreaux W. Taurine regulation of short term synaptic plasticity in fragile X mice. J Biomed Sci. 2010;17(Suppl 1):S15. | |
Obrenovitch TP. Amyotrophic lateral sclerosis, excitotoxicity and riluzole. Trends Pharmacol Sci. 1998;19(1):9–11. | |
Jahn K, Schlesinger F, Jin LJ, Dengler R, Bufler J, Krampfl K. Molecular mechanisms of interaction between the neuroprotective substance riluzole and GABA(A)-receptors. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(1):53–63. | |
Mantz J, Laudenbach V, Lecharny JB, Henzel D, Desmonts JM. Riluzole, a novel antiglutamate, blocks GABA uptake by striatal synaptosomes. Eur J Pharmacol. 1994;257(1–2):R7–R8. | |
Erickson CA, Weng N, Weiler IJ, et al. Open-label riluzole in fragile X syndrome. Brain Res. 2011;1380:264–270. | |
Reilly MT, Lobo IA, McCracken LM, et al. Effects of acamprosate on neuronal receptors and ion channels expressed in Xenopus oocytes. Alcohol Clin Exp Res. 2008;32(2):188–196. | |
Mann K, Kiefer F, Spanagel R, Littleton J. Acamprosate: recent findings and future research directions. Alcohol Clin Exp Res. 2008;32(7):1105–1110. | |
Boismare F, Daoust M, Moore N, et al. A homotaurine derivative reduces the voluntary intake of ethanol by rats: are cerebral GABA receptors involved? Pharmacol Biochem Behav. 1984;21(5):787–789. | |
Harris BR, Prendergast MA, Gibson DA, et al. Acamprosate inhibits the binding and neurotoxic effects of trans-ACPD, suggesting a novel site of action at metabotropic glutamate receptors. Alcohol Clin Exp Res. 2002;26(12):1779–1793. | |
Erickson CA, Mullett JE, McDougle CJ. Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord. 2010;40(11):1412–1416. | |
Erickson CA, Wink LK, Ray B, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl). 2013;228(1):75–84. | |
Nohria V, Giller E. Ganaxolone. Neurotherapeutics. 2007;4(1):102–105. | |
Pacey LKK, Tharmalingam S, Hampson DR. Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J Pharmacol Exp Ther. 2011;338(3):897–905. | |
Henderson C, Wijetunge L, Kinoshita MN, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012;4(152):152ra128. | |
Berry-Kravis EM, Hessl D, Rathmell B, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4(152):152ra127. | |
Bilousova TV, Rusakov DA, Ethell DW, Ethell IM. Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J Neurochem. 2006;97(1):44–56. | |
Bilousova TV, Dansie L, Ngo M, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46(2):94–102. | |
Paribello C, Tao L, Folino A, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10(1):91. | |
Leigh MJ, Nguyen DV, Mu Y, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr. 2013;34(3):147–155. | |
Dziembowska M, Pretto DI, Janusz A, et al. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am J Med Genet A. 2013;161A(8):1897–1903. | |
Schneider A, Leigh MJ, Adams P, et al. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J Psychopharmacol. 2013;27(10):956–963. | |
Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics. 1995;95(5):744–752. | |
Hagerman RJ, Murphy MA, Wittenberger MD. A controlled trial of stimulant medication in children with the fragile X syndrome. Am J Med Genet. 1988;30(1–2):377–392. | |
Van Oudheusden LJ, Scholte HR. Efficacy of carnitine in the treatment of children with attention-deficit hyperactivity disorder. Prostaglandins Leukot Essent Fatty Acids. 2002;67(1):33–38. | |
Torrioli MG, Vernacotola S, Peruzzi L, et al. A double-blind, parallel, multicenter comparison of L-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in fragile X syndrome boys. Am J Med Genet A. 2008;146A(7):803–812. | |
Pomponi MG, Neri G. Butyrate and acetyl-carnitine inhibit the cytogenetic expression of the fragile X in vitro. Am J Med Genet. 1994;51(4):447–450. | |
Torrioli M, Vernacotola S, Setini C, et al. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A. 2010;152A(6):1420–1427. | |
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276(39):36734–36741. | |
Tabolacci E, De Pascalis I, Accadia M, et al. Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet Genomics. 2008;18(8):738–741. | |
Kronk R, Bishop EE, Raspa M, Bickel JO, Mandel DA, Bailey DB Jr. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep. 2010;33(5):679–687. | |
Wirojanan J, Jacquemont S, Diaz R, et al. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med. 2009 15;5(2):145–150. | |
Romero-Zerbo Y, Decara J, el Bekay R, et al. Protective effects of melatonin against oxidative stress in Fmr1 knockout mice: a therapeutic research model for the fragile X syndrome. J Pineal Res. 2009;46(2):224–234. | |
Reiss AL, Hall SS. Fragile X syndrome: assessment and treatment implications. Child Adolesc Psychiatr Clin N Am. 2007;16(3):663–675. | |
Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A. Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci U S A. 2010;107(9):4389–4394. | |
Hollander E, Novotny S, Hanratty M, et al. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology. 2003;28(1):193–198. | |
Heinrichs M, Baumgartner T, Kirschbaum C, Ehlert U. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry. 2003;54(12):1389–1398. | |
Parker KJ, Buckmaster CL, Schatzberg AF, Lyons DM. Intranasal oxytocin administration attenuates the ACTH stress response in monkeys. Psychoneuroendocrinology. 2005;30(9):924–929. | |
Windle RJ, Kershaw YM, Shanks N, Wood SA, Lightman SL, Ingram CD. Oxytocin attenuates stress-induced c-fos mRNA expression in specific forebrain regions associated with modulation of hypothalamo-pituitary-adrenal activity. J Neurosci. 2004;24(12):2974–2982. | |
Hall SS, Lightbody AA, McCarthy BE, Parker KJ, Reiss AL. Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology. 2012;37(4):509–518. | |
Steingard RJ, Zimnitzky B, DeMaso DR, Bauman ML, Bucci JP. Sertraline treatment of transition-associated anxiety and agitation in children with autistic disorder. J Child Adolesc Psychopharmacol. 1997;7(1):9–15. | |
Indah Winarni T, Chonchaiya W, Adams E, et al. Sertraline may improve language developmental trajectory in young children with fragile x syndrome: a retrospective chart review. Autism Res Treat. 2012;2012:104317. | |
Hagerman RJFM, Leaman A, Riddle J, Hagerman K, Sobesky W. A survey of fluoxetine therapy in fragile X syndrome. Dev Brain Dysfunct. 1994;7:155–164. | |
Berry-Kravis E, Potanos K. Psychopharmacology in fragile X syndrome – present and future. Ment Retard Dev Disabil Res Rev. 2004;10(1):42–48. | |
Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther. 2002;302(1):381–389. | |
Jordan S, Koprivica V, Chen R, Tottori K, Kikuchi T, Altar CA. The antipsychotic aripiprazole is a potent, partial agonist at the human 5-HT1A receptor. Eur J Pharmacol. 2002;441(3):137–140. | |
Erickson CA, Stigler KA, Wink LK, et al. A prospective open-label study of aripiprazole in fragile X syndrome. Psychopharmacology (Berl). 2011;216(1):85–90. | |
Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4(2):147–153. | |
Greicius MD, Boyett-Anderson JM, Menon V, Reiss AL. Reduced basal forebrain and hippocampal activation during memory encoding in girls with fragile X syndrome. Neuroreport. 2004;15(10):1579–1583. | |
Reiss AL, Dant CC. The behavioral neurogenetics of fragile X syndrome: analyzing gene-brain-behavior relationships in child developmental psychopathologies. Dev Psychopathol. 2003;15(4):927–968. | |
Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in fragile X syndrome and potential intervention: a preliminary 1H MRS study. Am J Med Genet A. 2009;149A(3):403–407. | |
Sahu JK, Gulati S, Sapra S, et al. Effectiveness and safety of donepezil in boys with fragile x syndrome: a double-blind, randomized, controlled pilot study. J Child Neurol. 2013;28(5):570–575. | |
Cartagena CM, Phillips KL, Williams GL, et al. Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3. Neuromolecular Med. 2013;15(3):504–514. | |
Bickerdike MJ, Thomas GB, Batchelor DC, et al. NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke. J Neurol Sci. 2009;278(1–2):85–90. | |
Weiss SM, Wadsworth G, Fletcher A, Dourish CT. Utility of ethological analysis to overcome locomotor confounds in elevated maze models of anxiety. Neurosci Biobehav Rev. 1998;23(2):265–271. | |
Kramvis I, Mansvelder HD, Loos M, Meredith R. Hyperactivity, perseveration and increased responding during attentional rule acquisition in the Fragile X mouse model. Front Behav Neurosci. 2013;7:172. | |
Levenga J, Hayashi S, de Vrij FM, et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42(3):311–317. | |
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76(2):325–337. | |
Westmark CJ, Westmark PR, O’Riordan KJ, et al. Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS ONE. 2011;6(10):e26549. | |
Berry-Kravis E, Hessl D, Abbeduto L, Reiss AL, Beckel-Mitchener A, Urv TK; Outcome Measures Working Groups. Outcome measures for clinical trials in fragile X syndrome. J Dev Behav Pediatr. 2013;34(7):508–522. | |
Russo-Ponsaran NM, Yesensky J, Hessl D, Berry-Kravis E. Feasibility, reproducibility, and clinical validity of the pediatric anxiety rating scale-revised for fragile X syndrome. Am J Intellect Dev Disabil. 2014;119(1):1–16. | |
Chudley AE, Knoll J, Gerrard JW, et al. Fragile (X) X-linked mental retardation I: relationship between age and intelligence and the frequency of expression of fragil (X)(q28). Am J Med Genet. 1983;14(4):699–712. | |
Steyaert J, Borghgraef M, Legius E, Fryns JP. Molecular-intelligence correlations in young fragile X males with a mild CGG repeat expansion in the FMR1 gene. Am J Med Genet. 1996;64(2):274–277. | |
Lokanga RA, Entezam A, Kumari D, et al. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum Mutat. 2013;34(1):157–166. | |
Berry-Kravis E, Sumis A, Kim OK, Lara R, Wuu J. Characterization of potential outcome measures for future clinical trials in fragile X syndrome. J Autism Dev Disord. 2008;38(9):1751–1757. | |
Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS ONE. 2007;2(9):e931. | |
Weng N, Weiler IJ, Sumis A, Berry-Kravis E, Greenough WT. Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1253–1257. | |
Erickson CA, Ray B, Maloney B, et al. Impact of acamprosate on plasma amyloid-β precursor protein in youth: A pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res. 2014;59:220–228. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.