")
Back to Journals » ImmunoTargets and Therapy » Volume 3
Emerging options for the treatment of melanoma – focus on ipilimumab
Received 1 February 2013
Accepted for publication 1 May 2013
Published 17 March 2014 Volume 2014:3 Pages 67—78
DOI https://doi.org/10.2147/ITT.S43522
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Claire Roddie, Karl S Peggs
UCL Cancer Institute, Department of Hematology, London, UK
Abstract: Ipilimumab is a fully human immunoglobulin subclass G1 anticytotoxic-T-lymphocyte-antigen-4 monoclonal antibody. It has been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency for use in advanced melanoma following clear evidence of survival benefit in randomized Phase III studies. It is also under investigation as a treatment for other solid tumors such as renal cell, lung, and prostate cancers. The purported mechanism of antitumor activity of ipilimumab is through T-cell activation, and the side effect profile reflects this. Immune-related adverse events (irAEs) affect 60% of treated patients and 15% are defined as severe. Fortunately, most irAEs are reversible with early diagnosis and correct management. FDA approval of ipilimumab is dependent on the careful execution of a risk evaluation and mitigation strategy, with the aim of increasing awareness amongst patients and clinicians of the immunological risks of treatment, and providing algorithms for management of irAEs as they develop. Ipilimumab is one of the first immunotherapies to become widely available in the setting of solid tumors, and ongoing research aims to elucidate optimal dosing, optimal scheduling, and expanded access to ipilimumab as an adjuvant or maintenance therapy where appropriate. The identification of clinical correlates or biomarkers to identify those likely to benefit from this high-cost therapy is a top priority.
Keywords: ipilimumab, cytotoxic-T-lymphocyte-antigen-4, advanced melanoma, metastatic, overall survival
Introduction to ipilimumab, a CTLA-4 blocking agent for the treatment of melanoma
Cutaneous melanoma has an annual global incidence of 200,000 cases and comprises 4% of all skin cancers.1,2 Most patients present with stage 1 or 2 disease (84%), but 13% present with regional spread (stage 3) or distant metastases (stage 4) (See Table 1).3 Survival correlates with stage at diagnosis and a recent meta-analysis reports a disappointing 25% survival rate at year 1,4 and a 15.1% 5-year relative survival.3
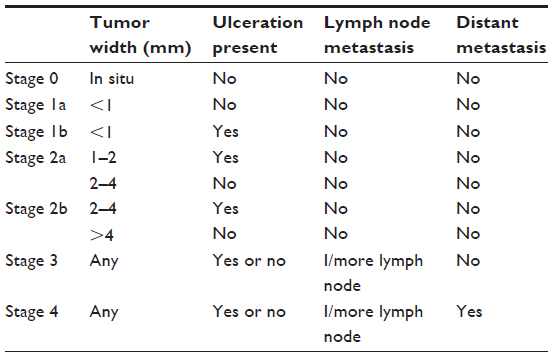 | Table 1 Clinical staging for melanoma |
Therapeutic options for unresectable and metastatic melanoma include systemic chemotherapy, radiotherapy, and best supportive care, but melanoma is refractory to most current systemic therapies.5,6 The treatment of choice over recent years has been dacarbazine (DTIC),7 or its oral analogue temozolamide,8 but objective response rates (ORR) have been consistently lower than 20% with unclear survival benefit when used either as single agents or in combination with other drugs.5,7,9,10
Interleukin-2 (IL-2) therapy has been evaluated in several Phase II studies and has been associated with durable remissions of 5 years or more in a small percentage (4%) of responding patients. Widespread clinical use of IL-2 is somewhat limited by its toxicity and is restricted to use in patients with good performance status and normal cardiorespiratory function.9,11 Crucially, however, the apparent impact of IL-2 has demonstrated the possible role of immunotherapy in this disease and led to the evaluation of other immunomodulatory compounds such as ipilimumab.
Ipilimumab (MDX-010; Yervoy®; Bristol-Myers-Squibb [BMS], New York, NY, USA and Medarex Inc, Princeton, NJ, USA) is a fully human immunoglobulin (Ig)Gκ monoclonal antibody directed against the T-cell receptor (TCR), cytotoxic-T-lymphocyte-antigen-4 (CTLA-4). It has been approved by the US Food and Drug Administration (FDA) for use in advanced melanoma following data from several Phase III studies in which it was shown to be the first drug to confer improved overall survival (OS) in this disease.12,13
Review of pharmacology, mode of action, pharmacokinetics
Immunological mode of action
CTLA-4 is a coinhibitory receptor of the immunoglobulin superfamily that is expressed following T-cell activation on the surface of CD4 and CD8 T cells and is constitutively expressed by CD4 T-regulatory cells. It is a negative regulator of CD28-dependent T cell immune responses and is thought to be crucial in the maintenance of peripheral immunological tolerance.14,15
Full T-cell activation requires TCR binding of antigen-bound major histocompatibility complex (MHC) on antigen-presenting cells in conjunction with a costimulatory signal generated through the engagement of the T-cell surface receptor CD28 with B7.1 or B7.2, expressed on antigen-presenting cells. Without this second signal through CD28, the TCR–MHC interaction leads to T-cell anergy.15
CTLA-4 is a closely related homologue of CD28, capable of binding both B7.1 and B7.2 with higher avidity than CD28. Following T-cell activation, CTLA-4 is upregulated at the immunological synapse and competes with CD28 for B7.1 and B7.2, thereby interrupting the costimulatory CD28 signal and acting as an immunological brake. Blockade of CTLA-4 via an antibody-dependent mechanism is thought to enhance and prolong T-cell activation by allowing CD28-B7 signaling to proceed unchallenged and in the context of malignancy, to promote an antitumor immune response.15–17
Preclinical investigation of anti-CTLA-4 antibodies has been extensively performed in murine models of cancer and has been shown to augment both T-cell-mediated immune function and antitumor effect, with a reduced risk of tumor recurrence in some solid tumors.16,18 The proposed mechanisms underlying these effects include anti-CTLA-4-dependent enhancement of effector T-cell proliferation and function, including interferon gamma (IFNγ) production by cytotoxic T-cells and a resultant upregulation of MHC expression on tumor cells, making them more “visible” targets to the immune system. Other mechanisms include enhanced tumor cell apoptosis and a reduction in tumor-associated angiogenesis.19
In the same way, ipilimumab is thought to enhance T-cell responses to tumor-associated antigens, resulting in immune-mediated antitumor efficacy.20
Pharmacokinetics of ipilimumab
Pharmacokinetic information on ipilimumab is derived from several Phase I–II studies, incorporating data from 499 patients with advanced melanoma who received one of three doses (0.3 mg/kg, 3 mg/kg, or 10 mg/kg) at intervals of 3 weeks, for a total of four doses. Peak (Cmax) and trough (Cmin) ipilimumab concentrations and area under the curve were proportional to the dosage administered within the dose range analyzed; steady-state drug concentration was reproducibly achieved by the third of four doses in all studies. The mean elimination half-life of ipilimumab was 14.7 days, with a systemic clearance of 15.3 mL/hour and a low volume of distribution of 7.21 L.21,22 Interestingly, 30% of patients receiving doses of 3 mg/kg achieved target trough levels of 20 mcg/mL, defined as the concentration at which ipilimumab confers maximal CTLA-4 blockade.23
Ipilimumab clearance increases with increasing body weight, but no dose adjustment has been recommended in this setting. Drug clearance is not affected by renal function (baseline creatinine clearance >29 mL/minute), hepatic dysfunction, gender, age (range 26–86 years), performance status, concurrent use of budesonide or prior use of systemic chemotherapies, HLA-A2*0201 status, baseline lactate dehydrogenase levels, or even the presence of circulating anti-ipilimumab antibodies. For this reason, no dose adjustment in these settings is deemed necessary. Any effect of race on drug clearance has not yet been evaluated.21,22
Ipilimumab has not been extensively investigated in pregnancy. However, due to the capacity of IgG1 to cross the placenta, it has the potential to be transmitted to the fetus. It is currently unknown whether it is secreted into human milk.21
Pharmacodynamics and efficacy of ipilimumab
The pharmacodynamics of ipilimumab are best illustrated through examination of the available clinical trial data. To evaluate the responses described with ipilimumab, we should remind ourselves of the poor median survival of <1 year for stage 4 melanoma when conventional treatments are used.4,9
Phase II studies of ipilimumab
In one early nonrandomized Phase II study of patients with metastatic melanoma failing at least one prior line of systemic therapy, O’Day et al24 instituted treatment with single agent ipilimumab at a dose of 10 mg/kg every 3 weeks for four doses, followed by a maintenance dose every 12 weeks beginning at week 24. This resulted in overall response rates (ORR) of 5.8%, a mean OS of 10.2 months, and a 1 year OS of 47.2%.
In a separate study of previously treated and untreated patients with advanced disease, the combination of ipilimumab 10 mg/kg and fotemustine resulted in an ORR of 29.1% and a 1-year OS of 51.8%.25 In a cohort of previously untreated patients with stage 3 and stage 4 disease, Patel et al26 report on the outcomes of ipilimumab 10 mg/kg as described above, in combination with temozolamide. They demonstrate an ORR of 28.1% including ten (15.6%) complete responses and eight (12.5%) partial responses.
The combination of ipilimumab (0.1–3 mg/kg) and IL-2 (720,000 U/kg every 8 hours for 15 doses) in previously untreated patients with advanced disease conferred objective tumor responses in 22% of patients, but there was no evidence of synergistic antitumor activity between ipilimumab and IL-2; for this reason, the combination is not recommended outside the clinical trial setting.27,28
Most early trials administered ipilimumab at a dose of 10 mg/kg, but optimal dosing had not been fully explored. Wolchok et al23 specifically compared three ipilimumab doses (10 mg/kg versus 3 mg/kg versus 0.3 mg/kg) in untreated stage 3 and stage 4 patients, and found that higher doses conferred a significantly better ORR (11.1% versus 4.2% versus 0%, respectively), progression free survival (PFS) (18.9% versus 12.9% versus 2.7%, respectively) and OS at 12 months (48.6% versus 39.3% versus 39.6%, respectively), but that the incidence of immune-related adverse events (irAEs) rose in proportion with increasing ipilimumab dose. Hamid et al30 also explored the optimal dose question in previously treated patients and found that 10 mg/kg compared with 3 mg/kg gave a better ORR of 11.9% versus 7.5%, respectively, but demonstrated that this did not translate into a survival benefit with OS at 12 months of 44.2% in the 10 mg/kg cohort compared with 60.9% in the 3 mg/kg cohort.
To summarize, Phase I and II data indicated that different doses of ipilimumab in different dosing schedules and in combination with other agents in advanced melanoma confer a (durable) antitumor effect and could be safely combined with other immunomodulators or chemotherapies.
Phase III studies
Definitive evaluation of the safety and efficacy of ipilimumab occurred in two large, randomized Phase III studies. In the first, a cohort of pretreated patients with stage 3 or stage 4 melanoma were entered into one of three treatment arms: ipilimumab 3 mg/kg + gp100 vaccine; ipilimumab 3 mg/kg + placebo; or gp100 vaccine + placebo.12 The investigational vaccine gp100 (control arm) was comprised of HLA-A*0201-restricted gp100 peptides (immunogenic peptides expressed on melanoma cells and recognized by T lymphocytes) and incomplete Freund’s adjuvant. Previous trials of gp100 monotherapy in metastatic melanoma have shown a potent induction of immune response towards tumor, but no demonstrable antitumor effect;31 however, it was shown that combination with IL-2 could improve antitumor efficacy.32 The implications of using this vaccine as an active control include the restriction of eligible patients to those with positive HLA-A*0201 status, although this allele is present in 50% of the Caucasian population. Crucially, it has been reported that ipilimumab can block CTLA-4 in an HLA-independent manner.33
Both ipilimumab groups conferred similar statistically significant benefits in terms of the primary endpoint of OS (see Table 2) and did not adversely affect quality of life assessments relative to single agent gp100 vaccine. The side effects of treatment were consistent with those described in Phase I and II trials, with 10%–15% of patients developing grade 3 or grade 4 irAEs.
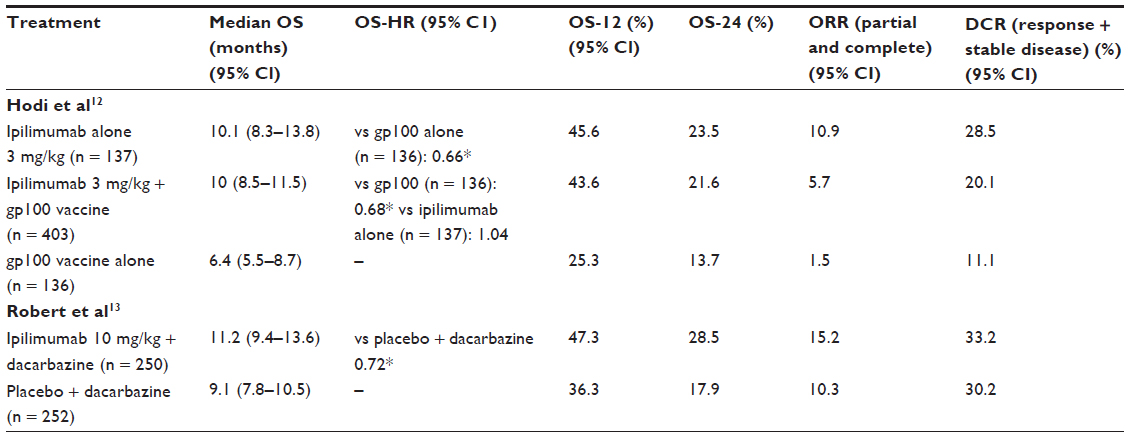 | Table 2 Summary of outcome measures for two pivotal Phase III trials of ipilimumab |
In the second large Phase III investigation (see Table 2), previously untreated patients were randomized to receive DTIC + ipilimumab 10 mg/kg or DTIC + placebo.13 The mean OS at 1-year in the combination arm was better than the DTIC alone arm (11.2 versus 9.1 months), and was independent of age, gender, performance status, baseline lactate dehydrogenase, and substage of metastatic disease. This benefit was consistently observed over years 2 and 3 of follow-up.13
Brain metastasis
Brain metastasis is a reasonably common manifestation of metastatic melanoma, affecting 45%–50% of patients with advanced disease. Autopsy data reveal that 75% of patients who die from melanoma have brain metastases.34–36
The treatment of choice is surgery or stereotactic radiotherapy, but beyond this, brain metastasis is refractory to all other standard treatments. Retrospective studies of ipilimumab in this setting have shown some promise. In a study by Margolin et al, two distinct patient groups with brain metastases were treated with single agent ipilimumab at a dose of 10 mg/kg at three weekly intervals for four doses, followed by maintenance from 24 weeks with 12 weekly dosings. Cohort A comprised patients who were neurologically asymptomatic and were not receiving concurrent corticosteroids, whereas Cohort B had neurological symptoms requiring corticosteroid treatment. Using the immune-related-response-criteria (irRC) assessment tool devised by Wolchok et al,37 described later in this article, disease control at 12 weeks was achieved in 26% of patients in Cohort A compared with 10% in Cohort B. Despite concerns that immune-modulating agents may induce further cerebral edema or brain inflammation in these patients, this trial reported no unexpected toxic neurological side effects of ipilimumab. The authors suggest that ipilimumab has a role in the management of recurrent brain metastasis postsurgery or sterotactic radiotherapy, but that its potency may be lost in symptomatic patients requiring corticosteroid treatment and that its use in this subgroup should undergo further evaluation.34–36
There is limited information on the subgroup of patients who develop uveal melanoma, as most are excluded from Phase III trials; it is rare, and approximately 50% of patients develop metastatic disease.38 Experience at Memorial Sloan-Kettering Cancer Centre (New York, NY, USA) of 20 patients with uveal disease who had received ipilimumab revealed a median OS of 8.6 months with an immune-related (ir)ORR of 5%.39 In a separate study, Danielli et al40 examined ipilimumab 10 mg/kg in pretreated patients with uveal melanoma, and the best outcome obtained was stable disease (SD) in three patients. Another small study reports SD in two of five treated patients and suggests that ipilimumab is a reasonable treatment option for these patients.38
Overall, trials have demonstrated clear clinical benefit and acceptable toxicity of ipilimumab in patients with stage 3 and 4 melanoma and it is the first drug to have shown improved OS in this therapeutically challenging patient cohort. Ipilimumab was approved at a dose of 3 mg/kg by the FDA in March 2011 for use in advanced (unresectable or metastatic) melanoma, and by the European Medicines Agency (EMA) as a second-line agent for advanced disease.
New evaluation criteria
It is important to recognize that traditional tools for the assessment of the efficacy of cytotoxic chemotherapeutic agents such as Response Evaluation Criteria in Solid Tumors (RECIST) or World Health Organization (WHO) criteria, utilized in earlier trials of ipilimumab, are often not adequate to assess the unconventional pattern of clinical responses to immunotherapy. For instance, some patients develop new lesions or tumor enlargement in the initial stages of immunotherapy, prior to demonstrating a clinically meaningful antitumor response. These patients would be defined by RECIST or WHO criteria as treatment failures. In fact, analysis of Phase II response data defined four patterns of response to ipilimumab which were all associated with improved survival: reduction in baseline lesions without new lesions; durable SD (and in some cases a slow shrinkage in index lesions); initial growth of index lesions; or development of new lesions followed by a reduction in tumor burden.37
Due to the often atypical pattern of clinical responses with these agents, the irRC assessment tool, devised by Wolchok et al,37 has been developed in an attempt to include additional response patterns observed with immune therapy in advanced melanoma which fall outside the boundaries of the RECIST or WHO systems. The irRC is currently being investigated in ongoing clinical trials.
Using this tool, antitumor responses are based on total measurable tumor burden. At baseline assessment, the sum of the products of the two largest perpendicular diameters (SPD) of all index lesions (five lesions per organ, up to ten visceral lesions and five cutaneous index lesions) is calculated. At subsequent assessments, the sum of the SPDs of the index lesions and of any new measurable lesions (≥5 × 5 mm; up to 15 new lesions per organ – five new cutaneous lesions and ten visceral lesions) is calculated to give the tumor burden reading. All responses must be confirmed in two consistent, discrete readings, taken at least 4 weeks apart. Table 3 defines the exact criteria for response assessment using the irRC model and directly compares these with conventional WHO definitions of response.37
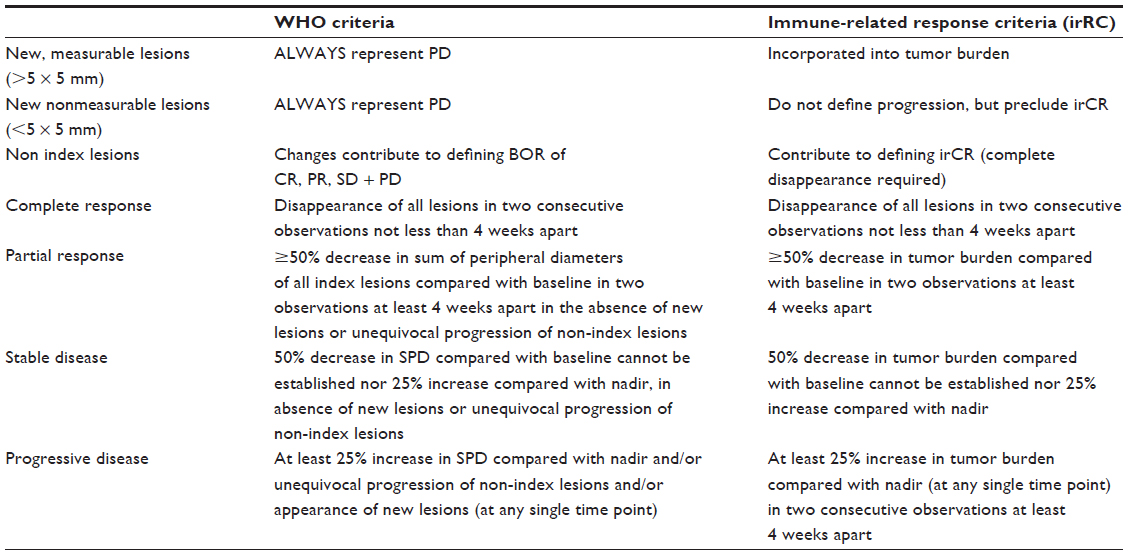 | Table 3 Comparison of WHO versus irRC staging for metastatic melanoma |
It is clear that responses to ipilimumab may be delayed by several months and it is recommended that patients should be evaluated for initial responses after 12 weeks of treatment, coinciding with the end of the treatment cycle. Repeated dosing of ipilimumab is not currently recommended.21
Interactions and adverse effects
No formal drug–drug interaction studies have been conducted with ipilimumab.21 However, metabolism of ipilimumab does not appear to involve the cytochrome P450 enzyme system and so, the potential for drug interaction is low.41
It should be noted that irAEs is the name given to the constellation of “autoimmune” side effects which can emerge following CTLA-4 blockade during treatment with ipilimumab. This important physiological role of CTLA-4 in maintenance of immune tolerance is most clearly demonstrated in experimental models where the receptor is absent. To illustrate, CTLA-4 knockout mice develop a polyclonal CD4-dependent lymphoproliferative disorder, resulting in premature death at 3–4 weeks after birth, often due to inflammatory myocarditis.42 What was not observed in these animals, or in preclinical animal toxicology studies of ipilimumab, was colitis, uveitis, hepatitis, or endocrinopathy, which are all commonly encountered side effects of CTLA-4 blockade in clinical studies in humans. In fact, these side effects only became apparent in early phases of clinical testing.43
For patients receiving FDA-approved dosing of ipilimumab, the incidence of irAEs is 60%, and approximately 15% will experience severe grade 3 or grade 4 complications.12 Moreover, irAEs of skin, gastrointestinal (GI) tract, liver, and endocrine system are most common. The development of symptoms follows a classic pattern in timing of onset, such that skin related irAEs can be reliably expected early post-treatment, at a median of 3 weeks, followed by GI and hepatic irAEs at a median of 6–7 weeks and endocrinopathies at a median of 9–11 weeks post treatment.43
The incidence of dose-limiting irAEs in patients receiving ipilimumab increases with dose, such that 25% of patients who receive 10 mg/kg will develop grade 3 or grade 4 irAEs, compared with 7% of those who receive 3 mg/kg. However, most of the immune-related side effects are reversible with early diagnosis and correct management.43
In view of the high incidence of drug toxicity, federal approval of ipilimumab is dependent on the concurrent dissemination and use of the communication-based risk evaluation and mitigation strategy to increase awareness amongst patients of irAEs and to provide investigators with an algorithm for investigation and treatment thereof. The algorithms include detailed management guidelines according to the clinical severity of the irAE and comprise measures to gain symptom control in conjunction with definitive high dose systemic corticosteroid, as required.44
Skin
Approximately 45% of treated patients will develop a pruritic macropapular rash, which is severe in 2.6% of cases. For milder presentations, topical moisturizers and antihistamines will often suffice; for moderate cases it is prudent to withhold ipilimumab and to administer topical or low-dose systemic steroids in the event of symptoms persisting beyond 1 week. Ipilimumab can be restarted once symptoms have abated, provided the steroid dose has been weaned. In the presence of Stevens–Johnson syndrome, toxic epidermal necrolysis, or other severe and life-threatening dermatitides, it is crucial to permanently discontinue ipilimumab and to commence high dose systemic corticosteroids.43,45
GI tract
GI toxicity is observed in approximately 33% of patients treated with ipilimumab at 3 mg/kg, of which 9% are grade 3 or grade 4 in severity. All patients with new GI symptoms should be screened for possible infectious causes; in those with mild symptoms, dietary modification and antidiarrheals should be prescribed as necessary. In moderate cases, where diarrhea, abdominal pain, mucus, or blood are present in stool, ipilimumab should be withheld and antidiarrheals given. If symptoms persist beyond 1 week, intermediate dose systemic corticosteroids should be administered. Ipilimumab can be restarted once symptoms have abated, provided the steroid dose has been weaned. In severe and life-threatening cases of GI toxicity, ipilimumab should be permanently discontinued and bowel perforation should be excluded.46 In the event of perforation, corticosteroids should be avoided and surgical intervention sought. In other circumstances, steroids should be given at high dose.43,45
Prophylaxis of colitis using budesonide – an orally active corticosteroid with limited systemic exposure as a result of the first pass effect – was tested in a double-blind, placebo-controlled Phase II study, but was ineffective in reducing the rate of grade >2 diarrhea. It can have a beneficial effect in mild presentations of ipilimumab-induced GI toxicity, and can be prescribed for this purpose.
Endocrinopathy
Endocrine irAEs are less common, with an overall incidence of 4.5%, of which 2.3% are severe presentations. Immune-related hypophysitis can present with symptoms of headache, nausea, vertigo, behavior change, and visual disturbance, but brain metastasis must first be excluded in all patients. Investigation should include imaging of the pituitary and serum testing of all pituitary axis hormones prior to the initiation of treatment. For symptomatic panhypopituitarism and for any other grade 3 or grade 4 endocrinopathy, ipilimumab should be held and systemic corticosteroids given. Permanent pituitary dysfunction has been reported in some patients. Ipilimumab can be resumed in these patients, provided symptoms have resolved and the patient is stable on hormone replacement therapy and a minimal dose of systemic corticosteroid.43,45
Hepatotoxicity
Hepatic irAEs are reported in 1.6% of all treated patients and are categorized as severe in 1.1%. It is important to consider and exclude infectious and malignant causes for deranged liver function tests in treated patients, but in cases where ipilimumab is clearly the underlying cause of moderate hepatic dysfunction, it should be withheld until the liver function tests return to more normal levels. For severe or life-threatening hepatotoxicity, namely transaminase levels exceeding >5 × the upper limit of normal or serum bilirubin exceeding >3 × the upper limit of normal, ipilimumab should be discontinued and high-dose systemic steroids considered.43,45 Of note, a recent publication reports a higher than expected incidence of hepatotoxicity in the context of ipilimumab therapy, where six of eleven treated patients developed grade ≥1 liver function test derangement, and three were classified as grade 3. Fortunately, resolution occurred upon cessation of ipilimumab without recourse to immunosuppressive agents, but this report underlines the importance of close monitoring of patients receiving ipilimumab and recognizes that toxicity rates may show marked variation across different patient populations.29
Neurological toxicity
Neurological complications occur in approximately 0.1% of all patients and often manifest as sensory or motor neuropathies. In the event of moderate neurologic symptoms that do not adversely impact upon activities of daily living, ipilimumab can be transiently discontinued until function returns to baseline. For severe events, ipilimumab must be discontinued and high-dose systemic steroids commenced.43,45 Unusual clinical presentations requiring hospital admission, such as Guillain–Barré syndrome and myasthenia gravis, have also been associated with ipilimumab therapy.47
Ophthalmological toxicity
Ipilimumab-related episcleritis or uveitis are more commonly described in patients with concurrent GI toxicity. Grade 1 and grade 3 uveitis can be treated with 1% corticosteroid eye drops and iridocyclics, which can restore vision to normal in almost all patients. In the event of grade 3 or grade 4 symptoms, or other eye-related complications which fail to respond to topical immunosuppression, ipilimumab should be permanently discontinued and systemic corticosteroids considered.43,45
The time course to recovery from irAEs depends on the end organ involved. Skin, GI, and liver toxicity recover promptly (2–4 weeks) whereas endocrinopathies can be slow, and in some cases permanent. During this period of recovery, corticosteroid treatment should be slowly weaned, over at least 6 weeks, to prevent relapse.44,45
Concerns regarding the possible adverse impact of systemic corticosteroids on the immunotherapeutic, antitumor benefits of ipilimumab are unfounded, as evidence suggests that antitumor responses are unaffected by corticosteroid use.48
Retreatment with ipilimumab has been safely executed in patients with grade 1 and grade 2 irAEs in whom symptoms have been downgraded to grade 1 or less following symptom control and corticosteroid use. Some investigators report retreatment in cases of grade 2 endocrinpathy on steroid replacement, in cases of mild colitis and in cases of grade 2 sarcoidosis, without complication. However, any grade 3 or grade 4 irAEs (excluding grade 3 skin toxicity) preclude the future use of ipilimumab.43
It has been argued that there is a correlation between the development of irAEs and of an antitumor effect in recipients of ipilimumab for metastatic melanoma and renal cell carcinoma. Data from several Phase II studies have demonstrated a positive trend towards higher rates of disease control and OS in those patients with grade >2 irAEs. It has also been reported that patients with resected high-grade melanoma receiving ipilimumab as an adjuvant therapy have an extended time to relapse in the event of clinically significant irAEs. This area is being further explored in ongoing large Phase III registrational studies.43
In the event of disease relapse in prior responders, several patients have been rechallenged with ipilimumab; with this process, an impressive 70% regain disease control.12 At present, BMS do not advocate retreatment with ipilimumab.21
Immunohistochemical staining of sections of skin and bowel from patients with treatment-related dermatitis or colitis reveals infiltration by activated CD4 and CD8 T-cells. Infiltration correlates with clinical severity and it has been proposed that cytokine release from inflammatory T-cells induces the development of irAEs, as the tumor necrosis factor alpha blocking antibody (infliximab) has been shown to induce rapid resolution of symptoms.49,50
In conclusion, ipilimumab should be administered at a dose of 3 mg/kg intravenously over 90 minutes at three weekly intervals for a total of four doses. Discontinuation of this drug is recommended in the event of severe or life threatening reactions, if moderate reactions persist or recur, if the corticosteroid dose cannot be lowered below 7.5 mg of prednisolone (or equivalent) per day, and in the circumstance where the patient fails to complete all four recommended doses within a 16-week window from the onset of therapy.21
Further research in use of ipilimumab as active immunotherapy for cancer
Ipilimumab has been shown to induce durable tumor regression in metastatic melanoma. Its role in the management of other solid tumors and even in some hematological malignancy is less clear than in melanoma, but is compelling and worthy of further evaluation nonetheless.
Renal cell cancer
Renal cell cancer is an immunogenic cancer which is responsive to immunotherapy with IL-2. Rosenberg et al51 conducted a Phase II trial of single agent ipilimumab in patients with metastatic disease and demonstrated cancer regression in 27% of patients receiving doses of 3 mg/kg, irrespective of prior response or lack of response to IL-2. Clinical responses were strongly associated with development of other immune-mediated complications such as enteritis, dermatitis, and endocrinopathy.
Lung cancer
Stage 3/stage 4 non-small cell lung cancer (NSCLC) has a median survival of 8–12 months following conventional treatment with platinum-based chemotherapy combinations. Recent interest in the potential of unlocking an immune response against the disease has prompted the investigation of ipilimumab in this condition. A recent Phase II study of previously untreated patients with advanced stage NSCLC demonstrated an improved irPFS (5.7 months versus 4.6 months) with phased ipilimumab plus carboplatin-paclitaxel chemotherapy. As expected, 15% of patients developed grade 3 or grade 4 irAEs.52
Reck et al52,53 also explored combination chemotherapy and phased ipilimumab in extensive disease small cell lung cancer and found similar improvements in irPFS in patients receiving ipilimumab, with grade 3 and grade4 irAEs comparable to the NSCLC data.
Overall, lung cancer physicians are encouraged by the improvements seen in PFS and suggest that ipilimumab be further evaluated in larger clinical trials.
Prostate cancer
Hormone refractory prostate cancer (HRPC) is one of the few tumor types where immunotherapy is arguably the current standard of care. Sipuleucel-T, an autologous cellular vaccine, is one such treatment; it was approved by the FDA in 2011.54 Ipilimumab also shows promise in this condition and in a small pilot study of HRPC, a single dose of ipilimumab at 3 mg/kg resulted in two of 12 patients developing a >50% reduction in serum prostate specific antigen levels, implying disease control. Significant clinical autoimmunity was not observed in these patients.55
A Phase I study by van den Eertwegh et al,56 in which safety was the primary study endpoint, reported that granulocyte/macrophage colony-stimulating factor-secreting cellular vaccine (GVAX®; Aduro Biotech, Inc, Berkeley, CA, USA) combined with ipilimumab in an escalating dose strategy (0.3–5.0 mg/kg), was tolerable and safe for patients with HRPC. The median OS was 29.2 (9.6–48.8) months and reported toxicities comprised transaminitis, colitis, rash, and endocrine-related toxicities including adrenal insufficiency and hypophysitis. Patients with hypophysitis were started on hormone replacement therapy and allowed to continue GVAX® plus ipilimumab therapy. Seven patients (25%) who received either 3 mg/kg or 5 mg/kg ipilimumab had prostate-specific antigen (PSA) declines of 50%. Regression of bone metastases occurred in two patients and stable bone metastases lasting 3–7 months were seen in 15 patients. Complete regression of abdominal lymphadenopathy was seen in one of four patients with measurable disease at baseline.56
A Phase I study by Madan et al57 explored the utility of combination therapy with the poxviral vaccine PSA–Tricom (PROSTVAC®; Bavarian Nordic, Mountain View, CA, USA) and ipilimumab at doses of 1–10 mg/kg, in 30 patients with HRPC, of whom 24 were chemotherapy naive. Treatment-comprised fixed doses of monthly PROSTVAC® with plans for a total of six doses of ipilimumab. In fact, 14 patients discontinued ipilimumab because of disease progression and 13 patients discontinued ipilimumab due to the development of irAEs. Only three patients completed all six planned doses of therapy. For all patients, the investigators reported a median PFS of 3.9 (3.3–6.3) months and a median OS of 34.4 (29.6–41) months. The most common toxicities were similar to those reported by van den Eertwegh et al56 in the GVAX® plus ipilimumab study.
In summary, the efficacy of ipilimumab alone or in combination with other immunotherapy or chemotherapy requires further investigation in the context of larger-scale clinical trials, several of which are currently ongoing. It is clear from the Phase I data cited here that immune-related side effects are not inconsequential in these HRPC patients, and that careful multidisciplinary strategies for managing these problems should be established for this disease.
Miscellaneous
Ipilimumab has also been investigated in the context of ovarian carcinoma, pancreatic adenocarcinoma, and in a Phase I study of diffuse large B-cell lymphoma, but with less clear cut results to date.58,59,66
Patient-focused perspectives such as quality of life
Metastatic melanoma has an impact on health-related quality of life (HRQL) which is comparable with other cancers.60 Studies have demonstrated that melanoma impacts adversely on psychological wellbeing, promoting anxiety, depression, and feelings of vulnerability.61,62 In addition, patients receiving treatment for metastatic melanoma report compromised physical function and increased fatigue.61,63
Several clinical trials of therapy for metastatic melanoma have addressed the issues around HRQL including the Phase III clinical trial by Hodi et al,12 in which patients with stage 3 or stage 4 melanoma were randomized to receive either ipilimumab 3 mg/kg + gp100 vaccine, ipilimumab 3 mg/kg + placebo, or gp100 vaccine + placebo. This trial conducted a European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30), allowing assessment of function, overall health status, and symptom scores at baseline and at week 12 of therapy; the trial compared ipilimumab ± vaccine with gp100 vaccine alone. Overall results conclude that ipilimumab alone or in combination with vaccine does not confer a negative HRQL impact during treatment induction, relative to gp100 vaccination alone.
Conclusions, place in therapy
Ipilimumab is a major breakthrough in the treatment of advanced stage melanoma as it is the first agent to have definitively shown improved OS in this difficult to treat condition.
The FDA approved its use in the USA as first or second line therapy for unresectable, advanced-stage melanoma. The EMA approved its use in Europe as a second line option for patients with advanced disease. In the UK, Britain’s National Centre for Pharmacoeconomics concluded that BMS, the manufacturers of ipilimumab, had “failed to demonstrate cost-effectiveness” and that they could not recommend “reimbursement at the submitted price.” The incremental cost-effectiveness ratio per quality-adjusted-life-year (QALY) gained with ipilimumab over best supportive care was £60,737/QALY.64,65
The National Institute for Health and Clinical Excellence (NICE) refused to recommend ipilimumab for the treatment of advanced (unresectable or metastatic) malignant melanoma in people who have received prior therapy, despite acknowledging that it met the criteria for being a “life-extending, end-of-life treatment” and a “significant innovation for a disease with a high unmet clinical need.” The NICE committee considered that the “magnitude of additional weight that would need to be assigned to the QALY gains for people with advanced (unresectable or metastatic) melanoma would be too great for ipilimumab to be considered a cost-effective use of NHS resources.” For now, most patients in the UK will therefore only be able to access ipilimumab through registration in an ongoing clinical trial.
A broader interpretation of the Hodi et al12 Phase III study, which looked at ipilimumab 3 mg/kg in previously treated patients, is that it is clinically ineffective or only marginally effective for the majority of eligible patients, with only 11% of treated patients achieving complete response or partial response. For this reason, one of the biggest research priorities should be the investigation of predictive factors or biomarkers to enable selection of patients who are most likely to benefit from ipilimumab, restricting exposure of the majority of patients to possible toxicities and helping to manage the cost implications. Possible markers include those related to analysis of tumor-infiltrating populations prior to therapy such as cytotoxic T-cell to regulatory T-cell ratios, though a marker based on flow-cytometric analysis of a blood sample following initiation of therapy has clear commercial appeal. The intratumor ratio of cytotoxic T-cells to regulatory T-cells has been shown to correlate with tumor rejection in murine models following therapy,67 though whether a baseline measurement prior to initiation of therapy in human cancers can predict outcome remains untested.
Other T-cell activation markers are under investigation as potential biomarkers. One example is outlined in a study by Sharma et al,68 who report a higher incidence of Inducible Costimulator (ICOS)-expressing T-cells in tumor and blood in the context of ipilimumab treatment. Further work by the same group using ICOS-sufficient and ICOS-deficient mice bearing B16 melanoma and treated with anti-CTLA-4 antibody showed that the antitumor effect was diminished in ICOS-deficient mice. In contrast, ICOS-sufficient T-cells were defined as Th1 cytokine-secreting, tumor–antigen-specific effector cells when exposed to anti-CTLA-4 antibody.69 They conclude that the ICOS/ICOS–ligand pathway is necessary for optimal therapeutic effect of anti-CTLA-4 antibody and that ICOS+FoxP3 T-cells warrant consideration as a potential predictive biomarker of clinical outcome with anti-CTLA-4 therapy. This view is supported by a clinical study of patients with metastatic melanoma receiving ipilimumab in which improved disease control and survival was associated with a higher frequency of ICOS+ T-cells in the tumor at week 7 of ipilimumab treatment.70
Future directions for ipilimumab
Many studies are currently underway exploring different dosing (3 mg/kg versus 10 mg/kg) of ipilimumab, both in advanced melanoma and in the adjuvant setting (cases of fully resected stage 3 melanoma), where it has been proposed that CTLA-4 blockade may prolong PFS and OS. A randomized Phase III trial of ipilumumab 3 mg/kg versus 10 mg/kg versus high-dose IFN-α-2b treatment in patients with completely resected, high risk melanoma is also ongoing.
Combinatorial strategies of ipilimumab plus other agents (adoptively transferred cytotoxic lymphocytes; IFN-α-2b; vemurafenib; aldesleukin; temozolamide; and cisplatin, to name but a few) are being investigated in Phase I and II trials for efficacy and safety.
Disclosure
The authors report no conflicts of interest in this work. The authors have no relevant affiliations or financial involvement with any organization or entity having a conflict with or financial interest in the subject matter contained in this manuscript.
References
Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med. 2006;355(1):51–65. | |
International Agency for Research on Cancer. The GLOBOCAN project [homepage on the Internet]. Geneva, Switzerland: World Health Organization; 2010. Available from: http://globocan.iarc.fr. Accessed January 12, 2013. | |
National Cancer Institute. SEER stat fact sheets: melanoma of the skin [webpage on the Internet]. Bethesda, MD: National Cancer Institute. Available from: http://seer.cancer.gov/statfacts/html/melan.html. Accessed January 12, 2013. | |
Korn EL, Liu PY, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26(4):527–534. | |
National Cancer Institute. Melanoma treatment (PDQ) [webpage on the Internet]. Bethesda, MD: National Cancer Institute; August 2011. Available from: http://www.cancer.gov. Accessed on January 12, 2013. | |
National Comprehensive Cancer Network. NCCN practice guidelines in oncology: Melanoma (version 3.2012) [webpage on the Internet]. Fort Washington, PA: National Comprehensive Cancer Network; 2011. Available from: http://www.nccn.com. Accessed on January 12, 2013. | |
Fricker J. New era in metastatic melanoma. Mol Oncol. 2010;4(1):91–97. | |
Quirbt I, Verma S, Petrella T, Bak K, Charette M; for Members of the Melanoma Disease Site Group of Cancer Care Ontario’s Program in Evidence-Based Care. Temozolomide for the treatment of metastatic melanoma. Curr Oncol. 2007;14(1):27–33. | |
Agarwala SS. Current systemic therapy for metastatic melanoma. Expert Rev Anticancer Ther. 2009;9(5):587–595. | |
Patel PM, Suciu S, Mortier L, et al; for EORTC Melanoma Group. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47(10):1476–1483. | |
Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363(8):711–723. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364(26):2517–2526. | |
Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily – CTLA-4. Nature. 1987;328(6127):267–270. | |
Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. | |
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. | |
Kirkwood JM, Tarhini AA, Panelli MC, et al. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26(20):3445–3455. | |
Hurwitz AA, Foster BA, Kwon ED, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60(9):2444–2448. | |
Paradis TJ, Floyd E, Burkwit J, et al. The anti-tumor activity of anti-CTLA-4 is mediated through its induction of IFN gamma. Cancer Immunol Immunother. 2001;50(3):125–133. | |
Graziani G, Tentori L, Navarra P. Ipilimumab: a novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol Res. 2012; 65(1):9–22. | |
Bristol-Myers Squibb Company. Ipilimumab US Prescribing Information 2011. Princeton, NJ: Bristol-Myers Squibb Company; 2012. Available from: http://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed on January 12, 2013. | |
YERVOY 5 mg/mL concentrate for solution for infusion – Summary of Product Characteristics (SPC) [webpage on the Internet]. Electronic Medicines Compendium; 2013. Available from: http://www.medicines.org.uk/emc/medicine/24779. Accessed on January 12, 2013. | |
Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010; 11(2):155–164. | |
O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21(8):1712–1717. | |
Di Giacomo AM, Ascierto PA, Pilla L, et al. Ipilimumab and fotemustine in patients with advanced melanoma (NIBIT-M1): an open-label, single-arm phase 2 trial. Lancet Oncol. 2012;13(9):879–886. | |
Patel SP, Hwu WJ, Kim KB, et al. Phase II study of the frontline combination of ipilimumab and temozolamide in patients with metastatic melanoma [abstract]. J Clin Oncol. 2012;(Suppl):8514. | |
Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. | |
Maker AV, Phan GQ, Attia P, et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol. 2005; 12(12):1005–1016. | |
Bernardo SG, Moskalenko M, Pan M, et al. Elevated rates of transaminitis during ipilimumab therapy for metastatic melanoma. Melanoma Res. 2013;23(1):47–54. | |
Hamid O, Schmidt H, Nissan A, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. | |
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. | |
Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. | |
Retsas S. Latest developments in the treatment of melanoma: ‘a penicillin moment for cancer’? J R Soc Med. 2011;104(6):269–272. | |
Margolin K. Ipilimumab in a Phase II trial of melanoma patients with brain metastases. Oncoimmunology. 2012;1(7):1197–1199. | |
Fonkem E, Uhlmann EJ, Floyd SR, et al. Melanoma brain metastasis: overview of current management and emerging targeted therapies. Expert Rev Neurother. 2012;12(10):1207–1215. | |
Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. | |
Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. | |
Khattak MA, Fisher R, Hughes P, Gore M, Larkin J. Ipilimumab activity in advanced uveal melanoma. Melanoma Res. 2013;23(1):79–81. | |
Khan KD, Callahan M, Postow MA, et al. Efficacy and safety of oral MEK162 in patients with locally advanced and unresectable or metastatic cutaneous melanoma harboring BRAFV600 or NRAS mutations [abstract]. J Clin Oncol. 2012;(Suppl):8511. | |
Danielli R, Ridolfi R, Chiarion-Sileni V, et al. Ipilimumab in pretreated patients with metastatic uveal melanoma: safety and clinical efficacy. Cancer Immunol Immunother. 2012;61(1):41–48. | |
Culver ME, Gatesman ML, Mancl EE, Lowe DK. Ipilimumab: a novel treatment for metastatic melanoma. Ann Pharmacother. 2011; 45(4):510–519. | |
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. | |
Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. | |
Bristol-Myers Squibb. Ipilimumab US prescribing information: risk evaluation and mitigation strategy [webpage on the Internet]. Princeton, NJ: Bristol-Meyers Squibb; 2012. Available from: http://www.yervoy.com/hcp/rems.aspx. Accessed January 12, 2013. | |
Trinh VA, Hagen B. Ipilimumab for advanced melanoma: A pharmacologic perspective. J Oncol Pharm Pract. Epub October 11, 2012. | |
Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591–5598. | |
Gaudy-Marqueste C, Monestier S, Franques J, Cantais E, Richard MA, Grob JJ. A severe case of ipilimumab-induced guillain-barré syndrome revealed by an occlusive enteric neuropathy: a differential diagnosis for ipilimumab-induced colitis. J Immunother. 2013;36(1):77–78. | |
Amin A, DePril V, Hamid O, et al. Evaluation of the effect of systemic corticosteroids for the treatment of immune-related adverse events (irAEs) on the development or maintenance of ipilimumab clinical activity [abstract]. J Clin Oncol. 2009;27(Suppl 15):9037. | |
Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372–8377. | |
Johnston RL, Lutzky J, Chodhry A, Barkin JS. Cytotoxic T-lymphocyte-associated antigen 4 antibody-induced colitis and its management with infliximab. Dig Dis Sci. 2009;54(11):2538–2540. | |
Yang JC, Hughes M, Kammula U, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–830. | |
Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. | |
Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24(1):75–83. | |
Kantoff PW, Higano CS, Shore ND, et al; for IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | |
Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13(6):1810–1815. | |
van den Eertwegh AJ, Versluis J, van den Berg HP, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):509–517. | |
Madan RA, Mohebtash M, Arlen PM, et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):501–508. | |
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. | |
Ansell SM, Hurvitz SA, Koenig PA, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2009; 15(20):6446–6453. | |
Cornish D, Holterhues C, van de Poll-Franse LV, Coebergh JW, Nijsten T. A systematic review of health-related quality of life in cutaneous melanoma. Ann Oncol. 2009;20 Suppl 6:vi51–vi58. | |
Trask PC, Paterson AG, Esper P, Pau J, Redman B. Longitudinal course of depression, fatigue, and quality of life in patients with high risk melanoma receiving adjuvant interferon. Psychooncology. 2004;13(8):526–536. | |
Boyle DA. Psychological adjustment to the melanoma experience. Semin Oncol Nurs. 2003;19(1):70–77. | |
Cohen L, Parker PA, Sterner J, De Moor C. Quality of life in patients with malignant melanoma participating in a phase I trial of an autologous tumor-derived vaccine. Melanoma Res. 2002;12(5):505–511. | |
Specenier P. Ipilimumab in melanoma. Expert Rev Anticancer Ther. 2012;12(12):1511–1121. | |
BMS single technology appraisal submission for ipilimumab. July 26, 2011. Available from: http://www.nice.org/uk/nicemedia/live/12092/56692/56692.pdf. Accessed January 12, 2013. | |
Hodi FS, Mihm MC, Soiffer RJ, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100(8):4712–4717. | |
Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116(7):1935–1945. | |
Liakou CI, Kamat A, Tang DN, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A. 2008; 105(39):14987–14992. | |
Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71(16):5445–5454. | |
Di Giacomo AM, Calabrò L, Danielli R, et al. Long-term survival and immunological parameters in metastatic melanoma patients who responded to ipilimumab 10 mg/kg within an expanded access programme. Cancer Immunol Immunother. Epub April 17, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.