")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Emerging drugs for partial-onset epilepsy: a review of brivaracetam
Received 2 December 2015
Accepted for publication 11 February 2016
Published 4 May 2016 Volume 2016:12 Pages 719—734
DOI https://doi.org/10.2147/TCRM.S90127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Lan Gao,1 Shuchuen Li2
1Deakin Population Health SRC, Faculty of Health, Deakin University, Burwood, Victoria, 2School of Biomedical Sciences and Pharmacy, The University of Newcastle, Callaghan, NSW, Australia
Abstract: There are more than 12 new antiepileptic drugs approved in the last 2 decades. Even with these newer agents, seizure remission is still unachievable in around 30% of patients with partial-onset seizures (POS). Brivaracetam (BRV) is chemically related to levetiracetam (LEV) and possesses a strong binding affinity for the synaptic vesicle protein 2A tenfold above that of LEV, and other possible modes of antiepileptic actions. BRV is now under Phase III development for POS, but data from one Phase III trial also suggested its potential efficacy for primary generalized seizures. The purpose of this review is to provide updated information on the mechanisms of action of the available antiepileptic drugs, with a focus on BRV to assess its pharmacology, pharmacokinetics, clinical efficacy, safety, and tolerability in patients with uncontrolled POS. To date, six Phase IIb and III clinical trials have been performed to investigate the efficacy, safety, and tolerability of BRV as an adjunctive treatment for patients with POS. Generally, BRV was well tolerated and did not show significant difference in safety profile, compared to placebo. The efficacy outcomes of BRV, although not consistent across trials, did indicate that BRV was a promising add-on therapy for patients with POS. In conclusion, the many favorable attributes of BRV, like its high oral efficacy, good tolerability, dosing regimen, and minimal drug interaction, make it a promising antiepileptic therapy for patients with uncontrolled partial-onset epilepsy.
Keywords: brivaracetam, partial-onset epilepsy, drug-resistant epilepsy, randomized controlled trial, review
Introduction
Epilepsy is a chronic neurological disorder characterized by recurrent seizures and is estimated to affect approximately 50 million people worldwide.1 Globally, an estimated 2.4 million people are diagnosed with epilepsy each year.1 Epilepsy is not only associated with detrimental effects on the health and quality of life (QoL) of the individual patient, but also places a huge psychological and economic burden on the family members of the sufferers. When the diagnosis of epilepsy is established, treatment will be initiated accordingly. The primary treatment objectives are to achieve lifelong seizure freedom,2,3 reduce morbidity and mortality, and improve QoL,4 ideally without intolerable adverse events (AEs).5 Nonpharmacological management modalities like surgery6 and vagus nerve stimulation7 are available for a selected proportion of patients, while the majority of individuals are managed with antiepileptic drugs (AEDs). Although the prognosis for the majority of patients is good, up to 30% of cases do not show complete remission despite appropriate therapy with AEDs. Normally, the AED is selected on the basis of the clinical efficacy, tolerability, drug interaction, and ease of use.8 It is reported that most patients could be successfully managed by monotherapy alone;8 however, up to 50% still need to be treated with combination therapy.9
Monotherapy is the best pharmacotherapeutic option when first starting AED treatment.10 If monotherapy is poorly tolerated or ineffective, the strategy is to switch to another drug; and if the first drug has partial efficacy and is well tolerated, it is worthwhile to try another drug in combination.11 However, add-on therapy has been shown to be more effective when started immediately after first drug failure rather than after a second drug has also failed.12 Nevertheless, when adequate trials of two tolerated, appropriately chosen, and administered AEDs (whether as monotherapy or in combination) fail to achieve sustained seizure freedom, a diagnosis of drug-resistant epilepsy could be subsequently made.3
Among the two major types of epilepsy, partial-onset seizures (POS) occur in more than 60% of patients and are the most commonly encountered type of seizure in the adult population.13 Often affected by comorbid disorders, these patients are difficult to treat, and approximately one-third of cases are resistant to AED management, including combination regimens.14 The introduction of a large number of newer AEDs with more attractive pharmacokinetic, safety, and tolerability profiles over the past 20 years has, indeed, increased the treatment options to clinicians, but has not had a substantial effect on the seizure remission for patients with drug-resistant epilepsy.14 A study assessing seizure freedom rates achieved with the newer AEDs including gabapentin, lamotrigine, topiramate, tiagabine, oxcarbazepine, levetiracetam (LEV), zonisamide, and pregabalin administered as an add-on therapy to patients with drug-resistant partial-onset epilepsy showed rather disappointing results.15 Among the included placebo-controlled randomized controlled trials (RCTs), the seizure-free rates for patients who completed the double-blind phase of the trials (between 12 and 21 weeks) ranged from 0.8% to a maximum of 7.1%.15 Although another study has reported a more promising seizure-free rate over 6 months, as high as 14%, with the use of the newer AEDs in real-life clinical practice,16 it is obvious that a considerable proportion of patients with epilepsy are inadequately controlled with the available AEDs.17
The current review aimed to discuss the emerging and current treatment for POS and then focus on the newly invented AED, brivaracetam (BRV), with a thorough examination of its pharmacology, mode of action, pharmacokinetics, comparative safety, efficacy, and tolerability, as well as its impact on patient-centered outcomes (eg, QoL, patient satisfaction).
Outline of targets for therapy and treatments for POS
POS originate from an area of the brain that is abnormally hyperexcitable and is intrinsically capable of increased abnormal firing of individual neurons.18 This hyperexcitable environment is due to the augmentation of excitatory synaptic currents mediated by the neurotransmitters glutamate and aspartate.18,19 This hyperexcitable state is usually quickly terminated by inhibitory currents related to the neurotransmitter gamma-aminobutyric acid (GABA).17 In order to produce a seizure, three conditions are needed:19 first, the existence of hyperexcitable pacemaker area able to develop intrinsic bursts of action potentials that are usually brief and abolished by inhibitory mechanisms related to the neurotransmitter GABA; second, the absence of inhibition that allows this hyperexcitable state in the pacemaker area to be sustained; and the third condition is the ability of this pacemaker to recruit other brain areas.17 Therefore, the currently marketed AEDs or emerging AEDs are targeted at one or more of the above-described mechanisms that are engaged in initiating and sustaining seizures. It is worth mentioning that LEV appears to have a unique mode of action that, at this time, remains to be clearly characterized. It is believed to bind to a specific, as yet unidentified, site on the synaptic plasma membrane.20 The characteristics of the currently available AEDs are summarized in Table 1.
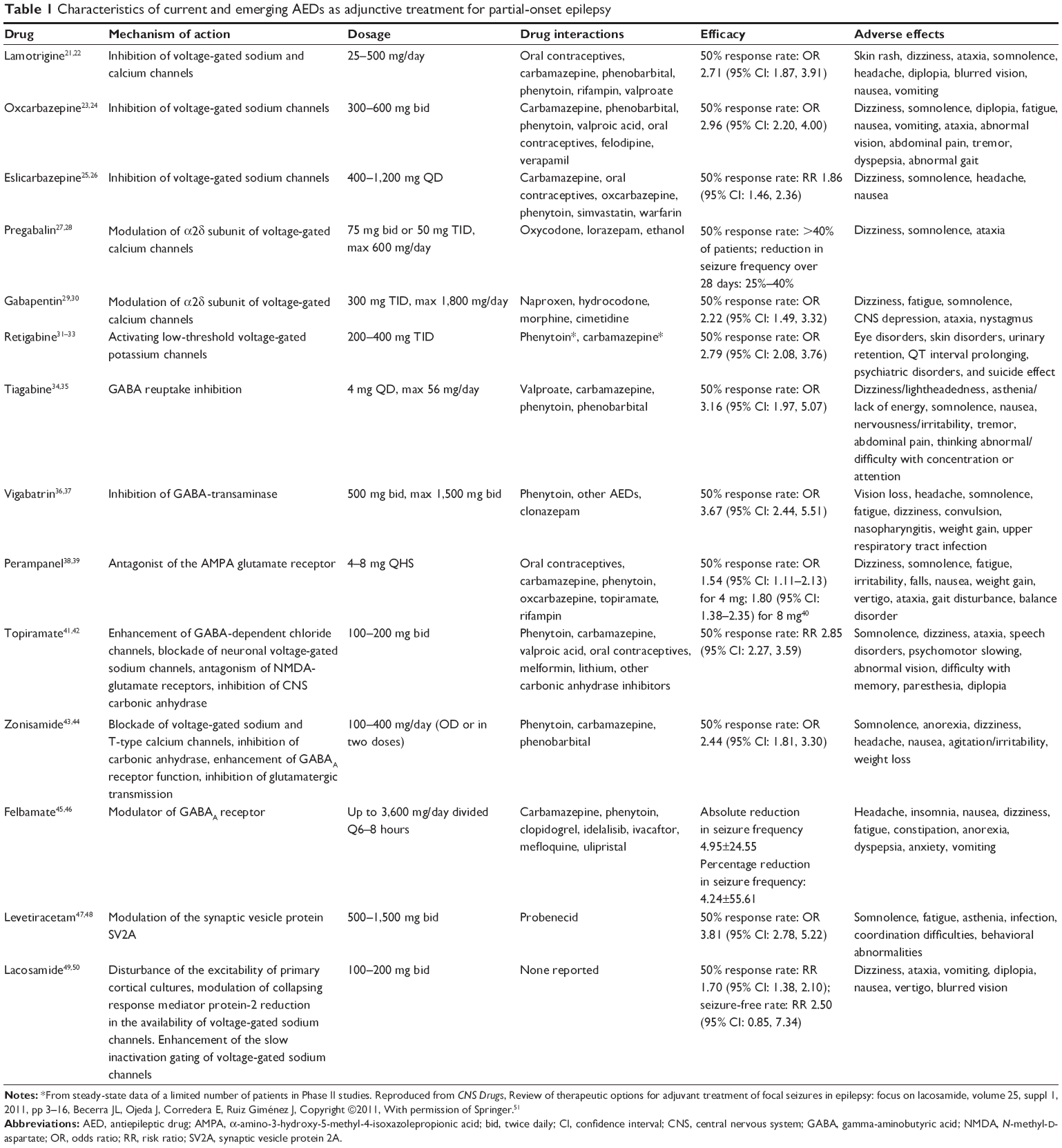 | Table 1 Characteristics of current and emerging AEDs as adjunctive treatment for partial-onset epilepsy |
AEDs that modulate voltage-gated channels
The most common target among AEDs is the sodium channel, which is responsible for the upstroke of the action potential in neurons and other excitable cells.52 Sodium channel blockade is the best characterized mechanism of currently available AEDs. These AEDs prevent the return of the channels to the active state by stabilizing the inactive form. In doing so, repetitive firing of the axons is prevented. Presynaptic and postsynaptic blockade of sodium channels of the axons causes stabilization of the neuronal membranes, blocks and prevents post-tetanic potentiation, limits the development of maximal seizure activity, and reduces the spread of seizures. Traditional sodium channel blockers include phenytoin, carbamazepine (and its derivatives), lamotrigine, and so on.
Another type of voltage-gated channel that AEDs usually bind is the calcium channel. These AEDs, for example, pregabalin and gabapentin, regulate the opening and closing of the α2δ subunit, thus controlling the entry of calcium ions into presynaptic neurons and decreasing the release of various presynaptic neurotransmitters.53,54
One novel ion channel that is targeted by AEDs is the neuronal potassium channel. It acts via enhancing the activity of Kv7.2/Kv7.3 potassium channels by binding within the pore region.55,56 These channels generate the M current, a non-inactivating potassium conductance that regulates the neuronal firing rate at a subthreshold voltage between -60 and -40 mV.57 Enhancing the M current hyperpolarizes the cell membrane toward the potassium equilibrium potential.58 Retigabine (ezogabine) is the AED that falls into this category.
AEDs that enhance GABA-related inhibitory activity
A seizure reflects an imbalance between excitatory and inhibitory activity in the brain, with an increment of excitation over inhibition. The most important inhibitory neurotransmitter in the brain is GABA. It is the predominant inhibitory neurotransmitter in the central nervous system and is released in up to 40% of all synapses.59 Impairment of GABA function is widely recognized to provoke seizures, while facilitation has an anticonvulsant effect.60 GABA inhibitory activity can be achieved via the activation of GABAA receptor (phenobarbital), GABA reuptake inhibitor (tiagabine),20 and GABA transaminase inhibitor (vigabatrin).20
AEDs that modulate the glutamate-mediated excitation
Glutamate is the principal excitatory neurotransmitter in the mammalian brain. Focal injection of glutamate induces seizures in animals, and overactivation of glutamatergic transmission or abnormal glutamate receptor properties are observed in certain experimental seizure models and human epilepsy syndrome. A new AED (perampanel) has a new well-defined mechanism of action by selectively modulating excitatory glutamatergic transmission via modulation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor.
AEDs that have multiple mechanisms of action
Some AEDs may invoke multiple mechanisms of action to exert their anticonvulsive effects. Valproate, a broad-spectrum AED, is reported to block voltage-dependent sodium channel,20 T-type calcium channel,61 and may elevate whole brain GABA levels and potentiate GABA response.60 Topiramate may be involved in the inhibition of sodium and calcium currents, blockade of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainite subtype of glutamate receptor, and facilitation of GABA effects at the GABAA receptor.20 Zonisamide could act through blocking the voltage-gated sodium channel, inhibiting T-type calcium channels,62–64 and enhancing GABA release and inhibiting glutamate release.62,64 Felbamate is believed to be the first effective AED with a direct action on the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor,20 and it also inhibits NMDA/glycine-stimulated increases in intracellular calcium,65 reduces inward currents evoked by NMDA application to striatal neurons,66 and blocks NMDA receptor-mediated excitatory postsynaptic potentials.67
Brivaracetam
Pharmacology
Mode of action
Synaptic vesicle protein 2 (SV2) is a membrane glycoprotein common to all synaptic and endocrine vesicles. The synaptic vesicle protein 2A (SV2A), the primary SV2 isoform, is a widely distributed central nervous system protein, and is believed to participate in the coordination of synaptic vesicle exocytosis and neurotransmitter release.68 This is shown by the observation that SV2A knockout mice develop seizures, which leads to their demise within the first few weeks.68 In contrast, while heterozygous SV2A knockout mice do not demonstrate spontaneous seizures, they exhibit enhanced susceptibility to the convulsant effect of pilocarpine and kainite, a reduced 6 Hz seizure threshold, and an enhanced rate of seizure kindling.69
LEV is thought to exert its main action at a specific binding site – the presynaptically located SV2A, which modulates presynaptic transmitter release.70 BRV is a highly selective and reversible SV2A ligand with a 15- to 30-fold higher affinity than LEV in rat and human brain.71,72 The increased binding affinity of BRV, compared to LEV, to the presynaptically located SV2A corresponds to a higher efficacy in the animal models.73 In addition to this, the modulation of SV2A function by BRV is also believed to contribute to its anticonvulsant effect. Besides, BRV also displays inhibitory activity at neuronal voltage-dependent sodium channels.74 At any rate, the sodium channel modulation represents a distinct activity of BRV compared with LEV.75 Furthermore, compared to LEV, BRV is able to inhibit NMDA-gated currents by up to one third.74 All these modes of action may be associated with the wider spectrum of anticonvulsant mechanism of BRV.
The activity of BRV was studied in both focal and generalized seizure models. In the amygdala-kindled rat, BRV produced a more profound suppression of both motor seizure severity and after-discharge duration than LEV.76 In mice genetically susceptible to audiogenic seizures, BRV offered more potent protection from clonic convulsions than LEV, while in the Genetic Absence Epilepsy Rat from Strasbourg, it exerted a more complete suppression of spike–wave discharge than LEV.77 The anticonvulsant properties of BRV were also investigated in a partially drug-resistant self-sustaining status epilepticus rat model. The combination of diazepam and BRV was able to reduce the duration of active seizures to 3% of controls, which indicated potent anticonvulsant activity of the compound in status epilepticus.78
Pharmacokinetics
BRV is rapidly absorbed after oral administration, with a tmax around 1 hour (ranging from 0.5 to 1.75 hours) and Cmax between 10 and 1,400 mg, when applied in a single dose.76 However, a high fat meal is likely to delay tmax to 3 hours and decrease Cmax by about 28%.79 BRV has a linear fashion of pharmacokinetics over a dose range from 10 to 600 mg. It was evenly absorbed throughout the gastrointestinal tract, as evidenced by the relative area under the curve (AUC) (100% = stomach) of 101%, 98%, and 97% following delivery in the proximal jejunum, distal jejunum, and ascending colon, respectively.76 The metabolic clearance of BRV is increased in a time-dependent manner at supratherapeutic doses and a steady state is reached within a week of repeated administration.76 The terminal elimination half-life of BRV is approximately 8 hours and does not vary with the applied doses.80 Its protein binding is low with a volume of distribution of 0.6 L/kg, marginally lower than total body water.79,81 The primary metabolic pathways of BRV include hepatic hydrolysis of the acetamide group, CYP2C8-mediated hydroxylation, and a combination of these pathways,76 mainly by the CYPSC8 isoform of cytochrome P450 (CYP450) and, to a lesser degree, by the isoforms CYP3A4 and CYP2C19,82 but all metabolites in the human urine (2-[2-oxo-4-propylpyrrolidin-1-yl]butyric acid (35%) and 2-[2-oxo-4-propylpyrrolidin-1-yl]-4-hydroxy-butanamide [<10%]) are not pharmacologically active.79 Its elimination is reliant on metabolism, largely via the activity of CYP450 system; thus, the renal clearance of the parent drug is low at 0.06 mL/min/kg (whereas the metabolites have a high renal clearance). It was reported that in patients with chronic liver disease, total body clearance of BRV may be reduced by 25%–35% and plasma half-life is accordingly prolonged to 14–17 hours,76 while no major adjustment in dosage is required in patients with severe renal impairment without dialysis.83
Pharmacodynamics
Studies in healthy volunteers showed that in high-dose range, BRV was associated with dose-related sedation and decreased alertness.79,81 For subjects on 200, 400, 800 mg daily and placebo, the mean (standard deviation) AUC change from baseline (ΔAUC0–12 hours) on day 7 was 6 (13), 12 (42), 17 (21), and 3 (9), respectively, for the Pentobarbital–Chlorpromazine–Alcohol Group subscale of the Addictive Research Centre Inventory-49 questionnaire (it measures sedative drug effects). The mean (standard deviation) ΔAUC0–12 hours of the visual analog scale (VAS), an alertness scale with higher value indicative of reduced alertness, was 5 (22), 46 (109), 57 (95), and -2 (26), respectively, while no trends of changes on the calmness VAS scale, ataxia (International Cooperative Ataxia Rating Scale Examination), or other neurological examinations were observed.79,81
The potential pharmacodynamic interaction between alcohol and BRV was also investigated. Coadministration of BRV and ethanol in healthy subjects was associated with additive effects on most of the pharmacodynamic variables (eg, saccadic peak velocity, smooth pursuit, adaptive tracking performance, and VAS alertness) without relevant pharmacokinetic interaction between BRV and ethanol.84
Comparative safety, efficacy, and tolerability of BRV
Safety and tolerability
Generally, all studies did not report statistically significant difference between BRV and placebo groups in terms of treatment-emergent adverse events (TEAEs).85–89 Furthermore, no evident trend was observed in the incidence of TEAEs across all doses of BRV. The most commonly reported TEAEs were dizziness, headache, nasopharyngitis, nausea, fatigue, and somnolence from mild to moderate intensity. The TEAEs reported by greater than 5% of patients during the treatment period in each trial are shown in Table 2. A visual inspection of the incidence of TEAE identified that dizziness, somnolence, and influenza more frequently occurred in the BRV-treated group regardless of the doses administered.88 The meta-analysis of five BRV trials also identified no statistically significant association between the administration of BRV and the majority of safety endpoints, including at least one TEAE, drug-related TEAEs, and serious adverse events.90 However, in terms of individual TEAEs, another pooled analysis reported a significant difference between 20 mg BRV and placebo in the incidences of fatigue and nasopharyngitis (relative risk [RR]: 3.00 [95% confidence interval {CI}: 1.20–7.47] and 5.98 [95% CI: 1.36–26.34], respectively). Moreover, increased incidence of fatigue and irritability was also detected in the 50 mg BRV group compared to placebo (RR: 2.38 [95% CI: 1.16–4.88] and 2.95 [95% CI: 1.03–8.44], respectively). In addition, the risk of somnolence was significantly different between 150 mg BRV and placebo groups (RR: 2.21 [95% CI: 1.02–4.80]), while no significant differences were observed for either dose of 5 or 100 mg with respect to the TEAEs recorded in individual studies.91
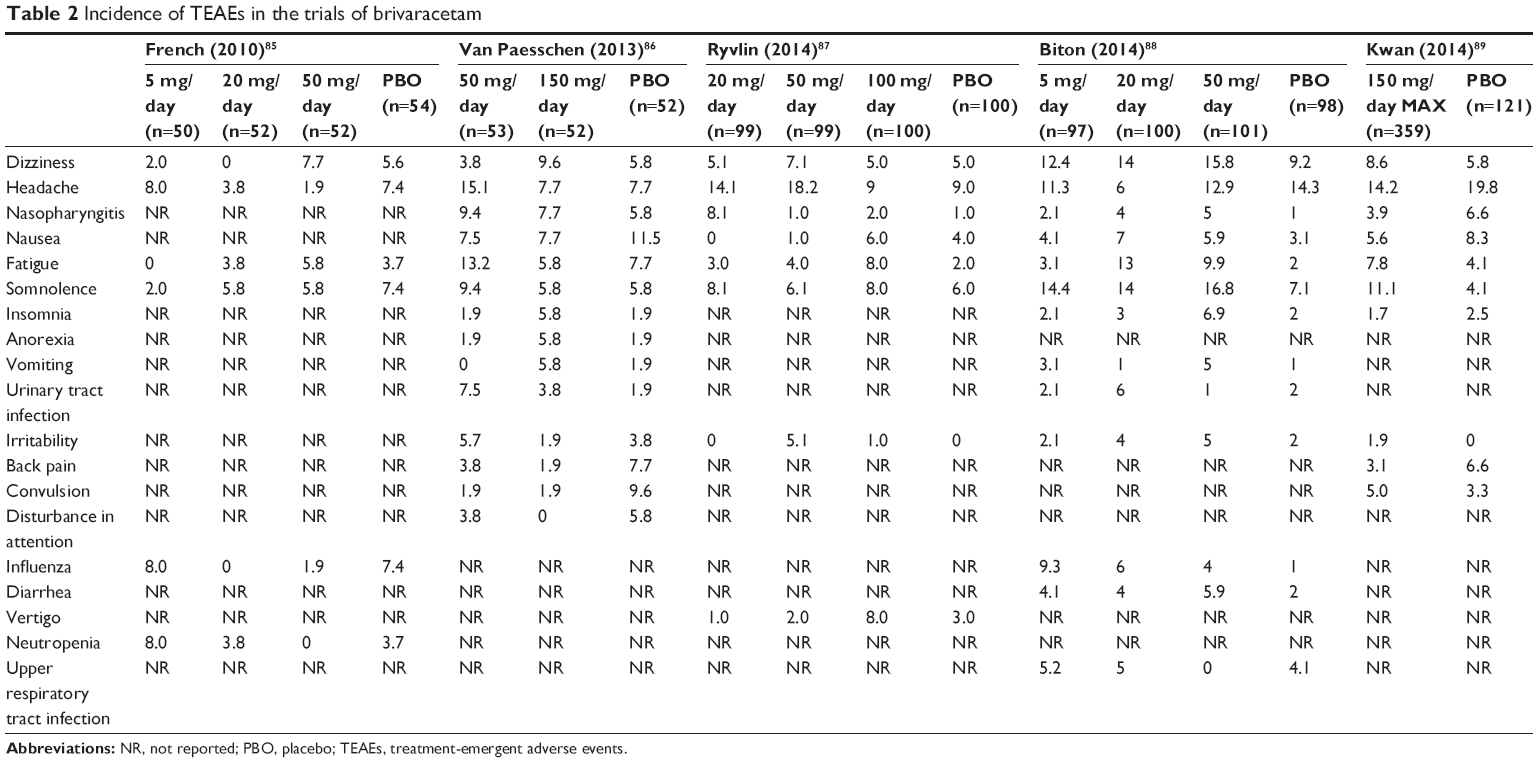 | Table 2 Incidence of TEAEs in the trials of brivaracetam |
Regarding treatment discontinuation, patients withdrew from each trial due to AEs, lack of efficacy, loss to follow-up, and withdrawal of consent due to personal reasons. However, all the RCTs did not show remarkable distinctions in withdrawal rates between BRV and placebo groups (Table 3). Nevertheless, the majority of discontinuation was accounted for by AEs caused by either BRV or concomitant AEDs. The results of meta-analysis on the overall withdrawal rate also indicated no statistical difference between BRV and placebo groups (RR: 1.08, 95% CI: 0.73–1.59; P=0.70).90 The reason for withdrawal as assessed by another meta-analysis also showed consistent and similar results.91
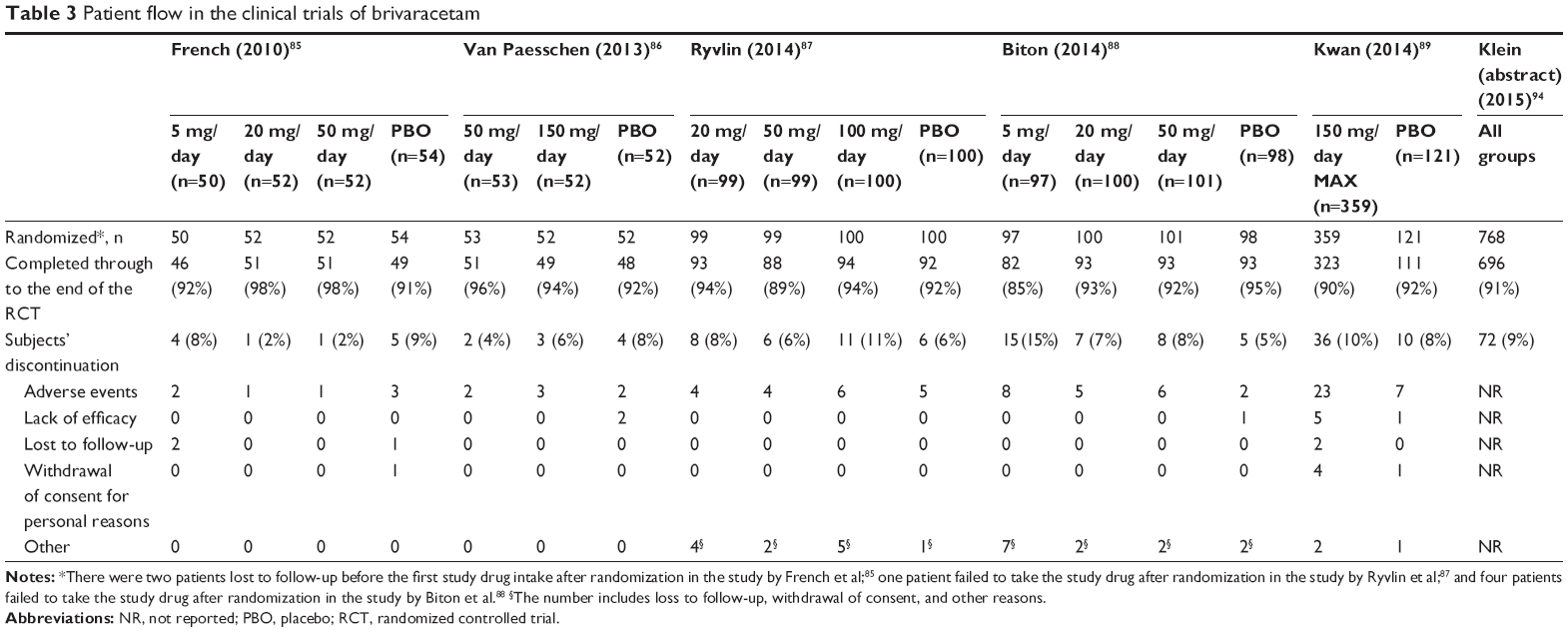 | Table 3 Patient flow in the clinical trials of brivaracetam |
Besides, the effects of BRV on cardiac repolarization and neurocognition were studied in healthy subjects. It was found that the QT interval did not increase with plasma concentration of BRV,92 and BRV did not alter the profile of cognitive, subjective, and electrophysiologic effects, compared to placebo.93
Efficacy
To date, six RCTs have reported the efficacy outcomes of various doses of BRV as adjunctive treatment in a population of patients with partial-onset epilepsies.85–89,94 The percentage reduction in baseline-adjusted POS frequency/week85–89 (or 28 days)94 over placebo was adopted as the primary efficacy outcome across all RCTs, while the responder rate (defined as the percentage of patients who achieved 50% reduction in seizure frequency compared to baseline) was employed as the secondary efficacy outcome. In general, higher dose of BRV is likely to produce significant efficacy outcome, compared to placebo. Among these studies, a Phase IIb trial86 and a Phase III trial89 failed to detect a significant difference in the primary efficacy outcome, while in the other four RCTs, 50 mg/day85,87,88 or 100 and 200 mg/day94 of BRV was found to be more effective than placebo in achieving the primary efficacy outcome. The percentage reduction in baseline-adjusted POS frequency/week ranged from -0.9%88 to 22.1%85 across all the doses. In comparison, a recently published study on two higher doses of BRV (100 and 200 mg/day) reported the most promising outcomes.94 The reduction in baseline-adjusted POS frequency/28 days over placebo was 22.8% (P<0.001) and 23.2% (P<0.001) for BRV 100 and 200 mg/day, respectively. Regarding the rate of responders, more studies reported significant difference for this outcome (Table 4). Nevertheless, the lower doses (ie, 5 and 20 mg/day) used in three RCTs did not show any statistical difference between BRV and placebo in the primary efficacy endpoint. Figures 1 and 2 summarize the ≥50% responder and seizure-free rates across all the RCTs of BRV.
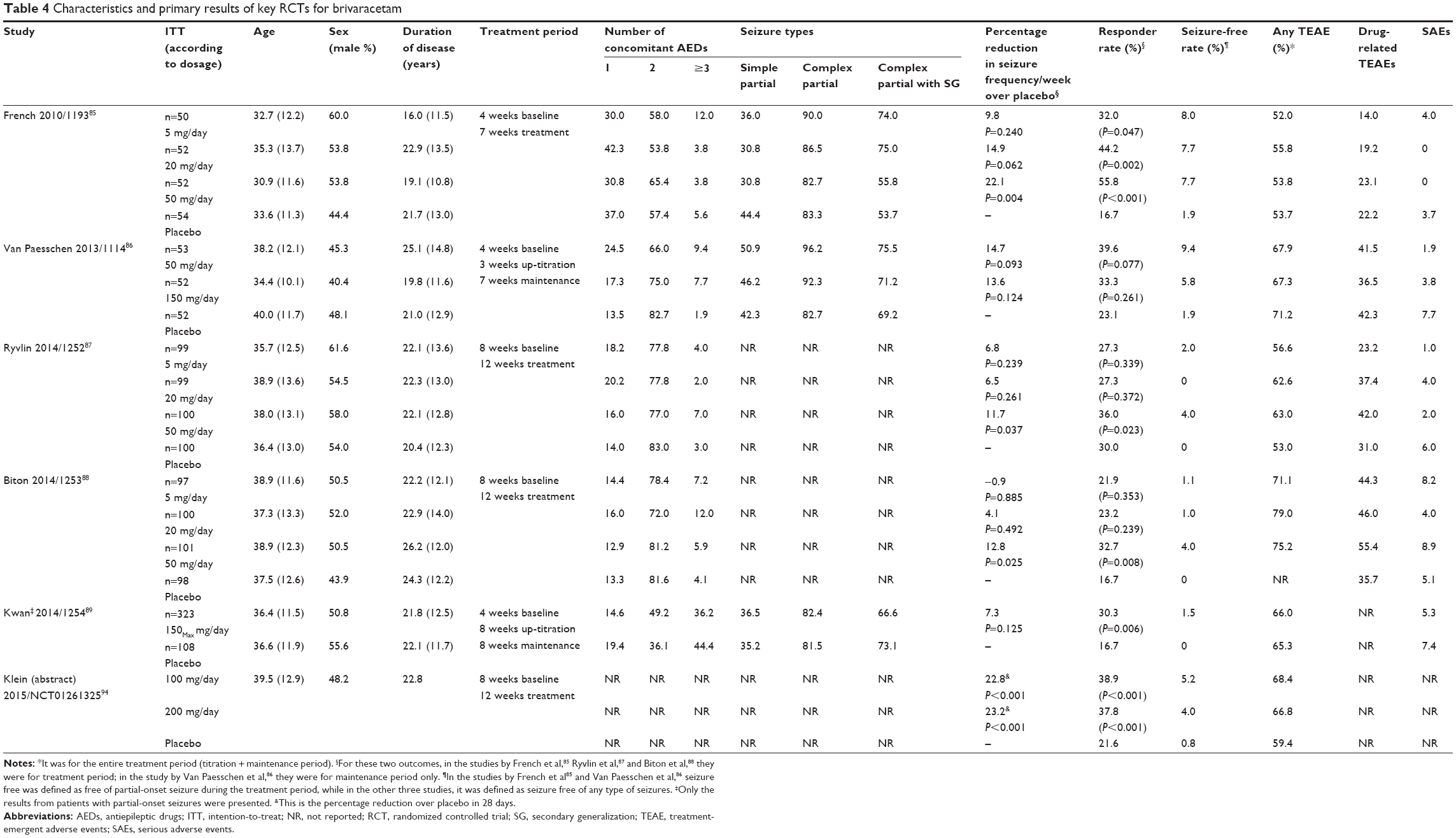 | Table 4 Characteristics and primary results of key RCTs for brivaracetam |
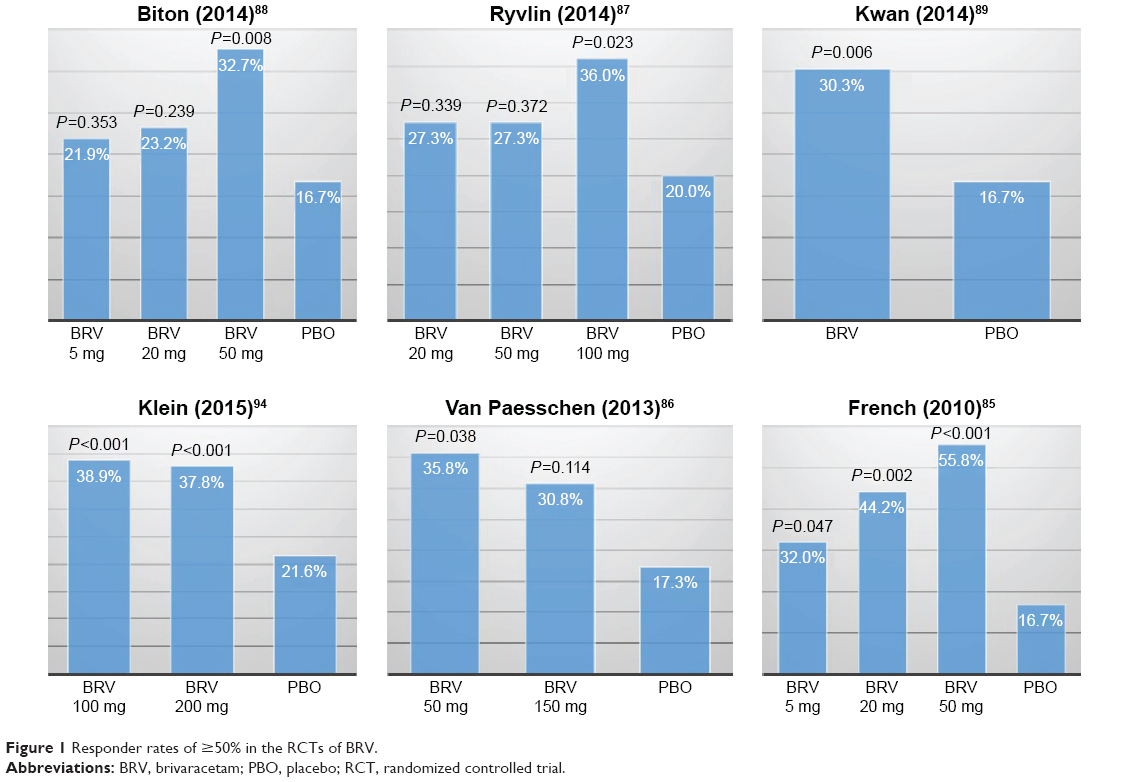 | Figure 1 Responder rates of ≥50% in the RCTs of BRV. |
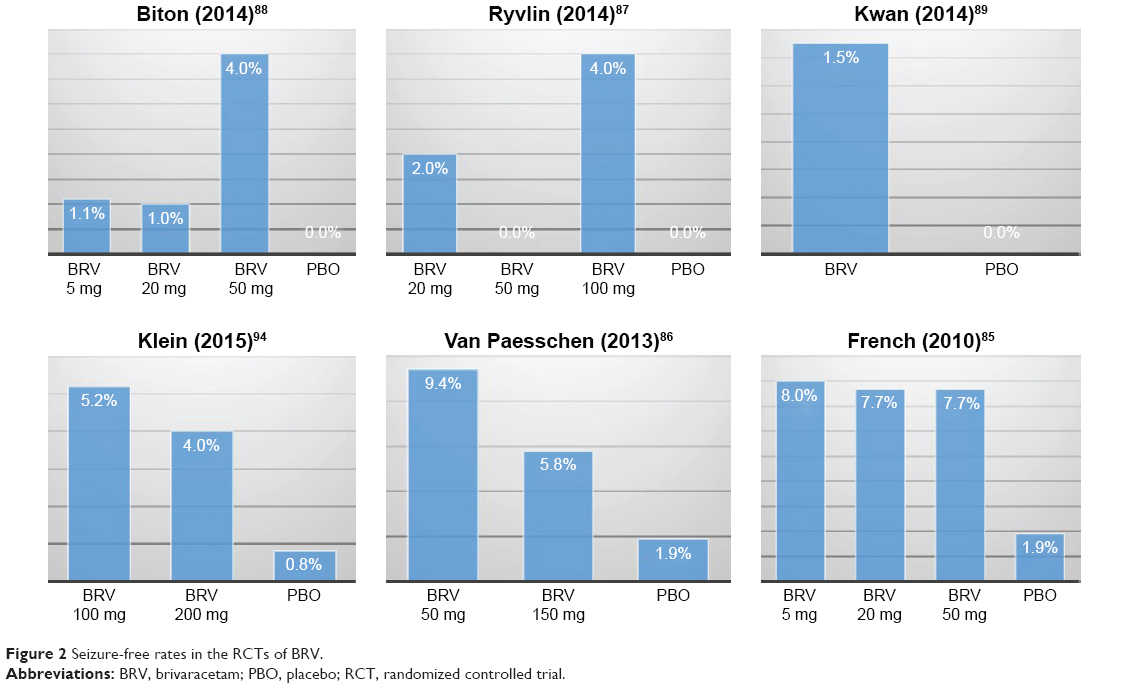 | Figure 2 Seizure-free rates in the RCTs of BRV. |
Two meta-analyses90,91 that synthesized the results of responder rate and seizure-free rate yielded similar results across all doses of BRV from five RCTs.85–89 In a fixed-effects model, the responder rate was significantly higher in BRV group compared with placebo group for doses of 20 mg/day (RR: 1.63, 95% CI: 1.18–2.27; P=0.003), 50 mg/day (RR: 2.00, 95% CI: 1.50–2.66; P<0.001), and 100 mg/day (RR: 1.80, 95% CI: 1.12–2.88; P=0.01).90 Whereas, regarding seizure-free rate reported in three RCTs,85,86,88 significant difference was detected for the dose of 50 mg/day (RR: 5.80, 95% CI: 1.54–21.84; P=0.009) in only one meta-analysis.90 However, it is worth noting that the seizure-free rate was defined as free from POS in two of those three studies.85,86
Patient profiles
Patients with similar epilepsy-specific and demographic characteristics were studied in the six Phase IIb and III RCTs. For recruitment, patients were required to experience at least four85,86,89 or eight 87,88 POS, depending on the length of the baseline period. However, the inclusion criteria across the studies varied slightly: the latest Phase III RCT enrolled subjects with the widest age range (16–80 years old)94 and three other RCTs recruited patients aged between 16 and 70 years, while subjects recruited in the two Phase IIb trials had the narrowest age range (16–65 years old).87–89 It was reported that compared to middle-aged patients with epilepsy, elderly patients were more likely to respond favorably to treatment even at lower doses.95 In fact, other studies have also shown better treatment outcome in elderly patients with epilepsy.96,97 This may give rise to heterogeneity in the study population across RCTs. With regard to the concomitant AEDs, except for the study by Kwan et al89 (with one to three AEDs allowed during the study period), only one or two concomitant AEDs were allowed in all the trials. This may lead to the enrollment of patients with varied levels of uncontrolled POS. Of note, the number of prior AEDs used by patients in the past 5 years in the reported trials varied substantially as well. For instance, in the study by Van Paesschen et al,86 34.6%–45.3% of patients had taken and discontinued ≥5 AEDs during the past 5 years compared to 2.0%–7.0% in the study by Ryvlin et al.87 It has been demonstrated that the rates of seizure freedom and patients with a >50% seizure frequency reduction after administration of new AED decrease as a function of the number of previously failed AEDs.98 So, this would be another source of heterogeneity in patient characteristics among trials. Nonetheless, from two meta-analyses,90,91 the statistical measures (χ2 and I2) did not show considerable heterogeneities across the included RCTs for outcomes such as responder rate and seizure-free rate. This might suggest that the aforementioned differences in the study populations were not significant in producing heterogeneity.
Patient-reported outcomes
Epilepsy, as a chronic disorder, has considerable negative effect on people’s day-to-day functioning. Meanwhile, seizures are still poorly controlled in around 30% of patients, even with multiple antiepileptic therapies. For these patients, traditional clinical outcomes that measure the treatment effect, such as seizure frequency, seizure-free days, and responder rate, might not be sufficient to capture all the benefits generated by treatment and are also incapable of reflecting the total impact of epilepsy on patient’s well-being and their own perception of treatment effect. Consequently, an increasing number of RCTs integrated the measurement of QoL of patients into the design of epilepsy trials to provide a more holistic evaluation of treatment effects and adverse effects of newly invented AEDs. To date, the QoL in patients with POS administered BRV has not been reported. Instead, the only study in patients with genetically ascertained Unverricht–Lundborg disease with action myoclonus reported a significant improvement in QoL as measured by Quality of Life in Epilepsy-30 at BRV 50 and 150 mg/day, despite its statistically significant effect on action myoclonus.99
At the time of this review, three Phase III, open-label, multicenter, flexible-dose (up to a maximum dose of 150 mg/day), long-term follow-up trials (NO1125, NCT00175916; NO1199, NCT00150800; and NO1315, NCT00761774) are still ongoing to evaluate the long-term safety/tolerability and maintenance of efficacy of BRV in patients with POS who had participated in previous trials. Although the proportion of patients from the BRV group who entered into the open phase of the individual trials did not generally show a favorable trend compared to the placebo group, a meta-analysis of trials of adjunctive AEDs in adults with drug-resistant focal epilepsy found the responder rates to placebo virtually double between 1989 and 2009.100 This may partially explain the unnoticeable difference between BRV and placebo groups. Anyway, when the results from those trials are released, a more comprehensive picture of BRV may become available.
Conclusion
Monotherapy is the first step to try for all patients with newly diagnosed epilepsy. Following an ineffective or intolerable initial monotherapy, the next step is to add or switch to another AED. Evidence has indicated that add-on therapy might be more effective when started immediately after the first drug failure rather than after a second drug has also failed.12 After failing to achieve sustained seizure freedom with two tolerated, appropriately chosen and administered AEDs (regardless of monotherapy or multitherapy) in a patient,3 the patient could be classified as having drug-resistant epilepsy. When add-on therapy is warranted (for those who either failed the first monotherapy or have drug-resistant epilepsy), seizure characteristics, drug and patient factors become an important consideration when implementing the management strategies.101 Selection of an AED is usually made based on the seizure type, spectrum of activity, tolerability, drug interaction, and patient’s personal circumstances.101
Similar to all the other newer generations of AEDs, BRV has been investigated as an adjunctive therapy for adult patients with uncontrolled partial-onset epilepsies (and in very limited number of patients with generalized epilepsies89). So far, BRV appears to be a useful new addition to adjunctive treatment option for partial-onset epilepsy, particularly for patients who had not achieved adequate seizure control with one or more other adjunctive therapies.85,87,88,94 In general, the newer group of AEDs has been associated with better safety and tolerability, fewer AEs, and improved QoL, compared with more traditional agents. However, with no direct head-to-head comparison among these newly invented drugs, it is extremely difficult for clinicians to choose from a dozen of these newer agents. It was even reported that clinicians often choose therapy according to their own comfort level with the particular AEDs, rather than weighing up all disadvantages and advantages of older versus newer, less-familiar drugs.8 Hence, drugs with simpler dosing regimens and uncomplicated titration schedules, undoubtedly, have an advantage. BRV, with a twice-daily dosing regimen and well tolerated by patients even at a dose of 200 mg/day without up-titration, would be an attractive option.94 But when LEV and BRV are compared, given the similarity in chemical structure and possible pharmacology between them, the choice is more complicated. The relationship between primary efficacy outcome in BRV pivotal trials and history of LEV administration (prior LEV, concomitant LEV, and LEV-naïve) was explored.88 Of interest, LEV-naïve patients and those with prior LEV histories achieved numerically (though statistically not significant) greater reduction in seizure frequency compared to placebo,85,86,88 while concomitant LEV might reduce the efficacy of BRV.
As BRV is still under review by different regulatory agencies (including US Food and Drug Administration and European Medicines Agency) for the treatment of POS in patients of age 16 years and above with epilepsy, no data about its use in real-life clinical practice is available at the moment. Meanwhile, it has gained an orphan drug status for development in progressive and symptomatic myoclonic seizures in Europe and US, respectively.102 In summary, the many favorable attributes of BRV, like its high oral efficacy, good tolerability, dosing regimen, and minimal drug interaction, make it a promising antiepileptic choice for patients with uncontrolled partial-onset epilepsy.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization [homepage on the Internet]. Epilepsy; 2016 [updated February, 2016]. Available from: http://www.who.int/mediacentre/factsheets/fs999/en/. Accessed August 18, 2015. | ||
Sander JW. The epidemiology of epilepsy revisited. Curr Opin Neurol. 2003;16:165–170. | ||
Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–1077. | ||
Perucca E, Beghi E, Dulac O, Shorvon S, Tomson T. Assessing risk to benefit ratio in antiepileptic drug therapy. Epilepsy Res. 2000;41:107–139. | ||
Kwan P, Brodie MJ. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure. 2002;11:77–84. | ||
Stefan H, Halasz P, Gil-Nagel A, et al. Recent advances in the diagnosis and treatment of epilepsy. Eur J Neurol. 2001;8:519–539. | ||
Fisher RS, Eggleston KS, Wright CW. Vagus nerve stimulation magnet activation for seizures: a critical review. Acta Neurol Scand. 2015;131:1–8. | ||
Brodie MJ, Kwan P. The star systems – overview and use in determining antiepileptic drug choice. CNS Drugs. 2001;15:1–12. | ||
Leppik IE. Monotherapy and polypharmacy. Neurology. 2000;55:S25–S29. | ||
Beghi E, Perucca E. The management of epilepsy in the 1990s. Acquisitions, uncertainties and priorities for future research. Drugs. 1995;49:680–694. | ||
Brodie MJ, French JA. Management of epilepsy in adolescents and adults. Lancet. 2000;356:323–329. | ||
Kwan P, Brodie MJ. Epilepsy after the first drug fails: substitution or add-on? Seizure. 2000;9:464–468. | ||
Schmitz B, Montouris G, Schauble B, Caleo S. Assessing the unmet treatment need in partial-onset epilepsy: looking beyond seizure control. Epilepsia. 2010;51:2231–2240. | ||
Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia. 2001;42:1255–1260. | ||
Gazzola DM, Balcer LJ, French JA. Seizure-free outcome in randomized add-on trials of the new antiepileptic drugs. Epilepsia. 2007;48:1303–1307. | ||
Callaghan BC, Anand K, Hesdorffer D, Hauser WA, French JA. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382–389. | ||
Nasreddine W, Beydoun A, Atweh S, Abou-Khalil B. Emerging drugs for partial onset seizures. Expert Opin Emerg Drugs. 2010;15:415–431. | ||
Dichter MA. Basic mechanisms of epilepsy: targets for therapeutic intervention. Epilepsia. 1997;38 Suppl 9:S2–S6. | ||
Najm IM, Janigro D, Babb TL. Mechanisms of epileptogenesis and experimental models of seizures. In: Elaine W, Ajay G, Deepak KL editors. The Treatment of Epilepsy: Principles and Practice. 3 ed. Philadelphia: Lippincott Williams & Wilkins; 2001:33–34. | ||
Kwan P, Sills GJ, Brodie MJ. The mechanisms of action of commonly used antiepileptic drugs. Pharmacol Ther. 2001;90:21–34. | ||
Ramaratnam S, Marson AG, Baker GA. Lamotrigine add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2001:CD001909. | ||
Lamotrigine (Lamictal). Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Lamictal/pdf/LAMICTAL-PI-MG.PDF. Accessed August 25, 2015. | ||
Castillo S, Schmidt DB, White S. Oxcarbazepine add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2000:CD002028. | ||
Oxcarbazepine (Trileptal) Prescribing Information. Available from: https://www.pharma.us.novartis.com/product/pi/pdf/trileptal.pdf. Accessed August 25, 2015. | ||
Chang XC, Yuan H, Wang Y, Xu HQ, Zheng RY. Eslicarbazepine acetate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2011:CD008907. | ||
Eslicarbazepine acetate (Aptiom) Prescribing Information. Available from: http://www.aptiom.com/Aptiom-Prescribing-Information.pdf. Accessed August 25, 2015. | ||
Uthman BM, Bazil CW, Beydoun A, et al. Long-term add-on pregabalin treatment in patients with partial-onset epilepsy: pooled analysis of open-label clinical trials. Epilepsia. 2010;51:968–978. | ||
Pregabalin (Lyrica) Prescribing Information. Available from: http://labeling.pfizer.com/ShowLabeling.aspx?id=561. Accessed August 25, 2015. | ||
Marson AG, Kadir ZA, Hutton JL, Chadwick DW. Gabapentin for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2000:CD001415. | ||
Gabapentin (Neutontin) Prescribing Information. Available from: http://labeling.pfizer.com/ShowLabeling.aspx?id=630. Accessed August 25, 2015. | ||
Craig D, Rice S, Paton F, Fox D, Woolacott N. Retigabine for the adjunctive treatment of adults with partial-onset seizures in epilepsy with and without secondary generalization: a NICE single technology appraisal. Pharmacoeconomics. 2013;31:101–110. | ||
Retigabine (Trobalt) Prescribing Information. Available from: https://www.medicines.org.uk/emc/medicine/24527. Accessed August 25, 2015. | ||
Splinter MY. Ezogabine (retigabine) and its role in the treatment of partial-onset seizures: a review. Clin Ther. 2012;34:1845–1856. e1841. | ||
Pereira J, Marson AG, Hutton JL. Tiagabine add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2002:CD001908. | ||
Tiagabine (Gabitril) Prescribing Information. Available from: http://www.gabitril.com/pdf/Gabitril_PI_GAB-012.pdf. Accessed August 25, 2015. | ||
Vigabatrin (Sabril) Prescribing Information. Available from: http://www.lundbeck.com/upload/us/files/pdf/Products/Sabril_PI_US_EN.pdf. Accessed August 25, 2015. | ||
Marson AG, Kadir ZA, Hutton JL, Chadwick DW. The new antiepileptic drugs: a systematic review of their efficacy and tolerability. Epilepsia. 1997;38:859–880. | ||
Faulkner MA. Perampanel: a new agent for adjunctive treatment of partial seizures. Am J Health Syst Pharm. 2014;71:191–198. | ||
Perampanel (Fycompa) Prescribing Information. Available from: http://ca.eisai.com/pdf/new/Fycompa-PM-April%203-13_EN_Eisai.pdf. Accessed August 25, 2015. | ||
Hsu WW, Sing CW, He Y, Worsley AJ, Wong IC, Chan EW. Systematic review and meta-analysis of the efficacy and safety of perampanel in the treatment of partial-onset epilepsy. CNS Drugs. 2013;27:817–827. | ||
Pulman J, Jette N, Dykeman J, Hemming K, Hutton JL, Marson AG. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008:CD001417. | ||
Topiramate (Topomax) Prescribing Information. Available from: https://www.topamax.com/topamax/assets/topamax.pdf. Accessed August 25, 2015. | ||
Chadwick DW, Marson AG. Zonisamide add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2005:CD001416. | ||
Zonisamide (Zonegran) Prescribing Information. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020789s022s0251bl.pdf. Accessed August 25, 2015. | ||
Shi LL, Dong JC, Ni HJ, Geng JS, Wu TX. Felbamate as an add-on therapy for refractory epilepsy. Cochrane Database Syst Rev. 2014;7:CD008295. | ||
Felbamate (Felbatol) Prescribing Information. Available from: http://www.felbatol.com/. Accessed August 25, 2015. | ||
Chaisewikul R, Privitera MD, Hutton JL, Marson AG. Levetiracetam add-on for drug-resistant localization related (partial) epilepsy. Cochrane Database Syst Rev. 2001:CD001901. | ||
Levetiracetam (Keppra) Prescribing Information. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021035s078s080,021505s021s024lbl.pdf. Accessed August 25, 2015. | ||
Weston J, Shukralla A, McKay AJ, Marson AG. Lacosamide add-on therapy for partial epilepsy. Cochrane Database Syst Rev. 2015;6:CD008841. | ||
Lacosamide (Vimpat) Prescribing Information. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000863/WC500050338.pdf. Accessed August 25, 2015. | ||
Becerra JL, Ojeda J, Corredera E, Ruiz Giménez J. Review of therapeutic options for adjuvant treatment of focal seizures in epilepsy: focus on lacosamide. CNS Drugs. 2011;25(Suppl 1):3–16. | ||
Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. | ||
Rogawski MA, Bazil CW. New molecular targets for antiepileptic drugs: alpha 2 delta, SV2A, and K(v)7/KCNQ/M potassium channels. Curr Neurol Neurosci Rep. 2008;8:345–352. | ||
Beydoun A, Nasreddine W, Atweh S. Efficacy and tolerability of pregabalin in partial epilepsy. Expert Rev Neurother. 2008;8:1013–1024. | ||
Rundfeldt C, Netzer R. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells transfected with human KCNQ2/3 subunits. Neurosci Lett. 2000;282:73–76. | ||
Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. | ||
Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. | ||
Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. | ||
Olsen RW, Avoli M. GABA and epileptogenesis. Epilepsia. 1997;38:399–407. | ||
Loscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog Neurobiol. 1999;58:31–59. | ||
Kelly KM, Gross RA, Macdonald RL. Valproic acid selectively reduces the low-threshold (T) calcium current in rat nodose neurons. Neurosci Lett. 1990;116:233–238. | ||
Holder JL Jr, Wilfong AA. Zonisamide in the treatment of epilepsy. Expert Opin Pharmacother. 2011;12:2573–2581. | ||
Schulze-Bonhage A. Zonisamide in the treatment of epilepsy. Expert Opin Pharmacother. 2010;11:115–126. | ||
Biton V. Clinical pharmacology and mechanism of action of zonisamide. Clin Neuropharmacol. 2007;30:230–240. | ||
Taylor LA, McQuade RD, Tice MA. Felbamate, a novel antiepileptic drug, reverses N-methyl-D-aspartate/glycine-stimulated increases in intracellular Ca2+ concentration. Eur J Pharmacol. 1995;289:229–233. | ||
Pisani A, Stefani A, Siniscalchi A, Mercuri NB, Bernardi G, Calabresi P. Electrophysiological actions of felbamate on rat striatal neurones. Br J Pharmacol. 1995;116:2053–2061. | ||
Pugliese AM, Corradetti R. Effects of the antiepileptic drug felbamate on long-term potentiation in the CA1 region of rat hippocampal slices. Neurosci Lett. 1996;215:21–24. | ||
Crowder KM, Gunther JM, Jones TA, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci U S A. 1999;96:15268–15273. | ||
Leclercq K, Kaminski R, Dassesse D, Klitgaard H, Matagne A. Seizure susceptibility of SV2A heterozygous mice in models of temporal lobe epilepsy. Program No. 492.17. In: Neuroscience Meeting Planner. San Diego, CA: Society for Neurosciences; 2007. | ||
Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. PNAS. 2004;101:9861–9866. | ||
Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol. 2011;664:36–44. | ||
Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. 2008;154:1662–1671. | ||
Schulze-Bonhage A. Brivaracetam for the treatment of epilepsy. Expert Opin Pharmacother. 2011;12:1959–1966. | ||
Zona C, Pieri M, Carunchio I, Curcio L, Klitgaard H, Margineanu DG. Brivaracetam (ucb 34714) inhibits Na(+) current in rat cortical neurons in culture. Epilepsy Res. 2010;88:46–54. | ||
Margineanu DG, Klitgaard H. Brivaracetam inhibits spreading depression in rat neocortical slices in vitro. Seizure. 2009;18:453–456. | ||
von Rosenstiel P. Brivaracetam (UCB 34714). Neurotherapeutics. 2007;4:84–87. | ||
Matagne AC, Kenda BM, Michel P, Klitgaard H. ucb 34714, a new pyrrolidone derivative: comparison with levetiracetam in animal models of chronic epilepsy in vivo. Epilepsia. 2003;44:260. | ||
Wasterlain U, Suchomelova L, Matagne A, et al. Brivaracetam is a potent anticonvulsant in experimental status epilepticus. Epilepsia. 2005;46:219–220. | ||
Sargentini-Maier ML, Rolan P, Connell J, et al. The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after single increasing oral doses in healthy males. Br J Clin Pharmacol. 2007;63:680–688. | ||
Rolan P, Pigeolet E, Stockis A. UCB 34714: Single and multiple rising dose safety, tolerability, and pharmacokinetics in healthy subjects. Epilepsia. 2004;45:314–315. | ||
Rolan P, Sargentini-Maier ML, Pigeolet E, Stockis A. The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after multiple increasing oral doses in healthy men. Br J Clin Pharmacol. 2008;66:71–75. | ||
Whomsley R, Brochot A, Dell’Aiera S. Identification of the cytochrome P450 isoforms responsible for the hydroxylation of Brivaracetam. AAPS J. 2007;9:3408. | ||
Sargentini-Maier ML, Sokalski A, Boulanger P, Jacobs T, Stockis A. Brivaracetam disposition in renal impairment. J Clin Pharmacol. 2012;52:1927–1933. | ||
Stockis A, Kruithof AC, Van Gerven JM, De Kam ML, Watanabe S, Peeters PA. Interaction study between brivaracetam and ethanol in healthy subjects. Epilepsy Curr. 2015;15:332. | ||
French JA, Costantini C, Brodsky A, von Rosenstiel P, Group NS. Adjunctive brivaracetam for refractory partial-onset seizures: a randomized, controlled trial. Neurology. 2010;75:519–525. | ||
Van Paesschen W, Hirsch E, Johnson M, Falter U, von Rosenstiel P. Efficacy and tolerability of adjunctive brivaracetam in adults with uncontrolled partial-onset seizures: a phase IIb, randomized, controlled trial. Epilepsia. 2013;54:89–97. | ||
Ryvlin P, Werhahn KJ, Blaszczyk B, Johnson ME, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double-blind, randomized, placebo-controlled trial. Epilepsia. 2014;55:47–56. | ||
Biton V, Berkovic SF, Abou-Khalil B, Sperling MR, Johnson ME, Lu S. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: A phase III randomized, double-blind, placebo-controlled trial. Epilepsia. 2014;55:57–66. | ||
Kwan P, Trinka E, Van Paesschen W, Rektor I, Johnson ME, Lu S. Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: results of a phase III, double-blind, randomized, placebo-controlled, flexible-dose trial. Epilepsia. 2014;55:38–46. | ||
Ma J, Huang S, You C. Adjunctive brivaracetam for patients with refractory partial seizures: a meta-analysis of randomized placebo-controlled trials. Epilepsy Res. 2015;114:59–65. | ||
Tian X, Yuan M, Zhou Q, Wang X. The efficacy and safety of brivaracetam at different doses for partial-onset epilepsy: a meta-analysis of placebo-controlled studies. Expert Opin Pharmacother. 2015;16:1755–1767. | ||
Rosillon D, Astruc B, Hulhoven R, et al. Effect of brivaracetam on cardiac repolarisation – a thorough QT study. Curr Med Res Opin. 2008;24:2327–2337. | ||
Meador KJ, Gevins A, Leese PT, Otoul C, Loring DW. Neurocognitive effects of brivaracetam, levetiracetam, and lorazepam. Epilepsia. 2011;52:264–272. | ||
Klein P, Schiemann I, Sperling M, et al. A randomized double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adults patients with partial-onset seizures. In: 67th American Academy of Neurology Annual Meeting, AAN; 2015. Washington DC: Neurology; 2015. | ||
Ramsay R, Rowan A, Slater J, Collins J, Nemire R, Ortiz W. Effect of age on epilepsy and its treatment: results from the VA Cooperative Study. Epilepsia. 1994;35:91. | ||
Cockerell OC, Johnson AL, Sander JWAS, Shorvon SD. Prognosis of epilepsy: a review and further analysis of the first nine years of the British National General Practice Study of Epilepsy, a prospective population-based study. Epilepsia. 1997;38:31–46. | ||
Mattson RH, Cramer JA, Collins JF. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic clonic seizures in adults. The Department of Veterans Affairs Epilepsy Cooperative Study No. 264 Group. N Engl J Med. 1992;327:765–771. | ||
Schiller Y, Najjar Y. Quantifying the response to antiepileptic drugs: effect of past treatment history. Neurology. 2008;70:54–65. | ||
Kalviaiinen R, Genton P, Andermann E, et al. Brivaracetam in patients with Unverricht-Lundborg disease: Results from two randomized, placebo-controlled, double-blind studies. Epilepsia. 2009;50:47. | ||
Rheims S, Perucca E, Cucherat M, Ryvlin P. Factors determining response to antiepileptic drugs in randomized controlled trials. A systematic review and meta-analysis. Epilepsia. 2011;52:219–233. | ||
Sander JW. The use of antiepileptic drugs – Principles and practice. Epilepsia. 2004;45:28–34. | ||
Johannessen Landmark C, Johannessen SI. Pharmacological management of epilepsy: recent advances and future prospects. Drugs. 2008;68:1925–1939. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.