")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 2
Emergence of a metalloproteinase/phospholipase A2 axis of systemic inflammation
Authors Fernandez-Patron C, Leung D
Received 9 May 2015
Accepted for publication 2 July 2015
Published 13 August 2015 Volume 2015:2 Pages 29—38
DOI https://doi.org/10.2147/MNM.S48748
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor William Parks
Carlos Fernandez-Patron,1 Dickson Leung2
1Department of Biochemistry, Cardiovascular Research Centre, Mazankowski Alberta Heart Institute, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, Alberta, Canada, 2Department of Biochemistry, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, Alberta, Canada
Abstract: We review select aspects of the biology of matrix metalloproteinases (MMPs) with a focus on the modulation of inflammatory responses by MMP-2. MMP-2 is a zinc- and calcium-dependent endoprotease with substrates including extracellular matrix proteins, vasoactive peptides, and chemokines. Humans and mice with MMP-2 deficiency exhibit a predominantly inflammatory phenotype. Recent research shows that MMP-2 deficient mice display elevated activity of a secreted phospholipase A2 in the heart. Additionally, MMP-2 deficient mice exhibit abnormally high prostaglandin E2 levels in various organs (ie, the heart, brain and liver), signs of inflammation and exacerbated lipopolysaccharide-induced fever. We briefly review the biology of sPLA2 enzymes to propose the existence of a heart-centric MMP-2/sPLA2 axis of systemic inflammation. Moreover, we postulate that PLA2 activation is induced by chemokines, whose ability to signal inflammation is regulated in a tissue-specific fashion by MMPs. Thus, genetic and pharmacologically induced MMP deficiencies can be expected to perturb PLA2-mediated inflammatory mechanisms.
Keywords: MMPs, inflammation, chemokines, secreted phospholipase A2
Introduction
The 1960s saw the demonstration that mammalian systems make collagenases,1,2 a discovery followed by the cloning of a superfamily of extracellular matrix metalloproteinases (MMPs). Today, over 20 different zinc- and calcium-dependent MMPs are known, each capable of degrading numerous substrates including (but not limited to) extracellular matrix components, and each involved in numerous biological processes and disease conditions.3
The focal point of this review is MMP-2, a ubiquitous and constitutive 72 kDa type IV collagenase. MMP-2 binds collagen through the fibronectin type II repeat in its catalytic domain and cleaves numerous extracellular matrix components including native collagens (types IV, V, VII, and X), denatured collagens, elastin, and heparin-sulfate proteoglycans that are extracellular matrix receptors for signaling molecules. MMP-2-mediated cleavage of these extracellular components is important for the integrity of the matrix and for signaling events that impact cell behavior. An illustrative example is MMP-2-mediated cleavage of decorin. Decorin is a multifunctional proteoglycan with a core protein that has a serine covalently linked to a dermatan/chondroitin sulphate glycosaminoglycan chain. The integrity of extracellular decorin is important for normal collagen fibrillogenesis. Decorin is cleaved by MMP-2, which impairs decorin’s ability to sequester transforming growth factor-β, a master regulator of fibrosis signaling through the SMADs.4,5
A striking and most interesting feature of MMPs (and MMP-2 in particular) is its high promiscuity in terms of substrates, which include non-extracellular matrix proteins and peptides.6,7 A relevant example with potential pathophysiological significance is the discovery that MMP-2 binds through its hemopexin-like domain and cleaves the CC motif chemokine, monocyte chemoattractant protein-3 (MCP-3, ~9 kDa, encoded by Ccl7). Cleavage of MCP-3 at a Gly4/Ile5 bond converts it into a general CC-chemokine receptor antagonist, which dampens inflammation.8 Interestingly, MCP-3 is also cleaved at the Gly4/Ile5 bond by MMP-1, -3, -13, and -14. MCP-1 is cleaved at its Ala4/Leu5 bond by MMP-1 and MMP-3, while MCP-2 is cleaved at its Ser4/Val5 bond by MMP-3, and MCP-4 is cleaved at its Ala4/Leu5 bond by MMP-1 and MMP-3.9 In addition to MCPs, MMP-2 is also known to cleave other substrates relating to inflammation. For example, MMP-2 cleaves CX3CL1, a pro-inflammatory protein, and possibly allows for protective effects against rheumatoid arthritis through the suppression of macrophage influx. Cleavage of Ym1, S100A8 or S100A9 by MMP-2 or MMP-9 reduces the chemotactic activity of these proteins.7 Thus, MMPs and substrates such as chemokines likely have interrelated functions in the context of inflammation and innate immunity.9,10 In the context of cardiovascular regulation, the first MMP-2 substrates to be identified were vasoactive peptides, ie, big endothelin-1,11 calcitonin gene-related peptide, and adrenomedullin.12,13 A striking similarity between these peptides is that MMP-2 attacks them at Gly/Leu bonds, a peptide bond that is homologous to amino acid sequences targeted by MMP-2 in interstitial collagen (Gly-Leu or Gly-Ile), laminin-5 (Ala-Leu), and MCP-3 (Gly-Leu).
Another interesting observation is that the proteolytic cleavage of any of these peptides would be expected to impact vascular contractility. Possible mechanisms include promoting vasoconstriction or reducing vasodilation, although the promotion of vasodilation is a potential outcome in the case of endothelin-1.11–14 When experimental animals with spontaneous or agonist-induced hypertension are administered an MMP inhibitor such as doxycycline, the severity of their hypertension is consistently attenuated.15–19 MMP-2 activity may also impact vascular tone in normal physiological processes such as aging20–22 and pregnancy,14,23,24 and in hypertensive conditions such as pregnancy-induced hypertension.14,20–22,25–27 Whether MMP-2 acts solely through the cleavage and regulation of vasoactive substrates or other mechanisms such as inflammation dampening (eg, the cleavage of pro-inflammatory chemokines that exert indirect vascular tone alterations) warrants further research.
Although there is a paucity of mechanisms, there is an emerging link between lipid metabolic gene expression and activity of MMPs – a topic we reviewed previously.3 For instance, MMP-2 may mediate a cardioprotective mechanism involving inhibition of the sterol regulatory element binding protein-2 (SREBP-2) pathway in the heart with the absence of MMP-2, predisposing to hypertensive heart disease and resistance to statins.28 Mice lacking MMP-2 show resistance to high fat-induced obesity.29 Different metabolic phenotypes have been exposed in mice lacking MMP-2, MMP-9, or tissue inhibitors of MMPs.29–38 Lipoparticle receptor and apoplipoprotein cleavage have been demonstrated for MMP-2 and MMP-14, and could lead to lipoparticle dysregulation.39–46 We recently reported that MMP-2 can cleave pro-protein convertase subtilisin/kexin type 9 (PCSK9, Pcsk9, ~70 kDa) in the pro-peptide and C-terminal domains.47 An important function of PCSK9 is to bind the receptor for low-density lipoprotein (LDLR) and reroute it from the recycling to the lysosomal degradation pathway. Consequently, PCSK9 binding to LDLR reduces LDLR bioavailability and impairs hepatic uptake of plasma LDL cholesterol. Mutations in PCSK9 can either cause or protect from hypercholesterolemia.48–50 The observation that MMP-2 binds and cleaves PCSK9 suggests a role for MMP-2 as an inhibitor of PCSK9-induced LDLR degradation and as a modulator of the metabolism of plasma LDL cholesterol and atherosclerosis development.47 Overall, there are puzzling links between MMPs (including MMP-2) and metabolism, making metalloproteinases attractive targets for changing the course of cardiometabolic diseases, whose etiology is notoriously complicated by co-morbidities such as obesity, atherosclerosis, diabetes, and metabolic syndrome.3 The challenge lies in determining specific mechanisms of MMPs and in establishing their context-dependent contribution to systemic metabolism.
The number of MMP-2 substrates has kept increasing steadily, and the trend might continue over time. A recent proteomic approach revealed a number of new potential MMP-2 targets, including galectin-1 and insulin growth factor binding protein-4, among others involved in angiogenesis.6 MMP-2-mediated cleavage of plasminogen may also yield the angiogenesis inhibitor, angiostatin.51,52 The ever increasing number of substrates likely modulated by MMPs justifies posing the question, “What do MMPs not do?” rather than the question, “What do MMPs do?”6,7,51
Relevant aspects of MMP-2 deficiency in humans and mice
Although rare, MMP2 deficiency is a pan-ethnic disorder, first reported in Saudi Arabian, Indian, and Turkish families – all presenting multicentric osteolysis with nodulosis and arthropathy (a condition also known as MONA, Online Mendelian Inheritance in Man number 259600).53 Many arthritic syndromes (eg, Torg and Winchester) that co-develop with MMP-2 deficiency have since been reported.53–55 This autosomal recessive condition is characterized by “vanishing bone” syndrome, carpal and tarsal osteolysis, and interphalangeal joint erosions, facial dysmorphia, fibrocollagenous nodules, and congenic heart defects. Lack of MMP-2 in humans results from inactivating mutations. Two family-specific homoallelic MMP2 mutations, R101H and Y244X, result in deletion of the substrate-binding and catalytic sites, and the fibronectin type II-like and hemopexin/TIMP2 binding domains of MMP-2. Not surprisingly, the deletion of the terminal hemopexin domain of MMP-2 (composed of 83 C-terminal amino acids) also causes MONA and cardiac dysfunction. Human MMP-2 deficiency has no known mechanism, and remains without cure.53
Arguably, work with models of MMP deficiency can tell us much about the physiological and pathological functions of MMPs. Work with MMP-2-deficient mice has revealed a pronounced inflammatory phenotype, while extracellular matrix accumulation due to a proteolytic defect seems to be limited.56–58 Mmp2−/− mice exhibit craniofacial abnormalities,54,59 are relatively small at birth, and have delayed growth vs age-matched wild-type mice.29,36 These features are reminiscent of MMP-2-deficient humans who also exhibit dwarfism.53–55 Mmp2−/− mice also show propensity to hypertensive heart disease with cardiac inflammation and hugely elevated activity of sPLA2 in myocardium,28,60 a hydrolase that targets the carbon 2 position of membrane glycerophospholipids to release fatty acids, which are precursors of eicosanoids and other lipid mediators, as well as modulators of lipid metabolic gene transcription.61,62
Furthermore, absence of MMP-2 in mice impairs resolution of lung inflammation induced by allergens. This lack in MMP-2 causes the accumulation of eosinophils and TH2 cytokines in the lung, affecting lung fibroblasts and smooth muscle cells. Without the appropriate chemokines, the lung is unable to form a chemokine gradient that allows for luminal clearance of inflammatory cells.57 As a result, Mmp2−/− mice have a reduced ability to clear recruited immune cells from the lung, have robust asthmatic reactions, and asphyxiate more easily than wild-type mice when challenged with allergens.56–58 Therefore, MMP-2 deficiency in both humans and mice is pro-inflammatory in multiple organs and potentially has a metabolic component.
Why does MMP-2 deficiency cause inflammation?
In the current review, we develop a hypothetical answer to the question of why MMP-2 deficiency causes inflammation, departing from the known reasons: 1) the ability of MMP-2 to cleave extracellular matrix components and the chemokine, MCP-3; and 2) MMP’s role as an endogenous inhibitor of cardiac sPLA2. We propose that MMP-2, MCP-3, and sPLA2 activity are interrelated in a potentially novel pathway, with ramifications for modulation of systemic inflammation and metabolism.
MMP-2 deficiency may cause an extracellular matrix breakdown defect. Paradoxically, studies of humans and mice with MMP-2 deficiency show a common predisposition to inflammation and arthritis;53–59,63 MMP-2 deficiency also predisposes to cardiac hypertrophy and fibrosis in response to angiotensin II but may protect against infarction.64–66 One study examined the cardiac expression of 56 metabolic and inflammatory genes in MMP-2-deficient mice.28 Results showed high levels of SREBP-2 and 3-hydroxy-3-methylglutarylcoenzyme A reductase (HMGCR), and elevated levels of pro-inflammatory genes such as Ccl5, Ccl2, and Ccl6. MMP-2 was found to negatively regulate a mechanism involving SREBP-2 and HMGCR (the rate-limiting enzyme in the synthesis of mevalonate) – factors predisposing to cardiac hypertrophy.28 Thus, MMP-2 expression during the development of agonist-induced hypertension may be cardioprotective by preventing the upregulation of the SREBP-2/HMGCR pathway in the heart. Establishing how and when MMP-2 is protective vs deleterious for cardiac function warrants further investigation.2,28,64–68
MMP-2 deficiency also impacts adipose tissue metabolism. Studies in mice indicate the formation of a fibrous cap around adipose tissue that might cause adipocyte hypotrophy and delayed growth.33,67 The delayed growth in mice may be present in humans, where the phenotype of MMP-2 deficiency is not “fibrosis” but is primarily crippling arthritis and dwarfism.53–55,59
A very attractive hypothesis is that MMP-2 primarily acts by cleaving and regulating chemokines, a characteristic shared by multiple MMPs (eg, MMP-2 and MMP-9).8,9,68–70 Although in vivo data are limited, chemokine cleavage and conversion from agonist into general chemokine receptor antagonist (as originally proposed for MCP-3) is an elegant mechanism whereby MMP-2 deficiency could exacerbate inflammation.8 Supporting studies include one where synthetic peptides with amino acid sequences corresponding to fragments of MCP-1, MCP-3, or MCP-4, equivalent to those that would be generated by MMP-mediated proteolysis, reduced carrageenan-induced paw swelling in rat.9 Similarly, Mmp2−/− mice infected with coxsackievirus develop mortal endocarditis within 2 weeks of infection.70 This exacerbated endocarditis is associated with elevated cardiac infiltration of neutrophils, macrophages, and T cells (such as CD4+ and CD8+), and significantly reduced by MCP-3-neutralizing antibody treatment.70
Recently, we advanced the hypothesis that a heart-centric MMP-2-mediated mechanism may modulate blood pressure homeostasis, inflammatory responses, and the severity of fever.60 We found hugely increased levels of sPLA2 in myocardium of Mmp2−/− mice. Tissue distribution analysis showed much higher sPLA2 activity in the heart than in the liver, adipose tissue, or kidney of Mmp2−/− mice. The enzyme is readily secreted from cardiac specimens ex vivo and has similar size, requirement for calcium, and apparent Michaelis Menten constant for diheptanoyl thio-PC with plasma sPLA2, suggesting that the heart secretes sPLA2, which next acts on distal tissues (such as the liver) as well as on the heart to impact their inflammatory and metabolic states.
Linking MMP-2 with the biology of A2 phospholipases
To illustrate the emerging connection between MMP-2 and cardiac sPLA2 activity and its potential pathophysiological significance, we briefly review some aspects concerning the various groups and functions ascribed to the PLA2 family. Phospholipases are membrane glycerophospholipid hydrolases, which, depending on their site of action, are subdivided into types A1, A2, B, C, or D. Type A1 releases the fatty acid esterified at carbon-1 (C-1) leaving behind a lysophospholipid. Similarly, type A2 releases the fatty acid esterified at C-2, leaving behind a lysophospholipid, while type B can release the fatty acid esterified at either C-1 or C-2. Type C phospholipase hydrolyzes the bond between C-3 and the phosphate group to release diacylglycerol and a phosphorylated head group. Phospholipase D cleaves the bond after the phosphate group, releasing diacylglycerol phosphate and a head group.
Current data are consistent with MMP-2 deficiency upregulating a type A2 phospholipase, an enzyme type whose existence was demonstrated in the 19th Century, when it was found as a major component of snake venoms.61,71 The sPLA2 family has at least eleven isoforms in humans and mice:61,71,72 PLA2G1B, PLA2G2 (A, C, D, E, and F), PLA2G3, PLA2G5, PLA2G10, and PLA2G12 (A and B). Members of the sPLA2 family have a molecular mass of 16–18 kDa, require calcium, have six to eight disulphide bridges, and have a histidine-aspartate dyad that catalyzes the hydrolysis and the release of fatty acids from the C-2 position of membrane glycerophospholipids. As a reflection of the multiple isoforms, sPLA2s target a spectrum of membrane phospholipids, with each individual phospholipase having distinct substrate selectivity and biological functions.72 This notion is best supported by a studies with gene knock-out models and mass-spectrometry-based lipidomics to functionally characterize individual members of the sPLA2 family.72 The data gathered for PLA2G1B, PLA2G2 (A, D, and E), PLA2G3, PLA2G5, and PLA2G10 now enable a functional subdivision of these isoforms as “digestive”, “inflammatory or bactericidal”, “resolving”, “metabolic”, “reproductive or anaphylactic”, “TH2-prone or metabolic”, and “asthmatic, reproductive or gastrointestinal”.72
The identity and amino acid sequence of the PLA2 upregulated in Mmp2−/− mice remains elusive. Enzyme inhibition assays with indoxam suggest that cardiac sPLA2 in Mmp2−/− mice is a mixture of sPLA2s (excluding PLA2G2A, for which the gene is disrupted in C57BL mice) or a completely novel enzyme with very interesting characteristics. For instance, the specific activity is elevated in excess of 102-fold in the heart, regardless of sex, and in a wide variety of dietary conditions vs wild-type (Mmp2+/+ C57BL/6) mice. Haplo-insufficiency (Mmp2−/+) lessens the specific activity of cardiac sPLA2, while pharmacological MMP inhibition increases both plasma and cardiac sPLA2 activity in wild-type (Mmp2+/+) but not in Mmp2−/− mice. However, the activity is not elevated in many organs such as the liver or adipose tissue, even though the Mmp2−/− mice are a whole-body gene knock-out model. Therefore, a cardiac-specific mechanism (perhaps, an agonist), which is under MMP-2 control, must be responsible for the cardiac-specific upregulation of sPLA2 activity in MMP-2-deficient mice.60
The emerging picture is one where, given the same pathophysiological context, sPLA2s each have unique induction profiles and functions, despite catalyzing the same type of biochemical reaction. What then is the biological function of cardiac sPLA2 expressed in MMP-2 deficiency? Evidently, cardiac sPLA2 is both pro-inflammatory and metabolic in the heart.60 MMP-2 deficiency results in cardiac overexpression of inflammatory marker genes, eg, Tnfa and Il1b, which are downregulated by the pan-sPLA2 inhibitor, varespladib. Similarly, MMP-2 deficiency results in cardiac dysregulation of lipid metabolic genes, eg, elevated Srebf2 (encoding sterol regulatory binding protein-2) and target genes, which are enzymes of the mevalonate pathway. The expression of these genes is normalized by varespladib. Moreover, cardiac sPLA2 pro-inflammatory effects appear to be systemic. In one study, MMP-2-deficient mice exhibit increased levels of prostaglandin E2 (PGE2) at baseline in heart, brain, and liver. Keeping in mind that cyclooxygenase and PGE2 synthase are important in the regulation of PGE2, 8-isoprostanes (prostaglandins not produced directly by cyclooxygenase) were also measured, once again showing elevated levels in Mmp2−/− mice compared to wild-type mice.60 Further, MMP-2-deficient mice develop exacerbated fever in response to low-dose bacterial lipopolysaccharide. This fever response is completely blunted by systemic administration of varespladib. In fact, the effect of cardiac sPLA2 on fever appears to be so dominant that only MMP-2-deficient mice, but not wild-type mice, show blunted fever in response to varespladib. Thus, it is not surprising that MMP-2-deficient mice rely on prostanoids for blood pressure homeostasis and develop acute hypertension 4 hours after treatment with either varespladib or indomethacin.60 A corollary of these observations is that the inflammatory state of the heart impacts the inflammatory and metabolic state at systemic levels, and impacts blood pressure homeostasis, at least in part through the MMP-2/sPLA2 axis. These observations bear potential significance for the understanding and clinical management of human conditions associated with reduced MMP-2 expression, particularly, human MMP-2 deficiency.60
The MMP-2/cardiac sPLA2 regulatory system is not an artefact of life-long MMP-2 deficiency. Cardiac sPLA2 activity is readily activated in mice treated with the pharmacological broad-spectrum MMP inhibitor doxycycline – the only MMP inhibitor with US Food and Drug Administration (FDA) approval. In these mice treated with doxycycline, plasma sPLA2 is significantly increased in 2–3 days, and cardiac sPLA2 is hugely upregulated after 2 weeks. These observations also suggest mechanisms for cardiac-specific effects of MMP-2 inhibitors, such as doxycycline, which currently has FDA approval and is widely used for its many actions, including as an antibiotic and a non-antibiotic/disease modifier drug with the capacity to inhibit the expression and activity of MMPs.73–77
Unanswered research questions
Beyond surmounting the challenge of identifying cardiac sPLA2, there are research opportunities to explore the biology of MMP-2 and the phenotype induced by its deficiency. Questions that could be answered by further research include elucidating: 1) which agonist(s) induce(s) cardiac sPLA2 activity; 2) how MMP-2 inhibits such agonist(s); 3) what the mechanism elicited by the agonist(s) is; 4) why sPLA2 activity is so highly elevated in the MMP-2-deficient heart; 5) whether cardiac and plasma sPLA2 are the same enzyme (or enzyme mixtures); and 6) how the heart impacts the inflammatory status of distal tissues and organs. If the heart impacts the inflammatory status of distal tissues through the secretion of sPLA2 from myocardium, the further question of how cardiac sPLA2 affects inflammation on target sites remains to be answered. Many pathologies exhibit dysregulation of either MMP-2 or sPLA2, eg, arteriosclerosis, diabetes, obesity, arthritis, asthma, anaphylaxis, and pain. It is tempting to speculate that the interaction between MMP-2 and sPLA2, which is released from myocardium, may contribute to the pathophysiology of these conditions. What is the actual contribution of the MMP-2/sPLA2 axis in these pathologies relative to other established mechanisms? Does the emergent link between MMP-2 and cardiac sPLA2 imply that the heart plays a more central role in systemic physiology and disease than previously suspected?
Explaining inflammation in MMP-2 deficiency
Previous research has shown that monocyte chemotaxis is significantly influenced by MCP-1.78–80 It has been shown that MCP-1 acts, at least in part, by binding to CC-chemokine receptor 2 to induce the intracellular translocation and activation of cytosolic calcium-dependent PLA2 (cPLA2) and calcium-independent PLA2 (iPLA2) isoforms.79 Interestingly, MCP-1 is cleaved and regulated by MMP-1 and MMP-3.9 Therefore, we hypothesize that MMPs, such as MMP-1 and MMP-3, may be important in the process of monocyte chemotaxis by cleaving MCP-1.79,80
A similar mechanism may apply to other MMPs and MCPs. MMP-2 cleaves MCP-3, and it is conceivable that MCP-3 shares with MCP-1 the ability to induce PLA2 activity in target cells (be it calcium-dependent, calcium-independent, or secreted PLA2 activity).9 If MCP-3 triggered the activity of secreted PLA2 in target cells, then cardiac MCP-3 could be an agonist of cardiac sPLA2 maturation and secretion in MMP-2-deficient hearts, with MMP-2 acting as an inhibitor through MMP-2-mediated proteolysis of MCP-3.
We postulate (Figure 1 and Table 1) that, since various MCP chemokines are susceptible to MMP-mediated proteolysis,9 tissue-specific MMP/MCP/PLA2 mechanisms might exist, whereby MCP triggers PLA2 activity under the control of MMP-mediated proteolysis of MCP. The existence of such mechanisms is supported by reports of MCP-1 actions in monocytes and regulatory loops, where cytokines produced downstream of MCPs or PLA2s modify the expression of MCPs, PLA2s, and MMPs, and PLA2 activity impacts MMP expression.81–84 Focusing on MCP-1, cytokines like TNF-α and IL-1β are major regulators of MCP-1 production in many systems, at least in part via the nuclear factor-κB pathway.85,86 Similarly, cPLA2 activity via the prostanoid pathway is known to regulate MMP-9 production in macrophages, while sPLA2 activity induces MMP-2 and MMP-9, thus facilitating fibroblast proliferation and chondrocyte development, respectively.81–84 Thus, MMPs, MCPs, and PLA2s depict a signaling network with the potential to affect inflammatory and metabolic responses through local and distal actions.
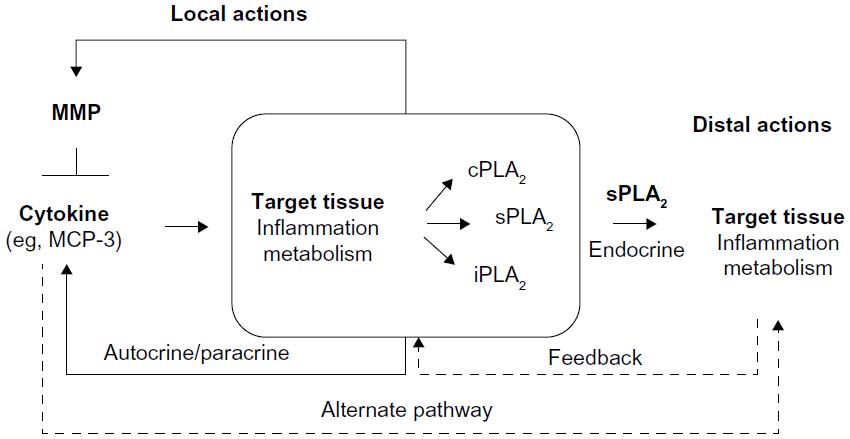 | Figure 1 The postulated mechanism of PLA2 regulation by MMPs via cytokines gives rise to many plausible PLA2-mediated signaling events that can impact target tissues at short and long distances. Alternate signaling pathways and feedback regulatory loops (dashed arrows) are likely part of the complex regulation of target tissues by MMPs. |
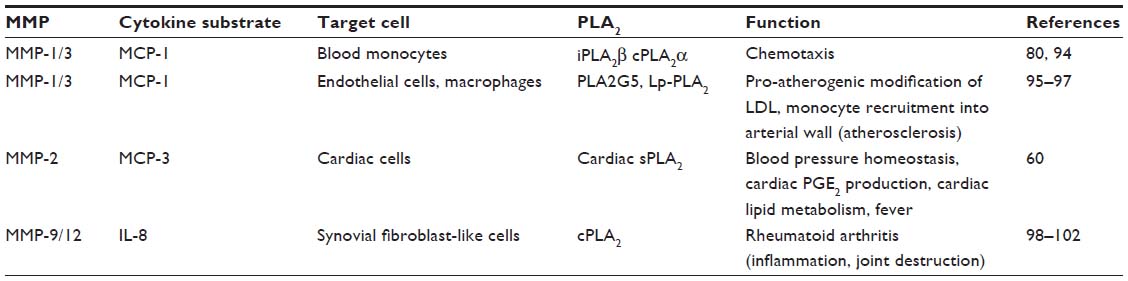 | Table 1 Possible MMP/cytokine/PLA2 axis subtypes |
Though this MMP/MCP/PLA2 axis is plausible, alternate mechanisms may be at play. Cytokines may act on distal organs directly to cause effects unrelated to the release of PLA2. Several MCPs can be cleaved by more than one subtype of MMP; for instance, MCP-3 cleavage can be brought about by MMP-1/3/13/14, not just MMP-2.9 Thus, in principle, absence of MMP-2 may be compensated for by the concurrent activity of other MMPs. However, given the overt inflammatory phenotype of MMP-2-deficient mice, one can only speculate that compensation is not a major mechanism at play in MMP-2 deficiency.
The MMP-2/cardiac sPLA2 system as a new endocrine function of the heart
In the 17th Century, William Harvey established the heart as a pump – a concept now familiar to all. However, through further research during the 17th Century, new exciting discoveries were made pointing to the heart as a gland. The mammalian heart was shown to be able to both store and release norepinephrine when stimulated, causing sympathetic peripheral effects.87 This concept of the heart possibly being a neuroendocrine organ was proven in 1983, when atrial natriuretic factor or atrial natriuretic peptide was first isolated and sequenced in rat. Similar to other endocrine organs, the heart was also found to contain a high number of developed endoplasmic reticulum, Golgi complex, and storage granules.88 This complex of systems allows the processing of peptides and then eventual release through the atrial-specific granules found in atrial cardiomyocytes.
Atrial natriuretic factor and brain natriuretic peptide are part of the family of cardiac natriuretic peptides. These peptides are found primarily in the heart, and when secreted, cause systemic effects such as hypotension, natriuresis, and diuresis. On the other hand, peptides such as adrenomedullin and big endothelin-1 are also found in the heart and have systemic effects, but are not truly endocrine products of the heart, as they are found mainly in blood vessels rather than cardiomyocytes.88
The proposed MMP-2/sPLA2 axis mechanism points again to the heart as an endocrine organ. We propose that this function is normally masked by the omnipresence of MMP-2 in the myocardium but is readily exposed when MMP-2 deficiency is induced by either gene deletion or pharmacological blockade.60 Cardiac sPLA2 activity in mice deficient in MMP-2 is elevated by orders of magnitude. In addition, there is no difference in sPLA2 activity between wild-type and Mmp2−/− mice in liver, kidney, and skeletal muscle. Thus, in addition to MMP-2, there must be heart-specific determinants of sPLA2 maturation and secretion from myocardium, whose discovery warrants further research. Similar to other heart-centric endocrine secretions where a peptide in the heart is activated by a protease and then secreted from storage granules to act on distal tissues and the heart itself, cardiac sPLA2 is negatively regulated by MMP-2 and is secreted from the heart through the conventional endoplasmic reticulum–Golgi pathway to affect fever, blood pressure homeostasis, and cardiac inflammatory/lipid metabolic gene expression.60 Notably, these functions of cardiac sPLA2 are different from those exerted by natriuretic peptides and other peptidic cardiac secretions.
MMP-2 overexpression and MMP-2 deficiency may cause inflammation
In principle, three scenarios can be distinguished: 1) normal physiology, 2) MMP-2 deficiency, and 3) MMP-2 overexpression, with the latter two scenarios contributing to inflammation. In MMP-2 deficiency, the likely cause of the inflammation is an effective excess of cytokines that would otherwise be kept at low levels through MMP-2-mediated proteolysis. This excess is a likely trigger of cardiac sPLA2 release, which may promote systemic inflammation (Figure 1).
In MMP-2 overexpression, the cause of the inflammation may be excessive or disproportionate cleavage of extracellular matrix, and perhaps, other cellular components (such as membrane receptors and growth factors) by MMP-2.3,7 Many conditions including arthritis, obesity, diabetes, tissue injury, cardiomyopathies, and hypertension have been associated with an upregulation of MMP-2 and inflammation.3,7,10 However, there is a paucity of reports on the exact mechanisms by which MMP-2 promotes inflammation in vivo. One suggestive example is, however, the setting of a vascular aneurysm where the overexpression of MMP-2 is deleterious.63,64 Purportedly, the pathological overexpression of MMP-2 creates an excess of collagen relative to elastin, thus also increasing stiffness, decreasing distensibility in the vasculature, and increasing the risk of aneurysm rupture.89,90 Additionally, a self-perpetuating loop may be created as smooth muscle cells release proteases that further break down the vascular intima. Chemoattractants attach on to the degraded intima and stimulate the migration of inflammatory and smooth muscle cells, ultimately exacerbating aneurysm development.89
In situations of MMP-2 overexpression, MMP-2 pharmacological inhibition may protect from inflammation if MMP-2 activity is restored to normal physiological levels. However, if MMP-2 activity were decreased beyond physiological levels, inflammatory signals mediated by chemokines and cardiac sPLA2 (Figure 1) may dominate the phenotype induced by MMP-2 inhibitor treatment. This could explain some of the contradictions between reports pertaining to the use of pharmacological MMP inhibitors, such as doxycycline, for therapeutic purposes, and may emphasize the need for titrating as opposed to completely inhibiting MMP-2 activity when treating inflammatory disease.60,91–93
Conclusion
Further research should refine the view of the heart as an endocrine organ that modulates systemic inflammatory responses through the secretion of sPLA2, a process that is under the control of MMP-2. The resultant knowledge should help researchers understand the mechanisms of MMP inhibitory drugs, such as doxycycline, and should enhance the clinical management of human MMP gene deficiencies, which are rare but very debilitating and ineffectively treated conditions.53
Acknowledgments
The current work was supported by summer studentships of the Natural Sciences and Engineering Council of Canada (NSERC) and the Alberta Innovates Health Solutions (granted to DL) and by operating grants from NSERC and the Canadian Institutes of Health Research (given to CFP).
Disclosure
The authors report no conflicts of interest in this work.
References
Gross J, Nagai Y. Specific degradation of the collagen molecule by tadpole collagenolytic enzyme. Proc Natl Acad Sci U S A. 1965; 54(4):1197–1204. | |
Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol. 2007;47:211–242. | |
Berry E, Bosonea AM, Wang X, Fernandez-Patron C. Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. J Vasc Res. 2013;50(1):52–68. | |
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. | |
Imai K, Hiramatsu A, Fukushima D, Pierschbacher MD, Okada Y. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J. 1997;322(Pt 3):809–814. | |
Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010;9(5):894–911. | |
Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803(1):39–54. | |
McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–1206. | |
McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100(4):1160–1167. | |
Morrison CJ, Butler GS, Rodriguez D, Overall CM. Matrix metalloproteinase proteomics: substrates, targets, and therapy. Curr Opin Cell Biol. 2009;21(5):645–653. | |
Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85(10):906–911. | |
Fernandez-Patron C, Stewart KG, Zhang Y, Koivunen E, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2-dependent cleavage of calcitonin gene-related peptide promotes vasoconstriction. Circ Res. 2000;87(8):670–676. | |
Martínez A, Oh HR, Unsworth EJ, et al. Matrix metalloproteinase-2 cleavage of adrenomedullin produces a vasoconstrictor out of a vasodilator. Biochem J. 2004;383(Pt 3):413–418. | |
Jeyabalan A, Novak J, Danielson LA, Kerchner LJ, Opett SL, Conrad KP. Essential role for vascular gelatinase activity in relaxin-induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res. 2003;93(12):1249–1257. | |
Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonist- induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res. 2004;94(1):68–76. | |
Friese RS, Rao F, Khandrika S, et al. Matrix metalloproteinases: discrete elevations in essential hypertension and hypertensive end-stage renal disease. Clin Exp Hypertens. 2009;31(7):521–533. | |
Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schonbein GW. Matrix metalloproteinases cleave the beta2-adrenergic receptor in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010; 299(1):H25–H35. | |
DeLano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage: mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52(2):415–423. | |
Castro MM, Rizzi E, Figueiredo-Lopes L, et al. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis. 2008;198(2):320–331. | |
Merchant SJ, Narumiya H, Zhang Y, Guilbert LJ, Davidge ST. The effects of preeclampsia and oxygen environment on endothelial release of matrix metalloproteinase-2. Hypertens Pregnancy. 2004; 23(1):47–60. | |
Lekontseva ON, Rueda-Clausen CF, Morton JS, Davidge ST. Ovariectomy in aged versus young rats augments matrix metalloproteinase-mediated vasoconstriction in mesenteric arteries. Menopause. 2010;17(3):516–523. | |
Lekontseva O, Jiang Y, Davidge ST. Estrogen replacement increases matrix metalloproteinase contribution to vasoconstriction in a rat model of menopause. J Hypertens. 2009;27(8):1602–1608. | |
Conrad KP, Davison JM. The renal circulation in normal pregnancy and preeclampsia: is there a place for relaxin? Am J Physiol Renal Physiol. 2014;306(10):F1121–F1135. | |
Jeyabalan A, Novak J, Doty KD, et al. Vascular matrix metalloproteinase-9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short-term administration of relaxin. Endocrinology. 2007;148(1):189–197. | |
Narumiya H, Zhang Y, Fernandez-Patron C, Guilbert LJ, Davidge ST. Matrix metalloproteinase-2 is elevated in the plasma of women with preeclampsia. Hypertens Pregnancy. 2001;20(2):185–194. | |
Myers JE, Merchant SJ, Macleod M, Mires GJ, Baker PN, Davidge ST. MMP-2 levels are elevated in the plasma of women who subsequently develop preeclampsia. Hypertens Pregnancy. 2005;24(2):103–115. | |
Brennan LJ, Morton JS, Davidge ST. Vascular dysfunction in preeclampsia. Microcirculation. 2014;21(1):4–14. | |
Wang X, Berry E, Hernandez-Anzaldo S, Takawale A, Kassiri Z, Fernandez-Patron C. Matrix metalloproteinase-2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element-binding protein-2/3-hydroxy-3-methylglutaryl-CoA reductase pathway in the heart. Hypertension. 2015;65(4):882–888. | |
Van Hul M, Lijnen HR. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. J Thromb Haemost. 2008;6(7):1198–1206. | |
Lijnen HR, Demeulemeester D, Van Hoef B, Collen D, Maquoi E. Deficiency of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) impairs nutritionally induced obesity in mice. Thromb Haemost. 2003;89(2):249–255. | |
Lee SW, Song KE, Shin DS, et al. Alterations in peripheral blood levels of TIMP-1, MMP-2, and MMP-9 in patients with type-2 diabetes. Diabetes Res Clin Pract. 2005;69(2):175–179. | |
Jaworski DM, Sideleva O, Stradecki HM, et al. Sexually dimorphic diet-induced insulin resistance in obese tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice. Endocrinology. 2011;152(4):1300–1313. | |
Van Hul M, Piccard H, Lijnen HR. Gelatinase B (MMP-9) deficiency does not affect murine adipose tissue development. Thromb Haemost. 2010;104(1):165–171. | |
Van Hul M, Lijnen HR. Effect of weight loss on gelatinase levels in obese mice. Clin Exp Pharmacol Physiol. 2011;38(9):647–649. | |
Van Hul M, Bauters D, Lijnen RH. Differential effects of a gelatinase inhibitor on adipocyte differentiation and adipose tissue development. Clin Exp Pharmacol Physiol. 2013;40(10):689–697. | |
Van Hul M, Bauters D, Himmelreich U, et al. Effect of gelatinase inhibition on adipogenesis and adipose tissue development. Clin Exp Pharmacol Physiol. 2012;39(1):49–56. | |
Lijnen HR, Silence J, Lemmens G, Frederix L, Collen D. Regulation of gelatinase activity in mice with targeted inactivation of components of the plasminogen/plasmin system. Thromb Haemost. 1998;79(6):1171–1176. | |
Bauters D, Van Hul M, Lijnen HR. Gelatinase B (MMP-9) gene silencing does not affect murine preadipocyte differentiation. Adipocyte. 2014; 3(1):50–53. | |
Park JY, Park JH, Jang W, et al. Apolipoprotein A-IV is a novel substrate for matrix metalloproteinases. J Biochem. 2011;151(3):291–298. | |
Park JH, Park SM, Park SH, Cho KH, Lee ST. Cleavage and functional loss of human apolipoprotein E by digestion of matrix metalloproteinase-14. Proteomics. 2008;8(14):2926–2935. | |
Park JH, Park SM, Park KH, Cho KH, Lee ST. Analysis of apolipoprotein A-I as a substrate for matrix metalloproteinase-14. Biochem Biophys Res Commun. 2011;409(1):58–63. | |
Marcel YL, Kiss RS. Structure-function relationships of apolipoprotein A-I: a flexible protein with dynamic lipid associations. Curr Opin Lipidol. 2003;14(2):151–157. | |
Mahley RW, Innerarity TL, Rall SC Jr, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. 1984; 25(12):1277–1294. | |
Kim SY, Park SM, Lee ST. Apolipoprotein C-II is a novel substrate for matrix metalloproteinases. Biochem Biophys Res Commun. 2006; 339(1):47–54. | |
Beisiegel U, Weber W, Ihrke G, Herz J, Stanley KK. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature. 1989;341(6238):162–164. | |
Aoki T, Sato D, Li Y, Takino T, Miyamori H, Sato H. Cleavage of apolipoprotein E by membrane-type matrix metalloproteinase-1 abrogates suppression of cell proliferation. J Biochem. 2005;137(1):95–99. | |
Wang X, Berry E, Hernandez-Anzaldo S, et al. MMP-2 inhibits PCSK9-induced degradation of the LDL receptor in Hepa1-c1c7 cells. FEBS Lett. 2015;589(4):490–496. | |
Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50 Suppl:S172–S177. | |
Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004; 279(47):48865–48875. | |
Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003; 34(2):154–156. | |
Doucet A, Overall CM. Protease proteomics: revealing protease in vivo functions using systems biology approaches. Mol Aspects Med. 2008; 29(5):339–358. | |
O’Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999;274(41):29568–29571. | |
Martignetti JA, Aqeel AA, Sewairi WA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28(3):261–265. | |
Mosig RA, Dowling O, DiFeo A, et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16(9):1113–1123. | |
Tuysuz B, Mosig R, Altun G, Sancak S, Glucksman MJ, Martignetti JA. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 2009; 17(5):565–572. | |
Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007; 87(1):69–98. | |
Corry DB, Rishi K, Kanellis J, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3(4):347–353. | |
Corry DB, Kiss A, Song LZ, et al. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004; 18(9):995–997. | |
Mosig RA, Martignetti JA. Loss of MMP-2 in murine osteoblasts upregulates osteopontin and bone sialoprotein expression in a circuit regulating bone homeostasis. Dis Model Mech. 2013;6(2):397–403. | |
Berry E, Hernandez-Anzaldo S, Ghomashchi F, et al. Matrix metalloproteinase-2 negatively regulates cardiac secreted phospholipase A2 to modulate inflammation and fever. J Am Heart Assoc. 2015; 4(4):e001868. | |
Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520. | |
Ou J, Tu H, Shan B, et al. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98(11):6027–6032. | |
Hartl D, Krauss-Etschmann S, Koller B, et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. J Immunol. 2008; 181(11):8053–8067. | |
Monden Y, Kubota T, Inoue T, et al. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2007; 293(1):H743–H753. | |
Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005;115(3):599–609. | |
Hayashidani S, Tsutsui H, Ikeuchi M, et al. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol. 2003; 285(3):H1229–H1235. | |
Lijnen HR, Collen D. Matrix metalloproteinase system deficiencies and matrix degradation. Thromb Haemost. 1999;82(2):837–845. | |
Pinto YM, Heymans S. Letter by Pinto and Heymans regarding article, “Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice”. Circulation. 2008;118(20):e697. | |
Cheung C, Marchant D, Walker EK, et al. Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation. 2008;117(12):1574–1582. | |
Westermann D, Savvatis K, Lindner D, et al. Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation. 2011;124(19):2082–2093. | |
Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011; 111(10):6130–6185. | |
Murakami M, Sato H, Miki Y, Yamamoto K, Taketomi Y. A new era of secreted phospholipase A2. J Lipid Res. 2015;56(7):1248–1261. | |
Seftor RE, Seftor EA, De Larco JE, et al. Chemically modified tetracyclines inhibit human melanoma cell invasion and metastasis. Clin Exp Metastasis. 1998;16(3):217–225. | |
Greenwald RA, Moak SA, Ramamurthy NS, Golub LM. Tetracyclines suppress matrix metalloproteinase activity in adjuvant arthritis and in combination with flurbiprofen, ameliorate bone damage. J Rheumatol. 1992;19(6):927–938. | |
Golub LM, McNamara TF, D’Angelo G, Greenwald RA, Ramamurthy NS. A non-antibacterial chemically-modified tetracycline inhibits mammalian collagenase activity. J Dent Res. 1987;66(8):1310–1314. | |
Golub LM, Wolff M, Lee HM, et al. Further evidence that tetracyclines inhibit collagenase activity in human crevicular fluid and from other mammalian sources. J Periodontal Res. 1985;20(1):12–23. | |
Gu Y, Lee HM, Sorsa T, et al. Non-antibacterial tetracyclines modulate mediators of periodontitis and atherosclerotic cardiovascular disease: a mechanistic link between local and systemic inflammation. Pharmacol Res. 2011;64(6):573–579. | |
Han KH, Hong KH, Park JH, et al. C-reactive protein promotes monocyte chemoattractant protein-1 – mediated chemotaxis through upregulating CC chemokine receptor 2 expression in human monocytes. Circulation. 2004;109(21):2566–2571. | |
Carnevale KA, Cathcart MK. Calcium-independent phospholipase A(2) is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J Immunol. 2001;167(6):3414–3421. | |
Mishra RS, Carnevale KA, Cathcart MK. iPLA2beta: front and center in human monocyte chemotaxis to MCP-1. J Exp Med. 2008; 205(2):347–359. | |
Ii H, Hontani N, Toshida I, Oka M, Sato T, Akiba S. Group IVA phospholipase A2-associated production of MMP-9 in macrophages and formation of atherosclerotic lesions. Biol Pharm Bull. 2008;31(3):363–368. | |
Gorovetz M, Schwob O, Krimsky M, Yedgar S, Reich R. MMP production in human fibrosarcoma cells and their invasiveness are regulated by group IB secretory phospholipase A2 receptor-mediated activation of cytosolic phospholipase A2. Front Biosci. 2008;13:1917–1925. | |
Choi YA, Kim DK, Bang OS, Kang SS, Jin EJ. Secretory phospholipase A2 promotes MMP-9-mediated cell death by degrading type I collagen via the ERK pathway at an early stage of chondrogenesis. Biol Cell. 2010;102(2):107–119. | |
Choi YA, Lim HK, Kim JR, et al. Group IB secretory phospholipase A2 promotes matrix metalloproteinase-2-mediated cell migration via the phosphatidylinositol 3-kinase and Akt pathway. J Biol Chem. 2004;279(35):36579–36585. | |
Ping D, Jones PL, Boss JM. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity. 1996;4(5):455–469. | |
Andoh A, Takaya H, Saotome T, et al. Cytokine regulation of chemokine (IL-8, MCP-1, and RANTES) gene expression in human pancreatic periacinar myofibroblasts. Gastroenterology. 2000;119(1):211–219. | |
Braunwald E, Harrison DC, Chidsey CA. The heart as an endocrine organ. Am J Med. 1964;36:1–4. | |
Ogawa T, de Bold AJ. The heart as an endocrine organ. Endocr Connect. 2014;3(2):R31–R44. | |
Chakraborti S, Chowdhury A, Alam MN, et al. Vascular aneurysms: a perspective. Indian J Biochem Biophys. 2014;51(6):449–456. | |
Jackson CL, Raines EW, Ross R, Reidy MA. Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler Thromb. 1993;13(8):1218–1226. | |
Zeng S, Zhou X, Tu Y, et al. Long-term MMP inhibition by doxycycline exerts divergent effect on ventricular extracellular matrix deposition and systolic performance in stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens. 2011;33(5):316–324. | |
Hori Y, Kunihiro S, Sato S, et al. Doxycycline attenuates isoproterenol-induced myocardial fibrosis and matrix metalloproteinase activity in rats. Biol Pharm Bull. 2009;32(10):1678–1682. | |
Vinet L, Rouet-Benzineb P, Marniquet X, et al. Chronic doxycycline exposure accelerates left ventricular hypertrophy and progression to heart failure in mice after thoracic aorta constriction. Am J Physiol Heart Circ Physiol. 2008;295(1):H352–H360. | |
Yen H, Zhang Y, Penfold S, Rollins BJ. MCP-1-mediated chemotaxis requires activation of non-overlapping signal transduction pathways. J Leukoc Biol. 1997;61(4):529–532. | |
Sonoki K, Iwase M, Ohdo S, Ieiri I, Takata Y, Kitazono T. Statin inhibits the expression of secretory phospholipase A2 and subsequent monocyte chemoattractant protein-1 in human endothelial cells. J Cardiovasc Pharmacol. 2014;64(6):489–496. | |
Hu MM, Zhang J, Wang WY, et al. The inhibition of lipoprotein-associated phospholipase A2 exerts beneficial effects against atherosclerosis in LDLR-deficient mice. Acta Pharmacol Sin. 2011;32(10):1253–1258. | |
Gonçalves I, Edsfeldt A, Ko NY, et al. Evidence supporting a key role of Lp-PLA2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol. 2012;32(6):1505–1512. | |
Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood. 2008;112(8):3455–3464. | |
Sommerfelt RM, Feuerherm AJ, Skuland T, Johansen B. Cytosolic phospholipase A2 modulates TLR2 signaling in synoviocytes. PLoS One. 2015;10(4):e0119088. | |
Kazantseva MG, Hung NA, Highton J, Hessian PA. MMP expression in rheumatoid inflammation: the rs11568818 polymorphism is associated with MMP-7 expression at an extra-articular site. Genes Immun. 2013;14(3):162–169. | |
McGarry T, Veale DJ, Gao W, Orr C, Fearon U, Connolly M. Toll-like receptor 2 (TLR2) induces migration and invasive mechanisms in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):153. | |
Silosi I, Cojocaru M, Foia L, et al. Significance of circulating and crevicular matrix metalloproteinase-9 in rheumatoid arthritis-chronic periodontitis association. J Immunol Res. 2015;2015:218060. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.