")
Back to Journals » Cancer Management and Research » Volume 12
ELL2 Is Required for the Growth and Survival of AR-Negative Prostate Cancer Cells
Authors Wang Z , Pascal LE , Chandran UR, Chaparala S, Lv S, Ding H, Qi L, Wang Z
Received 8 February 2020
Accepted for publication 1 May 2020
Published 10 June 2020 Volume 2020:12 Pages 4411—4427
DOI https://doi.org/10.2147/CMAR.S248854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Zhi Wang,1,2 Laura E Pascal,2 Uma R Chandran,3 Srilakshmi Chaparala,3 Shidong Lv,2,4 Hui Ding,2 Lin Qi,1 Zhou Wang2,5– 7
1Department of Urology, Xiangya Hospital of Central South University, Changsha, People’s Republic of China; 2Department of Urology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 3Department of Biomedical Informatics, University of Pittsburgh, Pittsburgh, PA, USA; 4Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 5UPMC Hillman Cancer Center, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 6Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 7Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Correspondence: Zhou Wang Email [email protected]
Background: Elongation factor for RNA polymerase II 2 (ELL2) was reported as a putative tumor suppressor in the prostate. ELL2 is frequently down-regulated in prostatic adenocarcinoma specimens, and loss of ELL2 induced murine prostatic intraepithelial neoplasia and enhanced AR-positive prostate cancer cell proliferation. However, the ELL2 gene appears to be amplified in AR-negative neuroendocrine prostate tumors, suggesting a potential oncogenic role for ELL2 in AR-negative prostate cancer cells. In this study, we explored the potential function of ELL2 in PC-3 and DU145, two AR-negative prostate cancer cell lines.
Materials and Methods: The role of ELL2 in PC-3 and DU145 cells was studied using siRNA-mediated ELL2 knockdown. Genes regulated by ELL2 knockdown in PC-3 cells were identified and analyzed using RNA-Seq and bioinformatics. The expression of representative genes was confirmed by Western blot and/or quantitative PCR. Cell growth was determined by BrdU, MTT and colony formation assays. Cell death was analyzed by 7-AAD/Annexin V staining and trypan blue exclusion staining. Cell cycle was determined by PI staining and flow cytometry.
Results: ELL2 knockdown inhibited the proliferation of PC-3 and DU145 cells. RNA-Seq analysis showed an enrichment in genes associated with cell death and survival following ELL2 knockdown. The interferon-γ pathway was identified as the top canonical pathway comprising of 55.6% of the genes regulated by ELL2. ELL2 knockdown induced an increase in STAT1 and IRF1 mRNA and an induction of total STAT1 and phosphorylated STAT1 protein. Inhibition of cell proliferation by ELL2 knockdown was partly abrogated by STAT1 knockdown. ELL2 knockdown inhibited colony formation and induced apoptosis in both PC-3 and DU145 cells. Furthermore, knockdown of ELL2 caused S-phase cell cycle arrest, inhibition of CDK2 phosphorylation and cyclin D1 expression, and increased expression of cyclin E.
Conclusion: ELL2 knockdown in PC-3 and DU145 cells induced S-phase cell cycle arrest and profound apoptosis, which was accompanied by the induction of genes associated with cell death and survival pathways. These observations suggest that ELL2 is a potential oncogenic protein required for survival and proliferation in AR-negative prostate cancer cells.
Keywords: DU145, PC-3, STAT1, IRF, CDK2
Introduction
Prostate cancer continues to be the most frequently diagnosed cancer and the second leading cause of cancer death in US males.1 Localized prostate cancer is treated and often cured by radical prostatectomy or radiation therapy. Androgen deprivation therapy (ADT) is the standard treatment for patients with metastatic prostate cancer; however, ADT is not curative and patients will relapse with castration-resistant prostate cancer (CRPC). The recently developed second-generation antiandrogens such as enzalutamide and abiraterone can prolong the life of CRPC patients for several additional months. Patients eventually develop resistance to second-generation antiandrogens, with some patients developing AR-negative prostate cancer with small-cell carcinoma or neuroendocrine phenotypes.2 There is no effective treatment for AR-negative prostate cancer and the biology of AR-negative prostate cancer is poorly understood. Defining the mechanisms regulating AR-negative prostate cancer growth may lead to new approaches to control this lethal disease.
Elongation factor for RNA polymerase II 2 (ELL2) is a member of the ELL family which consists of ELL1, ELL2, and ELL3.3–7 Human ELL1, also called eleven-nineteen lysine-rich leukemia, was initially identified as a gene located on chromosome 19p13.1 frequently translocated and fused with the Mixed-lineage leukemia (MLL) gene on chromosome 11q23 in acute myeloid leukemia.3,8 ELL family proteins increase RNA polymerase catalytical activity and facilitate RNA elongation by suppressing the transient elongation pause.4,9,10 The ELL family is one of the components constituting the Super Elongation Complex (SEC), which regulates the transcriptional elongation checkpoint control stage that is often dysfunctional and involved in the carcinogenesis of multiple malignancies.11 An SEC-like complex in Drosophila containing ELL was found to facilitate RNA Polymerase II (Pol II)-mediated transcription of small nuclear RNA (snRNA), the loss of which results in decreased production of snRNA.12 Downregulation of ELL2 influences processing to the secretion-specific poly(A) site as well as reduces histone H3K4 and H3K79 methylation on the IgH gene.13 These important functions of ELL2 suggest that it could play important roles in carcinogenesis.
ELL2 expression was found to be regulated by androgens in AR-positive prostate cancer cells.14,15 Knockdown of ELL2 can enhance cell proliferation, migration and invasion in AR-positive prostate cancer cell lines.14,16 Prostatic luminal epithelial cell-specific deletion of ELL2 results in prostatic intraepithelial neoplasia, a putative precursor of prostate cancer in the mouse model.15,16 ELL2 could co-immunoprecipitate with the tumor suppressor RB, and combined knockdown of ELL2 and RB induced an increase in proliferation, migration and invasion compared to knockdown of either ELL2 or RB alone.14 Taken together, these results indicate that ELL2 plays a potential tumor-suppressive role in the prostate.
ELL2 expression is down-regulated in prostatic adenocarcinoma specimens, consistent with its potential tumor-suppressive role in AR-positive prostate cancer cells.14,17 Unexpectedly, ELL2 gene appears to be amplified in AR-negative neuroendocrine prostate cancer specimens, based on in silico analysis.14,15 However, its biological role in AR-negative prostate cancer cells was not tested. In the present study, we investigated the role of ELL2 in PC-3 and DU145, two AR-negative prostate cancer cell lines that express neuroendocrine markers.18,19
Materials and Methods
Cell Culture and siRNA Knockdown
PC-3, DU145, LNCaP and 22Rv1 cells were obtained from American Type Culture Collection (Manassas, VA). C4-2 cells were a kind gift from Dr. Leland Chung (Cedars-Sinai Medical Center, Los Angeles, CA). All the cell lines were maintained in RPMI-1640 medium (10–040-CV, Corning Cellgro, Corning Inc., Corning, NY), supplemented with 10% fetal bovine serum (FBS) (S11150, Atlanta Biologicals, Flowery Branch, GA), 100 µg/mL streptomycin, 100 U/mL penicillin, and 1% glutamine (Invitrogen, Carlsbad, CA).
In knockdown assays, cells were transfected with nontarget siRNA control (D-001810-10-50, Dharmacon, Lafayette, CO) or siRNAs targeting ELL2 and/or STAT1 using transfection reagent (T-2001-03, Dharmacon) in 6-well plates, according to the manufacturer’s protocol. Two different siRNA sequences specific for each gene were utilized to exclude potential off-target effects. The total amount of siRNAs in different experimental groups was adjusted to the same by complementation with nontarget siRNA if needed. All ELL2 and STAT1 siRNA sequences are listed in Table 1. Cells were harvested at 48 hours or indicated time after transfection.
 |
Table 1 ELL2 and STAT1 siRNA Sequence |
RNA-Sequencing
RNA-seq was performed at the University of Pittsburgh HSCRF Health Sciences Sequencing Core at Children’s Hospital. Libraries were prepared with the TruSeq Stranded mRNA kit (Illumina, San Diego, CA) following the manufacturer’s protocol. Briefly, mRNA was purified using oligo-dT magnetic beads and fragmented. First-strand reverse transcription was performed with oligo(dT) primers. After second-strand synthesis, blunt-ended cDNA fragments were A-tailed followed by ligation of indexed sequencing adapters. PCR amplification provided selective enrichment of DNA with adapters ligated to both ends. Library quantity and quality were assessed by fluorometric assay (Qubit) and Agilent DNA 1000 TapeStation assay (Agilent, Santa Clara, CA, USA), respectively. Final libraries were normalized to 10 nM, pooled and diluted. Flowcells for the NextSeq 500 were seeded with 1.8 pM denatured library for automated cluster formation and 2 x 75 bp paired-end sequencing.
Next, we performed Gene Ontology Enrichment analysis through Ingenuity Pathway Analysis (IPA, Qiagen, Hilden, Germany) and gene sets enrichment analysis (GSEA) algorithm. IPA analysis was conducted based on the set of indirect relationships in the IPA® database. The algorithm of IPA computed a p value representing the probability of differentially expressed genes (DEGs) enriched in pathways and determined the most likely regulation-related set of function and pathways of the DEGs involved. DEGs with fold-changes >2 and differential expression p-values<0.05 were identified as well as pathways with p-values<0.05.
GSEA algorithm determined whether a set of the biological process were enriched in DEGs between the two groups. Reference gene sets were the genes listed in the Molecular Signatures Database (MSigDB). Benjamini-Hochberg adjusted p values and normalized enrichment score (NES) were applied to identify ontology enrichment function and pathways with significance (p<0.05). The datasets were deposited in the NCBI public database GEO with the following accession number: GSE134214.
RNA Isolation and Quantitative PCR
Total RNA was isolated with Trizol (15,596,018, Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. cDNA was synthesized using PrimeScript™ RT Master Mix (RR036A, Takara Bio USA, Inc., Mountain View, CA). Quantitative PCR (qPCR) was conducted to quantify ELL2, STAT1, and IRF1, with GAPDH as the reference, using real-time qPCR Master Mix (639,676, Takara Bio) on ABI Step-One Plus (Applied Biosystems, Foster City, CA). Primers are listed in Table 2.
 |
Table 2 Primers for Quantitative PCR |
Western Blot
Cell samples were lysed with RIPA lysis buffer, consisting of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40 (volume-to-volume), 0.1% sodium dodecyl sulfate (SDS), 0.25% sodium deoxycholate, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride, in the presence of protease inhibitor cocktail (catalog no. P8340, Sigma-Aldrich, St. Louis, MO) at a 1:100 dilution. The lysates were mixed with loading buffer, heated at 100°C for 10 minutes and then loaded on an SDS polyacrylamide gel. After electrophoresis, the proteins were transferred to polyvinylidene difluoride membranes. The membranes were incubated overnight with primary antibodies ELL2 (A302-505A, Bethyl Laboratories, Montgomery, TX), STAT1 (9172S, Cell Signaling Technology, Danvers, MA), p-STAT1 (9167S, Cell Signaling Technology), p-CDK2 (2561, Cell Signaling Technology), cyclin D1 (sc-8396, Santa Cruz Biotechnology, Dallas, Texas), cyclin E (sc-247, Santa Cruz Biotechnology), or GAPDH (sc-47724, Santa Cruz Biotechnology) overnight. Following incubation with an appropriate secondary antibody, the membranes were incubated in Clarity Max Western ECL Substrate (1,705,062, Bio-Rad Laboratories, Hercules, CA) for 2 minutes, and visualized using ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Hercules, CA).
Trypan Blue Exclusion Staining
Cells seeded in six-well plates were harvested by trypsin digestion after each experiment. The harvested cells were stained with 0.4% trypan blue solution (T8154, Sigma-Aldrich) and counted using a hemocytometer under the microscope. Each sample was counted four times.
MTT Assay
Cells were seeded in 96-well plates at a density of 5×10^3/well and incubated at 37°C in 95% humidity with 5% CO2 for 24 h. Cells were then transfected with siRNA. Subsequently, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide, 0.5 mg/mL) was added to each well at indicated time points and cells were incubated at 37 °C for 4 hours. Dimethyl sulfoxide was added to dissolve formazan crystals and the absorbance was measured at 490 nm using a spectrophotometer.
Colony Formation Assay
In colony formation assays, PC-3 and DU145 cells were digested 72 hours after siRNA transfection and re-seeded in 6 cm dishes at a density of 500 cells/dish before culture for 14 days. Colonies were then fixed for 10 minutes with 10% formaldehyde and stained with crystal violet (0.2%). The colonies in each dish were imaged using ChemiDoc (Bio-Rad) and counted by Image-Pro Plus (Media Cybernetics, Silver Spring, MD) software for each group of cells.
BrdU Assay
BrdU staining was performed as described previously.14,20 Each treatment condition was carried out in triplicate. Briefly, PC-3 and DU145 cells were seeded in 24-well plates at a density of 70,000 cells/well and treated with siRNA for 48 hours. At the end of each experiment, cells were cultured in the presence of 10 μM BrdU (B5002-250MG, Sigma) for the last 2 hours. Cells were immunostained with BrdU antibody (sc-51514, Santa Cruz) and subsequently with goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody (A10521, Invitrogen) and counterstained with DAPI (D9542, Sigma-Aldrich). The images were visualized with fluorescence microscopy (Nikon TE2000-U, Nikon, Tokyo, Japan).
Apoptosis and Cell Cycle Analysis
Apoptotic cells were detected using FITC Annexin V Apoptosis Detection Kit with 7-AAD (640922, BioLegend, San Diego, CA). Cells were washed twice with cold BioLegend’s Cell Staining Buffer and then resuspended in Annexin V Binding Buffer. For each staining reaction, 105 cells in 100 μL were incubated with 5 µL of FITC Annexin V, 5 µL of 7-AAD Viability Staining Solution or both for 15 minutes at room temperature in the dark before the addition of 400 µL Annexin V Binding Buffer to each tube.
Cell cycle was detected by flow cytometry with BD Cycletest™Plus DNA kit (340,242, BD Biosciences, Franklin Lakes, NJ) as described previously.21 Briefly, cells were harvested at 72 hours after knockdown, washed three times with buffer solution, and adjusted to a concentration of 106 cells/mL. Subsequently, 5×105 cells were incubated with solution A (Trypsin buffer), solution B (Trypsin inhibitor and RNase buffer), and Solution C (PI stain solution) for 10 minutes respectively in the darkroom before analysis. All flow cytometry analyses were performed using BD Accuri™ C6 Plus personal flow cytometer (BD Biosciences).
Statistical Methods
Statistical analyses were performed with GraphPad Prism (version 7.0 for Windows). Values were presented as Mean ± SD. Comparisons between groups were calculated using Student’s t-test. A p value <0.05 was considered to be significant statistically.
Results
Knockdown of ELL2 Inhibited Proliferation of PC-3 and DU145
Previous studies suggested that the ELL gene was amplified in AR-negative neuroendocrine prostate cancer cell datasets.14,15 However, according to a literature search, there were no functional studies of ELL2 in AR-negative prostate cancer cells. We examined the expression of ELL2 in prostate cancer cell lines using Western blot analyses. ELL2 protein was expressed in 22RV1, DU145, PC-3 and LNCaP prostate cancer cell lines, with higher levels in PC-3 and 22Rv1 as compared to DU145 and LNCaP cells (Supplemental Figure S1A). ELL2 expression levels in C4-2 were similar to that of LNCaP (Supplemental Figure S1B). ELL2 deletion was identified in prostate cancer specimens, and amplification was identified in castration-resistant and neuroendocrine prostate cancer specimens in several publicly available datasets through the cBioPortal for Cancer Genomics site (http://cbioportal.org),22,23 (Supplemental Figure S2). Prostate datasets with identified mutations and/or copy number alterations for ELL2 included: MICH24 NEPC (Multi-Institute 2016),2 The MPC Project (mpcproject.org/data-release), PRAD (MSKCC/DFCI 2018),25 Prostate (TCGA 2015),26 Prostate SU2C 2019,27 Broad/Cornell 2013,28 TCGA PanCan 2018,29–35 SU2C,36 MSKCC 2010,37 FHCRC 2016,38 and Broad/Cornell 2012.39 Data type shown is Events per Patient and is a summary including all patients in these studies. To explore the role of ELL2 in AR-negative prostate cancer cells, we tested the effect of ELL2 knockdown in PC-3 and DU145, two widely used AR-negative prostate cancer cell lines. Figure 1A and B are representative images and quantitative analysis showing a 2- to 3-fold inhibition of BrdU incorporation by ELL2 silencing using 2 different siRNAs in cultured PC-3 and DU145 cells. Knockdown of ELL2 was verified by Western blot analysis (Figure 1C).
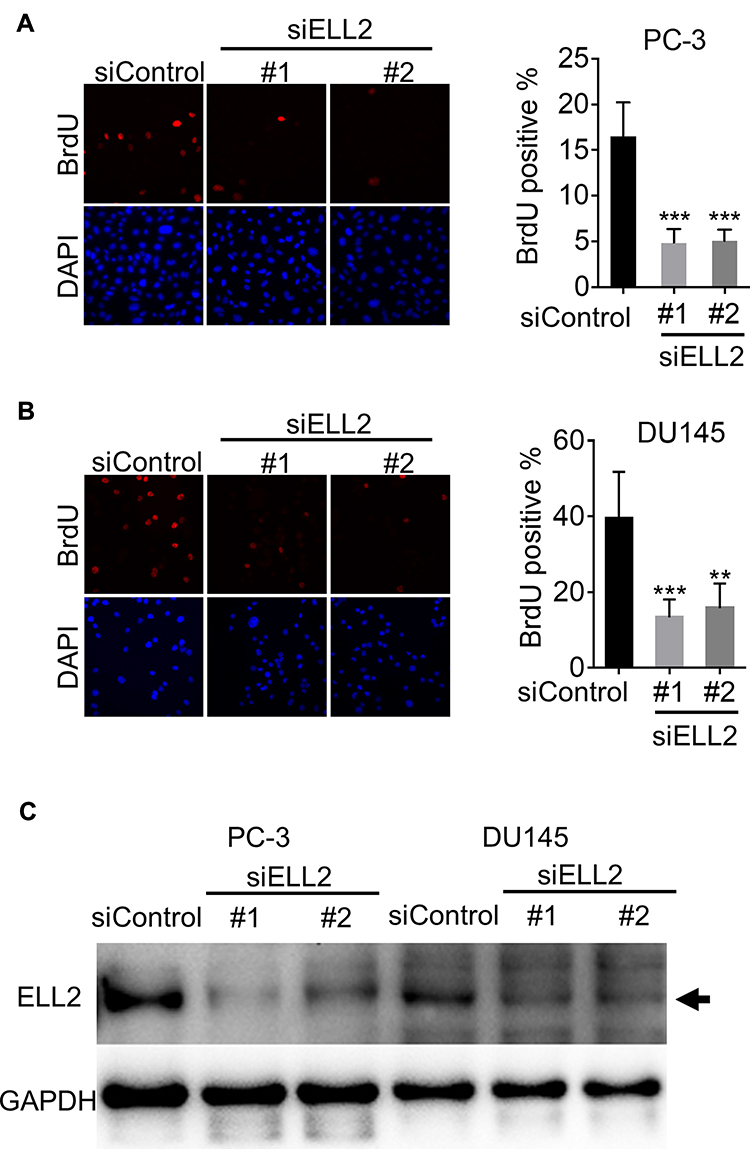 |
Figure 1 Impact of ELL2 knockdown on BrdU incorporation in AR-negative prostate cancer cells. Images shown are BrdU-positive nuclei in PC-3 cells (A) or DU145 (B) transfected with 25 nM nontarget control siRNA (siControl) or two different siRNAs targeting ELL2 (#1 or #2). DAPI staining shows all the nuclei. BrdU incorporation was quantified by determining the mean percentage ± SD of BrdU-positive cells relative to the total number of cells. Cells were counted from two different fields for each well from triplicate wells and 30–130 cells per field. (C) Efficiency of siELL2 knockdown in PC-3 cells was verified by Western blotting. ELL2 band denoted by the black arrow. GAPDH was used as a loading control. Results are representative of three individual experiments. **p < 0.01, ***p < 0.001. Abbreviations: AR, androgen receptor; BrdU, bromodeoxyuridine; DAPI, 4ʹ,6-diamidino-2-phenylindole; ELL2, elongation factor for RNA polymerase II 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; siRNA, small interfering RNA; SD, standard deviation. |
Identification of Genes and Pathways Regulated by ELL2
To explore the mechanisms of ELL2 action in AR-negative prostate cancer cells, RNA-seq was performed to identify genes that were up- or down-regulated by ELL2 knockdown in the PC-3 cell line. False discovery rate (FDR) combined with fold change (FC) was used as the criteria (|log2 FC|>2, FDR<0.05) for identifying differentially expressed genes (DEGs). This led to the detection of 1937 DEGs, consisting of 1091 (56.3%) up-regulated and 846 (43.7%) down-regulated genes after ELL2 knockdown. The dsRNA viral response pathway can be triggered by siRNA treatment,40,41 thus we examined the expression of several dsRNA pathway genes. TLR1, TLR2 and IFNb1 were up-regulated by ELL2 knockdown in PC-3 cells. However, no alterations in IFNa, IRF3, IRF7, ISG15, and OASL1 were detected in RNA-seq analysis of ELL2 knockdown in PC-3 cells. Through IPA analysis, interferon signaling was predicted as the top pathway, with 55.6% of these genes in this pathway as increased following ELL2 knockdown (Figure 2A). The Interferon (IFN) pathway involves STAT1 and IRF1 (Figure 2B), which play a critical role in regulating immune response, cell proliferation and cell death.42 Gene ontology enrichment analysis (GO) revealed that DEGs were involved in multiple biological process or diseases, with cancer and cell death/survival being ranked as the top disease or cellular function associated with ELL2 knockdown (Figure 2C and D). In Gene Set Enrichment Analysis (GSEA) analysis, genes up-regulated by ELL2 silencing appeared to be predominant over down-regulated genes and were associated with IFN and Tumor Necrosis Factor (TNF) pathways. Thus, ELL2 silencing may participate in the cell death and survival process, possibly through immune response pathways (Figure 2E and F). GSEA analysis also identified 131 genes that are associated with a set of genes up-regulated in cervical cancer tissue and named “Cervical Cancer Proliferation Cluster” (CCPC), the majority of which were related to cell cycle.43 ELL2 knockdown repressed the expression of 131 out of the 163 genes in CCPC (Figure 2G), suggesting that ELL2 loss may inhibit the proliferation of AR-negative prostate cancer cells in part through disruption of cell cycle regulation.
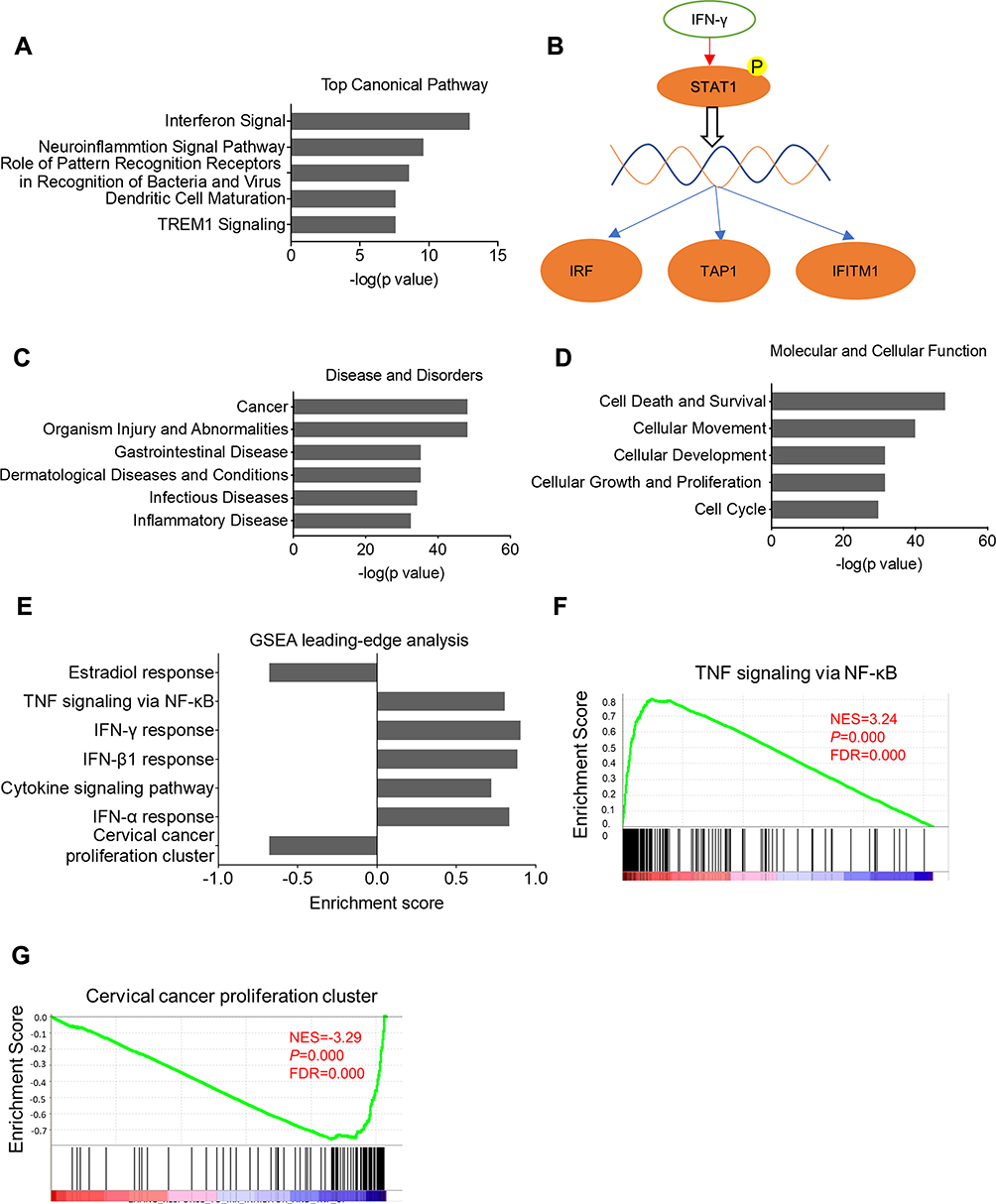 |
Figure 2 (A) Pathway identification by Ingenuity Pathway Analysis (IPA®) software based on differentially expressed genes (DEGs) upon ELL2 knockdown in PC-3 cells. This analysis shows DEGs mainly was enriched in the IFN-γ pathway, neuroinflammation signal pathway, and other indicated pathways. (B) IFN-γ pathway involving STAT1 phosphorylation and transcriptional activation of IRF1, TAP1, and IFITM1. (C) Ontological annotation of DEGs in disease and disorders. (D) Molecular function of ELL2-modulated genes. (E) GSEA of DEGs between ELL2-depleted and siControl treated PC-3 cells. Genes in the TNF-α and IFN-γ pathways were upregulated and genes in cervical cancer proliferation cluster were downregulated. (F and G) GSEA analysis of the DEGs enriched in the TNF-α pathway and cervical cancer proliferation cluster, respectively. Abbreviations: DEG, differentially expressed gene; IPA, Ingenuity Pathway Analysis; ELL2, elongation factor for RNA polymerase II 2; GSEA, Gene Set Enrichment Analysis; IFITM1, interferon-induced transmembrane protein 1; IFN, interferon; FDR, false discovery rate; NF-κB, nuclear factor kappa B; NES, normalized enrichment score; STAT1, signal transducer and activator of transcription 1; IRF, interferon regulatory factor; TAP1, transporter 1, ATP binding cassette subfamily B member; TREM1, triggering receptor expressed on myeloid cells 1; TNF, tumor necrosis factor. |
ELL2 Regulation of PC-3 and DU145 Cell Proliferation Is Mediated in Part Through STAT1
According to the RNA-seq analysis, STAT1 was one of the most dramatically up-regulated genes upon ELL2 knockdown in PC-3 cells. IFN-γ-STAT1 signaling was reported to be suppressive of the prostate tumor via the induction of multiple IFN-responsive genes, including the apoptosis-promoting IRF1.44–48 This led to our hypothesis that inhibition of AR-negative prostate cancer cell growth by ELL2 knockdown involves STAT1 activation. We first confirmed STAT1 upregulation by ELL2 knockdown using Western blot and qPCR. In addition, we showed an increased level of phosphorylated STAT1 (pSTAT1) and the increased expression of STAT1-downstream gene IRF1 (Figure 3). Previous studies have shown that LNCaP is unresponsive to the IFNγ-STAT1 pathway, while PC-3 and DU145 were responsive.47–50 Western blot analysis of STAT1 expression in C4-2 and LNCaP cells following ELL2 knockdown was compared to PC-3 cells. ELL2 knockdown induced a slight increase in STAT1 in C4-2 and LNCaP compared to PC-3 (Supplemental Figure S3). These results were also in agreement with our transcriptome analysis of the conditional Ell2 ko mouse prostate, which did not identify a difference in expression in STAT1 mRNA compared to wild-type control mouse prostate.15 Co-occurrence of genetic alterations in STAT1 and ELL2 was identified in 3 patients in the neuroendocrine prostate cancer dataset2 from the cBioPortal for Prostate Cancer Genomics22,23 (Supplemental Figure S4). Next, we tested the effect of siSTAT1 and siELL2 individually or in combination on short-term cell growth in PC-3 and DU145 cell line models using MTT assay. Silencing of STAT1 using two different siRNAs separately enhanced the growth of PC-3 and DU145 cells in MTT assays (Figure 4). In addition, STAT1 knockdown partially rescued siELL2 suppression on the growth of both PC-3 and DU145 cell line models (Figure 4). We also tested the effect of siSTAT1 and/or siELL2 on cell proliferation using BrdU assay and obtained similar results as MTT assay (Figure 5). Since STAT1 siRNA partially rescued the growth inhibition of ELL2 siRNA, ELL2 suppression of AR-negative prostate cancer cell proliferation is partially STAT1-dependent.
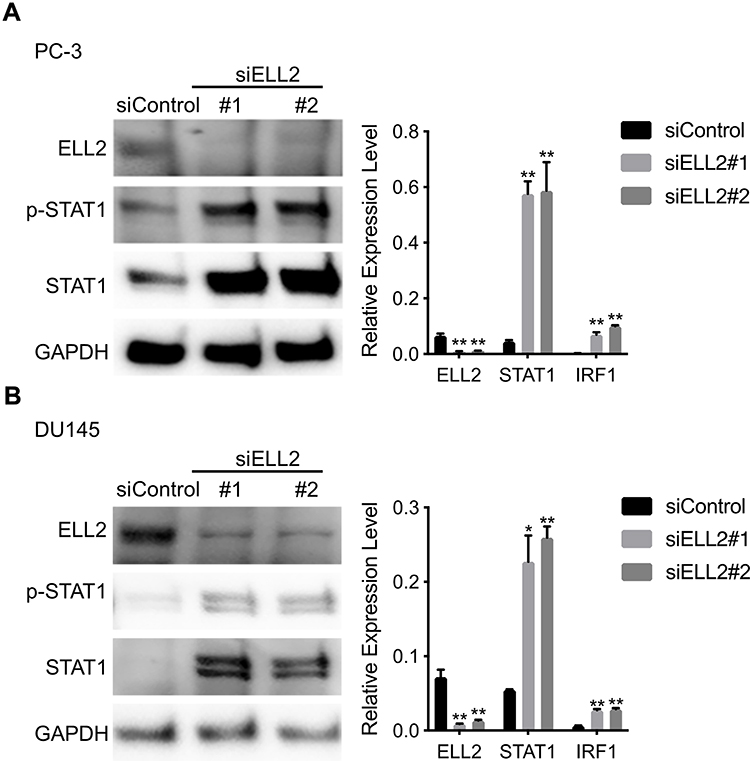 |
Figure 3 (A) Western blot analysis of ELL2, STAT1 and phospho-STAT1 (Tyr701) after ELL2 knockdown in PC-3 cells (left). Quantitive real-time PCR analysis of ELL2, STAT1, and IRF1 mRNA expression in ELL2 knockdown and control PC-3 cells (right). (B) Western blot analysis of ELL2, STAT1 and phospho-STAT1 (Tyr701) after ELL2 knockdown in DU145 cells (left). Quantitive real-time PCR analysis of ELL2, STAT1, and IRF1 mRNA expression in ELL2 knockdown and control DU145 cells (right). GAPDH was probed as the loading control in Western blot analysis. Western blots for PC-3 and DU145 were run on two different gels. Data were shown as mean ± SD; *p < 0.05; **p < 0.01. Abbreviations: ELL2, elongation factor for RNA polymerase II 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IRF1, interferon regulatory factor 1; mRNA, messenger RNA; STAT1, signal transducer and activator of transcription 1; p-STAT1, phosphorylated signal transducer and activator of transcription 1; PCR, polymerase chain reaction; si, small interfering RNA; SD, standard deviation. |
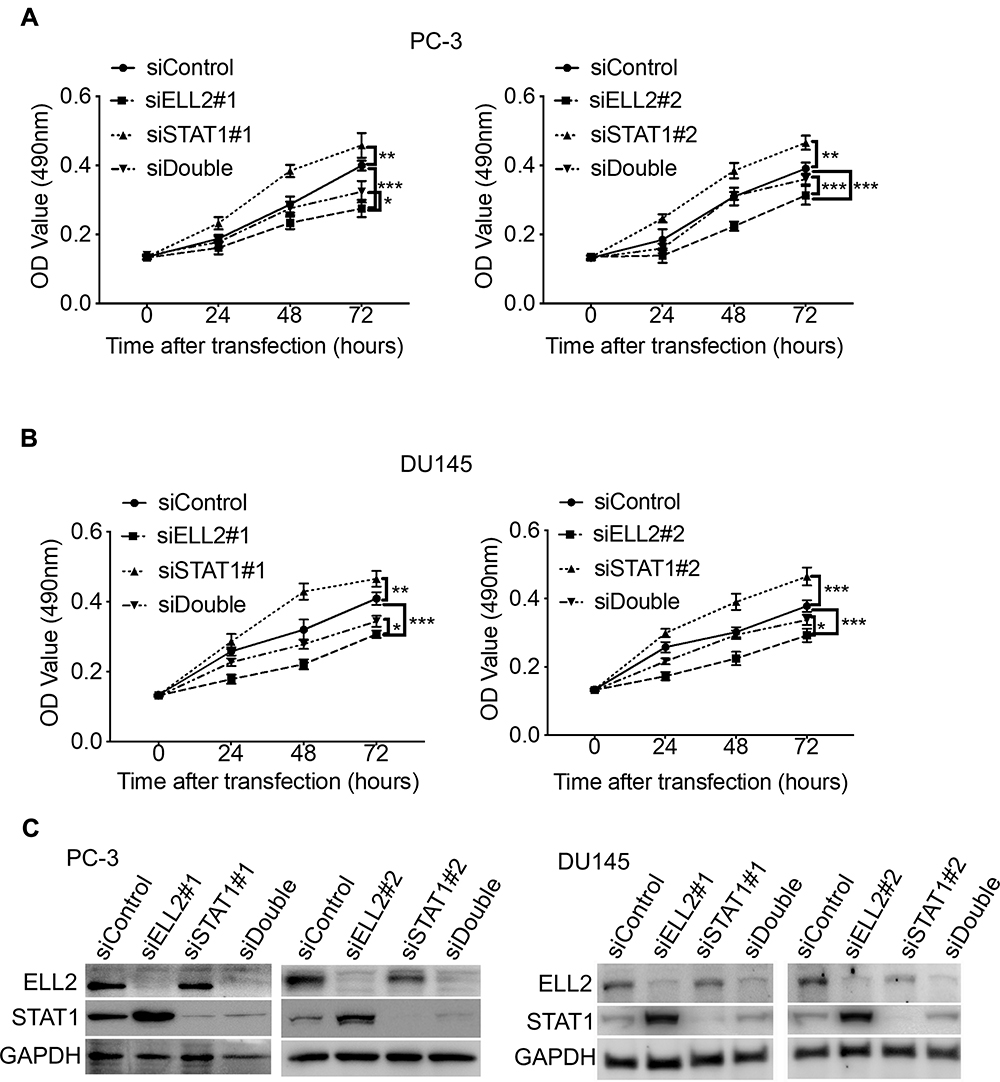 |
Figure 4 Effect of ELL2 and STAT1 knockdown individually or in combination (siDouble) on cell viability in (A) PC-3 and (B) DU145 cells. MTT assay was performed at indicated hours after transfection with indicated siRNAs. (C) Western blot analysis of ELL2 and STAT1 after knockdown in PC-3 and DU145 cells. GAPDH was probed as the loading control. Data were shown as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: ELL2, elongation factor for RNA polymerase II 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide; OD, optical density; si, small interfering RNA; STAT1, signal transducer and activator of transcription 1; SD, standard deviation. |
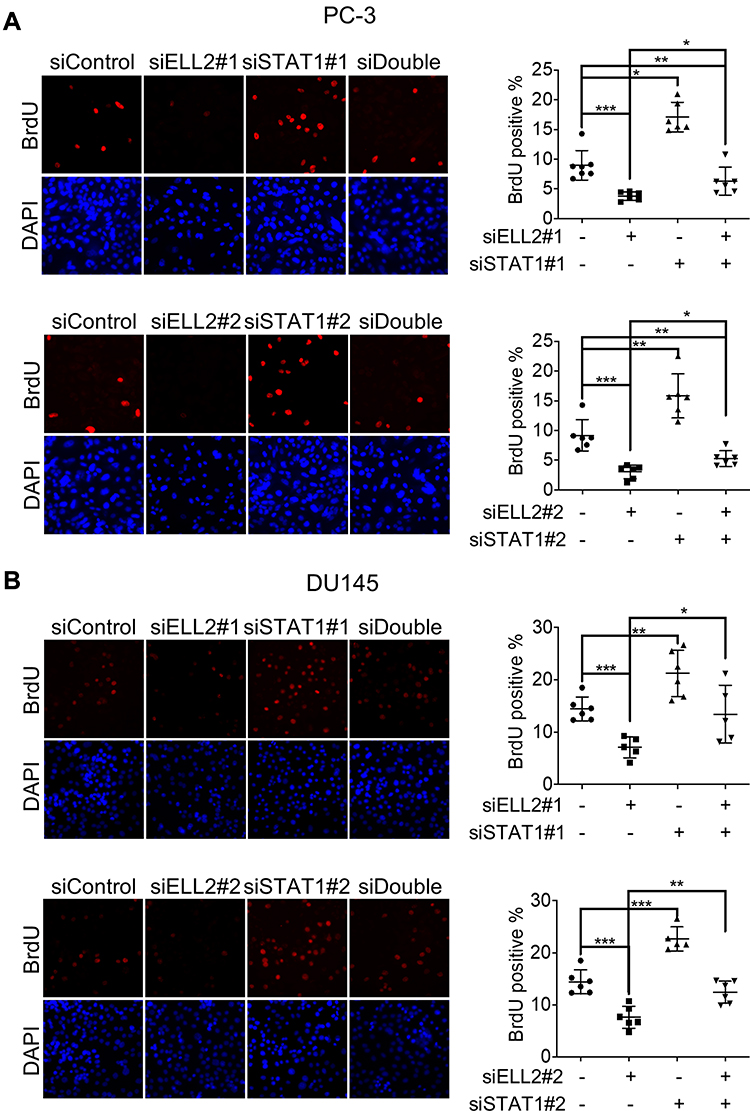 |
Figure 5 BrdU incorporation in PC-3 (A) and DU145 (B) cells transfected with nontargeted control (50 nM siControl) siRNA, targeted to ELL2 (25 nM siELL2+25 nM siControl), target to STAT1 (25 nM siSTAT1+25 nM siControl), and both (25 nM siELL2+25 nM siSTAT1) (siDouble). Upper panel shows BrdU-positive nuclei (red), and lower panel shows nuclear staining with DAPI (Blue). Images were acquired using a 40x objective. Quantification of BrdU incorporation is shown as mean percentage ± SD of BrdU-positive cells relative to the total number of cells. Cells were counted from two different fields for each well from triplicate wells and 50–170 cells per field. *p < 0.05; **p < 0.01, ***p < 0.001. Abbreviations: BrdU, bromodeoxyuridine; DAPI, 4ʹ,6-diamidino-2-phenylindole; ELL2, elongation factor for RNA polymerase II 2; si, small interfering RNA; STAT1, signal transducer and activator of transcription 1; SD, standard deviation. |
In addition to MTT and BrdU assays, we used colony formation assay to evaluate the effect of siSTAT1 and siELL2 individually or in combination in PC-3 and DU145 cells. Figure 6 shows that STAT1 siRNA enhanced colony formation while ELL2 siRNA profoundly inhibited colony formation. Unexpectedly, colony formation in siELL2 was similar to colony formation in the presence of both siELL2 and siSTAT1, suggesting that siSTAT1 did not rescue siELL2 suppression of colony formation (Figure 6).
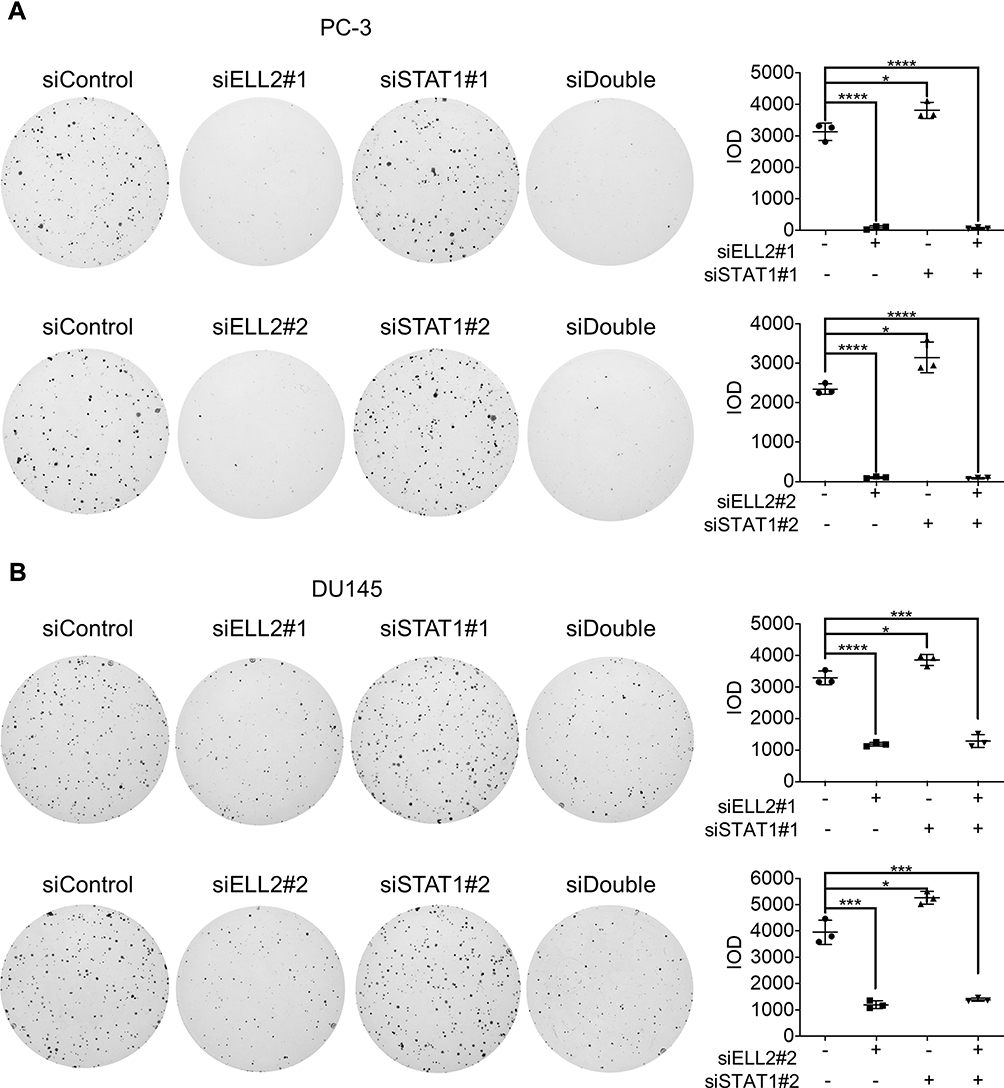 |
Figure 6 Effect of ELL2 and STAT1 knockdown individually or in combination (siDouble) on colony formation. PC-3 (A) and DU145 (B) cells were treated with indicated siRNAs as described in Figure 5, prior to colony formation assay. The colonies were visualized by crystal violet staining. The colonies in each dish were imaged by ChemiDoc™ Touch Imaging System and counted by Image-Pro Plus software for each group of cells. *p < 0.05, ***p < 0.001, ****p < 0.0001. Abbreviations: ELL2, elongation factor for RNA polymerase II 2; IOD, integrated option density; si, small interfering RNA; STAT1, signal transducer and activator of transcription 1. |
ELL2 Knockdown Caused S Phase Cell Cycle Arrest in PC-3 and DU145 Cells
Several genes identified by RNA-Seq analysis are involved in cell cycle regulation. Thus, one potential mechanism by which ELL2 knockdown suppresses PC-3 and DU145 cell proliferation may be the induction of cell cycle arrest. We tested the impact of ELL2 knockdown on cell cycles in PC-3 and DU145 cells 72 hours after treatment with siRNAs. As shown in Figure 7A, siELL2 caused a reduction of PC-3 cells in G0/G1 phase from 78.9% to 58.8% and an increase in S phase from 9.7% to 32.5%. Similar, but less dramatic results were obtained in DU145 cells (Figure 7B). However, STAT1 knockdown had no obvious effect on cell cycle distribution in both PC-3 and DU145 cells (Figure 7A and B). Since S phase cell cycle progression is orchestrated by expression and/or activation of stage-specific cyclins and cyclin-dependent kinase (CDK),51,52 we tested the effect of siELL2 and/or siSTAT1 on the levels of phosphorylated CDK2 (p-CDK2), cyclin D1, and cyclin E. Western blot analysis showed that siELL2 caused suppression of p-CDK2 level and cyclin D1, and up-regulated cyclin E level (Figure 7C). ELL2 knockdown in PC-3 cells dramatically repressed p-CDK2 level, while the repression of p-CDK2 by ELL2 knockdown in DU145 cells was weak but reproducible. In contrast, siSTAT1 did not influence the expression levels of p-CDK2, cyclin D1 and cyclin E (Figure 7C). These observations suggest that ELL2 knockdown can inhibit AR-negative prostate cancer cell proliferation via S phase cell cycle arrest and the S cell cycle arrest is independent of STAT1.
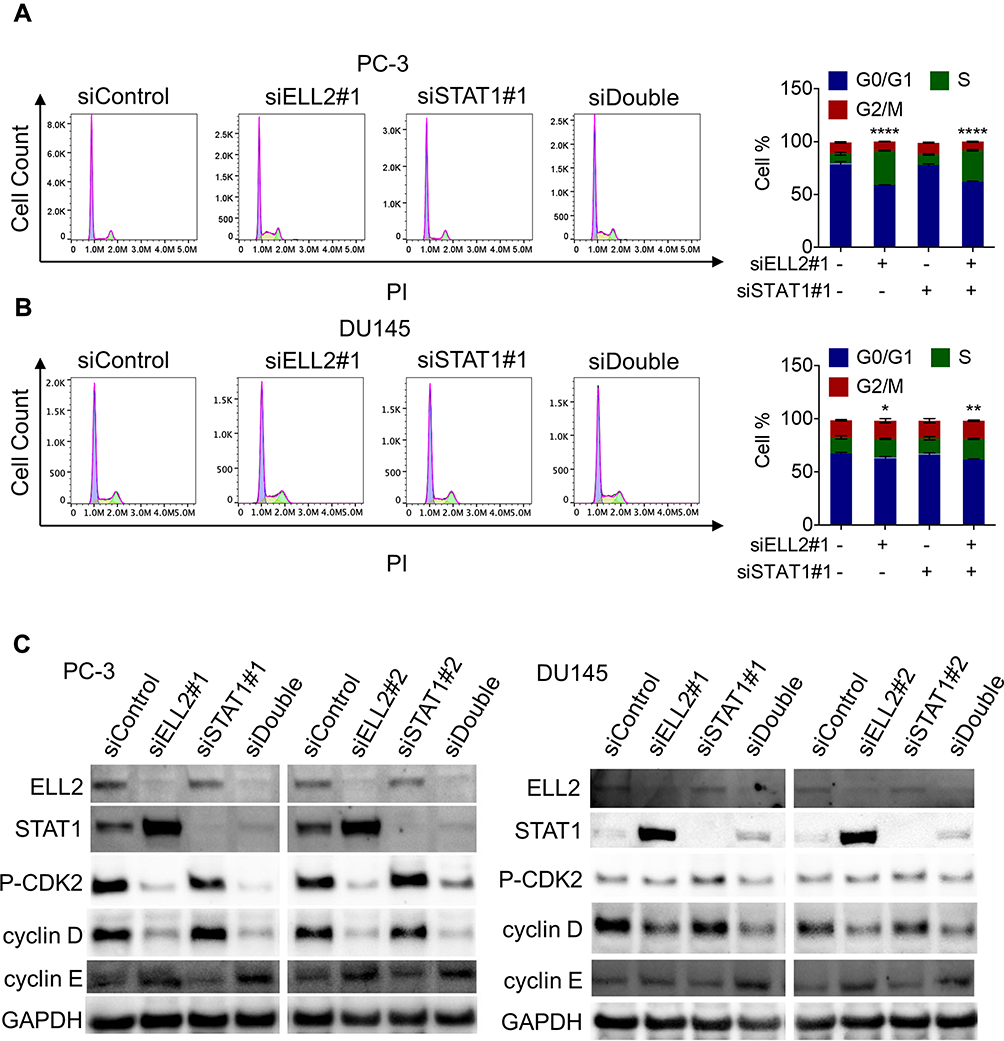 |
Figure 7 Flow cytometry cell cycle analysis of PC-3 (A) and DU145 (B) cells treated with indicated siRNAs as described in Figure 5. Western blot analysis of cyclin D1, cyclin E, p-CDK2 in PC-3 and DU145 cells (C) transfected with indicated siRNAs for 72 h. GAPDH was used as a loading control. *p < 0.05, **p< 0.01, ****p < 0.0001. Abbreviations: BrdU, bromodeoxyuridine; DAPI, 4ʹ,6-diamidino-2-phenylindole; PI, propidium iodide; ELL2, elongation factor for RNA polymerase II 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; p-CDK2, phosphorylated cyclin-dependent kinase 2; si, small interfering RNA; STAT1, signal transducer and activator of transcription 1; G0/G1, Gap0 phase/Gap1 phase; S, synthesis phase; G2/M, Gap2 phase/mitotic phase. |
ELL2 Knockdown Induced Cell Death of PC-3 and DU145 Cells
Since ELL2 knockdown profoundly suppressed colony formation in PC-3 and DU145 cells, this raised a possibility that ELL2 loss can effectively induce cell death in these cells. We thus evaluated the effect of ELL2 knockdown on cell death using trypan blue exclusion staining assay. As shown in Figure 8A, 34.2% of ELL2 knockdown PC-3 cells were stained with trypan blue whereas 9.0% of control siRNA treated PC-3 cells were positive with trypan blue staining. In the same experiment, siSTAT1 had no obvious influence on trypan blue staining of PC-3 cells, either in the absence or presence of siELL2. Similar results were observed in DU145 cell line model. We further performed flow cytometry analysis coupled with Annexin V-FITC and 7-Aminoactinomycin D (7-AAD) staining to evaluate apoptosis induction by ELL2 knockdown in both PC-3 and DU145 models. The results demonstrated a significant increase in early- and late-stage apoptosis in ELL2 knockdown groups compared to the control groups (Figure 8B and C), which are consistent with the findings using trypan blue staining assays. Knockdown efficiency was confirmed by Western blotting (Figure 8D). The above observations suggest that ELL2 is required for AR-negative prostate cancer cell survival and its down-regulation can lead to apoptosis.
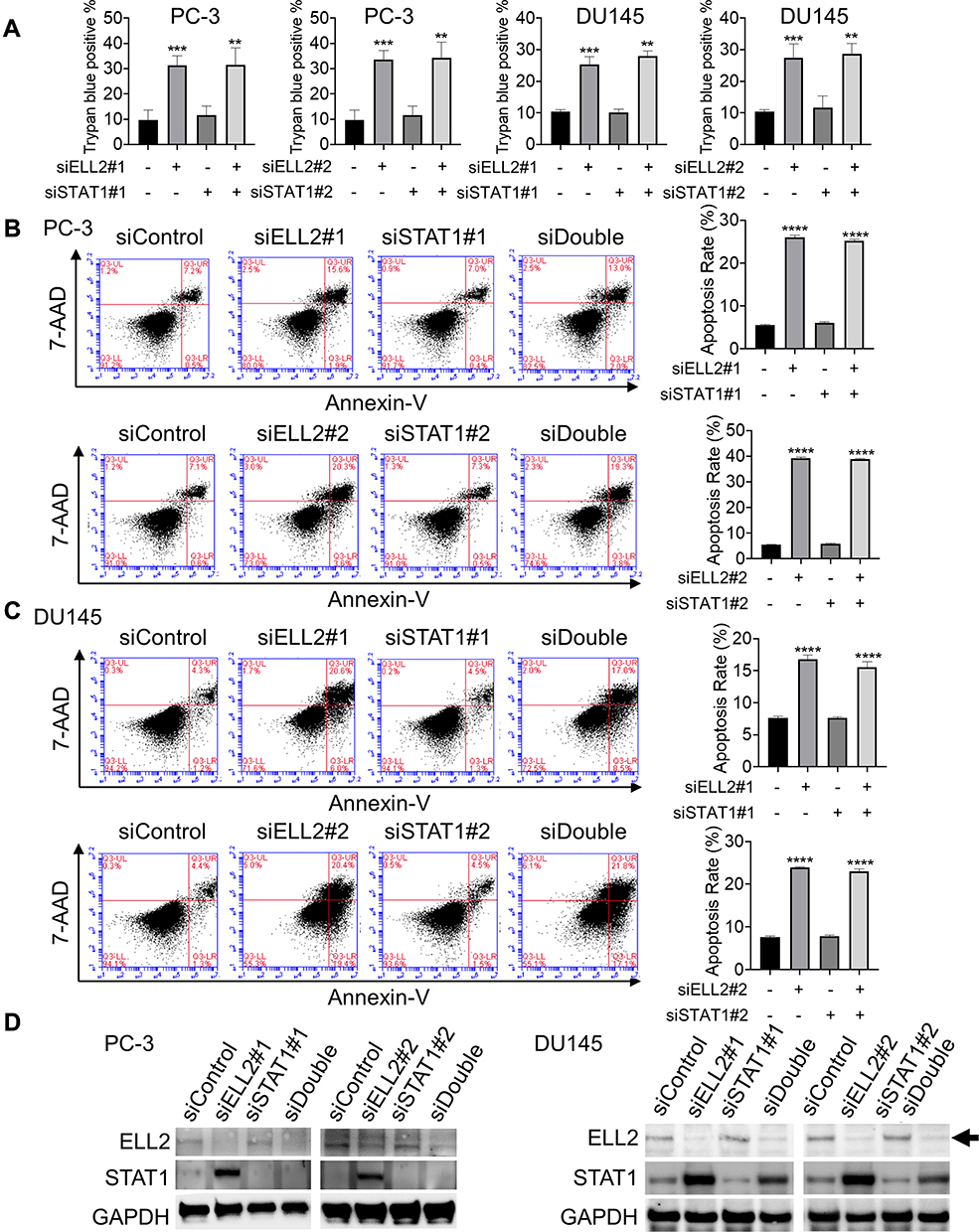 |
Figure 8 (A) Trypan blue exclusion assay of cell death of PC-3 and DU145 cells transfected with indicated siRNA as described in Figure 5. Data represent the means ± SD. Cell apoptosis was analyzed with Flow cytometry apoptosis analysis using Annexin V-FITC/7-AAD in PC-3 (B) and DU145 (C) cells transfected with indicated siRNAs. (D) PC-3 and DU145 cells were untreated or were transfected siELL2, siSTAT1or both for 72 h and analyzed for knockdown efficiency. ELL2 band denoted by the black arrow. GAPDH was used as a loading control. **p< 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: 7-AAD, 7-aminoactinomycin D; BrdU, bromodeoxyuridine; DAPI, 4ʹ,6-diamidino-2-phenylindole; ELL2, elongation factor for RNA polymerase II 2; STAT1, signal transducer and activator of transcription 1; FITC, fluorescein 5-isothiocyanate; PI, propidium Iodide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; si, small interfering RNA; SD, standard deviation. |
Discussion
The present study showed that ELL2 knockdown resulted in growth inhibition and apoptosis induction in PC-3 and DU145 cells. This indicates that ELL2 functions as a pro-survival and growth-stimulating protein in AR-negative prostate cancer cells, which is opposite to its growth-suppressive function in AR-positive LNCaP, C4-2, and 22Rv1 prostate cancer cells.14 The functional differences of ELL2 in AR-positive and AR-negative prostate cancer cell lines are consistent with its down-regulation in prostatic adenocarcinoma specimens and gene amplification in AR-negative prostate cancers.14,15 These observations suggest that ELL2 is a potential tumor suppressor in AR-positive and an oncogenic protein in AR-negative prostate cancer cells.
Differential functions of ELL2 in AR-negative and AR-positive prostate cancer cells suggest that ELL2 regulation of cell fate is context-dependent. Gene expression profiles in AR-positive prostate cancer cells should be very different from AR-negative prostate cancer cells, which could influence how genes are regulated by ELL2. In our previous transcriptome analyses of prostates from mice with conditional deletion of Ell2, Gene ontology-based analysis of Ell2 regulated genes using DAVID identified annotation clusters involving Golgi apparatus, intracellular transport, protein transport, protein localization, protein targeting, organelle membrane, and mitochondrion. Ingenuity Pathway Analysis (IPA) identified networks of interacting genes, with the most significant network associated with the differentiation of neurons and the generation of cells.15 In the present study, in AR-negative PC-3 cells, IPA analysis identified the interferon pathway as the top pathway regulated by ELL2. The down-stream genes regulated by ELL2 in the murine prostate and PC-3 cells appear to be very different and the differentially regulated genes upon ELL2 knockdown in these two cell lines are likely to play important roles in regulating cell proliferation and/or apoptosis of prostate cancer cells. Further studies will be needed to define how ELL2 regulates genes differentially in AR-positive and AR-negative prostate cancer cells and to identify key ELL2-regulated genes that can modulate cell proliferation and/or apoptosis.
We explored the mechanisms by which ELL2 regulates AR-negative prostate cancer cell fate by testing the role of STAT1. STAT1 was chosen in this initial mechanistic study because of its significant up-regulation upon ELL2 down-regulation in PC-3 and DU145 cells. STAT1 loss was reported to associate with prostate cancer recurrence.50 In both localized and advanced patient cohorts, low expression of STAT1 was associated with shorter time to disease recurrence. The authors also showed that STAT1 silencing enhanced PC-3 cell viability,50 which is confirmed by the present study. In our studies, PC-3 or DU145 cells with double knockdown of STAT1 and ELL2 grow faster than the cells with ELL2 knockdown alone but slower than the cells treated with STAT1 siRNA or control siRNA. Thus, ELL2 knockdown still inhibited growth when STAT1 was knockdown in these cells, indicating that STAT1 only partially mediates ELL2 regulation of PC-3 and DU145 proliferation. Our studies also showed S phase cell cycle arrest induction accompanied by suppression of p-CDK2 expression, inhibition of Cyclin D1, and up-regulation of Cyclin E, upon ELL2 knockdown but not STAT1 knockdown. This finding indicates that, in AR-negative cells, ELL2 can modulate proliferation via induction of S phase cell cycle arrest whereas STAT1 modulation of proliferation appears to be independent of cell cycle arrest. The above observations collectively suggest that ELL2 can stimulate AR-negative prostate cancer cell growth via suppressing STAT1-mediated, cell cycle-independent growth suppression as well as via preventing S phase cell cycle arrest involving p-CDK2, cyclins D, and E.
It is possible that STAT1 influences cell proliferation via other mechanisms such as influencing differentiation, but this will need to be elucidated in future studies. Other factors such as TGF-beta1 can inhibit cell survival and differentiation independent of cell-cycle.53 We examined the alteration of STAT1 and ELL2 identified in patients with neuroendocrine prostate cancer in the Neuroendocrine Prostate Cancer database published in the cBioPortal. No ELL2 down-regulation or deletion was observed in neuroendocrine prostate cancer specimens and only three out of 34 patients displayed alterations in both STAT1 and ELL2 (Supplemental Figure S4). The limited number of neuroendocrine prostate cancer specimens in the database makes it difficult to establish relationships between ELL2 and STAT1 in these clinical samples. Since STAT1 knockdown influenced AR-negative prostate cancer cell growth but not cell cycle progression and apoptosis, ELL2 regulation of neuroendocrine prostate cancer may be partially mediated through STAT1.
In the present study, the colony formation assay showed that ELL2 knockdown suppression of PC-3 and DU145 colony formation was not rescued by STAT1 knockdown, whereas MTT and BrdU assays suggested that STAT1 may partially mediate ELL2 regulation of PC-3 and DU145 cell growth. The inability of colony formation assay to detect partial rescue of ELL2 knockdown by siSTAT1 may be due to the low seeding density in the colony formation assay as compared to the MTT and BrdU assays. PC-3 and DU145 cells at low density may be highly susceptible to cell death/apoptosis induction by siELL2, which could result in profound suppression of colony formation. Since STAT1 knockdown does not appear to affect apoptosis, siSTAT1 may not influence cell death/apoptosis induced by ELL2 knockdown and therefore the siELL2 suppression of colony formation.
ELL2 appears to play a major role in the survival of AR-negative prostate cancer cells, because knockdown of ELL2 induced profound apoptosis in PC-3 and DU145 cell lines. PC-3 cells responded more dramatically to ELL2 knockdown than DU145 cells, which may be in part due to the higher expression of ELL2 in PC-3 cells compared to DU145. The data from ELL2 knockdown studies are consistent with the finding from RNA-seq and bioinformatics analyses that molecular functions of ELL2-modulated genes are mainly related to cancer, cell death, and survival in the PC-3 cell model. ELL2 knockdown appears to induce multiple genes related to cell death and survival in AR-negative prostate cancer cells.
ELL2 is not unique in that it can promote or suppress carcinogenesis depending upon cell types. Some proteins were reported to exhibit dual roles as tumor suppressors and oncogenic proteins in different assay systems. Examples of dual role proteins include TRAP154 and Myc.55 Simply inhibiting or activating this type of dual-functional proteins may not be desirable for the treatment and/or prevention of carcinogenesis, including prostate carcinogenesis. However, defining the signaling pathways regulated by this type of dual-function genes may lead to potential approaches to inhibit their oncogenic activities without suppressing their tumor-suppressive functions. Future studies aimed at elucidating the pro-survival and proliferative mechanisms of ELL2 in AR-negative prostate cancer cells may lead to novel approaches to target AR-negative prostate cancer specifically.
In summary, ELL2 is an important factor for the survival and proliferation of AR-negative prostate cancer cells, in contrast to its previously documented tumor-suppressive role in AR-positive prostate cancer cells. The oncogenic and tumor-suppressive roles of ELL2 in AR-negative and AR-positive prostate cancer cell lines, respectively, are consistent with its amplification in AR-negative prostate tumor specimens and down-regulation in AR-positive prostate tumor specimens. Future mechanistic studies of differential ELL2 actions in AR-negative and AR-positive prostate cancer cells may lead to new opportunities to target AR-negative and/or AR-positive prostate tumors specifically. The abstract of this paper was selected for presentation at the 2020 American Urological Association Annual Meeting as a poster presentation with interim findings. This meeting was canceled due to the COVID-19 outbreak. The poster’s abstract was published in “Poster Abstracts” in the Journal of Urology: https://www.auajournals.org/doi/abs/10.1097/JU.0000000000000841.06.
Acknowledgments
We thank Aiyuan Zhang and Jianhua Zhou for technical assistance. This project was supported in part by NIH grant R01 CA186780 (ZW) and R50 CA211242 (LEP). Zhi Wang received a scholarship from the China Scholarship Council. This study used the UPMC Hillman Cancer Center Cytometry Facility, Cancer Bioinformatics Services and University of Pittsburgh HSCRF Health Sciences Sequencing Core at Children’s Hospital which were supported in part by NCI award P30 CA047904 and the Senior Vice Chancellor’s Office at University of Pittsburgh.
Disclosure
Zhi Wang performed the experiments in Pittsburgh and is a visiting scholar to the University of Pittsburgh from Xiangya Hospital of Central South University, China. Dr Zhou Wang reports grants from NIH, during the conduct of the study. The authors declare no other conflicts of interests in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7.
2. Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi:10.1038/nm.4045
3. Thirman MJ, Levitan DA, Kobayashi H, Simon MC, Rowley JD. Cloning of ELL, a gene that fuses to MLL in a t(11;19)(q23;p13.1) in acute myeloid leukemia. Proc Natl Acad Sci U S A. 1994;91(25):12110–12114. doi:10.1073/pnas.91.25.12110
4. Shilatifard A, Duan DR, Haque D, et al. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci U S A. 1997;94:3639–3643. doi:10.1073/pnas.94.8.3639
5. Miller T, Williams K, Johnstone RW, Shilatifard A. Identification, cloning, expression, and biochemical characterization of the testis-specific RNA polymerase II elongation factor ELL3. J Biol Chem. 2000;275:32052–32056. doi:10.1074/jbc.M005175200
6. Johnstone RW, Gerber M, Landewe T, Tollefson A, Wold WS, Shilatifard A. Functional analysis of the leukemia protein ELL: evidence for a role in the regulation of cell growth and survival. Mol Cell Biol. 2001;21:1672–1681. doi:10.1128/MCB.21.5.1672-1681.2001
7. Alexander L-EMM, Watters J, Reusch JA, et al. Selective expression of the transcription elongation factor ELL3 in B cells prior to ELL2 drives proliferation and survival. Mol Immunol. 2017;91:8–16. doi:10.1016/j.molimm.2017.08.016
8. Mitani K, Kanda Y, Ogawa S, et al. Cloning of several species of MLL/MEN chimeric cDNAs in myeloid leukemia with t(11;19)(q23;p13.1) translocation. Blood. 1995;85(8):2017–2024. doi:10.1182/blood.V85.8.2017.bloodjournal8582017
9. Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271:1873–1876. doi:10.1126/science.271.5257.1873
10. Shilatifard A, Haque D, Conaway RC, Conaway JW. Structure and function of RNA polymerase II elongation factor ELL. Identification of two overlapping ELL functional domains that govern its interaction with polymerase and the ternary elongation complex. J Biol Chem. 1997;272(35):22355–22363. doi:10.1074/jbc.272.35.22355
11. Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13:543–547. doi:10.1038/nrm3417
12. Smith ER, Lin C, Garrett AS, et al. The little elongation complex regulates small nuclear RNA transcription. Mol Cell. 2011;44(6):954–965. doi:10.1016/j.molcel.2011.12.008
13. Milcarek C, Albring M, Langer C, Park KS. The eleven-nineteen lysine-rich leukemia gene (ELL2) influences the histone H3 protein modifications accompanying the shift to secretory immunoglobulin heavy chain mRNA production. J Biol Chem. 2011;286:33795–33803. doi:10.1074/jbc.M111.272096
14. Qiu X, Pascal LE, Song Q, et al. Physical and functional interactions between ELL2 and RB in the suppression of prostate cancer cell proliferation, migration, and invasion. Neoplasia. 2017;19:207–215. doi:10.1016/j.neo.2017.01.001
15. Pascal LE, Masoodi KZ, Liu J, et al. Conditional deletion of ELL2 induces murine prostate intraepithelial neoplasia. J Endocrinol. 2017;235:123–136. doi:10.1530/JOE-17-0112
16. Zhong M, Pascal LE, Cheng E, et al. Concurrent EAF2 and ELL2 loss phenocopies individual EAF2 or ELL2 loss in prostate cancer cells and murine prostate. Am J Clin Exp Urol. 2018;6(6):234–244.
17. Yang T, Jing Y, Dong J, et al. Regulation of ELL2 stability and polyubiquitination by EAF2 in prostate cancer cells. Prostate. 2018;78(15):1201–1212. doi:10.1002/pros.23695
18. Tai S, Sun Y, Squires JM, et al. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate. 2011;71(15):1668–1679. doi:10.1002/pros.21383
19. Leiblich A, Cross SS, Catto JW, Pesce G, Hamdy FC, Rehman I. Human prostate cancer cells express neuroendocrine cell markers PGP 9.5 and chromogranin A. Prostate. 2007;67(16):1761–1769. doi:10.1002/pros.20654
20. Wang Y, Pascal LE, Zhong M, et al. Combined loss of EAF2 and p53 induces prostate carcinogenesis in male mice. Endocrinology. 2017;158:4189–4205. doi:10.1210/en.2017-00409
21. Kotawong K, Chaijaroenkul W, Muhamad P, Na-Bangchang K. Cytotoxic activities and effects of atractylodin and beta-eudesmol on the cell cycle arrest and apoptosis on cholangiocarcinoma cell line. J Pharmacol Sci. 2018;136(2):51–56. doi:10.1016/j.jphs.2017.09.033
22. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
23. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
24. Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi:10.1038/nature11125
25. Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50(5):645–651. doi:10.1038/s41588-018-0078-z
26. Abeshouse A, Ahn J, Akbani R. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025. doi:10.1016/j.cell.2015.10.025
27. Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428–11436. doi:10.1073/pnas.1902651116
28. Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666–677. doi:10.1016/j.cell.2013.03.021
29. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291–304 e296. doi:10.1016/j.cell.2018.03.022
30. Ellrott K, Bailey MH, Saksena G, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6(3):271–281 e277. doi:10.1016/j.cels.2018.03.002
31. Taylor AM, Shih J, Ha G, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33(4):676–689 e673. doi:10.1016/j.ccell.2018.03.007
32. Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23(1):227–238 e223. doi:10.1016/j.celrep.2018.03.050
33. Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–416 e411. doi:10.1016/j.cell.2018.02.052
34. Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173(2):321–337 e310.
35. Bhandari V, Hoey C, Liu LY, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet. 2019;51(2):308–318. doi:10.1038/s41588-018-0318-2
36. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi:10.1016/j.cell.2015.05.001
37. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi:10.1016/j.ccr.2010.05.026
38. Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–378. doi:10.1038/nm.4053
39. Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. doi:10.1038/ng.2279
40. Reynolds A, Anderson EM, Vermeulen A, et al. Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA. 2006;12(6):988–993. doi:10.1261/rna.2340906
41. Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. 2005;23(11):1399–1405. doi:10.1038/nbt1161
42. Blazanin N, Cheng T, Carbajal S, DiGiovanni J. Activation of a protumorigenic IFNgamma/STAT1/IRF-1 signaling pathway in keratinocytes following exposure to solar ultraviolet light. Mol Carcinog. 2019;58(9):1656–1669. doi:10.1002/mc.23073
43. Rosty C, Sheffer M, Tsafrir D, et al. Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene. 2005;24(47):7094–7104. doi:10.1038/sj.onc.1208854
44. Park S-Y, Seol J-W, Lee Y-J, et al. IFN-γ enhances TRAIL-induced apoptosis through IRF-1. Eur J Biochem. 2004;271(21):4222–4228. doi:10.1111/j.1432-1033.2004.04362.x
45. Ni Z, Lou W, Lee SO, et al. Selective activation of members of the signal transducers and activators of transcription family in prostate carcinoma. J Urol. 2002;167(4):1859–1862. doi:10.1016/S0022-5347(05)65249-4
46. Patterson SG, Wei S, Chen X, et al. Novel role of Stat1 in the development of docetaxel resistance in prostate tumor cells. Oncogene. 2006;25(45):6113–6122. doi:10.1038/sj.onc.1209632
47. Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-gamma-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014;8(1):204–216. doi:10.1016/j.celrep.2014.05.045
48. Kominsky SL, Hobeika AC, Lake FA, Torres BA, Johnson HM. Down-regulation of neu/HER-2 by interferon-gamma in prostate cancer cells. Cancer Res. 2000;60(14):3904–3908.
49. Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005;65(8):3447–3453. doi:10.1158/0008-5472.CAN-04-4316
50. Hatziieremia S, Mohammed Z, McCall P, et al. Loss of signal transducer and activator of transcription 1 is associated with prostate cancer recurrence. Mol Carcinog. 2016;55(11):1667–1677. doi:10.1002/mc.22417
51. Yeo E-J, Ryu J-H, Chun Y-S, et al. YC-1 induces S cell cycle arrest and apoptosis by activating checkpoint kinases. Cancer Res. 2006;66(12):6345–6352. doi:10.1158/0008-5472.CAN-05-4460
52. Quemeneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003;170(10):4986–4995. doi:10.4049/jimmunol.170.10.4986
53. Pierelli L, Marone M, Bonanno G, et al. Modulation of bcl-2 and p27 in human primitive proliferating hematopoietic progenitors by autocrine TGF-beta1 is a cell cycle-independent effect and influences their hematopoietic potential. Blood. 2000;95(10):3001–3009.
54. Matassa DS, Agliarulo I, Avolio R, Landriscina M, Esposito F. TRAP1 regulation of cancer metabolism: dual role as oncogene or tumor suppressor. Genes. 2018;9(4). doi:10.3390/genes9040195
55. Uribesalgo I, Benitah SA, Di Croce L. From oncogene to tumor suppressor: the dual role of Myc in leukemia. Cell Cycle. 2012;11(9):1757–1764. doi:10.4161/cc.19883
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.