")
Back to Journals » International Journal of Nanomedicine » Volume 11
Elimination of the biphasic pharmacodynamics of 15d-PGJ2 by controlling its release from a nanoemulsion
Authors Abbasi S, Kajimoto K, Harashima H
Received 12 February 2016
Accepted for publication 14 April 2016
Published 8 June 2016 Volume 2016:11 Pages 2685—2694
DOI https://doi.org/10.2147/IJN.S106297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Saed Abbasi,* Kazuaki Kajimoto,* Hideyoshi Harashima
Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Hokkaido, Japan
*These authors contributed equally to this work
Abstract: 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) has a dual action of stimulating anti-inflammation and anti-proliferation when exogenously administered at high doses. However, at lower doses, it can be toxic inducing opposite actions, ie, stimulation of both inflammation and cell proliferation. This biphasic phenomenon of 15d-PGJ2 is believed to be due to its multitarget behavior. In this study, we provide a strategy for controlling such biphasic pharmacodynamics by separating its dual actions while retaining the beneficial one by using a nanoemulsion (NE). The 15d-PGJ2 was encapsulated in the NE composed of triolein/distearoyl phosphatidylcholine/Tween 80 at a high encapsulation ratio (>83%). Furthermore, NE enhanced drug retention by slowing down its release rate, which was, unconventionally, inversely dependent on the total surface area of the NE system. Next, focusing on the biphasic effect on cell proliferation, we found that the 15d-PGJ2-loaded slow-release NE showed only a dose-dependent inhibition of the viability of a mouse macrophage cell line, RAW264.7, although a fast-release NE as well as free 15d-PGJ2 exerted a biphasic effect. The observed slow-release kinetics are believed to be responsible for elimination of the biphasic pharmacodynamics of 15d-PGJ2 mainly for two reasons: 1) a high proportion of 15d-PGJ2 that is retained in the NE was delivered to the cytosol, where proapoptotic targets are located and 2) 15d-PGJ2 was able to bypass cell membrane-associated targets that lead to the induction of cellular proliferation. Collectively, our strategy of eliminating the 15d-PGJ2-induced biphasic pharmacodynamics was based on the delivery of 15d-PGJ2 to its desired site of action, excluding undesired sites, on a subcellular level.
Keywords: drug release, surface localization, anti-proliferative effect, cellular uptake, subcellular delivery
Introduction
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is a terminal product of the cyclooxygenase pathway, which is present in almost all human tissues.1 Contrary to its precursor (prostaglandin D2), 15d-PGJ2 plays an important role in the innate resolution of a once-initiated inflammatory response.2 The α,β-unsaturated carbonyl group in the cyclopentenone ring of 15d-PGJ2 gives rise to two electrophilic carbons that react readily with cellular nucleophiles, which account for the wide spectrum of its biological activity.3 15d-PGJ2 was first identified as the endogenous ligand of peroxisome proliferator-activated receptor (PPAR)γ,4,5 followed by its identification as a direct inhibitor of nuclear factor (NF)-κB.6 This unique mechanism of action attracted substantial attention to the possible use of exogenous 15d-PGJ2 as an anti-inflammatory as well as proapoptotic agent. Consequently, this naturally occurring compound holds a substantial advantage for use in treating cases where inflammation and abnormal cell proliferation are entangled, such as cancers of inflammatory origin (pancreatic and liver cancers)7,8 or rheumatoid arthritis.9
Unfortunately, since 15d-PGJ2 possesses biphasic pharmacodynamics, there are serious drawbacks to its use in the free form. When the effect of 15d-PGJ2 on cellular viability was measured, it was found to induce cell proliferation at low doses followed by apoptosis at higher doses.10 Intriguingly, the same pattern of biphasic action was observed when the inflammatory status was evaluated, in other words, proinflammation at low doses but stimulating anti-inflammatory response at higher doses.11,12 Such biphasic pharmacodynamics indicate that using 15d-PGJ2 at low doses, or if administered in an uncontrolled manner, could worsen a disease condition by inducing a reverse response.
Molecular mechanisms of the 15d-PGJ2-induced biphasic effect are believed to be due to the fact that it exerts its action on different cellular targets, each of which is located at a different location at the subcellular level. As described earlier, the beneficial effects of the drug are the activation of PPARγ, which leads to the activation of apoptosis and stimulating anti-inflammatory responses. Simultaneously, the inhibition of NF-κB plays a pivotal role in shutting down both proliferatory and inflammatory responses. Both these molecular targets are located in deeper regions of the cytosol or in the cell nucleus. Therefore, as governed by its lipophilicity, which makes it difficult to diffuse across the aqueous cytosol, a high dose of exogenously applied 15d-PGJ2 is necessary to achieve an effective concentration at the inner cytosol level. Unfortunately, before this drug diffuses to the inner cytosol, it encounters and activates an inner cell membrane-bound G-protein called H-Ras.13,14 The activation of H-Ras leads to Raf activation and the subsequent activation of the extracellular-signal-regulated kinases 1 and 2 (ERK1/2), which eventually activates a cascade of proliferatory and proinflammatory responses. Another possible route of the undesired effects induced by 15d-PGJ2 is its binding to a cell surface receptor known as the prostaglandin D2 receptor 2 (DP2), also known as the CRTH2.15,16 The binding of 15d-PGJ2 to DP2 increases the intracellular influx of calcium ions, which, in part, can activate ERK1/2.17,18 Therefore, a low dose of 15d-PGJ2 that accumulated largely around the cell membrane is associated with an undesired response.
The safe and efficient use of 15d-PGJ2 is contingent upon separating these two actions and then retaining only the beneficial one. Knowing that both induced actions are the result of drug action on different subcellular locations, we hypothesized that it is possible to control such biphasic pharmacodynamics by utilizing a drug delivery system. 15d-PGJ2 has been formulated in many nanoparticle platforms, including PLGA nanocapsules,19 liposomes,20 and albumin conjugates,21 to enhance its pharmacokinetics and tissue targeting. A nanoemulsion (NE) is also a widely used carrier for lipophilic drugs, since this formulation enhances their solubility, pharmacokinetics, and tissue targeting by utilizing both passive and active targeting.22 Moreover, its ease of preparation and composition of naturally occurring oils and nontoxic surfactants make it economical to use.23 Emulsions have been shown to be suitable carriers for antitumor prostaglandins24 and prostaglandin E1 (PGE1),25,26 which are structurally similar to 15d-PGJ2. Furthermore, PGE1 is commercially available for parenteral use in the form of NE (marketed in Japan as Palux® by Taisho Pharmaceuticals Co., Ltd., Tokyo, Japan). Herein, for the first time, we report on the formulation of 15d-PGJ2 in an NE type of drug delivery system and the successful control of 15d-PGJ2-induced biphasic pharmacodynamics by separating both actions and retaining only the beneficial one.
Materials and methods
Materials and reagents
15d-PGJ2 was purchased from Cayman (Ann Arbor, MI, USA). Triolein was purchased from Tokyo Chemical Industry (Tokyo, Japan). Tween 80, Dulbecco’s Modified Eagle’s Medium (DMEM), and fetal bovine serum were supplied by Sigma-Aldrich Co. (St Louis, MO, USA), and 1,2-distearoyl-sn-glycero-3 phosphocholine (DSPC) was obtained from NOF (Tokyo, Japan). A Cell Counting Kit-8 was obtained from Dojindo (Kumamoto, Japan). The 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) fluorescent probe was supplied by Thermo Fisher Scientific (Waltham, MA, USA).
NE preparation and characterization
Separate solutions of triolen (as the oil core) and DSPC (as an essential surfactant) were prepared in chloroform, while 15d-PGJ2 was dissolved in ethanol. The desired volume of each component was transferred, and the organic solvent was removed by evaporation using a stream of nitrogen gas. The remaining oil phase was placed in a water bath at 70°C for a few minutes. Then, 1 mL of preheated aqueous solution containing the desired amount of Tween 80 (cosurfactant) was added dropwise to the tube on a vortex mixer. After this step, an oil-in-water primary emulsion is formed, to reduce size of globules, probe sonication (Misonix, Farmingdale, NY, USA) was applied for 3 minutes at the power of 6 W. Finally, the NE was placed in a shaker at room temperature and allowed to cool down.
Particle size, polydispersity index, and ζ-potential were measured by using a Malvern Zetasizer (Malvern Instruments, Malvern, UK).
Determination of encapsulation ratio
15d-PGJ2-loaded NE was inserted into the donor chamber of Amicon tube (EMD Millipore, Billerica, MA, USA) having molecular weight cutoff value of 100 kDa, and centrifuged at 15,000× g at 25°C for 15 minutes to remove free drug. The remaining NE was then further washed with water, centrifuged, and resuspended in water. The amount of 15d-PGJ2 in the donor chamber after resuspension represents the amount of encapsulated drug in the NE particles. The encapsulation ratio was calculated as the ratio of encapsulated 15d-PGJ2 to the total amount of added 15d-PGJ2. NE samples, before and after free drug separation, were diluted in a mixture of dimethyl sulfoxide:chloroform (7:3), and the 15d-PGJ2 amount was determined by its light absorbance at λ=306 nm using a spectrophotometer (Beckman Coulter, Brea, CA, USA). Empty NEs were prepared in parallel to the loaded ones and used as blanks.
Drug release studies
15d-PGJ2-loaded NE was dialyzed against phosphate-buffered saline (PBS) using a dialysis membrane (molecular weight cutoff: 3 kDa, Spectrum Laboratories Inc., Rancho Dominguez, CA, USA). The volume of PBS was sufficiently large to not reach the saturation solubility of 15d-PGJ2, which ensures sink conditions. At specific time points (2 hours, 4 hours, 6 hours, and 24 hours), a sample of NE was withdrawn from the dialysis bag and dissolved in a mixture of dimethyl sulfoxide:chloroform (7:3), and the absorbance was measured as described earlier. The amount of 15d-PGJ2 in the NE at a specific time point divided by the total amount of 15d-PGJ2 represents the percentage of drug remaining in the formulation at that time point. To calculate the percentage of released drug, the values were subtracted from 100%.
Cell viability studies
RAW264.7 cells were cultured in DMEM supplemented with 10% fetal bovine serum for 24 hours in 96-well plates at a density of 0.75×104 cell/well. Treatment was then applied in a dose-dependent manner and was classified into two groups: free drug and NE treatment. In the NE treatment group, to normalize the interference of NE itself on cell viability, all the wells shared the same final concentration of NE, by adding empty NE, so the final NE concentration in each well equaled 630 μM of triolein content. After the addition of treatment, the plates were further incubated for 24 hours and a cell viability assay (water-soluble tetrazolium salt) was carried out. The absorbance was measured using a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA). Relative cell viability was calculated as the ratio of absorbance of the treated wells to the nontreated ones.
Quantitative assessment of NE cellular uptake
For fluorescence labeling of NE, DiD was added to the oil phase when NE was prepared, as 0.2 mol% of the triolein content. The cells (2.1×105 cells/well) were incubated in six-well plates for 24 hours, and then treated with the DiD-labeled NEs at a dose of 630 μM of triolein content for 2 hours, 4 hours, 6 hours, or 24 hours. The cells were washed with cold PBS followed by a cold heparin solution (20 U/mL in PBS). After washing, cells were collected, suspended in PBS containing 0.5% bovine serum albumin and 0.1% NaN3, and analyzed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Results
NE characterization and encapsulation ratio
To study the effect of each component on the characteristics of NE, the amount of the component in question was changed while other components were kept fixed (Table 1). By fixing the amount of triolein and increasing the amount of Tween 80, the particle size was decreased (NE 1–4). Likewise, increased amounts of DSPC were associated with a smaller particle size (NE 4–6). However, increasing the triolein content while fixing the surfactant content resulted in an increased particle size (NE 4, 7, and 8). Furthermore, fixing the ratio of the three components with respect to each other but increasing their total amounts was not associated with a notable change in particle size (NE 4, 9, and 10). In summary, increasing the surfactant content is associated with smaller particle size, while increasing the oil content is associated with larger particle size. In addition, the oil-to-surfactant ratio can be considered to be a determinant of the particle size regardless of the total amount of components. The encapsulation ratio of 15d-PGJ2 was quite high for all tested formulations (83%–99%), which indicates that NE is an efficient system for encapsulating15d-PGJ2.
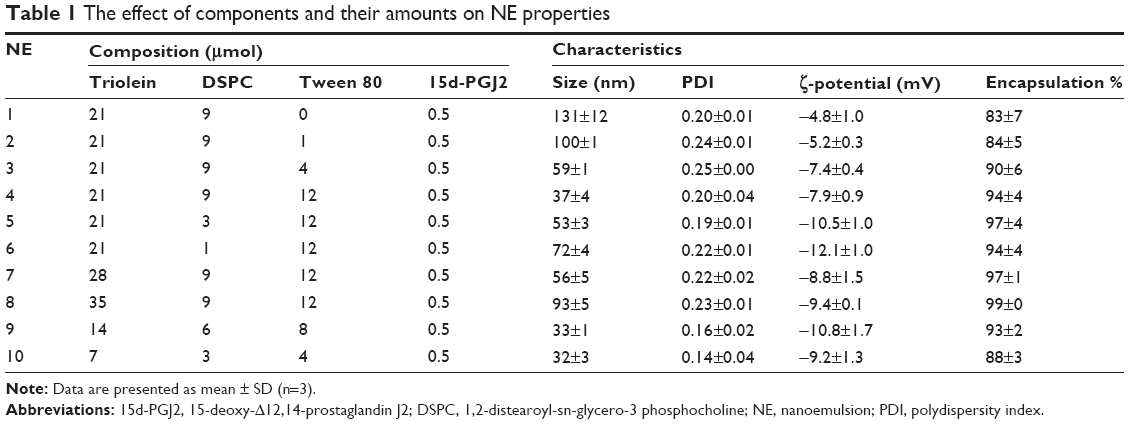 | Table 1 The effect of components and their amounts on NE properties |
Drug release studies
Obtaining a controlled release formulation with minimal release is important, since the drug is retained in a stable form in the particle phase until it is delivered to the cell. Accordingly, formulations listed in Table 1 were screened for their ability to control the rate of release of 15d-PGJ2. The NE formulations 1–4 showed a gradual decrease in release rates when the amount of Tween 80 was increased (Figure 1A). Similarly, but to a lesser extent, increasing the amount of DSPC in formulations 6–4 was associated with a slight decrease in release rate (Figure 1B). As described in the “NE characterization and encapsulation ratio” section, increased surfactant amounts result in a smaller particle size, and hence, a smaller particle size seems to be associated with a slower release. For a given system, it is generally known that a smaller particle size is related to a larger surface area and subsequently a faster drug release rate.27 However, interestingly, the rate of release of 15d-PGJ2 decreased with decreasing particle size. Furthermore, the effect of oil on drug release was evaluated. As shown in Figure 1C, no difference was found in the release profiles of formulations 4, 7, and 8, despite the clear difference in their particle size (Table 1). On the other hand, increasing the oil volume simultaneously with the amount of surfactant in a constant ratio slowed down the release rate (Figure 2), in spite of the fact that formulations 4, 9, and 10 show no significant change in their particle size.
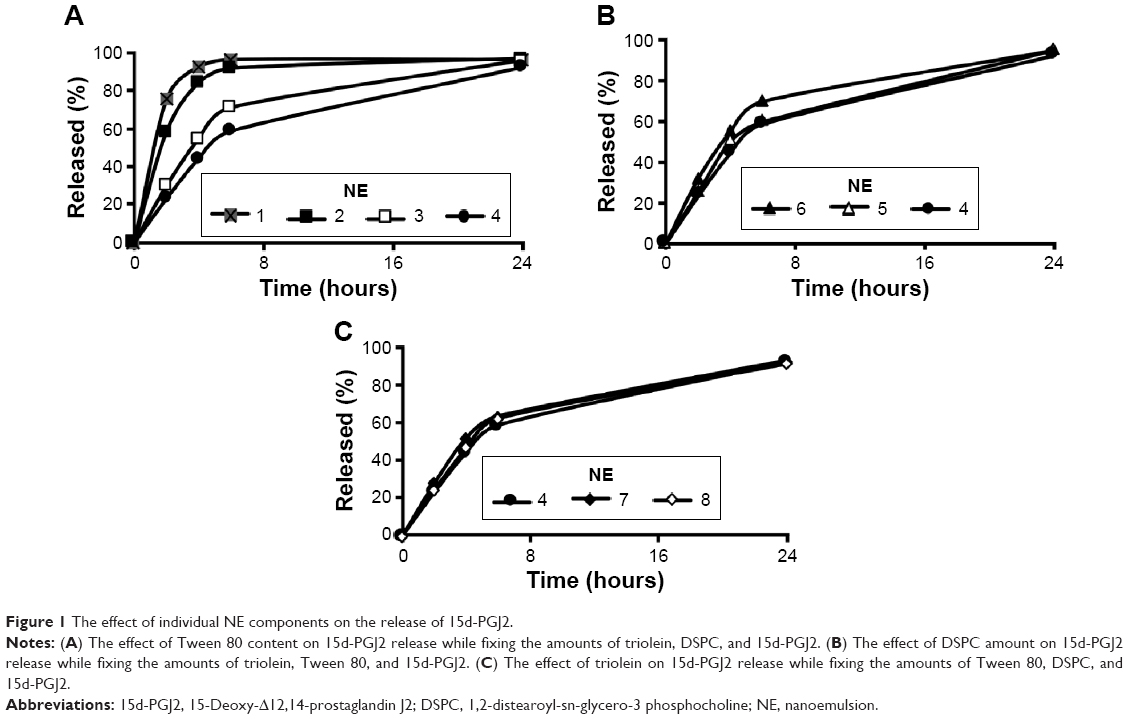 | Figure 1 The effect of individual NE components on the release of 15d-PGJ2. |
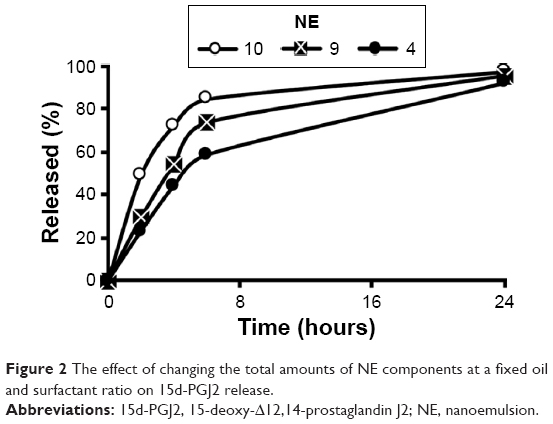 | Figure 2 The effect of changing the total amounts of NE components at a fixed oil and surfactant ratio on 15d-PGJ2 release. |
Cell viability studies
Treatment of RAW264.7 cells with free 15d-PGJ2 showed a biphasic pattern on cell viability, or in other words, enhanced cell viability at low doses (doses <20 μM, with a peak at ~7.5 μM), but promoted a reduction in cell viability at doses higher than that, 20 μM (Figure 3). In order to evaluate the effect of NEs characterized by rapid and slow drug release on cell viability, NE 1 and 4 were used, since they showed the fastest and slowest release profiles (Figure S1) and are referred to as fast-release NE (FRNE) and slow-release NE (SRNE), respectively. When the SRNE formulation was used, the biphasic pattern could be controlled and converted into a single inhibitory phase, which is a desired change (Figure 3). On the other hand, the FRNE failed to separate the biphasic pattern but maintained it to a lesser extent compared to the free drug (Figure 3).
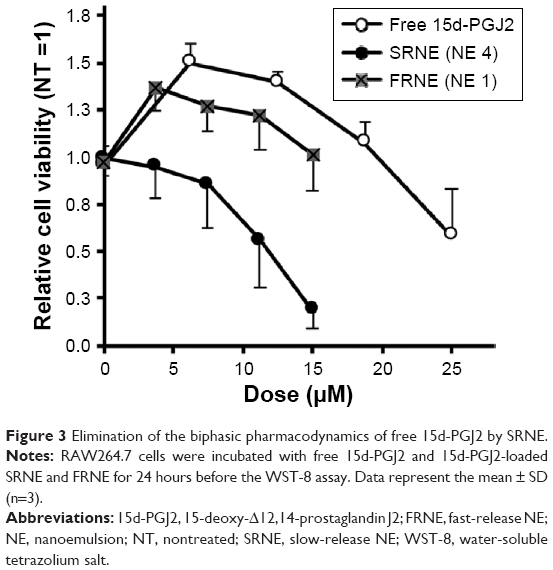 | Figure 3 Elimination of the biphasic pharmacodynamics of free 15d-PGJ2 by SRNE. |
FRNE and SRNE cellular uptake
To further elaborate the mechanism by which the SRNE, but not the FRNE, was able to eliminate the biphasic effect of 15d-PGJ2, a quantitative assessment of the cellular uptake of NEs was carried out. As a result, we found that the cellular uptake was higher in FRNE rather than that in SRNE (Figure 4), indicating that the difference in the cellular uptake between FRNE and SRNE is not the main reason to explain the elimination of the biphasic effect by SRNE.
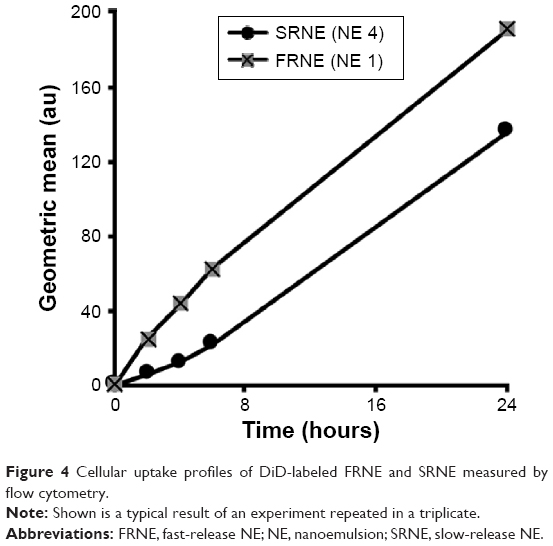 | Figure 4 Cellular uptake profiles of DiD-labeled FRNE and SRNE measured by flow cytometry. |
Discussion
The main focus of this study was to control the 15d-PGJ2-induced biphasic pharmacodynamics on cell proliferation. RAW264.7 cells were used as model cells for the important role of macrophages in the pathophysiology of many cancers and inflammatory disorders.28,29 Based on the molecular mechanisms of biphasic pharmacodynamics, 15d-PGJ2 has two main sites of action: a cytosolic one that comprises the desired target (proapoptotic) and the cell membrane that contains undesired targets (proliferative). Prostaglandins are lipophilic compounds that can passively diffuse across cellular membranes,30 in addition to the utilization of carrier transport systems (such as anionic carrier proteins). It has been reported that some cyclopentenone prostaglandins have specific transporters that permit them to move inward across cellular membranes,31,32 nonetheless, such a specific transporter for 15d-PGJ2 has not yet been identified, which leaves passive diffusion as the main possible route for its transport when added exogenously.
In cases where a drug response is only elicited after the drug arrival at the intracellular compartment, nanoparticles are overwhelmingly employed for this purpose.33 Both SRNE and FRNE have been shown to be taken up by RAW264.7 cells (Figure 4). FRNE showed a higher uptake that can be attributed to the lack of Tween 80. Tween 80 contains a polar head of a branched polyethylene glycol, which is widely known to hinder the cellular uptake of nanoparticles,34,35 and therefore, the presence of a polyethylene glycol moiety on the SRNE surface could explain its dampened uptake. In addition, to achieve successful intracellular delivery, it is also a fundamental task that the nanoparticle retains its cargo safe and intact until it reaches the intracellular compartment. Actually, FRNE was not able to convert the biphasic effect (Figure 3) because most of its 15d-PGJ2 was rapidly released from NE into the cell culture media, resulting in undesired effects by acting on cell membrane-associated targets such as DP2 and H-Ras. In other words, FRNE failed in the sufficient intracellular delivery of 15d-PGJ2 in spite of its large uptake by cells (Figure 4). On the other hand, SRNE was able to control the biphasic pattern by converting it into a single phase; a desirable occurrence (Figure 3). SRNE is characterized by minimal release outside the cell, meaning that a relatively high proportion of its cargo was retained in the NE particles, and that they bypassed the cell membrane and were delivered to the cytosol when NE was taken up, resulting in the effective activation of PPARγ and the inhibition of NF-κB. In order to prove our hypothesis mentioned earlier, further experiments to confirm a higher intracellular accumulation of 15d-PGJ2 in the case of SRNE are required, nevertheless, and since 97%–99% of the 15d-PGJ2 added to the cell culture medium was found to be inactivated,36 a NE is believed to increase the cellular internalization of 15d-PGJ2 compared to the bare form, which can be attributed to the function that NE provides in protecting 15d-PGJ2 from interacting with cell culture media and subsequent deactivation. Accordingly, the avoidance of cell-membrane-related targets achieved by a slow release combined with the direct intracellular delivery achieved by the uptake of NE is proposed as the main mechanism by which the SRNE was able to exclusively induce the desired effects (the mechanism is illustrated in Figure 5). In addition, further in vivo investigations are still needed to demonstrate the functionality of the 15d-PGJ2-loaded SRNE as a therapeutic agent.
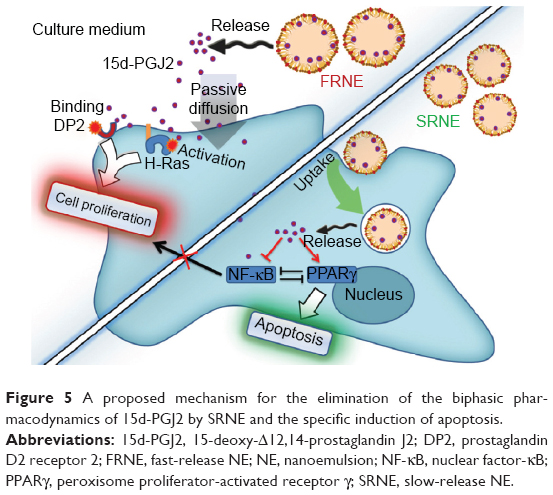 | Figure 5 A proposed mechanism for the elimination of the biphasic pharmacodynamics of 15d-PGJ2 by SRNE and the specific induction of apoptosis. |
From the drug release study, we found that an increase in the surfactant content was associated with a decrease in the rate of release of 15d-PGJ2 from NE (Figure 1A and B). The observed effect of DSPC on drug release was minimal compared to Tween 80 (Figure 1B). However, in NE 4–6, the content of Tween 80 (12 μmol) was higher than that of DSPC (1–9 μmol; Table 1). Therefore, it is likely that the effect of DSPC on drug release was masked due to the presence of high amounts of Tween 80. Figure S2 shows the clear effect of DSPC on slowing down the release of 15d-PGJ2 when added in the presence of low amounts of Tween 80. Based on this observation, and combined with the effect of Tween 80 on release, it is clear that the observed 15d-PGJ2 release rates are inversely proportional to the amount of surfactant used. In our experiments, increasing the surfactant content caused a decrease in particle size, and since the oil volume is fixed, the number of particles is supposedly increased, thus resulting in increased total surface area. However, in the case of increasing the oil content while fixing surfactant amounts, the increase in the surface area of a particle due to an increase in its diameter is compensated by a decrease in the number of particles, leaving the total surface area unchanged. Furthermore, in the case of increasing the total amounts of components while the particle size was unchanged by fixing the ratio of oil to surfactant, the increase in particle number is believed to be responsible for the increase in total surface area and explains the observed decrease in release rate. Consequently, from a geometrical viewpoint, 15d-PGJ2 release rates were slower in NE formulations that had a larger total surface area. Geometrical parameters were roughly estimated by mathematical calculations (Supplementary material) and were used to calculate the total surface area for each of the ten formulations (Table S1). To provide evidence of our proposed hypothesis that relates drug release to the total surface area of an NE system, the total surface area calculated for each formulation was plotted against its release half-life (R50), which is listed in Table S2. The plot shown in Figure 6 shows a directly proportional relationship between R50 and total surface area. Since the release rate and R50 are inversely dependent on each other, we predict an inversely proportional relationship between release rate and the calculated surface area for each formulation. Thus, we report on the unconventional release behavior of 15d-PGJ2 that is inversely dependent on the total surface area of the NE system.
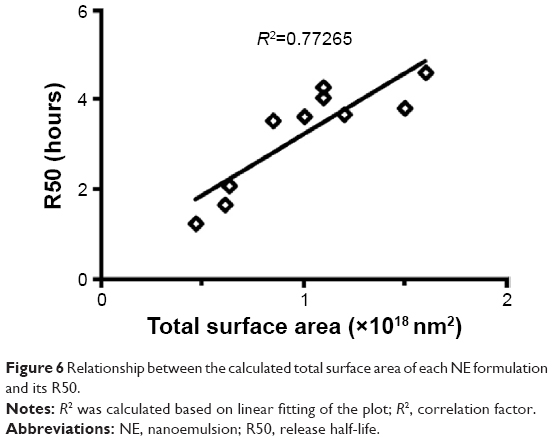 | Figure 6 Relationship between the calculated total surface area of each NE formulation and its R50. |
Conventionally, when a drug is homogeneously distributed in the oil core of NE, its release is governed by Fick’s diffusion across the interfacial membrane, meaning that the diffusional release rate is directly proportional to the surface area. The inverse dependency of the 15d-PGJ2 release rate on the NE total surface area could only be explained by the localization of 15d-PGJ2 on the interface between the oil and water. An increased total surface area results in a smaller drug surface density, or the number of 15d-PGJ2 molecules per unit surface area, which is accompanied with slower release rate. Additional supportive data that provide a clue about the 15d-PGJ2 location on an NE particle are the zeta potentials. All NE compositions formulated in this study were prepared in pairs: empty and drug-loaded particles. The drug-loaded NEs showed lower zeta potentials compared to empty particles (Table S3). This lowering is believed to be due to the ionization of the 15d-PGJ2 carboxyl group and its orientation to the NE exterior (Figure 7). Furthermore, NE size had a slight tendency to decrease after loading with 15d-PGJ2, which can be attributed to the surface-active properties possessed by the drug. Our observations and explanations are compatible with other published results, reporting that PGE1, which has a chemical structure similar to that of 15d-PGJ2, is localized on the surface of the emulsion.37,38
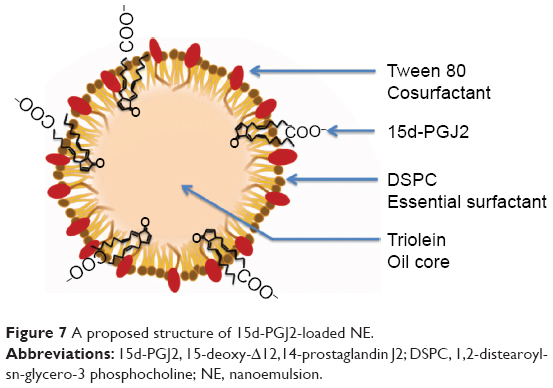 | Figure 7 A proposed structure of 15d-PGJ2-loaded NE. |
Conclusion
It is no longer a substantial problem to discover or synthesize potent therapeutic agents, but the real challenge is how to efficiently use the currently existing ones. In this study, NE was able to control the release of 15d-PGJ2, and NE, characterized by a slow release, could be employed to convert the biphasic pharmacodynamics of 15d-PGJ2 into a single, desirable phase. This can be attributed to the targeted delivery of 15d-PGJ2 to the desired site of action, excluding undesired sites, at a subcellular level. Our experiments were focused on the biphasic effect on cell proliferation; however, and since they share common molecular mechanisms, SRNE would be expected to be useful for controlling the inflammatory biphasic effect in a similar fashion.
Acknowledgments
This work was partially supported by grants from the Special Education and Research Expenses of the Ministry of Education, Culture, Sports, Science and Technology of Japan and by Japan Society for the Promotion of Science under the Japan–Korea Basic Scientific Cooperation Program. Finally, the authors thank Doctor MS Feather for his helpful advice in writing the manuscript in the English language.
Disclosure
The authors report no conflicts of interest in this work.
References
Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114(2):100–109. | ||
Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5(6):698–701. | ||
Kim EH, Surh YJ. 15-deoxy-Delta12,14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochem Pharmacol. 2006;72(11):1516–1528. | ||
Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83(5):803–812. | ||
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. | ||
Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000; 403(6765):103–108. | ||
Chen S, Liu C, Wang X, Li X, Chen Y, Tang N. 15-Deoxy-Delta(12,14)-prostaglandin J2 (15d-PGJ2) promotes apoptosis of HBx-positive liver cells. Chem Biol Interact. 2014;214:26–32. | ||
Farrow B, Sugiyama Y, Chen A, Uffort E, Nealon W, Mark Evers B. Inflammatory mechanisms contributing to pancreatic cancer development. Ann Surg. 2004;239(6):763–769; discussion 769–771. | ||
Kawahito Y, Kondo M, Tsubouchi Y, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106(2):189–197. | ||
Emi M, Maeyama K. The biphasic effects of cyclopentenone prostaglandins, prostaglandin J(2) and 15-deoxy-Delta(12,14)-prostaglandin J(2) on proliferation and apoptosis in rat basophilic leukemia (RBL-2H3) cells. Biochem Pharmacol. 2004;67(7):1259–1267. | ||
Bureau F, Desmet C, Mélotte D, et al. A proinflammatory role for the cyclopentenone prostaglandins at low micromolar concentrations: oxidative stress-induced extracellular signal-regulated kinase activation without NF-kappa B inhibition. J Immunol. 2002;168(10):5318–5325. | ||
Martinez AE, Sanchez-Gomez FJ, Diez-Dacal B, Oeste CL, Perez-Sala D. 15-Deoxy-delta(12,14)-prostaglandin J2 exerts pro- and anti-inflammatory effects in mesangial cells in a concentration-dependent manner. Inflamm Allergy Drug Targets. 2012;11(1):58–65. | ||
Powell WS. 15-Deoxy-delta12,14-PGJ2: endogenous PPARgamma ligand or minor eicosanoid degradation product? J Clin Invest. 2003; 112(6):828–830. | ||
Oliva JL, Pérez-Sala D, Castrillo A, et al. The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc Natl Acad Sci U S A. 2003;100(8):4772–4777. | ||
Ueki S, Kato H, Kobayashi Y, et al. Anti- and proinflammatory effects of 15-deoxy-delta-prostaglandin J2(15d-PGJ2) on human eosinophil functions. Int Arch Allergy Immunol. 2007;143(suppl 1):15–22. | ||
Zhang X, Young HA. PPAR and immune system – what do we know? Int Immunopharmacol. 2002;2(8):1029–1044. | ||
Chuderland D, Seger R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun Integr Biol. 2008;1(1):4–5. | ||
Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal. 2002;14(8):649–654. | ||
Napimoga MH, da Silva CA, Carregaro V, et al. Exogenous administration of 15d-PGJ2-loaded nanocapsules inhibits bone resorption in a mouse periodontitis model. J Immunol. 2012;189(2):1043–1052. | ||
Homem de Bittencourt PI Jr, Lagranha DJ, Maslinkiewicz A, et al. LipoCardium: endothelium-directed cyclopentenone prostaglandin-based liposome formulation that completely reverses atherosclerotic lesions. Atherosclerosis. 2007;193(2):245–258. | ||
Hagens WI, Mattos A, Greupink R, et al. Targeting 15d-prostaglandin J2 to hepatic stellate cells: two options evaluated. Pharm Res. 2007;24(3):566–574. | ||
Chime SA, Kenechukwu FC, Attama AA. Nanoemulsions – advances in formulation, characterization and applications in drug delivery. In: Sezer AD, editor. Application of Nanotechnology in Drug Delivery. Rijeka: InTech; 2014:77–126. | ||
McClements DJ. Nanoemulsions versus microemulsions: terminology, differences, and similarities. Soft Matter. 2012;8(6):1719–1729. | ||
Fukushima S, Kishimoto S, Takeuchi Y, Fukushima M. Preparation and evaluation of o/w type emulsions containing antitumor prostaglandin. Adv Drug Deliv Rev. 2000;45(1):65–75. | ||
Yamaguchi T, Fukushima Y, Itai S, Hayashi H. Rate of release and retentivity of prostaglandin E1 in lipid emulsion. Biochim Biophys Acta. 1995;1256(3):381–386. | ||
Mizushima Y, Hoshi K. Review: recent advances in lipid microsphere technology for targeting prostaglandin delivery. J Drug Target. 1993; 1(2):93–100. | ||
zur Muhlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery – drug release and release mechanism. Eur J Pharm Biopharm. 1998;45(2):149–155. | ||
Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P. Cancer stem cells and tumor-associated macrophages: a roadmap for multitargeting strategies. Oncogene. 2016;35(6):671–682. | ||
Schultze JL. Reprogramming of macrophages-new opportunities for therapeutic targeting. Curr Opin Pharmacol. 2016;26:10–15. | ||
Schuster VL. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002;68–69:633–647. | ||
Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23(3):144–150. | ||
Narumiya S, Fukushima M. Site and mechanism of growth inhibition by prostaglandins. I. Active transport and intracellular accumulation of cyclopentenone prostaglandins, a reaction leading to growth inhibition. J Pharmacol Exp Ther. 1986;239(2):500–505. | ||
Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–499. | ||
Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36(6):892–899. | ||
Liu F, Liu D. Long-circulating emulsions (oil-in-water) as carriers for lipophilic drugs. Pharm Res. 1995;12(7):1060–1064. | ||
Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J. 2008;411(2):297–306. | ||
Teagarden DL, Anderson BD, Petre WJ. Determination of the pH-dependent phase distribution of prostaglandin E1 in a lipid emulsion by ultrafiltration. Pharm Res. 1988;5(8):482–487. | ||
Yamaguchi T, Tanabe N, Fukushima Y, Nasu T, Hayashi H. Distribution of prostaglandin E1 in lipid emulsion in relation to release rate from lipid particles. Chem Pharm Bull. 1994;42(3):646–650. |
Supplementary materials
Method
Calculation of geometrical parameters of NE
Using the hydrodynamic size provided by a Zetasizer and assuming that the NE particles are perfect spheres and their volume is only occupied by triolein, the volume of each particle, number, and the total surface area of particles in each formulation could be roughly calculated, using the following equations:
Volume of oil = Given value | (S1) |
|
|
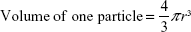
Surface area of one particle = 4πr2 | (S3) |
|
|
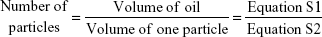
|
|
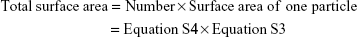
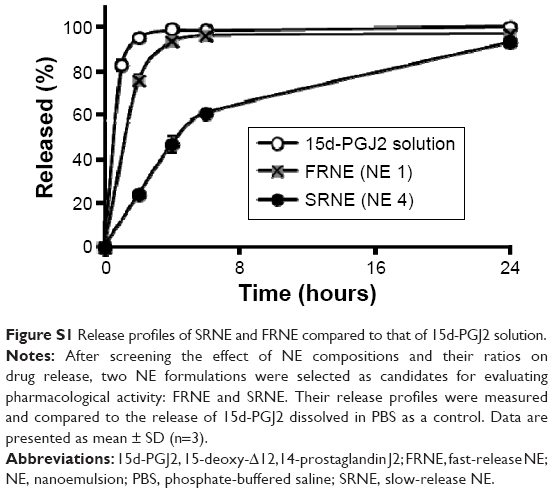 | Figure S1 Release profiles of SRNE and FRNE compared to that of 15d-PGJ2 solution. |
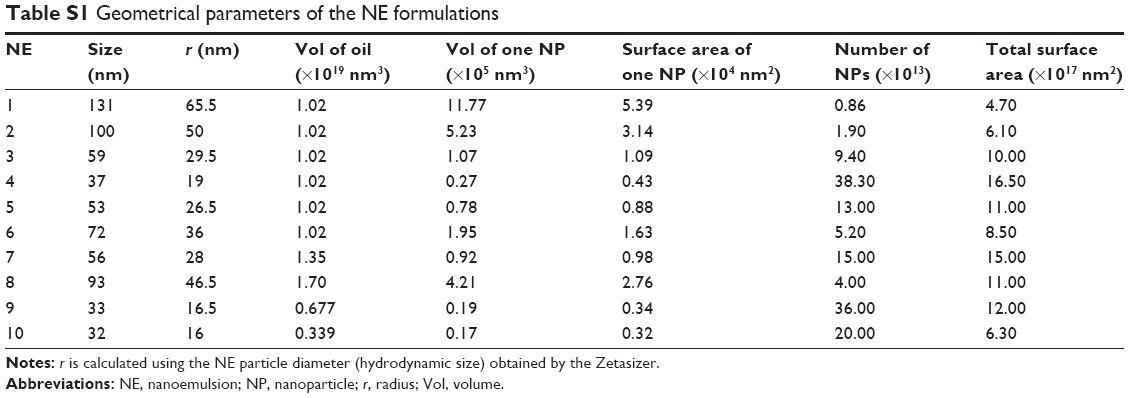 | Table S1 Geometrical parameters of the NE formulations |
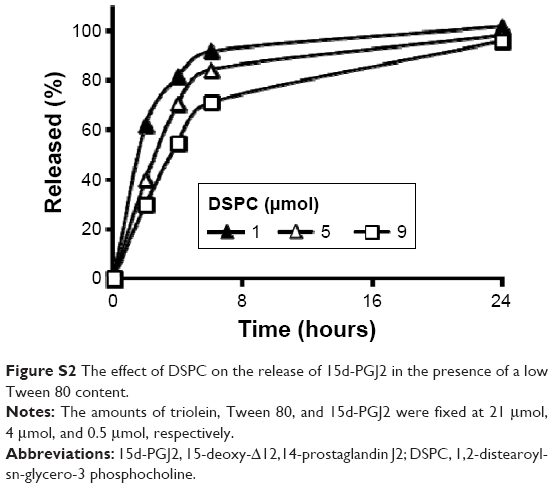 | Figure S2 The effect of DSPC on the release of 15d-PGJ2 in the presence of a low Tween 80 content. |
 | Table S2 Estimated R50 of the NE formulations |
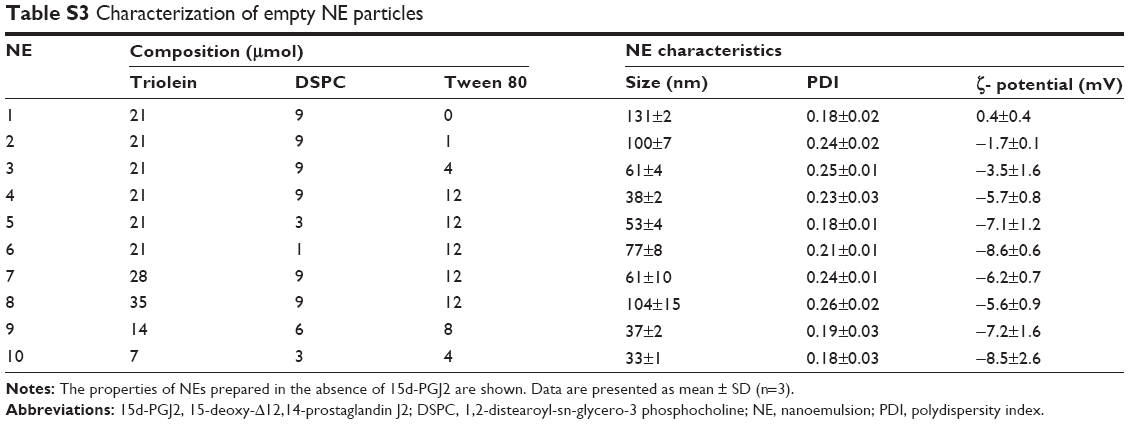 | Table S3 Characterization of empty NE particles |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.