")
Back to Journals » Vascular Health and Risk Management » Volume 13
Eligibility for alirocumab or evolocumab treatment in 1090 hypercholesterolemic patients referred to a regional cholesterol treatment center with LDL cholesterol ≥70 mg/dL despite maximal-tolerated LDL-cholesterol-lowering therapy
Authors Jetty V, Glueck CJ, Lee K, Goldenberg N, Prince M, Kumar A, Goldenberg M , Anand I , Wang P
Received 2 February 2017
Accepted for publication 8 June 2017
Published 6 July 2017 Volume 2017:13 Pages 247—253
DOI https://doi.org/10.2147/VHRM.S133690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Vybhav Jetty, Charles J Glueck, Kevin Lee, Naila Goldenberg, Marloe Prince, Ashwin Kumar, Michael Goldenberg, Ishan Anand, Ping Wang
Department of Internal Medicine, Cholesterol Center, Jewish Hospital of Cincinnati, Cincinnati, OH, USA
Background: Proprotein convertase subtilisin/kexin type 9 inhibitors, Praluent (alirocumab [ALI]) and Repatha (evolocumab [EVO]) have been approved as adjuncts to the standard-of-care maximal-tolerated dose (MTD) of low-density lipoprotein cholesterol (LDLC)-lowering therapy (LLT), statin therapy, in heterozygous (HeFH) (ALI or EVO) or homozygous (EVO) familial hypercholesterolemia, or clinical atherosclerotic cardiovascular disease (CVD) where LDLC lowering is insufficient (both). Since LDLC lowering has been revolutionized by ALI and EVO, specialty pharmaceutical pricing models will be applied to a mass market.
Methods: We applied US Food and Drug Administration (FDA) and insurance eligibility criteria for ALI and EVO to 1090 hypercholesterolemic patients serially referred over 3 years who then received ≥2 months maximal-tolerated dose of standard-of-care LDL cholesterol-lowering therapy (MTDLLT) with follow-up LDLC ≥70 mg/dL. MTDLLT did not include ALI or EVO, which had not been commercially approved before completion of this study.
Results: Of the 1090 patients, 140 (13%) had HeFH by clinical diagnostic criteria and/or CVD with LDLC >100 mg/dL despite ≥2 months on MTDLLT, meeting FDA insurance criteria for ALI or EVO therapy. Another 51 (5%) patients were statin intolerant, without HeFH or CVD.
Conclusion: If 13% of patients with HeFH-CVD and LDLC >100 mg/dL despite MTDLLT are eligible for ALI or EVO, then specialty pharmaceutical pricing models (~$14,300/year) might be used in an estimated 10 million HeFH-CVD patients. Whether the health care savings arising from the anticipated reduction of CVD events by ALI or EVO justify their costs in populations with HeFH-CVD and LDLC >100 mg/dL despite MTDLLT remains to be determined.
Keywords: proprotein convertase subtilisin/kexin type 9 inhibitors, Praluent (alirocumab), Repatha (evolocumab), hyperlipidemia, statin, PCSK9, cholesterol, heterozygous familial hypercholesterolemia, atherosclerotic cardiovascular disease
Introduction
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzyme expressed throughout the body,1,2 which functions to promote lipoprotein homeostasis, and has been implicated in many other disease processes.1,3,4 PCSK9 binds low-density lipoprotein (LDL) receptors, promoting LDL receptor degradation. This process prevents the receptor from returning to the cell surface, where the receptor could remove more LDL particles. However, if the PCSK9 molecule is blocked, LDL receptors are recycled, returned to the surface of cells, and remove more LDL particles from circulation. Further, PCSK9 inhibitors have been shown to reduce lipoprotein (a) [Lp(a)], which has been associated independently with cardiovascular risk.5–7 PCSK9 inhibition has shown great promise to lower LDL concentrations and decrease cardiovascular risk.8,9
The newly approved PCSK9 inhibitors, alirocumab (ALI) and evolocumab (EVO), are the most powerful agents currently available for lowering low-density lipoprotein cholesterol (LDLC).10–17 EVO facilitates regression of coronary atherosclerotic plaque, as shown by the GLAGOV Trial.16,18 ALI or EVO is approved for patients with heterozygous familial hypercholesterolemia (HeFH), EVO for homozygous familial hypercholesterolemia, and both for patients with atherosclerotic cardiovascular disease (CVD) unable to achieve LDLC goals despite maximal-tolerated standard-of-care dose (MTD) LDL cholesterol-lowering therapy (LLT), specifically statins, including zero-dose tolerance (statin intolerance).13,15,19 Preliminary results of safety- and efficacy-controlled clinical trials, although not powered or designed to definitively assess CVD events, revealed a 50% risk reduction in CVD events.13–15 Building on this preliminary data, Sabatine et al documented a 15% reduction of CVD events.20
Priced as a specialty drug for a common disease, insurance companies have implemented prior authorizations to limit use to high-risk patients who meet approved specifications. The process of obtaining prior authorization is arduous, requiring significant amount of uncompensated time and effort. This process requires an estimated 4–6 hours per patient to navigate the prior authorization system.21 In our center the prior authorization process takes ~6 hours of uncompensated staff time per patient. Despite this considerable amount of work, access to valuable PCSK9 inhibitors is still denied at times. The insurance-imposed barrier requires a streamlined process of appropriate selection and documentation to ensure medication approval without significant amounts of patient, staff, and clinician frustration. To meet this need, frameworks have been developed to identify appropriate patients for PCSK9 inhibitor therapy. Saeed et al have published an algorithm to specifically identify eligible patients and facilitate the prior authorization process for PCSK9 inhibitors.21
If the annual cost of the ALI or EVO were to remain ~$14,000–14,600 per patient, then specialty pharmaceutical pricing models previously reserved for drugs that benefited limited patient populations will collide with prospective treatment cohorts in the tens of millions of patients with HeFH and/or CVD, or at high risk for CVD, optimally treated with ALI or EVO.13–15,22–24 We have previously applied US Food and Drug Administration (FDA)-approved and commercial insurance eligibility criteria for PCSK9 inhibitor use in 734 patients serially referred to our cholesterol center and receiving ≥2 months maximal-tolerated dose of standard-of-care LDL cholesterol-lowering therapy (MTDLLT) with follow-up LDLC ≥70 mg/dL.22 We reported that 30% of these 734 hypercholesterolemic patients had clinically defined HeFH and/or CVD and retained LDLC >100 mg/dL despite MTDLLT,22 thus meeting FDA and commercial insurance eligibility for ALI or EVO treatment using LDLC goal-based guidelines.25,26 From our initial assessment,22 we speculated that ALI or EVO might be given to an estimated 13–23 million patients, costing $185–342 billion/year, but with an estimated 50% reduction in CVD events,13,15 resulting in $245 billion/year in savings to society, between the ALI or EVO costs of $185–342 billion.22 Using a simulation model of US adults aged 35–94 years, Kazi et al concluded that “PCSK9 inhibitor use in all eligible patients was estimated to reduce cardiovascular care costs by $29 billion over 5 years, but drug costs increased by $592 billion (a 38% increase over 2015 prescription drug expenditures)”.27
In a new, previously unstudied group of 1090 hypercholesterolemic patients serially referred over 3 years for diagnosis and treatment of high LDLC, who then received ≥2 months MTDLLT, but maintained LDLC ≥70 mg/dL, our specific aim was to apply FDA and insurance eligibility criteria for ALI and EVO, and estimate costs of therapy and costs to society.
Materials and methods
The study followed a protocol (12-03) approved by the Jewish Hospital Institutional Review board, with signed informed consent.
We assessed 1090 previously unstudied and unreported hypercholesterolemic patients serially referred to our Cholesterol Center over 3 years up to June 2016, who subsequently received ≥2 months of MTDLLT, with last follow-up LDLC ≥70 mg/dL. All patients were instructed in a cholesterol-lowering diet by a registered dietitian, and received MTDLLT, predominantly with statins; a small percentage took ezetimibe and/or colesevelam, either with or without statins. None of these patients received PCSK9 inhibitors ALI or EVO because they received MTDLLT before commercial release of ALI or EVO.
After an overnight fast, lipids and lipoprotein cholesterols were serially measured by LabCorp with direct measurement of LDLC if triglycerides were >400 mg/dL. Some but not all patients had initial measurement of Lp(a). HeFH was defined by LDLC ≥190 mg/dL and the presence of tendon xanthomas and/or by hypercholesterolemic first-degree relatives (Simon Broome criteria28). CVD included documented coronary, carotid, aortic, or peripheral artery atherosclerosis, as well as transient ischemic attack and ischemic stroke. While statin intolerance is a controversial issue and appropriate documentation of statin intolerance is difficult,29 we accepted patients intolerant to ≥3 statin medications because of development of myalgia–myositis within 2 weeks after initiation of statin therapy as “statin intolerant.”
Statistical methods
Data were processed using SAS version 9.4. Comparisons of entry LDLC and levels after 2 months on MTDLLT were made with paired Wilcoxon nonparametric tests.
Results
The 1090 hypercholesterolemic patients had LDLC ≥70 mg/dL despite ≥2 months MTDLLT. Mean±SD and median LDLC of the 1090 patients were 121±42 and 113 mg/dL at study entry, and 101±29, 94 mg/dL after ≥2 months on MTDLLT. Of the 1090 patients, 138 (13%) had HeFH by clinical diagnosis, 245 (22%) had CVD, and 96 (9%) were statin intolerant (Figure 1). Of the 245 patients who had CVD, their first CVD event was at age 62±13, median 63 years. After ≥2 months on MTDLLT, mean±SD and median LDLC in patients with HeFH were 126±45 and 121 mg/dL, 94±24 and 86 mg/dL in those with CVD events, and 118±42 and 111 mg/dL in statin-intolerant patients (Figure 1).
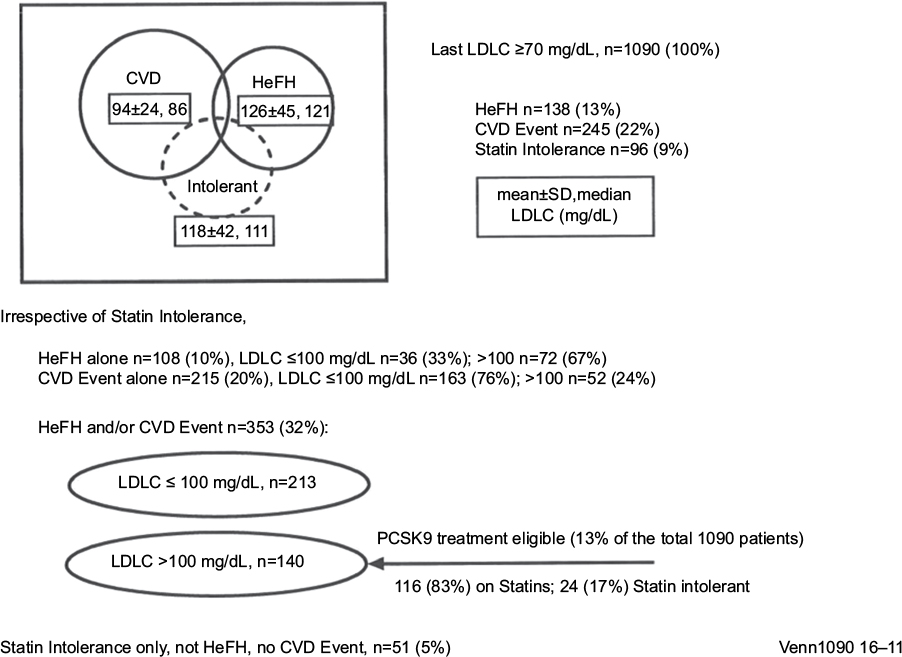 | Figure 1 HeFH and atherosclerotic CVD in 1090 patients with LDLC ≥70 mg/dL after ≥2 months maximal-tolerated cholesterol-lowering therapy. Notes: One hundred forty patients with HeFH and/or CVD (13% of the referred cohort) eligible for PCSK9 inhibitor therapy by US Food and Drug Administration and commercial insurance guidelines by virtue of LDLC on maximal-tolerated therapy >100 (dosage was MTDLLT, which included zero dose statin for those with statin intolerance). Abbreviations: CVD, cardiovascular disease; HeFH, heterozygous familial hypercholesterolemia; LDLC, low-density lipoprotein cholesterol; MTDLLT, maximal-tolerated dose of standard-of-care LDL cholesterol-lowering therapy; PCSK9, proprotein convertase subtilisin/kexin type 9. |
Of the 1090 patients, 353 (32%) had HeFH and/or a CVD event (Figure 1). Mean±SD and median LDLC at entry in these 353 patients were 137±53 mg/dL and 127 mg/dL, and after 2 months of MTDLLT, had fallen to 105±36 and 93 mg/dL, p<0.0001. Of these 353 patients, 213 (60%) had LDLC ≤100 mg/dL on MTDLLT, while 140 (13% of the original 1090 patients) had LDLC >100 mg/dL on MTDLLT (Figure 1). Mean±SD Lp(a) for these 353 patients was 40±48, median 21 mg/dL. Patients with elevated Lp(a), defined as ≥35 mg/dL, comprised 37% of this population (Table 1).
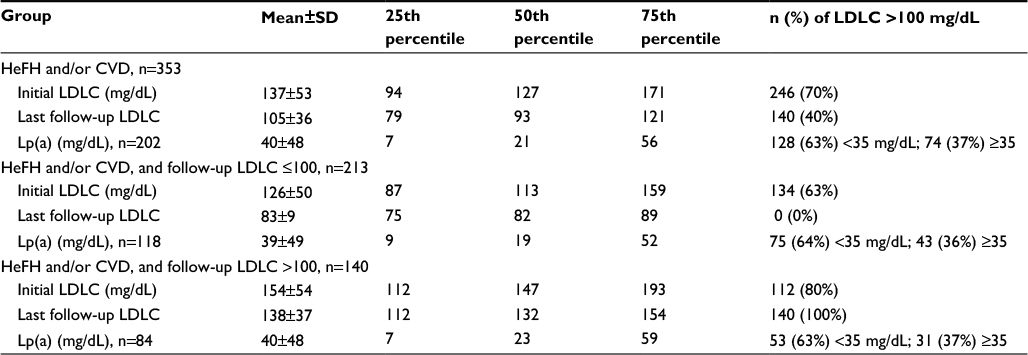 | Table 1 LDLC in 353 patients with HeFH and/or CVD at entry and after 2 months on maximal-tolerated LDLC-lowering therapy Notes: Lp(a) was available for 202 patients. Abbreviations: CVD, cardiovascular events; HeFH, heterozygous familial hypercholesterolemi; LDLC, low-density lipoprotein cholesterol; Lp(a), lipoprotein (a). |
In the 353 patient population, 140 patients were eligible by both FDA and commercial insurance criteria for ALI or EVO treatment, by virtue of having HeFH and/or CVD (Figure 1). Within these 140 patients, 116 (83%) were taking statins, and 24 (17%) were unable to take any statins, being intolerant to ≥3 different statins (Figure 1). In these 140 patients, median LDLC at entry was 147 mg/dL, and at 2 months on MTDLLT, 132 mg/dL, p=0.002 (Table 1). Mean±SD Lp(a) for this group (n=84) was 40±48, median 23 mg/dL. Lp(a) ≥35 mg/dL was found in 37% of these patients (Table 1). In the 213 patients whose follow-up LDLC on MTDLLT was ≤100 mg/dL (Figure 1; Table 1), mean±SD and median initial LDLC was 126±50 and 113 mg/dL. On follow-up these values decreased to 83±9 and 82 mg/dL, respectively, p<0.0001 (Table 1).
In the full cohort of 1090 patients, statin intolerance was present in 96 (9%) patients, and in those with neither HeFH nor CVD, in 51 (5%) (Figure 1).
Of the 140 patients having HeFH and/or a CVD event with LDLC >100 mg/dL on MTDLLT (Figure 1), 72 had HeFH alone, with LDLC on therapy 150±44, median 144 mg/dL, 52 had CVD events alone, with LDLC on therapy 121±19, median 113 mg/dL, and 16 had both HeFH and CVD, with LDLC on therapy 140±24, median 139 mg/dL. In the 68 patients with CVD, the first CVD event occurred at age 61±12, median 60 years.
Discussion
ALI and EVO are the most powerful agents currently available for lowering LDLC.10,12–16 Further studies have shown that the drugs are also able to decrease Lp(a),6,7 a molecule implicated to work synergistically with LDLC to cause CVD events.30–33 PCSK9 inhibitors were previously thought to decrease CVD events by 50%13–15,24; more recent outcomes data have shown a 15%–20% reduction.24 Priced as a specialty drug for a common disease, $14,000–14,600 per patient annually, the cost of the drug class has been highly controversial as the treatment cohort may reach in the tens of millions of patients, thereby significantly increasing the cost of cardiovascular care.
Our current analysis is focused on those high-risk patients for whom ALI or EVO is specifically indicated,13–15 restricted by medical insurance carriers, as an adjunct to MTDLLT in adults with HeFH or CVD who require additional lowering of LDLC. The population cost of ALI or EVO therapy largely depends on the number of subjects judged to be at high risk by virtue of HeFH with high LDLC,34–36 and/or by CVD with LDLC above target goals,25 despite MDTLLT. At the bottom end of the number of patients with HeFH eligible for ALI or EVO, genetic testing within a single US health care system provided an estimated prevalence of HeFH of 0.39% in unselected patients, and 0.84% in those recruited from the cardiac catheterization laboratory.37 Highlighting the undertreatment of HeFH variant carriers,38 only 46% of HeFH carriers had LDLC on statin therapy <100 mg/dL,37 the on-therapy cutpoint used in our current study to identify candidates with HeFH and/or CVD for ALI or EVO treatment. Retrospectively applying clinical criteria for diagnosis of HeFH to the same cohort, probable HeFH was diagnosed in 24% of familial hypercholesterolemic variant carriers.37 Large-scale, whole-exome sequencing studies suggest that HeFH has a prevalence of ~1 in 217 (0.46%) northern European individuals.39 At the upper end of the number of patients with HeFH eligible for ALI or EVO, in 313 patients with severe hypercholesterolemia (LDLC >194 mg/dL), Wang et al40 reported that 47.3% had monogenic familial hypercholesterolemia, 53.7% if polygenic scores were present, and 67.1% with extreme polygenic scores.
Of our 1090 patients in the current study referred for diagnosis and treatment of hypercholesterolemia, 140 (13%) were eligible for ALI or EVO treatment by virtue of HeFH and/or CVD events and LDLC >100 mg/dL after 2 months of MTDLLT. In a previously reported cohort of 734 patients referred for diagnosis and treatment of hyperlipidemia,22 30% would be eligible for ALI or EVO therapy by virtue of HeFH and/or CVD and LDLC >100 mg/dL despite MTDLLT. The previously reported cohort of 734 patients had much higher LDLC after ≥2 months MTDLLT, with LDLC 25th, 50th, and 75th percentiles 89, 105, and 141, significantly higher than 80, 94, and 114 mg/dL in the current 1090 patients. The previously reported cohort22 had 48% of patients with HeFH and/or CVD compared to 32% in the current study. These differences in sequentially analyzed referral cohorts emphasize that the percentage of hypercholesterolemic patients eligible for PCSK9 inhibitor therapy depends on the severity of hypercholesterolemia40 in the referred population.
In the NHANES 2003–2006 general population data,36 27% of subjects had high LDLC. In a 1999–2010 general population survey, LDLC was ≥130 mg/dL35 in 28% of adults. If 21%–36.7%19,35,36,41 of the general adult US population is judged to have high LDLC, then a high LDLC cohort would include an estimated 45–78 million subjects. Extrapolating from our referral cohort where 13% of hypercholesterolemic patients had HeFH and/or CVD with LDLC ≥100 mg/dL despite MTDLLT, it is possible that 5.9–10 million patients (13% of 45–78 million) might be candidates for treatment with ALI or EVO.
As for the general population, hypercholesterolemia is undertreated.19,42 Thirty-seven percent (78 million) adults (>21 years) were eligible for cholesterol-lowering medication, but of this group, only 55% were actively taking a cholesterol-lowering medication (~90% a statin drug).19
The advent of large-scale genetic identification37,40 of hypercholesterolemia, particularly in a population referred for diagnosis and therapy of hypercholesterolemia like ours22 and others,40 might increase the treatment population, particularly for HeFH, adding to our projected 10 million adults identified clinically.
Large numbers of subjects with elevated LDLC fail to achieve treatment targets,43–46 for many reasons.43,47,48 Beyond statin effects, ALI and EVO should optimize LDLC in most patients with HeFH, CVD, and statin intolerance.10,14,15,18,24,26,49 The paradigm-shifting improvement in LDLC lowering by ALI or EVO,13,15,50 however, comes at an annual price of $14,000–14,600 per patient.
Using a simulation model of US adults aged 35–94 years, Kazi et al27 concluded that “…reducing annual drug costs to $4536 per patient or less would be needed for PCSK9 inhibitors to be cost-effective at <$100,000 per QALY.” Rather than a simulation model, our estimates of candidates for ALI or EVO are based on real-world referrals for diagnosis and therapy of hypercholesterolemia. For the 10 million eligible patients based on the characteristics of our current hypercholesterolemic referral group, at $14,000/patient, yearly costs of ALI or EVO might total $140 billion. In 2010, CVD-related direct medical costs in the USA were estimated to be $273 billion, and indirect costs $172 billion, $445 billion total.51 If ALI or EVO therapy reduced CVD event rates by 15%,13,15,20,50 then using 2010 direct and indirect medical cost estimates, ~$67 billion would be saved. If using the secondary endpoint data (decrease of cardiovascular death, myocardial infarction, or stroke by 20%) reported by Sabatine et al20 then ~$89 billion would be saved. The researchers also reported an increase in the magnitude of risk reduction in both their primary endpoints and secondary endpoints – 12%–19% and 16%–25%, respectively, from the first year to the second year. If these risk reductions continue with prolonged use of PCSK9 inhibitors, we may see further cost savings. Programs targeted to prevention of CVD should provide substantial overall cost savings.52,53 Studies with statins54 have previously reported that prevention or reduction in CVD events results in overall cost savings, and it is possible that PCSK9 inhibitors could reduce CVD events a further 15%–20% beyond statin therapy.13,15,20
Estimating from our data, if CVD were reduced by 15%20 then CVD care would save $67 billion. For 10 million patients, PCSK9 inhibitors cost should be less than $6,700 per patient per year for the savings to outweigh the costs. Likewise, if CVD is reduced by 20%,20 CVD care is reduced by ~$89 billion. From our data we estimate that treating ~10 million patients, the cost of PCSK9 inhibitors should be <$8,900 per patient per year for care savings to outweigh the costs of the expensive, specialty drug.
Overall costs to society also need to include analysis of any potential adverse effects arising from ALI or EVO use. In a meta-analysis of 25 randomized controlled trials with PCSK9 inhibitors, Zhang et al50 “detected largely no significant difference in major adverse events rates between PCSK9 inhibitor therapy and placebo and no difference between different dosages of EVO.” Within this frame of reference,50 we speculate that costs to society arising from adverse effects specifically attributed to ALI or EVO may be minimal.
Strengths of our study include documentation that, of patients referred to a regional center for therapy of high LDLC, ≥70 mg/dL despite MTDLLT, 32% had HeFH and/or CVD, and 13% retained LDLC >100 mg/dL despite MTDLLT, meeting both FDA indications14 and commercial insurance coverage eligibility.
Limitations of our study include bias toward higher LDLC and CVD by virtue of referral to a regional cholesterol treatment center, which would increase the percentage of hypercholesterolemic patients with HeFH40 and/or CVD eligible for insurance coverage of ALI or EVO.
Conclusions
- Of our 1090 patients in the current study referred for diagnosis and treatment of hypercholesterolemia, 140 (13%) were eligible for ALI or EVO treatment by virtue of HeFH and/or CVD events and LDLC >100 mg/dL after 2 months of MTDLLT.
- For the 10 million eligible patients based on the characteristics of our current hypercholesterolemic referral group, at $14,000/patient, yearly costs of ALI or EVO might total $140 billion. In 2010, CVD-related direct medical costs in the United States were estimated to be $273 billion, and indirect costs $172 billion, $445 billion total.51 If ALI or EVO therapy reduced CVD event rates by 15%,13,15,20,50 then using 2010 direct and indirect medical cost estimates, ~$67 billion would be saved. If using the secondary endpoint data (decrease of cardiovascular death, myocardial infarction, or stroke by 20%) reported by Sabatine et al20 then ~$89 billion would be saved.
- Whether the health care savings arising from the anticipated reduction of CVD on the ALI or EVO justify the broad use of these specialty priced agents remains to be determined.
Acknowledgments
The study followed a protocol approved by the Jewish Hospital Institutional Review Board. The data sets used and analyzed during the current study are available from the corresponding author upon request ([email protected]). Support was provided from the Lipoprotein Research Fund of the Jewish Hospital of Cincinnati.
Authors’ contribution
All authors made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. All were involved in the drafting, revising, and finalization of the article. Further, all authors agree to be accountable for the accuracy and integrity of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100(3):928–933. | ||
Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res. 2016;112(1):429–442. | ||
Ibarretxe D, Girona J, Plana N, et al. Circulating PCSK9 in patients with type 2 diabetes and related metabolic disorders. Clin Investig Arterioscler. 2016;28(2):71–78. | ||
Zhang L, Song K, Zhu M, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in lipid metabolism, atherosclerosis and ischemic stroke. Int J Neurosci. 2016;126(8):675–680. | ||
Ferdinand KC, Nasser SA. PCSK9 Inhibition: discovery, current evidence, and potential effects on LDL-C and Lp(a). Cardiovasc Drugs Ther. 2015;29(3):295–308. | ||
Raal FJ, Giugliano RP, Sabatine MS, et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. J Lipid Res. 2016;57(6):1086–1096. | ||
Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63(13):1278–1288. | ||
Joseph L, Robinson JG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition and the future of lipid lowering therapy. Prog Cardiovasc Dis. 2015;58(1):19–31. | ||
Weinreich M, Frishman WH. Antihyperlipidemic therapies targeting PCSK9. Cardiol Rev. 2014;22(3):140–146. | ||
Everett BM, Smith RJ, Hiatt WR. Reducing LDL with PCSK9 inhibitors–the clinical benefit of lipid drugs. N Engl J Med. 2015;373(17):1588–1591. | ||
Lepor NE, Kereiakes DJ. The PCSK9 inhibitors: a novel therapeutic target enters clinical practice. Am Health Drug Benefits. 2015;8(9):483–489. | ||
Lipinski MJ, Benedetto U, Escarcega RO, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J. 2016;37(6):536–545. | ||
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–1499. | ||
Robinson JG, Kastelein JJ. PCSK9 inhibitors and cardiovascular events. N Engl J Med. 2015;373(8):774. | ||
Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–1509. | ||
Reiner Z. PCSK9 inhibitors–past, present and future. Expert Opin Drug Metab Toxicol. 2015;11(10):1517–1521. | ||
Pecin I, Reiner Z. Alirocumab: targeting PCSK9 to treat hypercholesterolemia. Drugs Today (Barc). 2015;51(12):681–687. | ||
Nicholls SJ, Puri R, Anderson T, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316(22):2373–2384. | ||
Mercado C, DeSimone AK, Odom E, Gillespie C, Ayala C, Loustalot F. Prevalence of cholesterol treatment eligibility and medication use among adults - United States, 2005-2012. MMWR Morb Mortal Wkly Rep. 2015;64(47):1305–1311. | ||
Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. | ||
Saeed A, Virani SS, Jones PH, Nambi V, Zoch D, Ballantyne CM. A simplified pathway to proprotein convertase subtilisin/kexin type 9 inhibitor prior authorization approval: a lipid clinic experience. J Clin Lipidol. 2017;11(3):596–599. | ||
Glueck CJ, Shah P, Goldenberg N, et al. Eligibility for PCSK9 treatment in 734 hypercholesterolemic patients referred to a regional cholesterol treatment center with LDL cholesterol >/=70 mg/dl despite maximal tolerated cholesterol lowering therapy. Lipids Health Dis. 2016;15(1):55. | ||
Roth EM. Alirocumab for hyperlipidemia: ODYSSEY phase III clinical trial results and US FDA approval indications. Future Cardiol. 2016;12(2):115–128. | ||
Sabatine MS, Wasserman SM, Stein EA. PCSK9 inhibitors and cardiovascular events. N Engl J Med. 2015;373(8):774–775. | ||
Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the national cholesterol education program adult treatment panel III guidelines. Circulation. 2004;110(2):227–239. | ||
Shrank WH, Barlow JF, Brennan TA. New therapies in the treatment of high cholesterol: an argument to return to goal-based lipid guidelines. JAMA. 2015;314(14):1443–1444. | ||
Kazi DS, Moran AE, Bibbins-Domingo K. Cost-effectiveness of PCSK9 inhibitor therapy-reply. JAMA. 2016;316(20):2152. | ||
Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303(6807):893–896. | ||
Rosenson RS, Baker SK, Jacobson TA, Kopecky SL, Parker BA, The national lipid association’s muscle safety expert P. An assessment by the statin muscle safety task force: 2014 update. J Clin Lipidol. 2014;8(3 Suppl):S58–S71. | ||
Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease. Meta-analysis of prospective studies. Circulation. 2000;102(10):1082–1085. | ||
Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Lipoprotein(a) and risk of myocardial infarction–genetic epidemiologic evidence of causality. Scand J Clin Lab Invest. 2011;71(2):87–93. | ||
Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. | ||
Schreiner PJ, Morrisett JD, Sharrett AR, et al. Lipoprotein[a] as a risk factor for preclinical atherosclerosis. Arterioscler Thromb. 1993;13(6):826–833. | ||
Elshazly MB, Martin SS, Blaha MJ, et al. Non-high-density lipoprotein cholesterol, guideline targets, and population percentiles for secondary prevention in 1.3 million adults: the VLDL-2 study (very large database of lipids). J Am Coll Cardiol. 2013;62(21):1960–1965. | ||
Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999–2010. NCHS Data Brief. 2012;(103):1–8. | ||
Toth PP, Potter D, Ming EE. Prevalence of lipid abnormalities in the United States: the national health and nutrition examination survey 2003-2006. J Clin Lipidol. 2012;6(4):325–330. | ||
Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016;354(6319):aaf7000-1–aaf7000-7. | ||
Reiner Z. Management of patients with familial hypercholesterolaemia. Nat Rev Cardiol. 2015;12(10):565–575. | ||
Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the copenhagen general population study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37(17):1384–1394. | ||
Wang J, Dron JS, Ban MR, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol. 2016;36(12):2439–2445. | ||
Kuklina EV, Carroll MD, Shaw KM, Hirsch R. Trends in high LDL cholesterol, cholesterol-lowering medication use, and dietary saturated-fat intake: United States, 1976-2010. NCHS Data Brief. 2013(117):1–8. | ||
Knickelbine T, Lui M, Garberich R, Miedema MD, Strauss C, VanWormer JJ. Familial hypercholesterolemia in a large ambulatory population: statin use, optimal treatment, and identification for advanced medical therapies. J Clin Lipidol. 2016;10(5):1182–1187. | ||
Hsia SH, Desnoyers ML, Lee ML. Differences in cholesterol management among states in relation to health insurance and race/ethnicity across the United States. J Clin Lipidol. 2013;7(6):675–682. | ||
Mann D, Reynolds K, Smith D, Muntner P. Trends in statin use and low-density lipoprotein cholesterol levels among US adults: impact of the 2001 national cholesterol education program guidelines. Ann Pharmacother. 2008;42(9):1208–1215. | ||
Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120(1):28–34. | ||
Zimmerman MP. How do PCSK9 inhibitors stack up to statins for low-density lipoprotein cholesterol control? Am Health Drug Benefits. 2015;8(8):436–442. | ||
Glueck CJ, Budhani SB, Masineni SS, et al. Vitamin D deficiency, myositis-myalgia, and reversible statin intolerance. Curr Med Res Opin. 2011;27(9):1683–1690. | ||
Khayznikov M, Hemachrandra K, Pandit R, Kumar A, Wang P, Glueck CJ. Statin intolerance because of myalgia, myositis, myopathy, or myonecrosis can in most cases be safely resolved by vitamin D supplementation. N Am J Med Sci. 2015;7(3):86–93. | ||
Stein EA, Raal FJ. Lipid-lowering drug therapy for CVD prevention: looking into the future. Curr Cardiol Rep. 2015;17(11):104. | ||
Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med. 2015;13:123. | ||
Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933–944. | ||
Milani RV, Lavie CJ. Impact of worksite wellness intervention on cardiac risk factors and one-year health care costs. Am J Cardiol. 2009;104(10):1389–1392. | ||
Arena R, Guazzi M, Briggs PD, et al. Promoting health and wellness in the workplace: a unique opportunity to establish primary and extended secondary cardiovascular risk reduction programs. Mayo Clin Proc. 2013;88(6):605–617. | ||
Grover SA, Ho V, Lavoie F, Coupal L, Zowall H, Pilote L. The importance of indirect costs in primary cardiovascular disease prevention: can we save lives and money with statins? Arch Intern Med. 2003;163(3):333–339. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.