")
Back to Journals » International Journal of Nanomedicine » Volume 11
Electrospinning of calcium phosphate-poly(D,L-lactic acid) nanofibers for sustained release of water-soluble drug and fast mineralization
Authors Fu QW, Zi YP, Xu W, Zhou R, Cai ZY, Zheng WJ, Chen F , Qian QR
Received 3 June 2016
Accepted for publication 28 July 2016
Published 6 October 2016 Volume 2016:11 Pages 5087—5097
DOI https://doi.org/10.2147/IJN.S114224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Qi-Wei Fu,1,* Yun-Peng Zi,1,* Wei Xu,1 Rong Zhou,1 Zhu-Yun Cai,1 Wei-Jie Zheng,1 Feng Chen,2 Qi-Rong Qian1
1Department of Orthopedics, Changzheng Hospital, Second Military Medical University, 2State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Calcium phosphate-based biomaterials have been well studied in biomedical fields due to their outstanding chemical and biological properties which are similar to the inorganic constituents in bone tissue. In this study, amorphous calcium phosphate (ACP) nanoparticles were prepared by a precipitation method, and used for preparation of ACP-poly(D,L-lactic acid) (ACP-PLA) nanofibers and water-soluble drug-containing ACP-PLA nanofibers by electrospinning. Promoting the encapsulation efficiency of water-soluble drugs in electrospun hydrophobic polymer nanofibers is a common problem due to the incompatibility between the water-soluble drug molecules and hydrophobic polymers solution. Herein, we used a native biomolecule of lecithin as a biocompatible surfactant to overcome this problem, and successfully prepared water-soluble drug-containing ACP-PLA nanofibers. The lecithin and ACP nanoparticles played important roles in stabilizing water-soluble drug in the electrospinning composite solution. The electrospun drug-containing ACP-PLA nanofibers exhibited fast mineralization in simulated body fluid. The ACP nanoparticles played the key role of seeds in the process of mineralization. Furthermore, the drug-containing ACP-PLA nanofibers exhibited sustained drug release which simultaneously occurred with the in situ mineralization in simulated body fluid. The osteoblast-like (MG63) cells with spreading filopodia were well observed on the as-prepared nanofibrous mats after culturing for 24 hours, indicating a high cytocompatibility. Due to the high biocompatibility, sustained drug release, and fast mineralization, the as-prepared composite nanofibers may have potential applications in water-soluble drug loading and release for tissue engineering.
Keywords: calcium phosphate, water-soluble drug, nanofibers, drug release, electrospinning
Introduction
Bone defects originating from trauma and other diseases are common clinical cases.1 The use of biodegradable porous scaffolds may be one of the most promising therapeutic methods for bone defect, compared with autograft/allograft transplantation and use of nondegradable metal scaffolds. In the past decades, the calcium phosphate (CaP)-based biomaterials have attracted much attention in the research field of biomedical scaffolds.2,3 The CaP materials have similar chemical properties to the inorganic constituents of hard tissue in vertebrate, and are considered as ideal biomaterials for bone regeneration.4,5 Therefore, the synthetic CaP-based biomaterials (such as bone cements and porous ceramics) have been widely used in treatment of bone and tooth defects.6–8
Electrospinning is a simple method of fabricating polymer/composite nanofibers and bioceramics which are widely used in drug delivery and tissue engineering.9–11 Electrospun nanofibers can provide porous structure which biomimics the natural extracellular matrix constructed by native protein nanofibers (eg, collagen fibrils).12 Meanwhile, the functional molecules (eg, drugs and growth factors) and bioactive elements can also be electrospun into the composite nanofibers to achieve high bioactivity and cell affinity.13–15 Nanostructured CaP materials with high biocompatibility are usually added in the polymer solution to modify the electrospun composite nanofibers.16–18 For example, hydroxyapatite (HA) and anti-inflammatory drugs can be incorporated into polycaprolactone fibers by electrospinning, achieving sustained drug release and mineralization.19 The CaP material with amorphous calcium phosphate (ACP) phase exists in native bone tissue, which is an intermediate phase in the HA formation process and displays good bioactivity.20–22 Therefore, the electrospun ACP-containing nanofibers may have potential applications in the studies of mineralization and bone repair/tissue engineering.17,23
As a major category of drugs, water-soluble drugs can be easily dissolved in aqueous solution and utilized by patients, and play important roles in the biomaterial studies. Many strategies and technologies have been developed to promote cell adhesion/growth, osteointegration with the host tissue, and bone growth using water-soluble molecules (eg, growth factors and cytokines).24 For example, different growth factors have been chosen for preparation of functional biomaterials to adjust the differentiation of stem cells and promote the cell growth.25,26 However, due to the good solubility and fast diffusion in water or body fluid, it is necessary to improve the effectiveness of the loading and sustained release of water-soluble drugs. Therefore, different materials including liposomes,27 surfactants,28 microcapsules,29 and polymer nanofibers30 have been developed and used as carriers of water-soluble drugs. The electrospun hydrophobic polymers (eg, poly[D,L-lactic acid] [PLA] nanofibers) are usually used as drug carriers.31 However, the hydrophobic polymer solutions cannot have the drug dissolved in water. This reduces the potential of electrospinning hydrophobic polymer nanofibers containing water-soluble drugs. Therefore, emulsion electrospinning32,33 and coaxial electrospinning34 have been developed for encapsulating water-soluble drugs. The electrospinning of water-soluble drug into a stable nanofibrous matrix well controls the drug diffusion into tissue and prolongs the effects of the drug. However, it still suffers from the low encapsulation efficiency in the process of drug loading and further functionalization.
Herein, the ACP nanoparticles which were prepared by a precipitation method in aqueous solution were hybridized with PLA polymer and electrospun into ACP-PLA composite nanofibers. Then, the water-soluble drug-containing ACP-PLA composite nanofibers were also prepared using the native biomolecule of lecithin as a surfactant. The use of lecithin overcame the difficulty that the water-soluble drugs are difficult to be mixed in hydrophobic polymers solution. The resulting electrospun structure, in situ mineralization, in vitro drug release, and cytotoxicity of the water-soluble drug-containing ACP-PLA nanofibers were investigated.
Experimental
Preparation of ACP nanoparticles
The ACP nanomaterial was prepared according to our previous report with modification.35 For preparing ACP nanoparticles, the PO43− ions-containing solution was obtained through dissolving 0.355 g of Na2HPO4·12H2O and 0.025 g of PLA-block-monomethoxy(polyethyleneglycol) (PLA-mPEG) in 60 mL of deionized water. Meanwhile, Ca2+ ions-containing solution was prepared by dissolving 0.166 g of CaCl2, 0.025 g of PLA-mPEG, and 10 mL ammonia (25%) in 50 mL of deionized water. Then, Ca2+ ions-containing solution was slowly added into PO43− ions-containing solution (20 mL min−1). Then, the product of ACP nanoparticles was collected by centrifugation after stirring for 1 hour at room temperature. The as-prepared product was washed using deionized water and ethanol repeatedly and then kept in acetone solution. The polymers of PLA-mPEG (molecular weight [Mw] =8,000 Da, MwPLA =3,000 Da) of medical grade were obtained from Jinan Daigang Biomaterials Co. Ltd (Jinan, People’s Republic of China). All other chemicals used in the preparation process were of analytical reagent grade and obtained from Sinopharm Chemical Reagent Co. (Shanghai, People’s Republic of China), and used as received without further purification.
Fabrication of water-soluble drug-containing ACP-PLA nanofibers
To 5 mL of mixed solvent (chloroform and N, N-dimethylformamide, 1:1), 0.300 g of ACP and 0.300 g of PLA were separately added, and the resulting solution was stirred for 12 hours to obtain a homogeneous solution. The racemic PLA (Mw =500,000 Da) of medical grade was obtained from Jinan Daigang Biomaterials Co. Ltd. The concentration of ACP or PLA in the resulting solution was 6% (w/v). For preparation of water-soluble drug-containing ACP-PLA composite nanofibers, 0.05 g of soya bean lecithin (>98%; Shanghai Aladdin Bio-Chem Technology Co., LTD., Shanghai, People’s Republic of China) was added into the prepared solution under stirring for 1 hour. Then, 0.2 mL aqueous solution containing 2 mg bovine serum albumin (BSA) (molecular biology grade; Sangon Biotech [Shanghai] LTD., Shanghai, People’s Republic of China) was added into the mixed solution, and the resulting solution was stirred for 12 hours. The resulting solutions were then taken in a 10 mL syringe for electrospinning using a electrospinning equipment (Changsha Nayi Equipment Technology Co., Ltd., Changsha, People’s Republic of China) under a high voltage of 15 kV. The experiment was performed at room temperature (~25°C) and a relative humidity of ~60%. The solution feeding rate was 1 mL h−1. The inner diameter of stainless steel needle was 0.2 mm. The needle tip was fixed at 15 cm above the aluminum foil collector. Figure 1 shows an illustration of electrospinning process for preparation of water-soluble drug-containing ACP-PLA nanofibers.
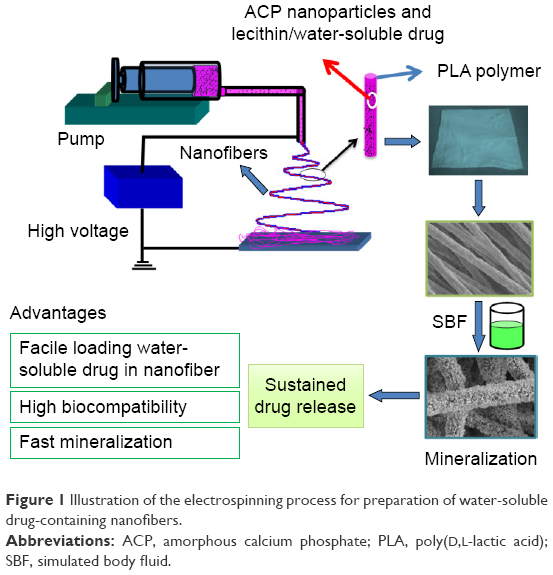 | Figure 1 Illustration of the electrospinning process for preparation of water-soluble drug-containing nanofibers. |
Characterization of the samples
The structures and morphologies of the as-prepared samples were observed using a field-emission transmission electron microscope (JEM-2100F; JEOL, Tokyo, Japan) at an accelerating voltage of 200 keV and a field-emission scanning electron microscope (JSM-6700; JEOL). The X-ray powder diffraction (XRD) of the samples was characterized by a Rigaku D/max 2550V X-ray diffractometer (Rigaku, Tokyo, Japan) with high-intensity Cu Kα radiation (λ=1.54178 Å) and a graphite monochromator. The Fourier transform infrared (FTIR) spectra of the samples were taken on a FTIR spectrometer (FTIR-7600; Lambda Scientific, Edwardstown, Australia). The hydrophilic properties were studied by an Automatic Contact Angle Meter (SL200A/B/D; Solon Tech., Shanghai, People’s Republic of China).
Mineralization of ACP-PLA and water-soluble drug-containing ACP-PLA nanofibers
The PLA, ACP-PLA, and water-soluble drug-containing ACP-PLA nanofibrous round mats (diameter, 14 mm) were immersed in simulated body fluid (SBF) (20 mL) in round culture dishes (diameter, 60 mm) at 37°C, and 10 mL of SBF was replaced with same volume of fresh SBF every 24 hours. The standard SBF solution was obtained according to the reported method.36 The SBF solution has definite ion concentrations which are almost equal to those in the human blood plasma (ie, Na+ 142.0 mM, K+ 5.0 mM, Mg2+ 1.5 mM, Ca2+ 2.5 mM, Cl− 148.8 mM, HCO3− 4.2 mM, and HPO42− 1.0 mM).36 Then, ACP-PLA nanofibrous mats for characterization were taken out and washed several times with deionized water on days 1, 3, and 7.
In vitro BSA release
BSA was used as a water-soluble drug to prepare drug-containing ACP-PLA nanofibers. The BSA-loaded ACP-PLA nanofibrous mats (0.2 g) were used for the in vitro drug release studies, which were immersed into 50 mL of SBF at 37°C under shaking (160 rpm) in a desk incubator. The release solution (2 mL) was taken out at a given time for the UV–vis analysis at 562 nm, using a bicinchoninic acid kit (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China). Then, 2 mL of fresh SBF (37°C) was used to replace the withdrawn solution. The cast membrane of BSA-containing ACP-PLA was used as a control sample, which was prepared by directly drying the solution at 60°C for 2 hours.
Cell viability
The PLA, ACP-PLA, and water-soluble drug-containing ACP-PLA nanofibrous round membranes (14 mm) were prepared and used for cell viability study, using the osteoblast-like cells (MG63). The cells were cultured in α-minimum essential medium with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C under 5% CO2 in a humidified incubator. The nanofibrous membranes were fixed in 24-well plates (Corning Incorporated, Corning, NY, USA) after sterilization by 75% ethanol aqueous solution, and 1.0 mL cell suspension (1×105 cells mL−1) was seeded onto the samples. The microplate reader (MK3; Thermo Fisher Scientific, Waltham, MA, USA) was used to determine the absorbance of MTT kit at an excitation wavelength of 492 nm on the given days.
The scanning electron microscopy (SEM) images were obtained to study the morphologies of cells on the water-soluble drug-containing ACP-PLA nanofibers. After culturing for 24 hours, unattached cells were rinsed out using phosphate-buffered saline, and then fixed with 2.5% glutaraldehyde for 30 minutes. Then, the cells on the fibrous membranes were subsequently dehydrated by ethanol solutions (30, 50, 75, 95, and 100 vol%) for 10 minutes, and by absolute ethanol twice for final dehydration, before being observed by SEM.
Results and discussion
Characterization of the as-prepared ACP nanoparticles
The ACP nanoparticles were synthesized by a modified coprecipitation method using PLA-mPEG as a soft template. As the TEM micrograph in Figure 2A shows, the ACP product displayed a nanoparticle structure with a small diameter (20–30 nm). The PLA-mPEG molecules which could self-assemble to form micelles in aqueous solution played the role of soft template in the preparation process of ACP product. Then, CaP sample with nanospherical structure was formed by the reaction between Ca2+ ions and PO43− ions.37,38 The selected-area electron diffraction pattern indicated an amorphous crystal phase of the product (Figure 2B). The XRD pattern of the nanosphere product showed no obvious diffraction peaks but a broad characteristic peak of the amorphous phase at around 2θ=30° (Figure 2C), indicating an amorphous crystal phase which agrees well with the result of selected-area electron diffraction.39
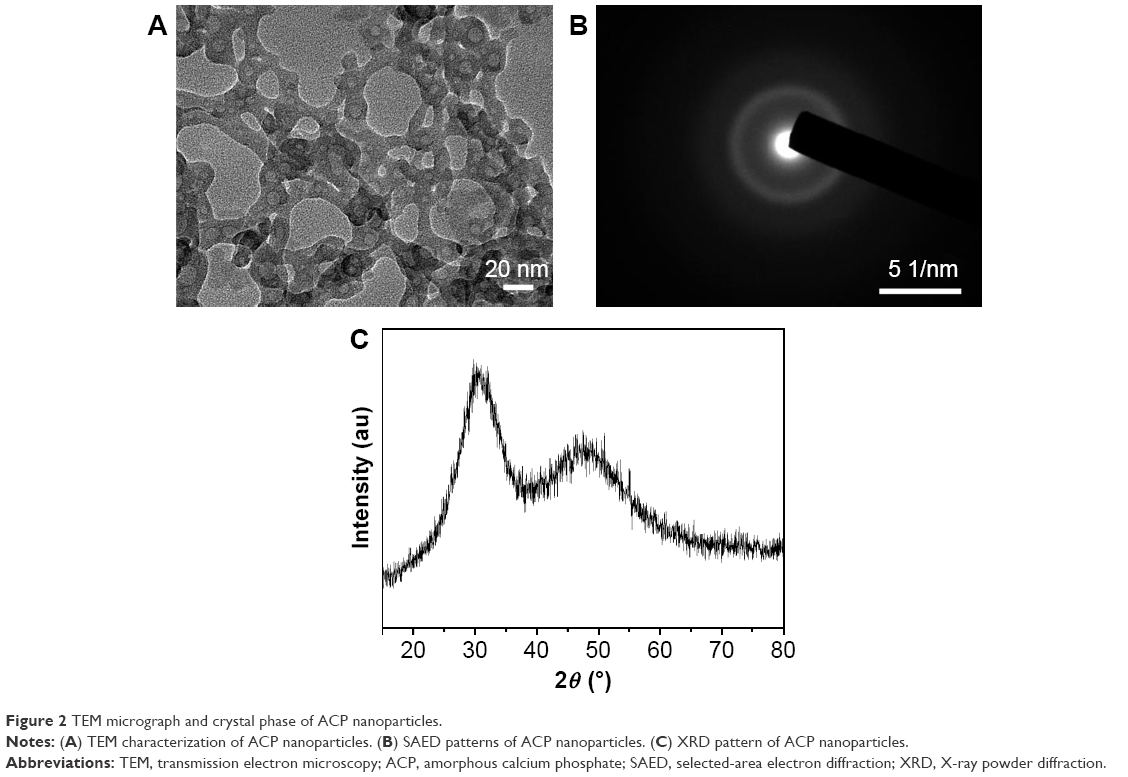 | Figure 2 TEM micrograph and crystal phase of ACP nanoparticles. |
Characterization of water-soluble drug-containing ACP-PLA nanofibers
The ACP sample can be easily hybridized with the PLA polymer, and the mixed solution can be electrospun into ACP-PLA nanofibers. However, the water-soluble drug cannot be directly blended into the PLA and ACP-PLA solutions due to the incompatibility between water and PLA molecule. The stratification phenomenon exists in the mixed solution when the water-soluble drug aqueous solution is directly added into the PLA and ACP-PLA solutions. For the preparation of uniform water-soluble drug-containing ACP-PLA solution, the lecithin is added into the prepared solution before adding aqueous solution containing water-soluble drug of BSA. The resulting solution is homogeneous even when kept for overnight. If the aqueous solution containing BSA and lecithin are added into the PLA solution without ACP nanoparticles, the resulting solution can maintain the homogeneous feature for only a short time, and the stratification phenomenon appears after a prolonged time period. Therefore, both lecithin and ACP nanoparticles are important for the fabrication of BSA-containing solution for electrospinning.
Then, the BSA-containing ACP-PLA solution is easily electrospun into the nanofibers. As the SEM micrographs in Figure 3 show, the as-prepared BSA-containing ACP-PLA nanofibers displayed a fibrous structure. The average diameter of these nanofibers was ~650 nm with a rough surface. The ACP-PLA nanofibers and pure PLA nanofibers were separately prepared as control samples. The ACP-PLA nanofibers also displayed good nanofibrous morphology, and the average diameter was ~300 nm. Meanwhile, the pure PLA nanofibers with an average diameter of ~550 nm and smooth surface were observed. The diameter of BSA-containing ACP-PLA nanofibers was the largest among the three sample types. The result indicated that the addition of water-soluble drug and lecithin has an effect on the morphology of the nanofibers.
 | Figure 3 SEM characterization of different nanofibers. |
FTIR spectra of three nanofiber samples are shown in Figure 4. For the PLA nanofibers, the characteristic peaks were observed, such as the C–O stretching at ~1,089 cm−1 and carbonyl (−C=O) band at 1,762 cm−1. For the ACP sample, the distinctive peak which can be indexed to PO43− group was observed at 565 cm−1. The peak at 2,927 cm−1 revealed the presence of C–H bonding of lecithin. For the ACP-PLA and BSA-containing ACP-PLA nanofibers, the peaks of C–O stretching, −C=O, and PO43− group were found. Because there was just a small quantity of lecithin and BSA in the nanofibers, the characteristic peaks of lecithin and BSA were not obvious in the FTIR spectra. Perhaps, some of the peaks might have been hidden due to overlapping of other peaks of the composite nanofibers.
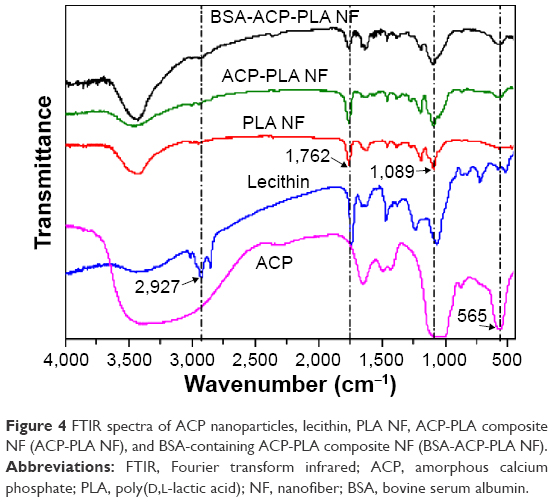 | Figure 4 FTIR spectra of ACP nanoparticles, lecithin, PLA NF, ACP-PLA composite NF (ACP-PLA NF), and BSA-containing ACP-PLA composite NF (BSA-ACP-PLA NF). |
Mineralization of water-soluble drug-containing ACP-PLA nanofibers
Figure 5 displays the SEM micrographs of the mineralized pure PLA, ACP-PLA, and BSA-containing ACP-PLA composite nanofibers. Compared with the morphologies shown in Figure 3, the morphologies of samples were largely changed after immersing in SBF for only 1 day, except for the pure PLA nanofibers. From the magnified SEM micrographs, we found a large number of small nanoparticles deposited on the surfaces of ACP-PLA and BSA-containing ACP-PLA composite nanofibers. Then, more inorganic matter with nanosheet structure was observed on the surface of composite nanofibers after extending the mineralization time to 3 days. Sequentially, these nanosheets continued to form eventually a three-dimensional porous network on the surface of composite nanofibers when the mineralization time was extended to 7 days. However, as for the pure PLA nanofibers, it was clearly seen that the surface of the nanofibers did not display obvious change after immersed in SBF for 1 and 7 days.
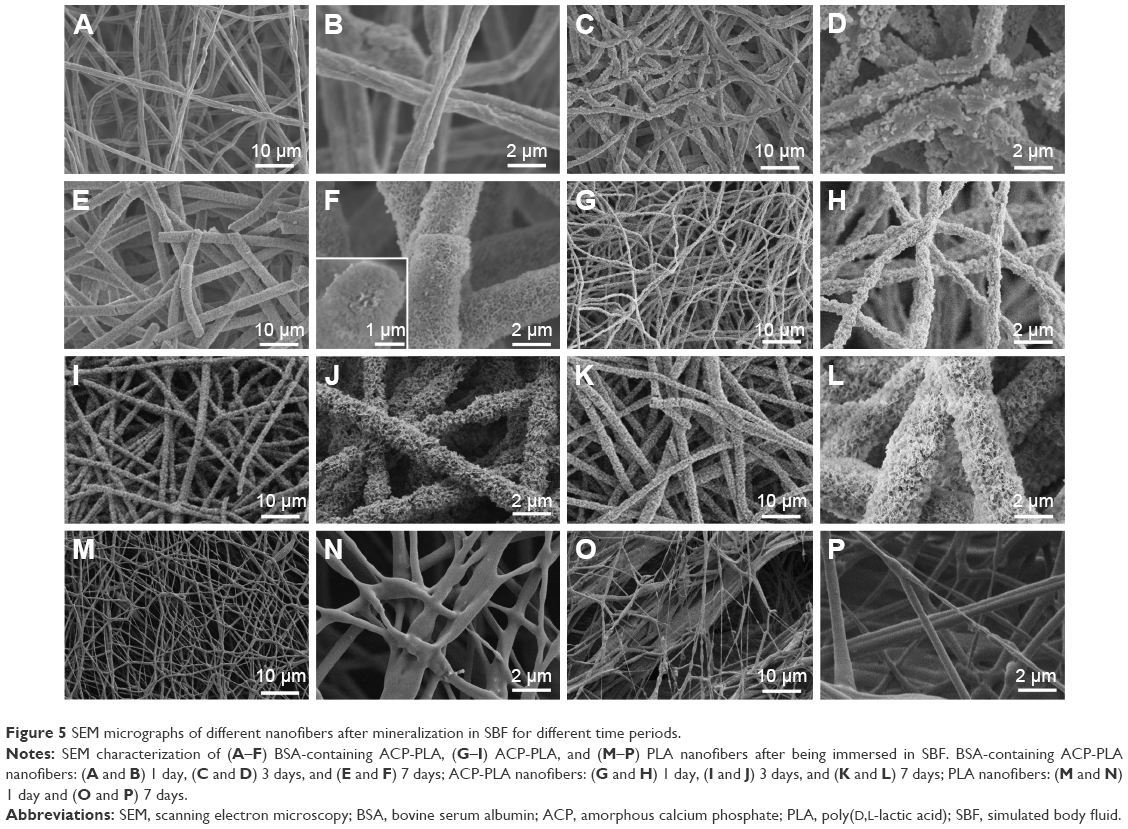 | Figure 5 SEM micrographs of different nanofibers after mineralization in SBF for different time periods. |
The contact angles on the surfaces of the fresh and mineralized samples were measured to characterize hydrophilic property. As the results in Figure 6 show, there was a sharp decrease in the values of contact angle of BSA-containing ACP-PLA and ACP-PLA composite nanofibers after 1 and 7 days of mineralization, compared with the fresh samples. After 1 day of mineralization, the surfaces of BSA-containing ACP-PLA and ACP-PLA composite nanofibers became completely hydrophilic. The results may be explained by the change of the surface structure on the composite nanofibers. There was clearly an increase in porous nanosheet formation on the surface of composite nanofibers after being immersed in SBF. For the pure PLA naofibers, the contact angles slightly decreased to 14° and 30° after 1 and 7 days. The results may be explained by the mineralization process of different samples in SBF. The HA nanosheets formed on the surface of composite nanofibers in SBF by a process of mineralization. The porous HA nanosheets might have contributed toward the spreading and adsorption of water. Therefore, the mineralization process can lead to hydrophilic modification of both composite nanofibers.
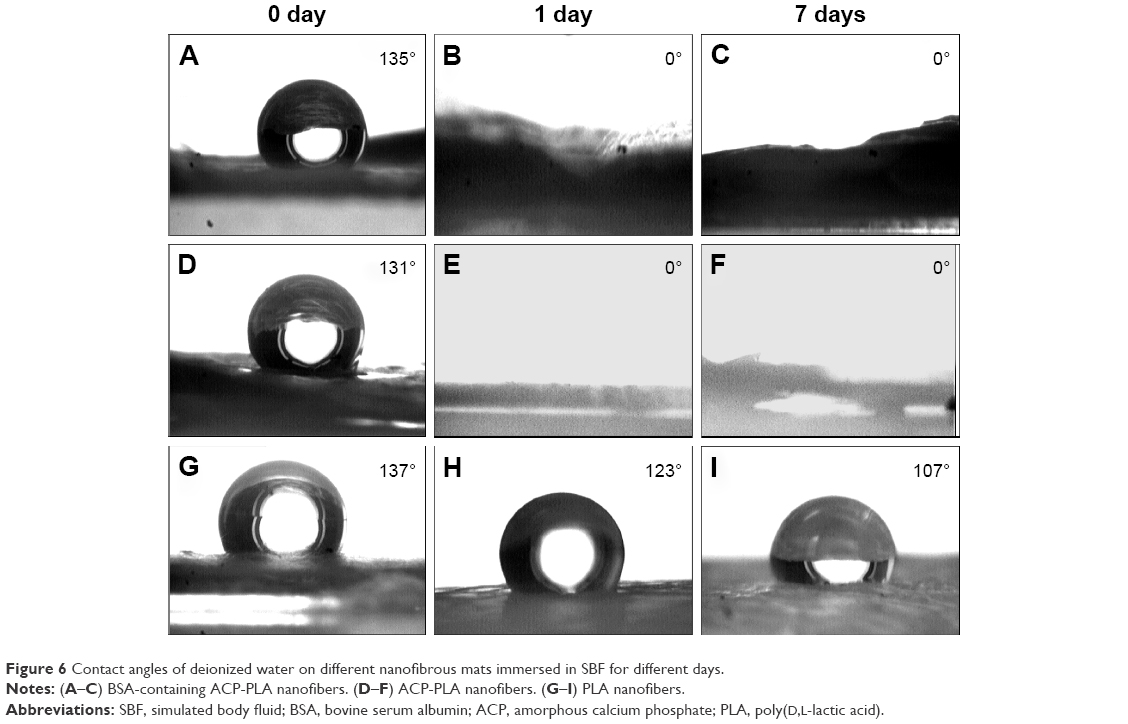 | Figure 6 Contact angles of deionized water on different nanofibrous mats immersed in SBF for different days. |
Mineralization is a common method to prepare HA-based biomaterials by immersing the macromolecules and some inorganic materials in SBF. But the process always takes up a lot of time, especially for hydrophobic polymers. For example, it usually requires several weeks to form a mineralized HA layer on the surface of pure PLA polymer.40,41 The result can be explained by the hydrophobic properties of PLA polymer which cannot supply active groups or sites for the mineralization.23,42 The result obtained for PLA nanofibers also indicated poor mineralization. However, the surface morphologies of BSA-containing ACP-PLA and ACP-PLA composite nanofibers were largely changed in the process of mineralization. The addition of ACP showed obvious effect on the formation of nanosheets structure on the surface of BSA-containing ACP-PLA and ACP-PLA composite nanofibers. It is well known that the ACP can easily crystallize to form stable HA crystal in SBF. Therefore, the transformation from ACP to HA occurs when the BSA-containing ACP-PLA and ACP-PLA nanofibers are in contact with SBF solution. Sequentially, the surfaces of these composite nanofibers can become rough, which improve the hydrophilicity of the nanofibers. Then, the rough surface may promote the reaction of Ca2+ and PO43− ions in SBF for further formation of HA on the surface of composite nanofibers. The ACP nanoparticles play a key role of seeds in mineralization, promoting the formation of HA on the BSA-containing ACP-PLA nanofibers and ACP-PLA nanofibers in SBF.
The in vitro BSA release of BSA-containing ACP-PLA nanofibers
The BSA-containing ACP-PLA nanofibers were prepared and used for studying drug release in vitro. The cast membrane of BSA-containing ACP-PLA was used as a control sample, which was prepared by directly drying the BSA-containing ACP-PLA solution. The amount of BSA in the composite nanofiber or the control sample was 3.3 mg g−1. The cumulative release amounts of BSA versus release time are shown in Figure 7A. The cumulative release amounts of BSA from BSA-containing ACP-PLA nanofibers in SBF reached 1.8%, 6.5%, 16.8%, and 35.3% on 1, 3, 7, and 14 days, respectively. The release was slow in the first 2 days, and then became sustained from 2 to 14 days. The cumulative release amounts of BSA from the control sample in SBF reached 1.7%, 2.8%, 8.2%, and 20% on 1, 3, 7, and 14 days, respectively. Compared with the membrane, the cumulative release amounts of BSA from BSA-containing ACP-PLA nanofibers were higher. The results can be explained by the small size of the nanofibers. Both samples showed sustained BSA release properties.
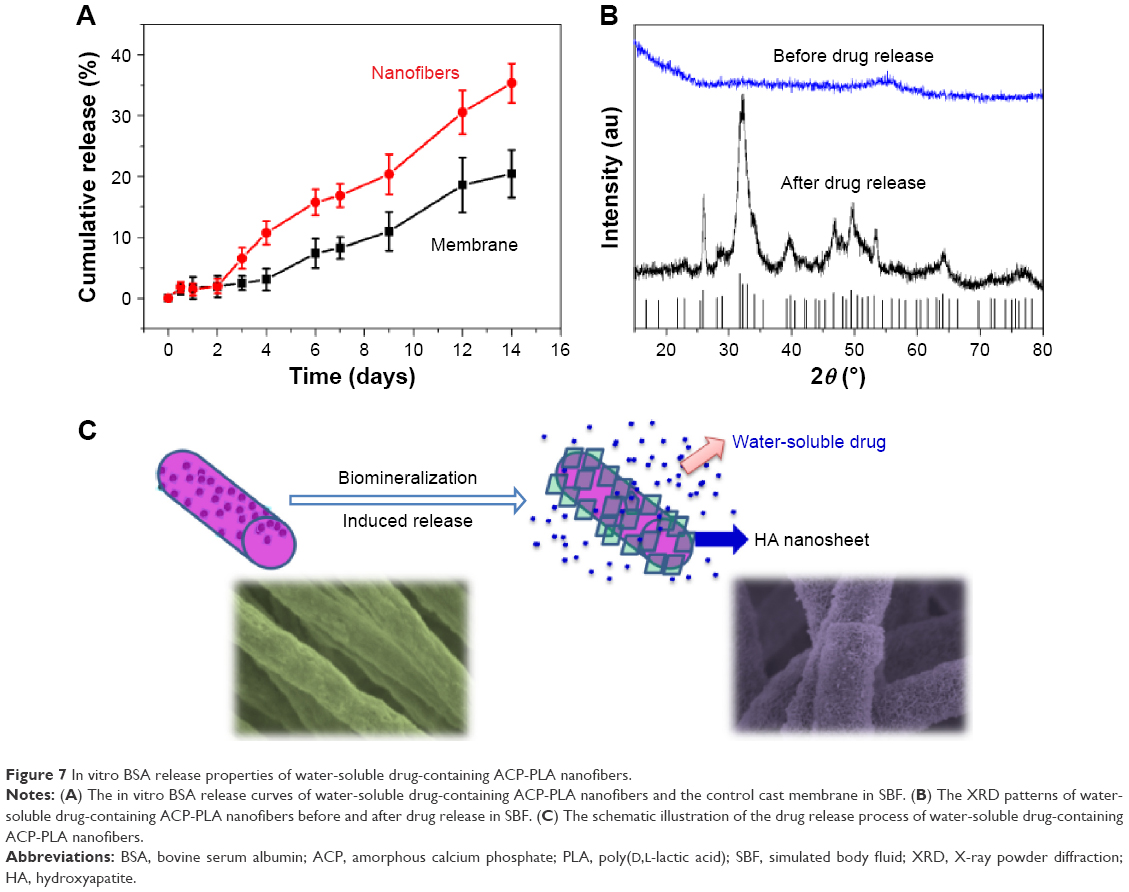 | Figure 7 In vitro BSA release properties of water-soluble drug-containing ACP-PLA nanofibers. |
The XRD patterns of the BSA-containing ACP-PLA nanofibers before and after release of BSA in SBF are displayed in Figure 7B. The results indicate that the crystal phase of ACP nanoparticles in BSA-containing ACP-PLA nanofibers transformed from amorphous phase to crystalline HA phase (JCPDS No 09-0432) after the in vitro release process in SBF. The diffraction peaks of crystal HA at around 2θ≈25.8°, 31.7°, 34.0°, 40.5°, 46.7°, 49.5°, 53.1°, and 64.1° can be well indexed to (002), (211), (202), (221), (222), (213), (004), and (304) crystal faces of HA, respectively. These results indicate that the HA crystal can be well formed in mineralization and display a good bioactivity of the BSA-containing ACP-PLA nanofibers. The supposed release process of BSA-containing ACP-PLA nanofibers is illustrated in Figure 7C. The main change of the nanofibers was the crystal phase transformation from ACP to HA during the mineralization process of the BSA-containing ACP-PLA nanofibers. The surface structures of the BSA-containing ACP-PLA nanofibers were largely changed in SBF and led to a significant improvement of hydrophilicity which is favorable for the release of BSA from the composite nanofibers.
Cell viability of BSA-containing ACP-PLA nanofibers
The cytocompatibility of BSA-containing ACP-PLA, ACP-PLA, and PLA nanofibers was measured using MG63 cells. As shown in Figure 8, the results indicate that MG63 cells can grow well on the different nanofibrous membranes from 1 to 7 days. These growth curves display high cytocompatibility of the cells cultured on the as-prepared BSA-containing ACP-PLA, ACP-PLA, and PLA nanofibers. The high compatibility can be explained by the favorable chemical properties of the BSA, lecithin, ACP nanoparticles, and PLA polymer which are common biomaterials or biocompatible molecules.
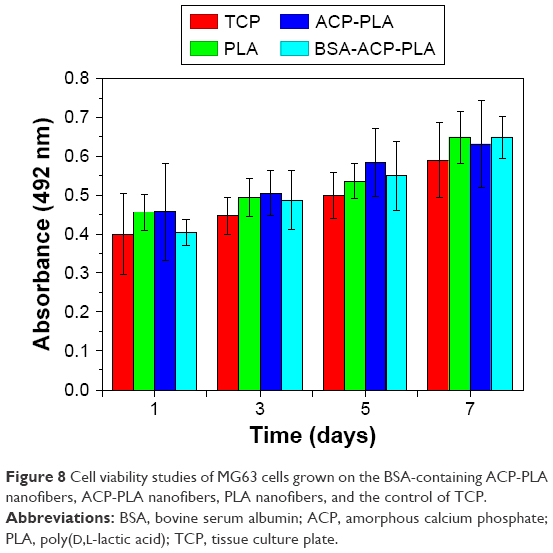 | Figure 8 Cell viability studies of MG63 cells grown on the BSA-containing ACP-PLA nanofibers, ACP-PLA nanofibers, PLA nanofibers, and the control of TCP. |
In addition, the cell adhesion and spreading behavior on BSA-containing ACP-PLA nanofibrous membranes were also studied by SEM. As the micrographs in Figure 9 show, many cells with spreading filopodia were well observed on the nanofibrous membranes after culturing for 24 hours, which is the normal morphology of the osteoblast cells. Therefore, these results display that the BSA-containing ACP-PLA nanofibrous membranes have good cytocompatibility and favorable properties for cell adhesion and spreading.
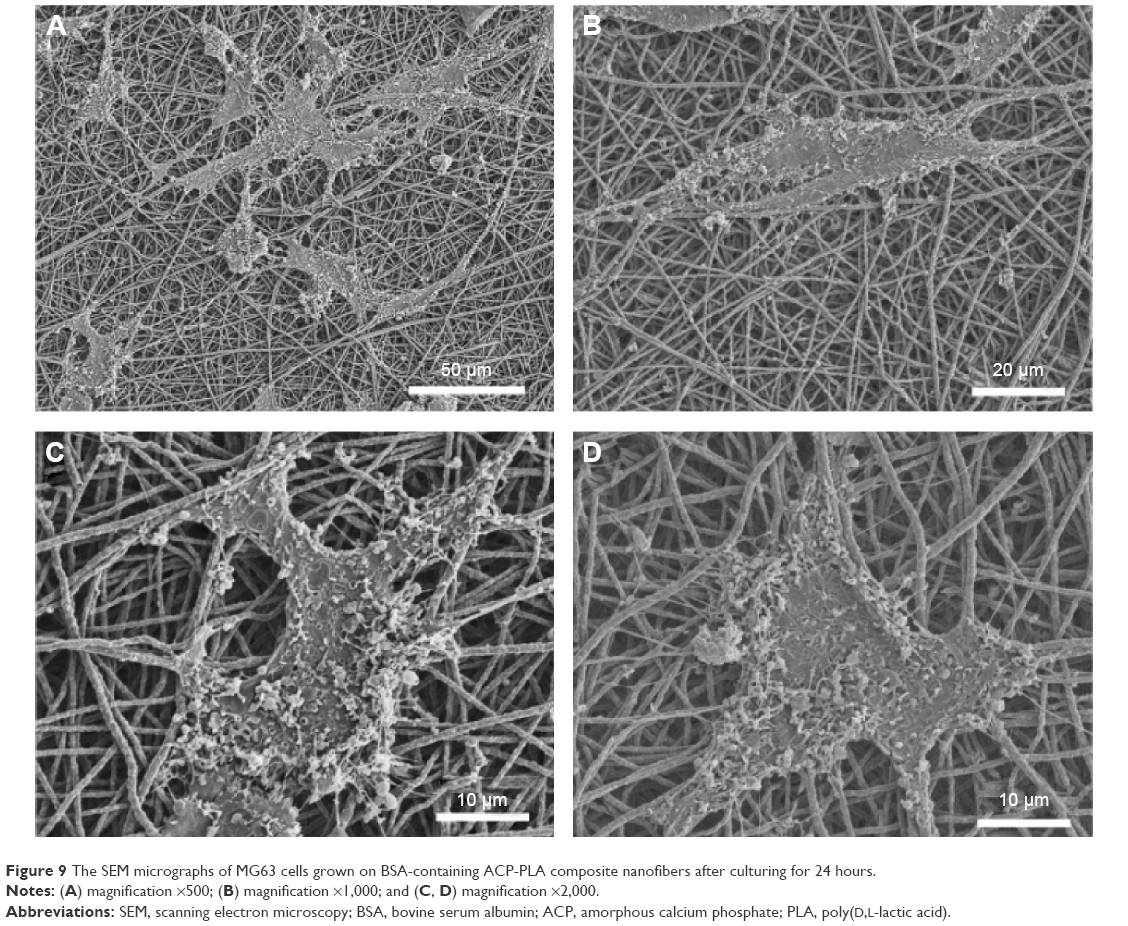 | Figure 9 The SEM micrographs of MG63 cells grown on BSA-containing ACP-PLA composite nanofibers after culturing for 24 hours. |
Conclusion
In summary, water-soluble drug-containing ACP-PLA nanofibers with favorable mineralization and sustained drug release properties were achieved. ACP nanoparticles were synthesized through a precipitation route using PLA-mPEG as a soft template. Then, ACP sample was blended into PLA polymer solution for electrospinning of composite nanofibers. Meanwhile, the water-soluble drug-containing ACP-PLA composite nanofibers were also prepared. The water-soluble protein molecule BSA was mixed into hydrophobic PLA solution for electrospinning, using the native biomolecule of lecithin as a surfactant. The BSA-containing ACP-PLA nanofibers showed excellent mineralization properties. For mineralization, the ACP displayed a key role as seeding material on the surface of BSA-containing ACP-PLA nanofibers, and promoted the formation of HA. The drug-containing ACP-PLA nanofibers exhibited sustained drug release in SBF which simultaneously occurred with the process of mineralization. The result of in vitro cell viability test indicated that BSA-containing ACP-PLA has good biocompatibility, and the MG63 cells attached and spread onto the as-prepared nanofibrous membranes after culturing for 24 hours. Considering the facile fabrication process, sustained drug release property, fast mineralization, and high biocompatibility, the as-prepared BSA-containing ACP-PLA nanofibers may have potential applications in water-soluble drug delivery for tissue engineering.
Acknowledgments
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (81171727) and the Science and Technology Commission of Shanghai (13JC1407300).
Disclosure
The authors report no conflicts of interest in this work.
References
El-Ghannam A. Bone reconstruction: from bioceramics to tissue engineering. Expert Rev Med Devices. 2005;2(1):87–101. | ||
Surmenev RA, Surmeneva MA, Ivanova AA. Significance of calcium phosphate coatings for the enhancement of new bone osteogenesis – a review. Acta Biomater. 2014;10(2):557–579. | ||
Ginebra MP, Traykova T, Planell JA. Calcium phosphate cements as bone drug delivery systems: a review. J Control Release. 2006;113(2):102–110. | ||
Chen F, Zhu YJ. Multifunctional calcium phosphate nanostructured materials and biomedical applications. Curr Nanosci. 2014;10(4):465–485. | ||
Dorozhkin SV, Epple M. Biological and medical significance of calcium phosphates. Angew Chem Int Ed Engl. 2002;41(17):3130–3146. | ||
Zhou R, Xu W, Chen F, et al. Amorphous calcium phosphate nanospheres/polylactide composite coated tantalum scaffold: facile preparation, fast biomineralization and subchondral bone defect repair application. Colloids Surf B Biointerfaces. 2014;123:236–245. | ||
Chen XY, Sun XL, Yang XY, et al. Biomimetic preparation of trace element-codoped calcium phosphate for promoting osteoporotic bone defect repair. J Mater Chem B. 2013;1(9):1316–1325. | ||
Zhou CH, Zhang DL, Bai YX, Li S. Casein phosphopeptide-amorphous calcium phosphate remineralization of primary teeth early enamel lesions. J Dent. 2014;42(1):21–29. | ||
Braghirolli DI, Steffens D, Pranke P. Electrospinning for regenerative medicine: a review of the main topics. Drug Discov Today. 2014;19(6):743–753. | ||
Schiffman JD, Schauer CL. A review: electrospinning of biopolymer nanofibers and their applications. Polym Rev. 2008;48(2):317–352. | ||
Ahmad Z, Huang J, Thian ES, et al. Freeform fabrication of nano-biomaterials using 3D electrohydrodynamic print-patterning. J Biomed Nanotechnol. 2008;4(2):185–195. | ||
Stevens MM, George JH. Exploring and engineering the cell surface interface. Science. 2005;310(5751):1135–1138. | ||
Yu JS, Lee AR, Lin WH, Lin CW, Wu YK, Tsai WB. Electrospun PLGA fibers incorporated with functionalized biomolecules for cardiac tissue engineering. Tissue Eng Part A. 2014;20(13–14):1896–1907. | ||
Wang C, Wang M. Electrospun multifunctional tissue engineering scaffolds. Front Mater Sci. 2014;8(1):3–19. | ||
Mattanavee W, Suwantong O, Puthong S, Bunaprasert T, Hoven VP, Supaphol P. Immobilization of biomolecules on the surface of electrospun polycaprolactone fibrous scaffolds for tissue engineering. ACS Appl Mater Interfaces. 2009;1(5):1076–1085. | ||
Xie J, Peng C, Zhao QH, et al. Osteogenic differentiation and bone regeneration of iPSC-MSCs supported by a biomimetic nanofibrous scaffold. Acta Biomater. 2016;29:365–379. | ||
Zhang H, Fu QW, Sun TW, et al. Amorphous calcium phosphate, hydroxyapatite and poly(D,L-lactic acid) composite nanofibers: electrospinning preparation, mineralization and in vivo bone defect repair. Colloids Surf B Biointerfaces. 2015;136:27–36. | ||
Zhang YZ, Venugopal JR, El-Turki A, Ramakrishna S, Su B, Lim CT. Electrospun biomimetic nanocomposite nanofibers of hydroxyapatite/chitosan for bone tissue engineering. Biomaterials. 2008;29(32):4314–4322. | ||
Karavasili C, Bouropoulos N, Kontopoulou I, et al. Preparation and characterization of multiactive electrospun fibers: poly-epsilon-carpolactone fibers loaded with hydroxyapatite and selected NSAIDs. J Biomed Mater Res A. 2014;102(8):2583–2589. | ||
Yao YM, Shao HB, Zhang QF. Development and characterization of a novel amorphous calcium phosphate/multi (amino acid) copolymer composite for bone repair. J Biomater Tissue Eng. 2015;5(5):387–390. | ||
Chen Z, Cao SS, Wang HR, et al. Biomimetic remineralization of demineralized dentine using scaffold of CMC/ACP nanocomplexes in an in vitro tooth model of deep caries. PLoS One. 2015;10(1):e0116553. | ||
Stock SR. The mineral-collagen interface in bone. Calcif Tissue Int. 2015;97(3):262–280. | ||
Ma Z, Chen F, Zhu YJ, Cui T, Liu XY. Amorphous calcium phosphate/poly(D,L-lactic acid) composite nanofibers: electrospinning preparation and biomineralization. J Colloid Interface Sci. 2011;359(2):371–379. | ||
Agarwal R, Garcia AJ. Biomaterial strategies for engineering implants for enhanced osseointegration and bone repair. Adv Drug Deliv Rev. 2015;94:53–62. | ||
Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27(18):3413–3431. | ||
Porter JR, Ruckh TT, Popat KC. Bone tissue engineering: a review in bone biomimetics and drug delivery strategies. Biotechnol Progr. 2009;25(6):1539–1560. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Abdelkader H, Alani AWG, Alany RG. Recent advances in non-ionic surfactant vesicles (niosomes): self-assembly, fabrication, characterization, drug delivery applications and limitations. Drug Deliv. 2014;21(2):87–100. | ||
Song WX, He Q, Mohwald H, Yang Y, Li JB. Smart polyelectrolyte microcapsules as carriers for water-soluble small molecular drug. J Control Release. 2009;139(2):160–166. | ||
Zhou L, Zhu CH, Edmonds L, Yang HL, Cui WG, Li B. Microsol-electrospinning for controlled loading and release of water-soluble drugs in microfibrous membranes. RSC Adv. 2014;4(81):43220–43226. | ||
Cui WG, Li XH, Zhu XL, Yu G, Zhou SB, Weng J. Investigation of drug release and matrix degradation of electrospun poly(D,L-lactide) fibers with paracetanol inoculation. Biomacromolecules. 2006;7(5):1623–1629. | ||
Norouzi M, Soleimani M, Shabani I, Atyabi F, Ahvaz HH, Rashidi A. Protein encapsulated in electrospun nanofibrous scaffolds for tissue engineering applications. Polym Int. 2013;62(8):1250–1256. | ||
Xu XL, Zhuang XL, Chen XS, Wang XR, Yang LX, Jing XB. Preparation of core-sheath composite nanofibers by emulsion electrospinning. Macromol Rapid Commun. 2006;27(19):1637–1642. | ||
Ji W, Yang F, van den Beucken JJJP, et al. Fibrous scaffolds loaded with protein prepared by blend or coaxial electrospinning. Acta Biomater. 2010;6(11):4199–4207. | ||
Chen F, Huang P, Zhu YJ, Wu J, Cui DX. Multifunctional Eu3+/Gd3+ dual-doped calcium phosphate vesicle-like nanospheres for sustained drug release and imaging. Biomaterials. 2012;33(27):6447–6455. | ||
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27(15):2907–2915. | ||
Deng SG, Cao JM, Feng J, et al. A bio-inspired approach to the synthesis of CaCO3 spherical assemblies in a soluble ternary-additive system. J Phys Chem B. 2005;109(23):11473–11477. | ||
Horikoshi K, Hata K, Kawabata N, Ikawa SI, Konaka S. Vibrational-spectra and conformation of polyethylene-glycol complexed with calcium and magnesium chlorides. J Mol Struct. 1990;239:33–42. | ||
Xu GF, Aksay IA, Groves JT. Continuous crystalline carbonate apatite thin films. A biomimetic approach. J Am Chem Soc. 2001;123(10):2196–2203. | ||
Chen JL, Chu B, Hsiao BS. Mineralization of hydroxyapatite in electrospun nanofibrous poly(L-lactic acid) scaffolds. J Biomed Mater Res A. 2006;79A(2):307–317. | ||
He CL, Xiao GY, Jin XB, Sun CH, Ma PX. Electrodeposition on nanofibrous polymer scaffolds: rapid mineralization, tunable calcium phosphate composition and topography. Adv Funct Mater. 2010;20(20):3568–3576. | ||
Suh H, Hwang YS, Lee JE, Han CD, Park JC. Behavior of osteoblasts on a type I atelocollagen grafted ozone oxidized poly L-lactic acid membrane. Biomaterials. 2001;22(3):219–230. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.