")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Eldecalcitol Inhibits LPS-Induced NLRP3 Inflammasome-Dependent Pyroptosis in Human Gingival Fibroblasts by Activating the Nrf2/HO-1 Signaling Pathway
Authors Huang C, Zhang C, Yang P, Chao R, Yue Z, Li C, Guo J, Li M
Received 2 July 2020
Accepted for publication 23 October 2020
Published 13 November 2020 Volume 2020:14 Pages 4901—4913
DOI https://doi.org/10.2147/DDDT.S269223
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Cancan Huang, Chaotao Zhang, Panpan Yang, Rui Chao, Ziqi Yue, Congshan Li, Jie Guo, Minqi Li
Department of Bone Metabolism, School and Hospital of Stomatology, Cheeloo College of Medicine, Shandong University & Shandong Key Laboratory of Oral Tissue Regeneration & Shandong Engineering Laboratory for Dental Materials and Oral Tissue Regeneration, Jinan 250012, People’s Republic of China
Correspondence: Minqi Li
Department of Bone Metabolism, School and Hospital of Stomatology, Cheeloo College of Medicine, Shandong University & Shandong Key Laboratory of Oral Tissue Regeneration & Shandong Engineering Laboratory for Dental Materials and Oral Tissue Regeneration, Jinan 250012, China
Tel +86-531-88382095
Email [email protected]
Purpose: Periodontitis is a major chronic oral disease that is accelerated by activation of the NLRP3 inflammasome and the resulting pyroptosis. According to recent studies, active vitamin D and its analogs have been reported to have great anti-inflammatory effects. However, the anti-inflammatory mechanism of a newly found vitamin D analog, eldecalcitol (ED-71), is still unclear. This study investigates whether ED-71 could protect human gingival fibroblasts (HGFs) from LPS-induced pyroptosis and, if so, determine its underlying mechanism.
Methods: After HGFs were treated with LPS alone or with LPS and ED-71, their viability was measured by CCK8 assay. The degrees of inflammation and pyroptosis were measured via LDH assay, H2O2 assay, fluorescent staining, flow cytometry, and Western blots. Intracellular ROS, Hoechst 33,342, and PI stains were assessed with a fluorescence microscope. ROS inhibitor NAC, NLRP3 inhibitor MCC950, and Nrf2 inhibitor ML385 were added to further clarify the mechanism.
Results: LPS induced cytotoxicity in HGFs, as shown by CCK8 assay. LPS also increased intracellular ROS, H2O2 levels, release of LDH, and expression of the pyroptosis-related proteins NLRP3, caspase-1, and IL-1β. NAC and MCC950 reduced LPS-induced NLRP3, caspase-1, and IL-1β. Pretreatment with ED-71 effectively inhibited the LPS-induced pyroptosis and was associated with activation of the Nrf2/HO-1 signaling pathway. This beneficial effect of ED-71 was suppressed by ML385.
Conclusion: This study demonstrates the therapeutic effect of ED-71 on LPS-induced NLRP3 inflammasome-dependent pyroptosis in HGFs and further reveals that ED-71 can inhibit pyroptosis by activating the Nrf2/HO-1 pathway. Our results thus suggest that ED-71 is a potential candidate for the treatment of periodontitis.
Keywords: HGFs, pyroptosis, NLRP3, ED-71, Nrf2/HO-1
Introduction
Periodontitis is a progressive and chronic oral disease that leads to periodontal tissue damage. The host-mediated inflammatory response to pathogenic bacteria in periodontal pockets is a key factor in its occurrence and development.1 Human gingival fibroblasts (HGFs) are the most abundant cells in periodontal tissue, and their inflammatory response to pathogenic bacteria leads to the release of immunoregulatory factors and proteolytic enzymes. The former activated osteoclasts to cause alveolar bone resorption, and the latter directly destroy periodontal tissue, which ultimately leads to the progression of periodontitis.2–4 Therefore, inhibition of the inflammatory response of HGFs is of critical importance for the prevention of periodontitis.
Recent studies have shown that innate immunity, in which pyroptosis plays a vital role, is closely associated with periodontitis.5,6 Pyroptosis, a type of caspase-1-dependent inflammatory programmed cell death, was first observed in macrophages infected by Salmonella typhimurium.7 Compared with apoptosis, pyroptosis is characterized by cell swelling, perforation, lysis, and release of cell contents. During this process, the integrity of the cell membrane is reduced, and the permeability of the cell membrane is increased, which permits propidium iodide to enter the cells. In a recent study, activation of caspase-1 and the resultant pyroptosis were found to contribute to the progression of apical periodontitis.8 Active caspase-1 can lead to the production of interleukin-1β (IL-1β) and IL-18. The activation of caspase-1 is due to the assembly of inflammasomes, with the NLRP3 inflammasome playing an important role in periodontitis.9 In this process, NLRP3, ASC, and pro-caspase-1 are assembled into inflammasomes, then pro-caspase-1 is cleaved into active caspase-1 p20 to perform the appropriate function.
In periodontitis, lipopolysaccharide (LPS) from Porphyromonas gingivalis is the main pathogen-associated molecular pattern. As an important causative factor, LPS interacts with TLR4 to promote the release of inflammatory cytokines such as IL-6 and IL-8 to promote the expansion of inflammation.10 In addition, the generation of reactive oxygen species (ROS) in response to LPS is the key signaling event in the pro-inflammatory response in HGFs,11 and the ROS can also stimulate the activation of NLRP3 inflammasomes, which can further promote periodontitis.12 The expression of NLRP3 inflammasomes is upregulated in periodontitis,13 and the inhibition of the NLRP3 inflammasome can reduce the periodontitis in mice.14 Thus, inhibition of NLRP3 inflammasome activation and the resulting pyroptosis may be essential in improving periodontitis.
Nrf2 (nuclear factor-erythropoietin 2-related factor 2) is a basic leucine zipper redox-sensitive transcription factor with anti-oxidant and anti-inflammatory properties and is a major regulator of other cytoprotective genes. Under static conditions, Nrf2 interacts with Keap1 and is inactive. In response to oxidative and inflammatory stress, it is released from Keap1 and transported to the nucleus where it activates the expression of dozens of cytoprotective genes, such as hemeoxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase-1 (NQO1), to defend against oxidative stress and inflammation.15 HO-1 prevents the expansion of inflammation by degrading heme groups into products such as bilirubin, which plays a protective antioxidant role. The heme group is found in many cells, including gingival fibroblasts. Studies have found that the activation of Nrf2 and induction of HO-1 could inhibit the NLRP3 inflammasome and thus pyroptosis.16–18 Activation of the Nrf2 pathway with isorhamnetin reduces the inflammatory response of HGFs.19 These findings promote speculation that the activation of Nrf2/HO-1 may be a protection mechanism in periodontitis and reduce pyroptosis.
ED-71, a new kind of active vitamin D analog, has been widely used for the treatment of osteoporosis in Japan. Osteoporosis is a major risk factor for periodontal disease and seems to be related to alveolar bone loss in disease progression.20 Compared with calcitriol, ED-71 has a different mode of binding to vitamin D binding protein (DBP), the vitamin D receptor (VDR), and vitamin D 24-hydroxylase (CYP24A1). ED-71 consequently has a longer half-life, a lower clearance rate, and stronger VDR-mediated effects, resulting in greater efficacy than calcitriol.21 Recent research has shown that vitamin D plays a role in a variety of inflammatory diseases, such as inflammatory bowel disease.22 Vitamin D can activate the Nrf2/HO-1 pathway to ameliorate liver and kidney injuries, which are markedly promoted by oxidative stress and inflammation.23 Because ED-71 is an analog of active vitamin D, it is hypothesized that ED-71 may have a similar effect on other inflammatory diseases, including periodontitis.
The anti-pyroptotic effect of ED-71 on LPS was explored in the present study. In addition, potential mechanisms of pyroptosis involving the Nrf2/HO-1 signaling pathway were investigated.
Materials and Methods
Chemicals and Reagents
ED-71 was purchased from Chugai Pharmaceutical Co., Ltd (Japan). E.coli LPS was purchased from Solarbio (Beijing, China). The Nrf2 inhibitor (ML385) and ROS inhibitor (NAC) were purchased from MedChemExpress (Shanghai, China). The NLRP3 inhibitor MCC950 was bought from Tocris (Shanghai, China). Antibodies against Nrf2, Histone H, HO-1, NLRP3, ASC, IL-18, IL-1β, IL-6 and IL-8 were purchased from Abcam (Shanghai, China). Antibodies against GAPDH and TLR4 were bought from Proteintech (Wuhan, Hubei, China).
Cell Culture
This study was approved by the Ethics Committee of the School of Stomatology, Shandong University (No. 20,200,103), and all participants provided written informed consent before the collection of fresh tissue. All procedures were carried out according to the principles of the Declaration of Helsinki. Normal human gingival fibroblasts (HGFs) were cultured from four patients undergoing maxillofacial surgery who were entirely free from clinical periodontal disease. The HGFs were isolated and cultured as previously described.24 All HGFs were cultured in α-minimum essential medium (α-MEM; Hyclone, Logan, UT, USA) containing 10% FBS (Gibco, Grand Island, NY, USA) at 37°C with 5% CO2 in a cell incubator.
Detection of Cell Viability
In order to evaluate the cytotoxicity of ED-71 and LPS, cell viability was measured by CCK8 assay (MedChemExpress, China). HGFs were seeded in 96-well plates. After 24 h, cells in some of the wells were treated with LPS (at 0, 0.05, 0.5, 5, and 50 μg/mL) for 6 h, 12 h, 24 h, and 48 h. Cells in other wells were treated with ED-71 (at 0, 0.5, 5, and 50 nM) for 24 h and then stimulated with or without 5 μg/mL LPS for 6 h. CCK8 (10 μL) was added to each well and incubated for 3 h. Finally, the optical density at 450 nm was measured with a Bio-Rad Microplate Reader (Model 680, Bio-Rad, USA).
Lactate Dehydrogenase (LDH) Release Assay
To evaluate the cytotoxicity of LPS and curative effect of ED-71, LDH release was assessed by LDH Assay Kit (Beyotime, China). Briefly, HGFs were plated in 96-well plate with 3000/well. The cell supernatant was harvested after treatment with indicated drugs, and LDH release was evaluated with an LDH assay kit. The absorbance at 490 nm was measured with the microplate reader.
Detection of ROS Generation
The production of intracellular ROS was detected by DCFH-DA (Beyotime, China). Cells were seeded in 6-well plates and exposed to treatments as described before. DCFH-DA (10 μM) was added to each well, and the plates were then incubated at 37°C in the dark, then washed 3 times with PBS. ROS was detected by fluorescence microscopy (OLYMPUS IX73, Tokyo, Japan) and captured by camera and imaging software (OLYMPUS cellSens Standard 1.17) at room temperature in the dark.
Determination of H2O2 Content
Cells were seeded in 6-well plates at 1 × 106 cells/well. After drug treatments, the cells were collected and centrifuged. After discarding the supernatant, 1 mL acetone was added for every 5 million cells, and the cells were ultrasonically broken. After centrifuging at 8000 g at 4°C for 10 min, the supernatant was saved, and the content of H2O2 was determined following the protocol of the Micro Hydrogen Peroxide Assay Kit (Solarbio, Beijing, China).
Flow Cytometry
Cells were seeded in 6-well plates at 1 × 106 cells/well. After drug treatments, the cells were collected and washed twice in cool PBS, and then suspended in 100 µL 1× binding buffer. We added 5 µL Annexin V-FITC and 5 µL PI (Beyotime, Beijing, China) and incubated at room temperature in the dark for 20 min. Finally, 400 µL of 1× binding buffer was added to the cells. Cells were analyzed by flow cytometry using Accuri C6 plus software (Becton Dickinson) within an hour. Annexin-V binds to phosphatidylserine exposed on the external leaflet of the plasma membrane in apoptotic cells, but can also label pyroptotic cells because membrane rupture permits its entry into the cell and binding to phosphatidylserine on the inner leaflet.25
RNA Isolation and Quantitative Real‐time Polymerase Chain Reaction (qRT‐PCR)
After drug treatment, total RNA was extracted by Trizol (TaKaRa, Tokyo, Japan), then reverse transcribed to cDNA using a SuperScript TM II reverse transcriptase kit (TaKaRa, Tokyo, Japan) following the manufacturer’s instructions. Real-time quantitative PCR was measured using SYBR Premix Ex Taq (TaKaRa Bio, Inc., Otsu, Japan). The conditions of denaturation, annealing, and extension were as follows: 95°C for 30 sec, 45 cycles at 95°C for 5 sec, and 60°C for 20 sec. Relative gene levels were analyzed by the 2−ΔΔCt method and standardized by the GAPDH. The primers used were as follows: 5ʹ-GCCTGTTCCTGTGATGT GGAG-3ʹ (forward primer) and 5ʹ -TGCCCACAGACATTCATACAGTTTC-3ʹ (reverse primer) for the human Caspase-1 gene, 5ʹ -CCAGGGACAGGATATGGAGC-3ʹ (forward primer) and 5ʹ -TTCAACACGCAGGACAGGTACAG-3ʹ (reverse primer) for the human IL-1β gene, 5ʹ-GAT CTTCGCTGCGATCAACA-3ʹ (forward primer) and 5ʹ-GGGATTCGAAACACGTGCATTA-3ʹ (reverse primer) for the human NLRP3 gene, 5ʹ-GTATGCAACAGGACATTGAGCAAG-3ʹ (forward primer) and 5ʹ-TGGAACCATGGTAGTCTCAACCAG-3ʹ for the human NRF2 gene, 5ʹ-GGAACTTTCAGAAGGGCCAGGT-3ʹ (forward primer) and 5ʹ-TGCAGCTCTTCTGGGAAGT AGACA-3ʹ (reverse primer) for the human HO-1 gene. The internal reference control was GAPDH, 5ʹ-CCTGCACCACCAACTGCTTA-3ʹ (forward primer), 5ʹ-GGCCATCCACAGTCTT CTGAG-3ʹ (reverse primer).
Western-Blot Analysis
After the indicated treatments, HGFs were lysed by RIPA lysis buffer (Beyotime, China) and the concentration of the protein was detected by BCA Protein Assay Kit (Beyotime, P0010, China). One-fourth volume of 5x SDS loading buffer was added in proteins and heated at 95°C for 5 min. Nuclear protein was extracted by a Nuclear Protein Extraction kit (Boster, Wuhan, China) according to the manufacturer’s protocol. All of the proteins were then separated by 6–15% SDS-PAGE and transferred to PVDF membrane. 5% BSA was added to membranes for 1 h. Correspondent primary antibodies were added to the membranes and stayed for 12 h at 4°C. After washing by TBST for three times, the second antibodies were added to membranes. Washing three times by TBST, the ECL detection system (SmartChemi 420, Beijing, China) was used to measure the immune reaction zone. All experiments were repeated three times.
Statistical Analysis
After all the statistics were collected, Image J and Graphpad Prism 7 were used to analyze the statistics. We used Student’s t-test to calculate the differences between the control group and the experiment group. Data were all presented as average value ± SEM. P < 0.05 was considered as statistical significance.
Results
LPS-Induced NLRP3 Inflammasome-Dependent Pyroptosis by the Activation of ROS in HGFs
As showed in Figure 1A, the cytotoxicity of LPS to HGF is dose-dependent, with a significant effect at 5 μg/mL. This concentration was used to guide subsequent experiments. As noted above, pyroptosis is characterized by activation of caspase-1 and the release of IL-1β, IL-18, and LDH. As shown in Figure 1B–D. When cells were exposed to different concentrations of LPS for 6 hours, TLR4 expression was up-regulated. Subsequently, the expression of NLRP3 inflammasome-related proteins caspase-1 p20, IL-1β, and IL-18 increased, and the release of LDH also increased. The inflammatory factors IL-6 and IL-8 increased at the same time. To further verify whether pyroptosis is NLRP3-dependent, we introduced NLRP3 inhibitor MCC950 (100 nM). The results show that with NLRP3 inhibition, expression of caspase-1 p20 and IL-1β decreased rapidly (Figure 1H) while cell viability increased, and the release of LHD was attenuated (Figure 1F and G). ROS production is an important event in LPS-induced pyroptosis, which results in the activation of the NLRP3 inflammasome. As shown in Figure 1J and K, LPS caused the production of large quantities of intracellular ROS and H2O2, indicating that the cells were in a state of high oxidative stress. To confirm that LPS-induced ROS was closely associated with the NLRP3 inflammasome and subsequent pyroptosis, we added the ROS inhibitor NAC (20 mM), which decreased the relative expression levels of NLRP3, mature caspase-1, and IL-1β (Figure 1I). Correspondingly, cell viability was improved, and the release of LDH was decreased (Figure 1F and G). Together, these findings confirm that the LPS could induce pyroptosis in HGFs, which was primarily due to the NLRP3 inflammasome, with ROS playing a substantial role.
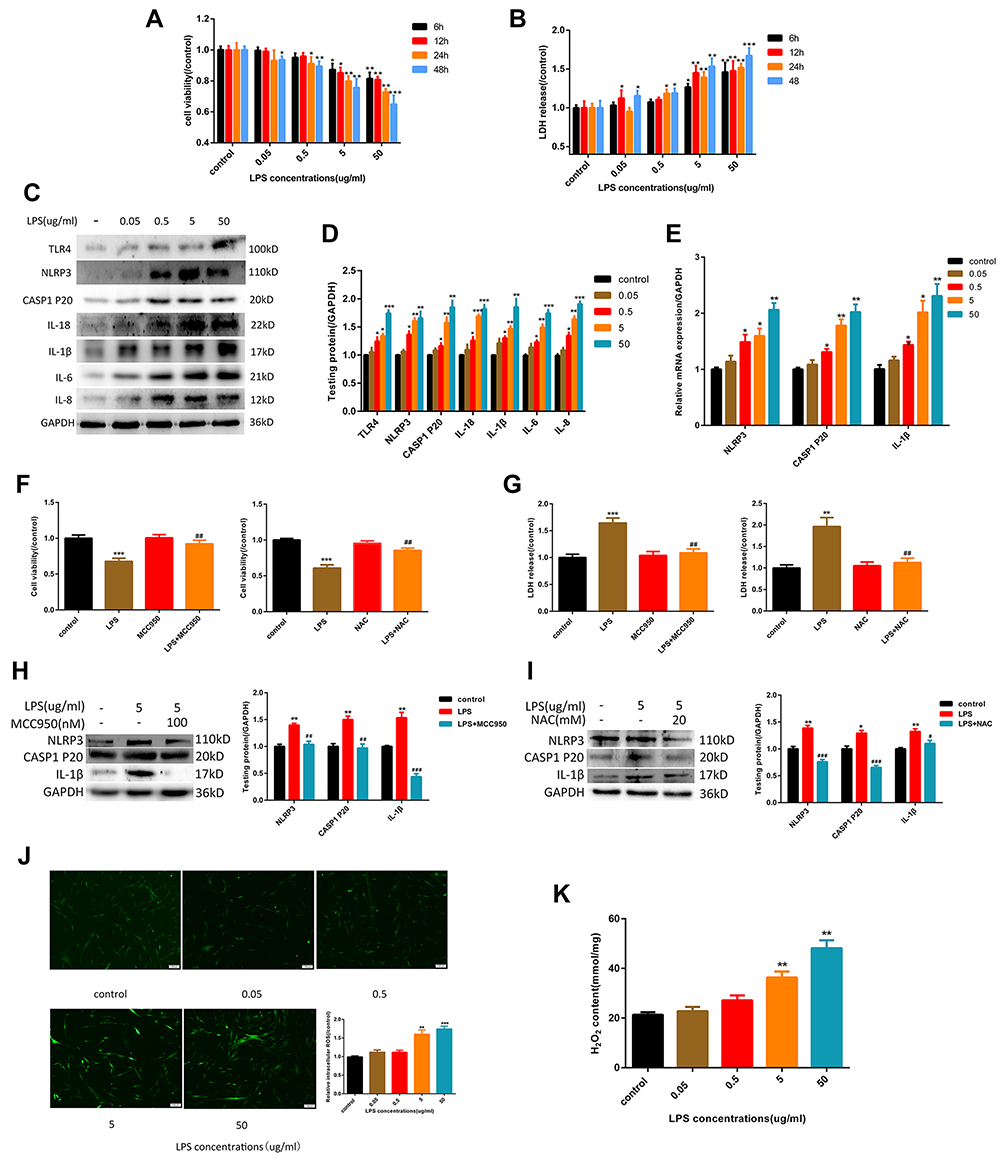 |
Figure 1 LPS-induced NLRP3 inflammasome-dependent pyroptosis by the activation of ROS in HGFs. (A) Cell viability and (B) relative LDH release in HGFs treated by 0, 0.05, 0.5, 5 and 50 μg/mL of LPS for 6 h, 12 h, 24 h and 48 h. (C and D) The protein content in HGFs treated with 0, 0.05, 0.5, 5, 50 μg/mL of LPS for 6 h. (E) Relative mRNA expression was shown. (F) Cell viability and (G) relative LDH release in HGFs treated with 5 μg/mL LPS for 6 h with or without MCC950 (100 nM) or NAC (20 mM) for 1h.(Hand I) The protein content was shown. (J) Relative ROS and (K) H2O2 content in HGFs treat with 0, 0.05, 0.5, 5, 50 μg/mL of LPS for 6 h. Data were shown as mean ± SEM from three experiments independently. ***P < 0.001, **P < 0.01 and *P < 0.05 compared with control group. ###P < 0.001, ##P < 0.01 and #P < 0.05 compared with the group only treated by 5 μg/mL LPS. |
ED-71 Reduces LPS-Induced Cell Death in HGFs
Figure 2A presents the chemical formula of ED-71. To assess the cytotoxicity of ED-71 and further investigate its effect on HGFs treated by LPS, CCK8 and the release of LDH were examined. When treated with ED-71 alone, the cells were not affected (Figure 2B). As shown above, LPS induced cell death in HGFs in a dose-dependent manner. After pretreatment with ED-71 (at 0, 0.5, 5, and 50 nM) for 24 h, cells were treated with 5 μg/mL LPS for 6 h. As shown in Figure 2C, ED-71 reduced cell death induced by LPS.
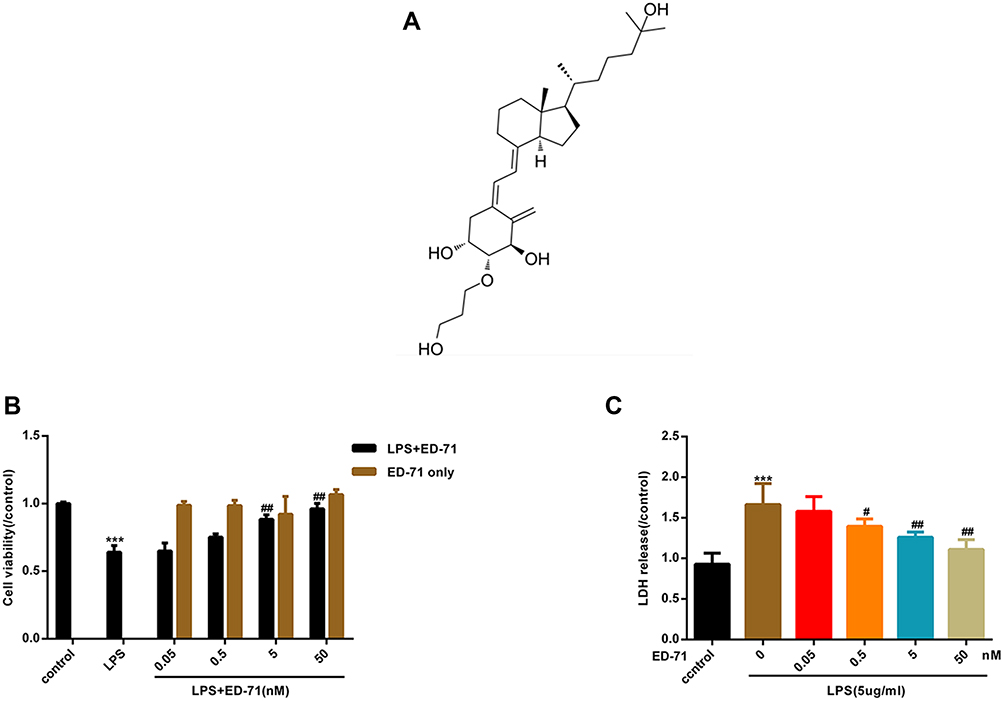 |
Figure 2 ED-71 reduces LPS-induced cell death in HGFs. Part of cells were only treated by ED-71 (0, 0.5, 5 and 50 nM) for 24 h. Another part of cells were pretreated by ED-71 (0, 0.5, 5 and 50 nM) for 24 h, followed by 5 μg/mL LPS for 6 h. (A) The chemical formula of ED-71. (B) Cell viability were detected by CCK8. (C) The LDH released in HGFs supernatant were detected by LDH assay kit. Data were demonstrated as mean ± SEM from three experiments independently. ***P < 0.001 compared with control group. ##P < 0.01, #P < 0.05 compared with the group only treated by 5 μg/mL LPS. |
ED-71 Suppressed LPS-Induced NLRP3 Inflammasome-Dependent Pyroptosis in HGFs
To assess the anti-pyroptotic ability of ED-71, cells were pretreated with 0, 0.5, 5, or 50 nM ED-71 for 24 h, then treated with 5 μg/mL LPS for 6 h. RNA was collected and analyzed by qPCR. The results show that NLRP3, caspase-1, and IL-1β showed a dose-dependent decrease with increasing concentration of ED-71 (Figure 3C). All proteins were collected and analyzed by Western blots. TLR4, NLRP3, caspase-1 p20, ASC, and GSDMD-N (Figure 3A and B) were all found to decrease in a dose-dependent manner as compared with the group treated with LPS alone. ED-71 pretreatment reduced the release of IL-1β and IL-18 to normal levels (Figure 3A and B). Moreover, ED-71 inhibited the production of IL-6, IL8 (Figure 3D and E). Intercellular ROS and H2O2 were decreased by ED-71 (Figure 3F and G). In addition, flow cytometry analyses of annexin V and propidium iodide staining showed that double-positive cells increased significantly after LPS treatment, whereas ED-71 pretreatment reduced the double-positive rate (Figure 3H). Combined with LDH release (Figure 2C), the nature of cell death was confirmed as pyroptosis. These results indicate that ED-71 can ameliorate the pyroptosis and inflammation caused by LPS.
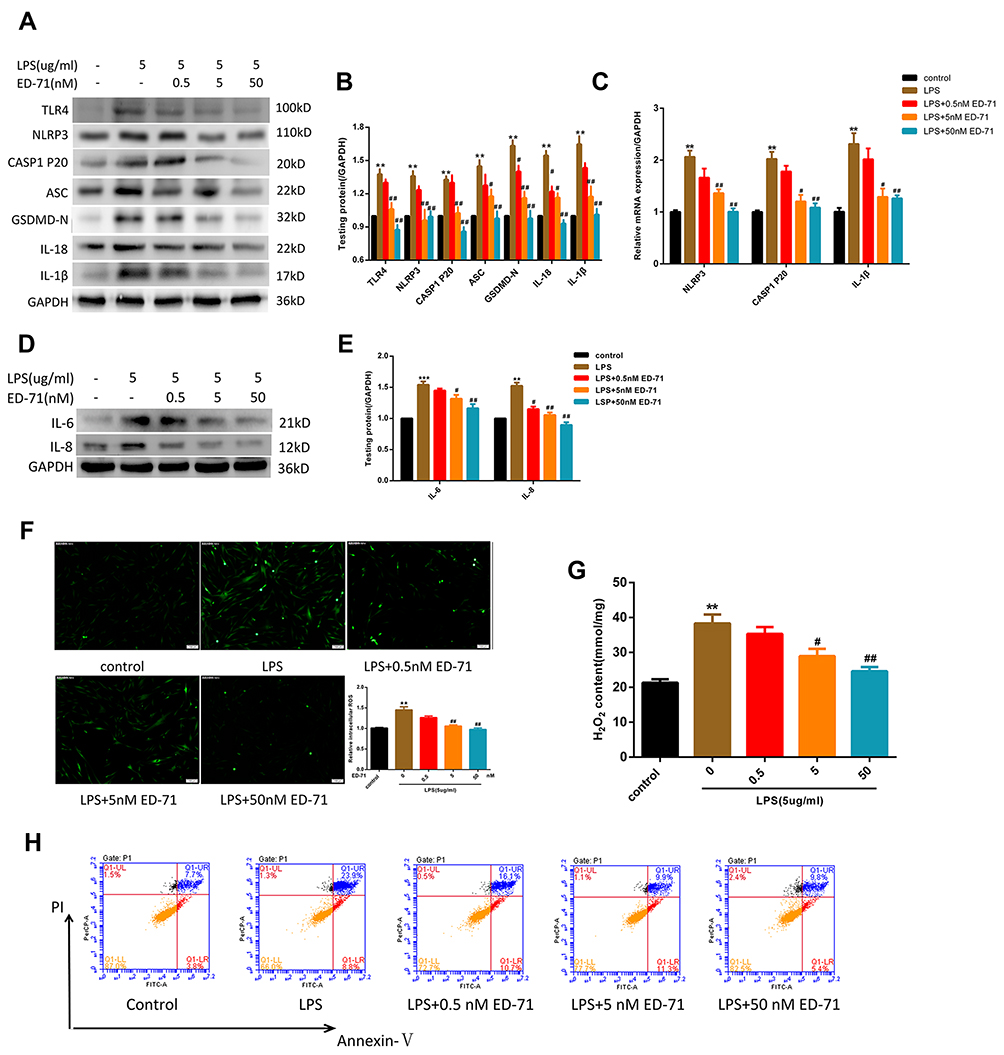 |
Figure 3 ED-71 suppressed LPS-induced NLRP3 inflammasome-dependent pyroptosis in HGFs. (A and B) The protein content in HGFs treated with various concentration ED-71 or 5 μg/mL LPS for 6 h. (C) Relative mRNA expression was shown. (D and E) The expression of IL-6 and IL-8 were evaluated by Western-blot. (F) Relative ROS and (G) H2O2 content in HGFs treated with various concentration ED-71 for 24 h or 5 μg/mL LPS for 6 h. (H) Flow cytometry of propidium iodide and annexin V fluorescein isothiocyanate (FITC)-stained cells. ***P < 0.001 and **P < 0.01 compared with control group. ##P < 0.01 and #P < 0.05 compared with the group only treated by 5 μg/mL LPS. |
ED-71-Activated Nrf2/HO-1 Suppressed by LPS in HGFs
As an essential factor for the response of cells to external stimuli, Nrf2 participates in LPS-induced cellular response. At the transcription level, after treatment with LPS up to a concentration of 5 μg/mL, Nrf2 and its effector molecule HO-1 were found to be significantly down-regulated (Figure 4C). In addition, the Nrf2 entering the nucleus was significantly reduced, and the protein expression of its effector molecule HO-1 was also significantly reduced (Figure 4A and B). At this time, pretreatment with ED-71 caused increased transcription of Nrf2 and HO-1 (Figure 4F), and simultaneously increased nuclear translocation of Nrf2 and expression of HO-1 (Figure 4D and E).
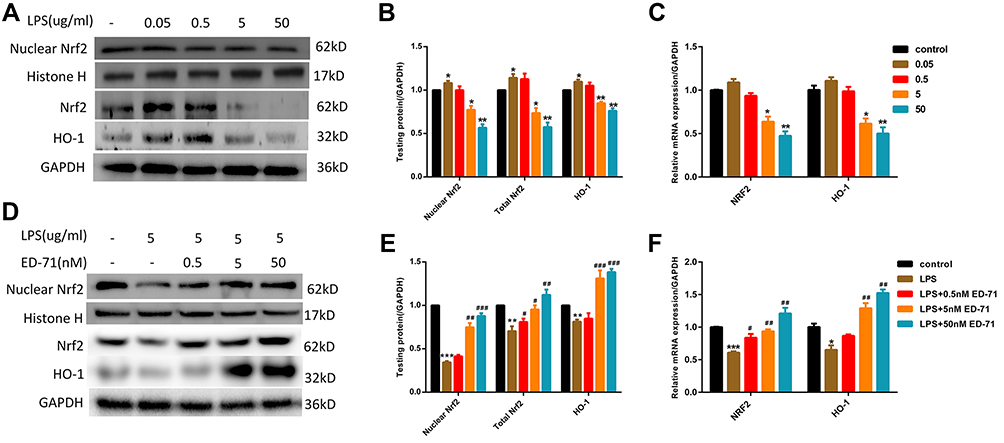 |
Figure 4 ED-71 activated Nrf2/HO-1 suppressed by LPS. (A and B) The protein content and (C) Relative mRNA expression in HGFs treated only by various concentration LPS. (D and E) The protein content and (F) Relative mRNA expression in HGFs treated with various concentration ED-71 for 24 h or 5 μg/mL LPS for 6 h. Values were exhibited as mean ± SEM from three experiments independently. ***P < 0.001, **P < 0.01 and *P < 0.05 compared with control group. ###P < 0.001, ##P < 0.01 and #P < 0.05 compared with the group only treated by 5 μg/mL LPS. |
The Nrf2 Signaling Pathway Plays a Role in the ED-71-Mediated Amelioration of Pyroptosis in HGFs
To further confirm that the anti-pyroptotic effect of ED-71 was caused by Nrf2 activation, the Nrf2 inhibitor ML385 was added. ML385 interacts with Nrf2 and affects the DNA binding activity of its protein complex, inhibiting the expression of its downstream target gene.26 Cells were first treated with 5 μM ML385 followed by 5 nM ED-71 for 24 h, then exposed to 5 μg/mL LPS. The effects on cell viability and LDH release are presented in Figure 5A and B. Compared with the LPS-only group, ED-71 reduced the pyroptosis-associated protein and activated the Nrf2/HO-1 pathway, as shown with Western blots (Figure 5C–H), consistent with the conclusion reached from the previous results. After blocking Nrf2, TLR4 increased, which led cells to respond more negatively to LPS (Figure 5C and D). The anti-pyroptotic ability of ED-71 decreased sharply (Figure 5F and G), and the number of IL-6, IL-8 increased rapidly (Figure 5I and J). The transcriptional results were consistent with the Western-blot (Figure 5E and H). Moreover, in cells in which Nrf2 was inhibited, ED-71 did not reduce intracellular ROS and H2O2 (Figure 5K and L). In this case, cells are under high oxidative stress, and caspase-1 can successfully cleave GSDMD and induce the release of IL-1β and IL-18, further resulting in the swelling and lysis of the cell. In addition, annexin V and PI double-staining were strongly positive (Figure 5M), indicating that the number of pyroptotic cells increased after Nrf2 was blocked. In summary, without Nrf2 activation, ED-71 could not fully protect cells from LPS.
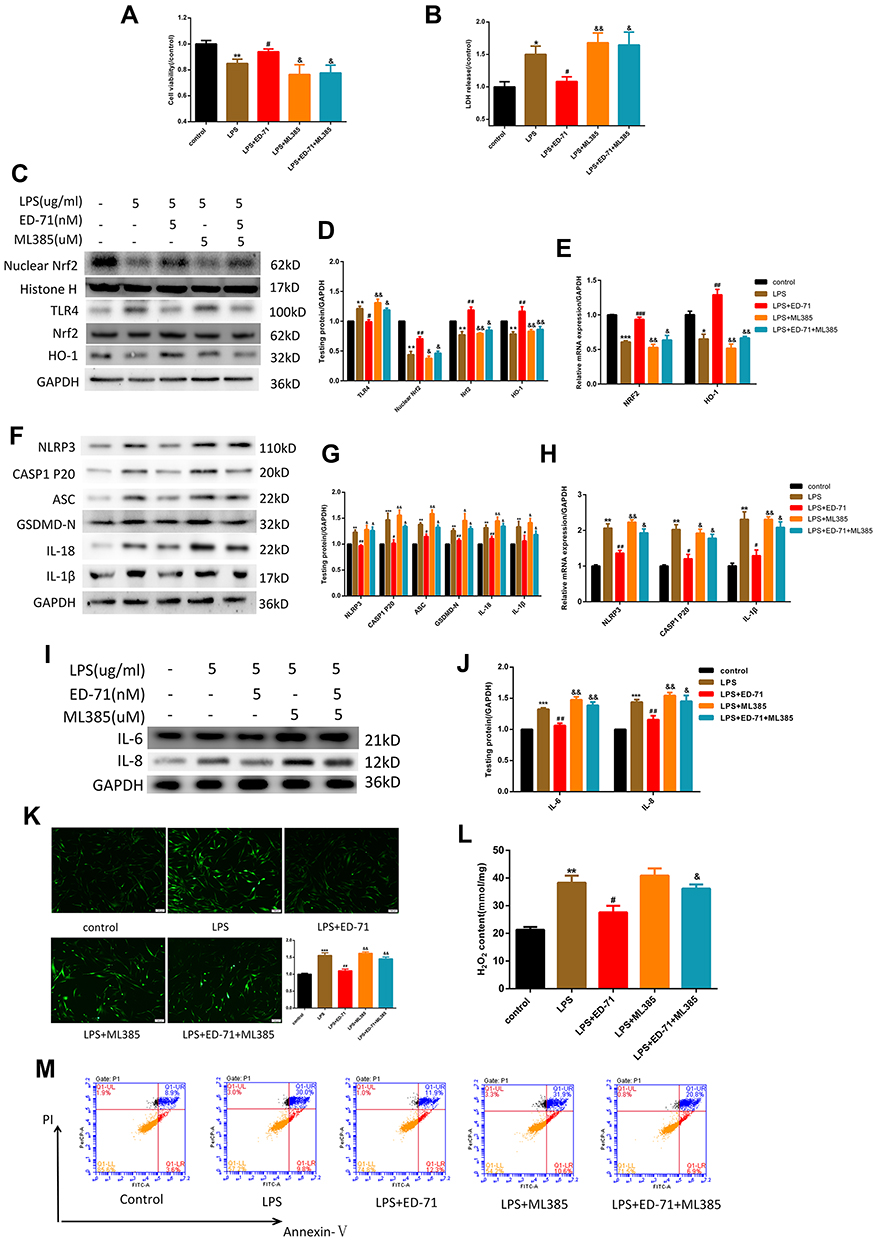 |
Figure 5 The anti-pyroptotic ability of ED-71 was Nrf2/HO-1-dependent. After treating by 5 μM ML385 for 3 h, cells were the treated by 5 nM ED-71 for 24 h and then incubated with 5 μg/mL LPS for 6 h. (A) Cell viability and (B) LDH release were detected by CCK8 and LDH assay kit respectively. (C and D) The relative proteins expression of TLR4, Nrf2, HO-1. (E) Relative mRNA expression in HGFs. (F and G) pyroptosis- associated proteins were detected by Western-blot. (H) Relative mRNA expression was shown. (I and J) The expression of IL-6 and IL-8. (K and L) The intercellular ROS and H2O2 content in HGFs. (M) Flow cytometry of propidium iodide and annexin V (FITC)-stained HGFs. The data collected from three independent experiments were presented as mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05 compared with control group. ###P < 0.001, ##P < 0.01 and #P < 0.05 compared with the group only treated by 5 μg/mL LPS. &&P < 0.01 and &P < 0.05 compared with 5 nM ED-71 treated and then stimulated by 5 μg/mL LPS group. |
Discussion
This study is the first to provide evidence that ED-71 can ameliorate pyroptosis via the activation of the Nrf2/HO-1 signaling pathway on HGFs, and that pyroptosis is induced via the activation of the NLRP3 inflammasome stimulated by LPS. Because pyroptosis is known to be closely associated with periodontitis, ED-71 may provide a new treatment for periodontitis, and its relevant mechanism has been preliminarily revealed.
Periodontitis is a serious oral disease characterized by the inflammation of the gingival tissue caused by infection; it results in the loss of connective tissue and bone support and is a major cause of tooth loss in adults.27 Because HGFs are an important component of gingival connective tissue, their inflammation contributes to the pathogenesis of periodontitis.4 Recent studies have suggested that innate immunity plays a crucial role in the development of periodontitis, and innate immunity is characterized by pyroptosis.5,6 Inflammasomes are important sensors/receptors in innate immunity and participate in the response to inflammatory diseases.28 Recent research has shown that NLRP3 plays an essential role in the progression of periodontitis and promotes the destruction of periodontal tissue.29 The overall intensity of NLRP3 expression is notably higher in gingival tissues with periodontitis than in healthy tissues.30 In addition, it has been found that blocking NLRP3 inhibits inflammation and the production of inflammatory cytokines in mouse periodontal ligament fibroblasts.12 Based on these results, it was posited that the activation of the NLRP3 inflammasome may trigger innate immunity and thus lead to the occurrence and development of periodontitis in vivo.
In this study, it is shown that the activation of the NLRP3 inflammasome in HGFs was strongly induced by LPS and further resulted in cell death that was subsequently identified as pyroptosis with the resulting release of IL-1β and IL-18 in a dose-dependent manner. It was also shown that pyroptosis could be reduced by MCC950, indicating that pyroptosis in HGF is a result of LPS-induced excessive activation of the NLRP3 inflammasome. Moreover, it was revealed that the expression of the marker proteins IL-6 and IL-8 in periodontitis was significantly increased in LPS-induced HGFs in a dose-dependent manner. Previous research indicated that the IL-18 release can induce the release of MMP-9, which causes tissue degeneration.31 IL-1β can further mediate the inflammatory activation in HGFs.32 IL-6 causes bone resorption,33 and IL-8 increases the number of local neutrophils via chemotaxis, further destroying the periodontal ligament and aggravating the progression of periodontitis.34 Pivotally, the release of pro-inflammatory factors (IL-1β, IL-8) in periodontal tissue will activate osteoclasts and eventually lead to alveolar bone resorption. The latter is a key indicator to distinguish periodontitis from gingivitis.35,36
Porphyromonas gingivalis has been confirmed to be the principal pathogen in periodontitis. Though P. gingivalis LPS is more relevant to periodontitis, it is not usually present. Moreover, the ligands of P. gingivalis LPS are TLR2 and TLR4; this makes the present data hard to interpret, as TLR4 is the study’s main target.37 An ultraportable preparation of E. coli LPS was used in this study, which is an identified TLR4 agonist as has been indicated in the previous research. LPS was presented with CD14, interacted with TLR4, and was then transported into the cells by TLR4 to cause a series of responses in the cell.38 One of the most important events is the generation of ROS, which is a vital element for NLRP3 inflammasome activation.16 The crystal structure of NLRP3 includes a highly conserved disulfide bond connecting the PYD domain and the nucleotide-binding site domain, which is very sensitive to altered redox states.39 In the present research, adding LPS to gingival cells led to a significant increase in mitochondrial respiration, but it led to the production and accumulation of a large amount of H2O2 rather than ATP. This change marks a fundamental change in the homeostasis of the cell’s mitochondria and will cause abnormal cell function. Therefore, the determination of H2O2 content is of great significance for further revealing whether cells are in a state of oxidative stress.40 Based on these studies, our study shows that LPS can interact with TLR4 and then induce the accumulation of total intracellular ROS, H2O2, and the NLRP3 inflammasome relative proteins and caspase-1 p20 in a dose-dependent manner, and further that the ROS inhibitor NAC can inhibit the activation of NLRP3 inflammasomes. These experimental data fully verify that the generation of oxidative stress is an important change in human gingival fibroblasts induced by LPS while further demonstrating that its consequence is pyroptosis. In summary, the results indicate that LPS was successfully recognized by TLR4 and activated the NLRP3 inflammasome in HGFs, and that the activation of the NLRP3 inflammasome was caused by ROS.
ED-71, a new active vitamin D analog, has been used for the treatment of osteoporosis in Japan. Research has found that 1α,25-dihydroxyvitamin D3 has a good therapeutic effect on various inflammatory diseases, such as rheumatoid arthritis and multiple sclerosis.41 The results of the present study indicate that pretreatment with ED-71 can effectively reduce the accumulation of ROS, NLRP3 inflammasome relative protein, and subsequent pyroptosis in LPS-induced HGFs in a dose-dependent manner. RT-PCR results showed a reduced transcription level of NLRP3. In addition, it reduced caspase-1 p20, which is the functional component of the activated NLRP3 inflammasome. Moreover, it reduces the secretion of inflammatory cytokines IL-1β, IL-18, IL-6, and IL-8 in a dose-dependent manner. Considering that the NLRP3 inflammasome and subsequent pyroptosis are of great significance for the course of periodontitis, and in the context of the experimental results, it is likely that ED-71 has a therapeutic effect on periodontitis. Moreover, 25-hydroxyvitamin D3 has been found to attenuate experimental periodontitis via the downregulation of TLR4,42 The results of the present study indicate that LPS-induced TLR4 was also inhibited by ED-71 in a dose-dependent manner, which may be an additional therapeutic benefit of ED-71. We hypothesize that the downregulation of TLR4 can reduce the susceptibility of HGFs to LPS, which may lead to the weakening of the connection between TLR4 and LPS to induced periodontitis; however, these results have not been expanded upon in this work, and the related mechanism requires further research.
The previous study shows that the Nrf2 signaling pathway is an important antioxidant response pathway, and it has been proven to be activated by vitamin D to reduce the inflammation reaction.23 The binding of active vitamin D to VDR, interaction with retinoid X receptor (RXR), and then binding to the vitamin D response element (VDRE) can activate many vitamin D-sensitive target genes, including Nrf2.43 Chen et al found that the VDR has the ability to physically bind the Nrf2 promoter using a ChIP approach.44 The activated Nrf2 is transported to the nucleus, activates the antioxidant response element (ARE), and increases the transcription of Nrf2 regulatory genes (such as HO-1, GST, NQO-1). Heme oxygenase 1 (HO-1) is to a degree imitating Nrf2; these proteins, in addition to removing toxic heme, also produce biliverdin, iron ions, and carbon monoxide. HO-1 and its products induce anti-oxidative damage and regulate inflammation.45 A previous observation showing association of Nrf2 with negative regulation of inflammasome revealed that Nrf2 induces the NQO1 expression that leads to the inhibition of NLRP3 inflammasome activation, caspase-1 cleavage, and IL-1β generation in macrophages. Furthermore, a well-known Nrf2 activator, tert-butylhydroquinone (tBHQ), negatively regulated NLRP3 transcription by activating the ARE in an Nrf2-dependent manner.46 In the present study, nuclear Nrf2 can be activated to promote cell survival after exposure to low treatment concentration with a certain time by LPS, which may be a self-protection mechanism in cells. However, nuclear Nrf2 was found to be notably inhibited by a high concentration (5 μg/mL) of LPS, resulting in the loss of self-protection and thus promoting the subsequent pyroptosis in HGFs. ED-71 pretreatment can effectively restore the inhibitory effect of LPS on Nrf2/HO-1 pathway and regulate the transcription of NLRP3. In addition, ED-71 inhibited LPS-induced ROS, NLRP3 inflammasome relative protein, caspase-1 p20, and subsequent pyroptosis. Furthermore, the inhibitory effect of ED-71 on the NLRP3 inflammasome and subsequent pyroptosis was eliminated with the Nrf2 inhibitor ML385. These results indicated that the therapeutic effect of ED-71 is achieved by activating the Nrf2/HO-1 pathway.
In conclusion, our study clarifies the possible protective effect of ED-71 on LPS-induced pyroptosis in HGFs via the Nrf2/HO-1 signaling pathway. The results indicate that the LPS-induced activation of the NLRP3 inflammasome and eventual pyroptosis on HGFs, which can be weakened by NAC or MCC950. ED-71 reduced LPS-induced ROS levels, the activation of the NLRP3 inflammasome, and eventual pyroptosis by activating the Nrf2/HO-1 pathway. In addition, Nrf2 inhibition by ML385 was shown to reduce the inhibitory effect of ED-71 on the NLRP3 inflammasome and subsequent pyroptosis. The possible mechanism of action of ED-71 is shown in Figure 6. The present study validates the anti-periodontitis effects of ED-71 and is a new possible clinical treatment of periodontitis.
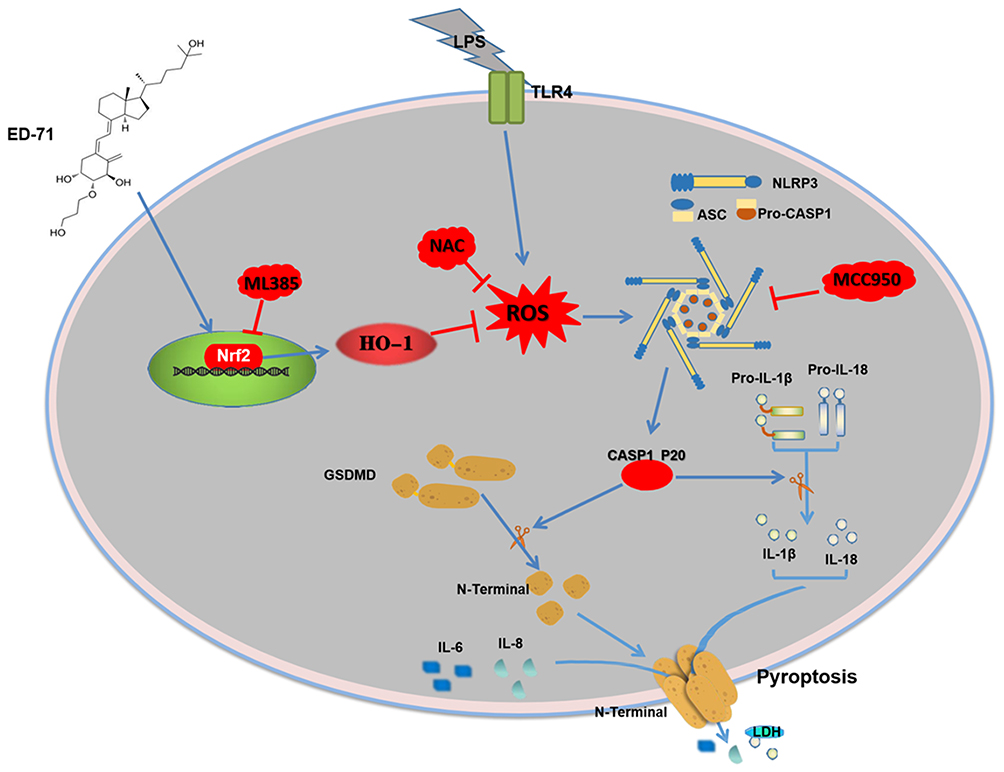 |
Figure 6 Mechanisms of ED-71 inhibiting NLRP3 inflammasome-dependent pyroptosis in HGFs. |
Acknowledgments
This study was supported by the National Nature Science Foundation of China (No. 81972072) and the Shandong Key Research and Development Project (No. 2018GSF118134) to Li M, and the National Nature Science Foundation of China (Nos. 81771108, 81970964) to Guo J.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. Conceptualization, C.H.; data curation, C.H. and C.Z.; formal analysis, C.H. and C.Z.; funding acquisition, J.G. and M.L.; investigation, P.Y., R.C., Z.Y., C.L. J.G. and M.L.; methodology, P.Y., R.C., Z.Y., C.L. J.G. and M.L.; project administration, J.G. and M.L.; resources, J.G. and M.L.; Software, R.C., Z.Y., C.L. and P.Y.; supervision, J.G. and M.L.; validation, C.H. and M.L.; visualization, C.H. and C.Z.; writing – original draft, C.H.; writing – review & editing, C.H.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44.
2. Binderman I, Gadban N, Yaffe A. Extracellular ATP is a key modulator of alveolar bone loss in periodontitis. Arch Oral Biol. 2017;81:131–135. doi:10.1016/j.archoralbio.2017.05.002
3. Tanabe S, Bodet C, Grenier D. Treponema denticola lipooligosaccharide activates gingival fibroblasts and upregulates inflammatory mediator production. J Cell Physiol. 2008;216:727–731. doi:10.1002/jcp.21447
4. Naruishi K, Nagata T. Biological effects of interleukin-6 on Gingival Fibroblasts: cytokine regulation in periodontitis. J Cell Physiol. 2018;233:6393–6400. doi:10.1002/jcp.26521
5. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–142. doi:10.1111/imr.12287
6. Meyle J, Dommisch H, Groeger S, Giacaman RA, Costalonga M, Herzberg M. The innate host response in caries and periodontitis. J Clin Periodontol. 2017;44:1215–1225. doi:10.1111/jcpe.12781
7. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A. 1999;96:2396–2401. doi:10.1073/pnas.96.5.2396
8. Cheng R, Feng Y, Zhang R, Liu W, Lei L, Hu T. The extent of pyroptosis varies in different stages of apical periodontitis. Biochimica Et Biophysica Acta Mol Basis Dis. 2018;1864:226–237. doi:10.1016/j.bbadis.2017.10.025
9. Xue F, Shu R, Xie Y. The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT-PCR study and immunohistochemistry. Arch Oral Biol. 2015;60:948–958. doi:10.1016/j.archoralbio.2015.03.005
10. Herath TD, Darveau RP, Seneviratne CJ, Wang CY, Wang Y, Jin L. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One. 2013;8:e58496.
11. Kim DY, Jun J-H, Lee H-L. N-acetylcysteine prevents LPS-induced pro-inflammatory cytokines and MMP2 production in gingival fibroblasts. Arch Pharm Res. 2007;30:1283–1292. doi:10.1007/BF02980269
12. Lian D, Dai L, Xie Z. Periodontal ligament fibroblasts migration injury via ROS/TXNIP/Nlrp3 inflammasome pathway with Porphyromonas gingivalis lipopolysaccharide. Mol Immunol. 2018;103:209–219. doi:10.1016/j.molimm.2018.10.001
13. García‐Hernández AL, Muñoz‐Saavedra ÁE, González‐Alva P, et al. Upregulation of proteins of the NLRP3 inflammasome in patients with periodontitis and uncontrolled type 2 diabetes. Oral Dis. 2019;25:596–608.
14. Li H, Zhong X, Li W, Wang Q. Effects of 1,25-dihydroxyvitamin D3 on experimental periodontitis and AhR/NF-κB/NLRP3 inflammasome pathway in a mouse model. J Appl Oral Sci. 2019;27:e20180713. doi:10.1590/1678-7757-2018-0713
15. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi:10.1074/jbc.R900010200
16. Hu Q, Zhang T, Yi L, Zhou X, Mi M. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. BioFactors (Oxford, England). 2018;44:123–136. doi:10.1002/biof.1395
17. Yan Z, Qi W, Zhan J, et al. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J Cell Mol Med. 2020. doi:10.1111/jcmm.15905
18. Gao Z, Sui J, Fan R, Qu W, Dong X, Sun D. Emodin protects against acute pancreatitis-associated lung injury by inhibiting NLPR3 inflammasome activation via Nrf2/HO-1 signaling. Drug Des Devel Ther. 2020;14:1971–1982. doi:10.2147/DDDT.S247103
19. Qi F, Sun J-H, Yan J-Q, Li C-M, Lv X-C. Anti-inflammatory effects of isorhamnetin on LPS-stimulated human gingival fibroblasts by activating Nrf2 signaling pathway. Microb Pathog. 2018;120:37–41. doi:10.1016/j.micpath.2018.04.049
20. Reynolds MA. Modifiable risk factors in periodontitis: at the intersection of aging and disease. Periodontol 2000. 2014;64:7–19. doi:10.1111/prd.12047
21. Kondo S, Takano T, Ono Y, Saito H, Matsumoto T. Eldecalcitol reduces osteoporotic fractures by unique mechanisms. J Steroid Biochem Mol Biol. 2015;148:232–238. doi:10.1016/j.jsbmb.2015.01.016
22. Barbalho SM, Goulart RDA, Gasparini RG. Associations between inflammatory bowel diseases and vitamin D. Crit Rev Food Sci Nutr. 2019;59:1347–1356. doi:10.1080/10408398.2017.1406333
23. Abo El-Magd NF, Eraky SM. The molecular mechanism underlining the preventive effect of vitamin D against hepatic and renal acute toxicity through the NrF2/BACH1/HO-1 pathway. Life Sci. 2020;244:117331. doi:10.1016/j.lfs.2020.117331
24. Bletsa A, Abdalla H, Løes S, Berggreen E. Lymphatic growth factors are expressed in human gingiva and upregulated in gingival fibroblasts after stimulation. J Periodontol. 2018;89:606–615. doi:10.1002/JPER.17-0400
25. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi:10.1038/nature22393
26. Singh A, Venkannagari S, Oh KH, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214–3225.
27. Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet (London, England). 2005;366:1809–1820. doi:10.1016/S0140-6736(05)67728-8
28. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687.
29. Olsen I, Yilmaz Ö. Modulation of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J Oral Microbiol. 2016;8:30385. doi:10.3402/jom.v8.30385
30. Aral K, Berdeli E, Cooper PR, et al. Differential expression of inflammasome regulatory transcripts in periodontal disease. J Periodontol. 2019.
31. Jablonska E, Jablonski J. Effect of IL-18 on the release of IL-6 and its soluble receptors: sIL-6R α and sgp130 by human neutrophils. Immunol Invest. 2002;31:159–167. doi:10.1081/IMM-120016237
32. Arancibia R, Maturana C, Silva D. Effects of chitosan particles in periodontal pathogens and gingival fibroblasts. J Dent Res. 2013;92:740–745. doi:10.1177/0022034513494816
33. Feng W, Liu B, Liu D. Long-term administration of high-fat diet corrects abnormal bone remodeling in the tibiae of interleukin-6-deficient mice. J Histochem Cytochem. 2016;64:42–53. doi:10.1369/0022155415611931
34. Bickel M. The role of interleukin-8 in inflammation and mechanisms of regulation. J Periodontol. 1993;64:456–460.
35. Cochran D. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79:1569–1576. doi:10.1902/jop.2008.080233
36. Graves D, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391–401. doi:10.1902/jop.2003.74.3.391
37. Li JP, Li FY, Xu A, et al. Lipopolysaccharide and hypoxia-induced HIF-1 activation in human gingival fibroblasts. J Periodontol. 2012;83:816–824.
38. Tsukamoto H, Takeuchi S, Kubota K. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKKϵ-IRF3 axis activation. J Biol Chem. 2018;293:10186–10201. doi:10.1074/jbc.M117.796631
39. Bae J, Park H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J Biol Chem. 2011;286:39528–39536. doi:10.1074/jbc.M111.278812
40. Napa K, Baeder A, Witt J, et al. P. gingivalisLPS from negatively alters gingival cell mitochondrial bioenergetics. Int J Dent. 2017;2017:2697210. doi:10.1155/2017/2697210
41. Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010;9:941–955.
42. Wang Q, Li H, Xie H. 25-Hydroxyvitamin D3 attenuates experimental periodontitis through downregulation of TLR4 and JAK1/STAT3 signaling in diabetic mice. J Steroid Biochem Mol Biol. 2013;135:43–50. doi:10.1016/j.jsbmb.2013.01.008
43. Berridge MJ. Vitamin D: a custodian of cell signalling stability in health and disease. Biochem Soc Trans. 2015;43:349–358.
44. Chen L, Yang R, Qiao W, et al. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell. 2019;18:e12951. doi:10.1111/acel.12951
45. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73:3221–3247.
46. Liu X, Zhang X, Ding Y, et al. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid Redox Signal. 2017;26:28–43. doi:10.1089/ars.2015.6615
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.