")
Back to Journals » OncoTargets and Therapy » Volume 12
EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC
Authors Meng J, Chang C, Chen Y, Bi F, Ji C, Liu W
Received 25 March 2019
Accepted for publication 26 June 2019
Published 26 July 2019 Volume 2019:12 Pages 6033—6043
DOI https://doi.org/10.2147/OTT.S209441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Jiao Meng,1,2,* Cuicui Chang,1,* Yuhua Chen,1,2 Fangfang Bi,1,2 Chen Ji,1 Wei Liu2
1Central Laboratory of Medicine School, Xi’an Peihua University, Xi’an 710100, People’s Republic of China; 2Department of Medical Science Research Center, Shaanxi Fourth People’s Hospital, Xi’an 710143, People’s Republic of China
*These authors contributed equally to this work
Background: Several EGFR-tyrosine kinase inhibitors (TKIs), such as gefitinib (Gef), have been used as effective clinical therapies for patients with non-small cell lung cancer (NSCLC). However, due to acquired resistance, the efficacy of Gef treatment is severely blocked. Our preliminary study found that epigallocatechin gallate (EGCG) in combination with Gef could work synergistically to increase the sensitivity to Gef in NSCLC, but the mechanisms responsible for this have not been completely defined.
Purpose: In our present study, we devoted to investigate the synergistic effects of combined EGCG and Gef treatment and the importance of autophagy and ERK signaling pathway in overcoming acquired drug resistance to Gef in NSCLC.
Methods: We evaluated the synergistic effects of combined EGCG and Gef treatment through in vitro cell proliferation/viability assays and in vivo xenograft studies, respectively. Autophagic flux was assessed by GFP-microtubule-associated protein 1 light chain 3 (LC3) plasmid transfection and western blot detection of autophagy-related proteins. Besides, the role of ERK on acquired resistance was validated with a ERK inhibitor.
Results: We discovered that EGCG can synergize with Gef to inhibit the proliferation of Gef-resistant NSCLC cells and suppress tumor growth in a xenograft mouse model. The underlying mechanisms of synergism were investigated, and the results showed that co-treatment with Gef and EGCG could inhibit Gef-induced autophagy and ERK phosphorylation. Consistently, the expression of LC3-II/I and ATG5 were inhibited, whereas the expression of p62 was enhanced in EGCG and Gef combination treatment groups. Further, inhibition of autophagy in Gef-resistant A549 cells could augment cell death.
Conclusion: In conclusion, EGCG overcomes Gef resistance by inhibiting autophagy and augmenting cell death through targeting ERK pathway in NSCLC. Gef and EGCG combination therapy may be an effective strategy to overcome acquired resistance in NSCLC.
Keywords: EGCG, gefitinib resistance, NSCLC, autophagy, ERK
Introduction
Lung cancer is one of the most common cancers in the world, causing a serious social burden.1,2 According to the statistics, the prevalence of non-small cell lung cancer (NSCLC) is over 80% in all lung cancers.3 It is well known that the EGFR plays a key role in NSCLC; EGFR-tyrosine kinase inhibitors (TKIs) such as gefitinib (Gef) have significantly improved the treatment outcome of NSCLC patients.4 Although treatment of TKIs has achieved some initial results, the tumor eventually relapses because of the emergence of acquired resistance to the TKIs because of EGFR mutation.5 In half of the patients, the reason for acquired resistance can be attributed to secondary EGFR mutations, mainly T790M point mutation.6
Though some genetic mutations severely influence the treatment of TKIs, metabolic alteration is also one of the causes of drug resistance. Researches have demonstrated that Gef-mediated ROS triggers drug resistance and mitochondrial dysfunction in lung cancer cells,7,8 and antioxidants may alleviate ROS-mediated resistance.7 Zhao et al discover that Gef can enhance the expression of microtubule-associated protein 1 light chain 3 (LC3), and the acquired drug resistance may be associated with autophagy.9 Chitosan nanoparticles entrapping Gef and chloroquine (CQ), an inhibitor of autophagolysosomes formation, can help to overcome Gef-acquired resistance and improve the efficacy of cancer treatment.9 Lv et al demonstrat that Honokiol shows enhanced antitumor effects through inhibiting autophagy and inducing cell death in NSCLC when delivered with CQ.10 Besides, increasing evidence indicate that autophagy is a conserved catabolic process activated under oxidative stress conditions.11 Therefore, we speculate that the inhibition of autophagy might be a potential mechanism to overcome acquired resistance.
In addition to autophagy, the ERK signaling pathway also has a critical role in cell proliferation, tumorigenesis, cell cycle arrest and drug-acquired resistance.12 ERK is activated via a pathway that involves GTP loading of Ras and the sequential phosphorylation and activation of Raf, MEKs and ERK.13 ERK activation is also found in various cancer cells which are resistant to EGFR or MEK kinase inhibitors.14,15 Abrams et al indicate that targeting the Raf/MEK/ERK pathway may be an effective approach to govern drug resistance.16 Therefore, there has been an increasing number of research interested in identifying novel therapies to target this signaling pathway.
Epigallocatechin gallate (EGCG) is a polyphenol natural product derived from green tea (Camellia sinensis). A large number of evidence indicate that EGCG exhibits a wide range of biological activities, including antioxidant, antimutagenic and anticancer activities.17–20 Studies have shown that EGCG affects multiple signaling pathways through binding to various molecular targets, including transmembrane receptors, kinases and other key proteins, and ultimately leading to apoptosis, proliferation inhibition and inhibition of invasion, angiogenesis and metastasis.21 Besides, Xiaokaiti et al suggeste that EGCG ameliorates the neutrophil elastase-induced migration of NSCLC cells.22 Then, we speculated that EGCG, an antioxidant polyphenol, may alleviate drug resistance to some extent.
Our preexperiment found that EGCG in combination with Gef could work synergistically to increase the sensitivity to Gef in NSCLC, but the mechanisms responsible for this have not been completely defined. In the present study, we investigated the synergistic effects of combined EGCG and Gef treatment and the importance of autophagy and ERK signaling pathway in overcoming acquired drug resistance to Gef in NSCLC.
Materials and methods
Reagents
Gef (s1025), EGCG (s2250) and SCH772984 (s7101) were purchased from Selleck Ltd. (Shanghai, China). CQ (c6628) and rapamycin (Rap) (553211) were from Sigma-Aldrich (St. Louis, MO, USA). The drugs were diluted in DMEM (Hyclone, Logan, UT, USA) to achieve the final desired concentration. Antibodies against ERK1/2 (5376), p-ERK1/2 (Thr202/Tyr204) (4370), MEK1/2 (4694), p-MEK1/2 (Ser217/221) (3958), LC3 (12741), ATG5 (12994), ATG12 (2011), p62 (5114) and ki67 (12075) were from Cell Signaling Technology Ltd. (Danvers, MA, USA).
Cell lines and culture
A549 cells and the human bronchial epithelial cell line 16HBE were bought from Chinese Academy of Medical Sciences Cancer Cell Bank (Beijing, China), A549 cells were maintained in DMEM containing 10% FBS (Gibco, Carlsbad, CA, USA), 100 μg/mL streptomycin (Gibco) and 100 μg/mL penicillin (Gibco) at 37°C with 5% CO2.
16HBE cells were maintained at 37°C in minimum essential medium (Gibco) supplemented with 10% (v/v) FBS (Gibco), 100 µg/mL penicillin (Gibco) and 100 µg/mL streptomycin (Gibco) in an incubator with 5% CO2.
Cell proliferation/viability assays
Cell viability was examined with a CellTiter96 Aqueous one solution Cell proliferation assay (MTS assay). Cells were seeded in 96-well tissue culture plates and treated with various concentrations of Gef, EGCG and combined treatment (Gef+EGCG) for 48 hrs. After treatment, 10 μL MTS assay solution was added to each well and incubated at 37°C for 3 hrs. Since MTS is light-sensitive, the procedure was performed in dark environment. Then, the optical density of each well was measured at 570 nm with a microplate reader (Molecular Device, Sunnyvale, CA, USA).
IC50 for each drug were calculated using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). The effects of different drug combinations were determined using CompuSyn software (Biosoft, Ferguson, MO, USA), which was based on the median effect model of Chou Talalay method.23 The combination index (CI) was calculated using the formula: CI = (D)1/(Dx)1+ (D)2/(Dx)2, where, Dx indicates the dose of one compound alone required to produce an effect, and (D)1 and (D)2 are the doses of compounds 1 and 2, respectively, necessary to produce the same effect in combination. The combined effects of two compounds can be interpreted as follows: CI <1, CI =1 and CI >1 indicate synergistic, additive and antagonistic effects, respectively.
TUNEL staining
TUNEL staining was performed using a TUNEL Apoptosis Assay Kit (T2190, Solarbio, Beijing, China). Apoptotic cell nuclei were stained with fluorescein-dUTP, and Hoechst (B8040, Solarbio) was used to stain all cell nuclei. The drug-treated cells were fixed with paraformaldehyde (PFA) for 15 mins, gently rinsed with PBS and mixed with Triton-X-100 (0.2%) (T8200, Solarbio) for 10 mins at room temperature. Add the newly prepared TUNEL assay solution and incubate at 37°C in the dark for 1 hr, observed using a fluorescence microscope.
GFP-LC3 plasmid transfection and confocal microscopy
For standard plasmid transfection, the cells were resuspended in serum and antibiotic-free DMEM at suitable densities (to achieve 30–50% density on the next day). Two hundred microliters of cell suspension were mixed with 5 μg GFP-LC3 plasmid (Hanheng, Shanghai, China), then transferred to a 0.4-cm electroporation cuvette (BioRad, Hercules, CA, USA), and electroporated using Gene Pulser Xcell™ electroporation system (BioRad).24 The pulse amplitude of electroporation varied between 80 and 120 V with a pulse duration of 20 ms/transfection. When the electroporation was over, the cells were incubated at room temperature for 5 mins and transferred to a 8-well LAB-Tek confocal CHAMBERCVG (LAB-Tek, 155411, Hatfield, PA, USA)
Forty-eight hours after transfection, cells were treated with 1.87 μL Gef, 34 μL EGCG and combined treatment (1.87 μL Gef +34 μL EGCG) for 48 hrs. Subsequently, cells were fixed with 4% PFA (P1110, Solarbio), permeabilized with Triton-X-100, and nuclei were stained with DAPI (C0065, Solarbio). Treated cells were then examined with a Leica TCS SP5 laser scanning confocal microscope equipped with a 40× objective lens.
Ki67 staining
The Ki67 cellular proliferation detection kit was obtained from Sangon Biotech Co., Ltd. (Shanghai, China). Briefly, cells were fixed with 4% PFA for 30 mins, gently rinsed with PBS for three times and then incubated with 0.2% Triton-X-100 for 10 mins. After being washed with PBS, the cells were incubated with the blocking buffer for 1 hr, followed by incubation with anti-ki67 rabbit antibody (CST, Danvers, MA, USA, 1:300) at 4°C overnight. Then, the cells were washed three times and incubated with secondary antibody at room temperature for 1 hr. After incubation with secondary antibodies, cells were counterstained with DAPI solution (C0065, Solarbio) and then analyzed with a fluorescence microscope.
Immunoblot analysis assay
Briefly, A549 cells were lysed, then protein concentrations were quantified using the BCA assay (23225, Thermo Scientific, Carlsbad, CA, USA). An equal amount of protein was subjected to SDS-PAGE and transferred to a 0.45-µm polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA, HATF09025). Then, the membranes were blocked with blocking buffer (5% nonfat dry milk and 0.05% Tween-20), followed by incubation with primary antibodies: p-ERK1/2 (CST, 1:1000), p-MEK1/2 (CST, 1:1000), LC3 (CST, 1:1000), ATG5 (CST, 1:1000), ATG12 (CST, 1:1000), p62 (CST, 1:1000) and anti-GAPDH (Abcam, Cambridge, MA, USA, 1:20,000) at 4°C overnight. Then, the cells were washed three times and incubated with secondary antibody at room temperature for 1 hr followed by triple 10-min wash in Tris-buffered saline and Tween 20. Bands were detected using an enhanced chemiluminescence system (BioRad). Data within a linear range were quantified using Quantity One software (BioRad).
Xenograft studies
All animal experiments were performed according to the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Fourth People’s Hospital of Shaanxi (China). BALB/C male nude mice weighing 18–20 g were purchased from the Experimental Animal Center of Xi’an Jiaotong University. The human lung A549 adenocarcinoma cells were cultivated in DMEM high-glucose medium supplemented with 10% FBS at 37°C with 5% CO2. A549 cells were trypsinized, centrifuged, washed in PBS twice and injected s.c. (1×106, 100 μL of PBS) into the left flanks of the 7–8-week-old mice (the animals were adapted to the lab environment for 7 days before experiments). When tumors grew to 100 mm3 in size, the animals were randomly divided into four groups (n=10 mice per group): treated with vehicle, Gef (10 mg/kg/day, p.o.), EGCG (200 mg/kg/day, p.o.) and Gef plus EGCG (10 mg/kg/day Gef +200 mg/kg/day EGCG, p.o.), respectively. The tumor volume was measured every 3 days, and the physical signs and weight of the animals were monitored as well. The tumor volume was determined according to the following formula: TV (mm3) =1/2×width2×length. To evaluate the effect of agents on the survival rate of A549 tumor-beard mice, we observed their effect on survival in a cycle of 60 days. The survival curves were performed with an n=10 for BALB/C nude mice and were analyzed using the Mantel–Cox test, which is used to test the null hypothesis that survival curves are not different between groups.
Statistical analysis
All data are expressed as the mean ± SEM, and statistical analysis was conducted with GraphPad Prism 5.0. Multiple comparisons among vehicle and treatment groups were performed using one-way ANOVA followed by Bonferroni correction. The in vivo study used two-way ANOVA statistic assay. A value of P<0.05 was considered statistically significant.
Results
EGCG and Gef synergized in inhibiting the proliferation of NSCLC cell
To exclude any cytotoxic effects of EGCG on normal lung cells, we treated 16HBE cells with different concentrations of EGCG for 48 hrs. The results showed that EGCG below 80 μM had no cytotoxicity (Figure 1A).
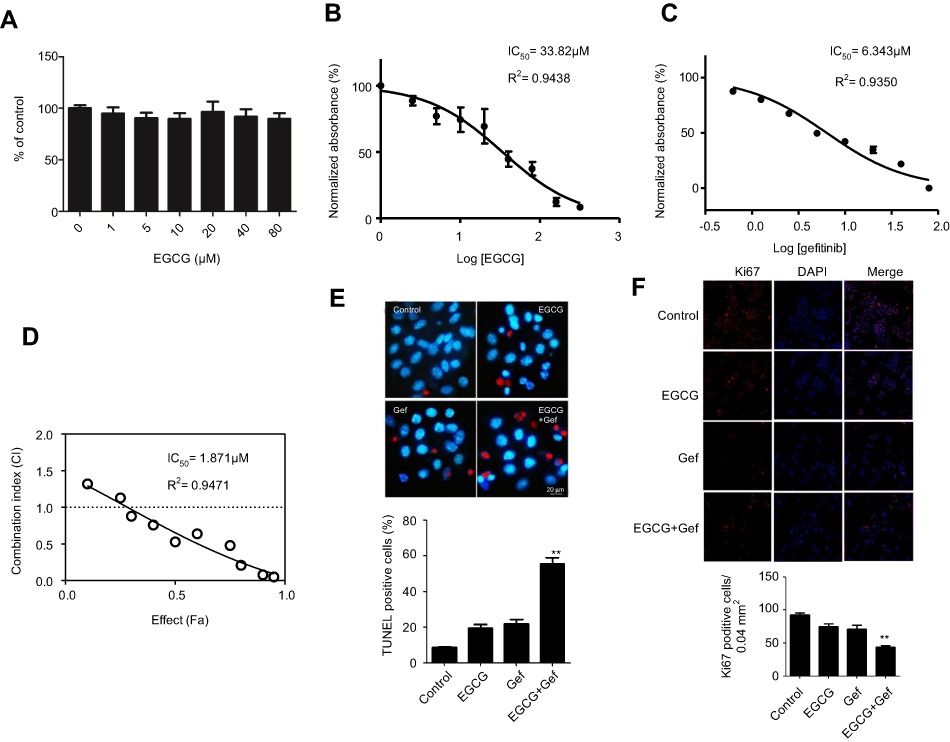 |
Figure 1 EGCG and Gef synergized in inhibiting the NSCLC cell viability. (A) Effect of EGCG on 16HBE cells. Data are shown as mean ± SEM of three independent experiments. A549 cells were cultured in 96-well plates and treated with the EGCG alone (B), Gef alone (C) and the IC50 was determined. (D) The CI values of different combinations of Res and Gef were determined using the Fa-CI plot. (E) TUNEL staining detected apoptosis of A549 cells with different treatments. (F) Ki67 staining of A549 cells with different treatments. **P<0.01, vs Gef-alone treatment group. Abbreviations: EGCG, epigallocatechin gallate; Gef, gefitinib; NSCLC, non-small cell lung cancer. |
To assess whether EGCG could sensitize A549 cells to Gef, we evaluated the effects of EGCG (Figure 1B), Gef (Figure 1C) and EGCG-Gef combinational treatment (Figure 1D) on the proliferation of A549 cells. Compared with Gef treatment alone, EGCG and Gef combination treatments showed a decrease in IC50 values (Figure 1D). As expected, after 48 hrs of treatment, EGCG presented IC50 equals 33.82 μM as well as IC50 of Gef equal to 6.343 μM, and then in the synergetic effect tests, EGCG (34 μM) combined with Gef (1.87 μM) treatment showed the greatest synergistic effect (P<0.05) and was chosen for the subsequent studies.
We also used TUNEL staining to detect apoptosis of Gef-resistant A549 cells. TUNEL-positive cells, which were stained with red fluorescence, were significantly increased in EGCG + Gef group. Nuclei were stained by Hoechst with blue fluorescence (Figure 1E).
In addition, the synergetic effect of EGCG and Gef was further confirmed using Ki-67 staining. Ki-67-positive cells were significantly decreased in EGCG + Gef group (P<0.05) (Figure 1F). All these results verified EGCG and Gef synergized in inhibiting the NSCLC cell proliferation.
EGCG and Gef synergized in suppressing tumor growth in A549 xenograft mouse model
We further evaluated the effect of combined Gef and EGCG on tumor progression in vivo, using an athymic nude mouse xenograft model. When tumors grew to 100 mm3 in size, the animals were treated with vehicle, Gef (10 mg/kg/day, p.o.), EGCG (200 mg/kg/day, p.o.) and Gef plus EGCG (10 mg/kg/day Gef +200 mg/kg/day EGCG, p.o.), respectively. Tumor volumes were observed in every 3 days during the 21 days of treatment. Animal mass and individual tumor curve of the xenograft mouse model are shown in Figure S1. Figure 2A shows that although Gef and EGCG treatment individually demonstrated inhibitory effects on tumor volume in comparison to the vehicle-treated group, the combination of Gef and EGCG demonstrated a higher inhibition compared to Gef- or EGCG-alone treatment groups, as indicated by a statistically significant decrease in tumor volume.
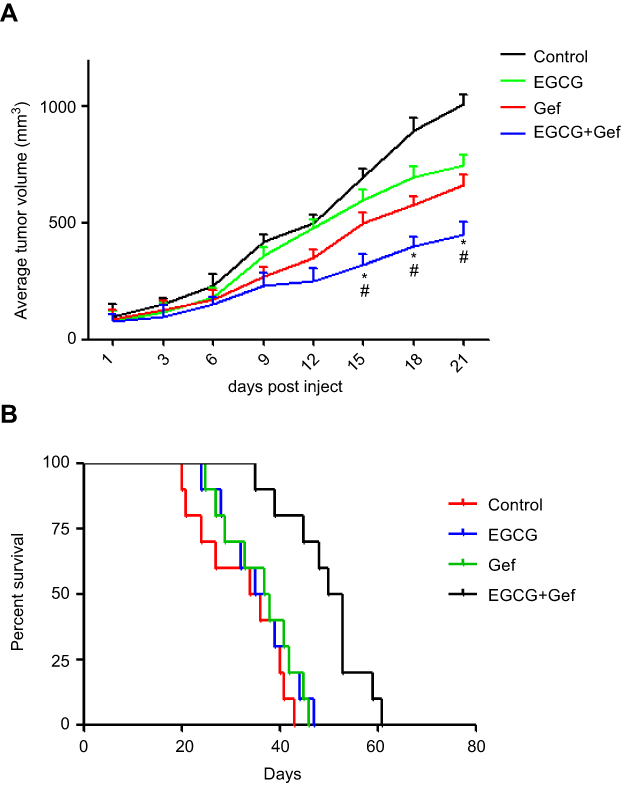 |
Figure 2 EGCG and Gef synergized in suppressing tumor growth in the xenograft mouse model. (A) Inhibitory effects of different treatments on tumor volume in a xenograft mouse model. Data are shown as mean ± SEM. *P<0.05 vs Gef-alone treatment group, #P<0.05 vs EGCG-alone treatment group (n=10). (B) Illustration of the survival periods of xenograft-bearing mice treated with different treatments. Control (normal saline), EGCG (200 mg/kg/day, p.o.), Gef (10 mg/kg/day, p.o.), Gef plus EGCG (10 mg/kg/day Gef +200 mg/kg/day EGCG, p.o.). Day 0 was used to represent the starting of treatment (n=10).Abbreviations: EGCG, epigallocatechin gallate; Gef, gefitinib. |
To determine the long-term survival-prolongation effect of the drug combination, we investigated the survival rate of the above four groups in a cycle of 60 days approximately. As illustrated in Figure 2B, compared with Gef or EGCG-alone treatment groups, the survival rate of A549 tumor-beard nude mice treated with EGCG plus Gef was significantly increased.
EGCG inhibited Gef-induced autophagy in A549 cells
To estimate the regulation of autophagy by EGCG in A549 cells, we transfected A549 cells with GFP-LC3. The green LC3 puncta mainly indicated the dynamic autophagosomes formation. In our study, the green puncta number and intensity were significantly higher in Gef (1.87 μM) treatment group compared with the control group, whereas, EGCG (34 μM) treatment reversed the autophagy induced by Gef (Figure 3A). These results suggested that autophagic flux was inhibited in EGCG and Gef co-treatment group.
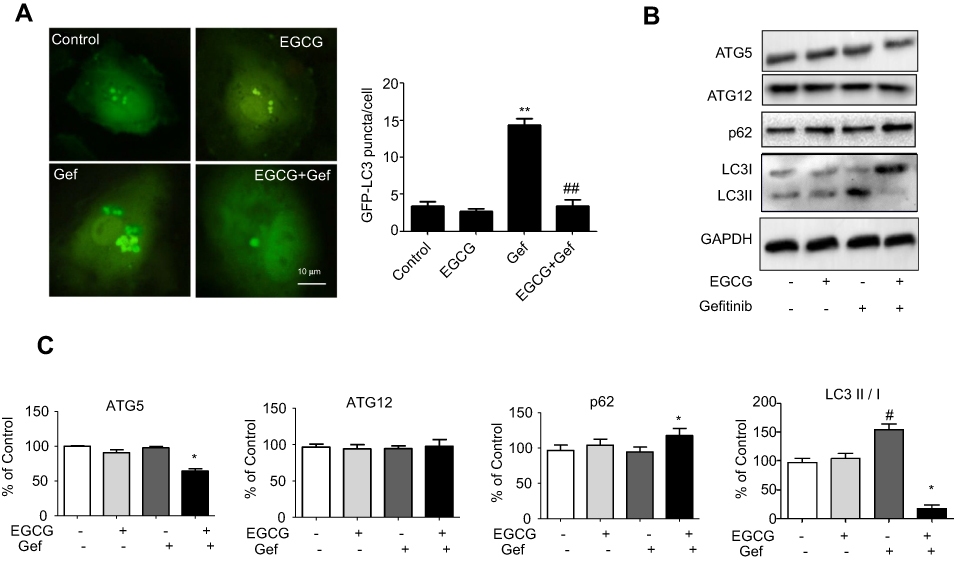 |
Figure 3 Combined EGCG and Gef inhibited Gef-induced autophagy in Gef-resistant A549 cells. (A) Representative images and statistical result of GFP-LC3 puncta in A549 cells treated with EGCG, Gef and their combinations. The green LC3 puncta primarily represent autophagosomes. (B) Western blot analysis assessed the expression of ATG5, ATG12, LC3-II/I and p62 in A549 cells treated with EGCG, Gef and their combinations. (C) Bar charts show the quantification of ATG5, ATG12, LC3-II/I and p62. Values are expressed as the mean ± SEM, *P<0.05, vs Gef-alone treatment group, #P<0.05, vs control group. Abbreviations: EGCG, epigallocatechin gallate; Gef, gefitinib; LC3, microtubule-associated protein 1 light chain 3. |
Next, we detected the level of LC3-II, which is an autophagosomal marker.25 LC3-II/I ratio obviously rises when autophagy occurs in mammalian cells. Western blot assay revealed that the ratio of LC3-II/I was significantly increased in Gef-alone treatment group, while addition of EGCG decreased the ratio of LC3-II/I (Figure 3B). Besides, EGCG and Gef combination treatment also decreased the expression of ATG5 and increased expression of p62 which functions as the adaptors of autophagy (Figure 3B and C), but did not affect the expression of ATG12. Overall, EGCG inhibited autophagy induced by Gef in A549 cells.
Inhibition of autophagy in A549 cells promoted cell death
We next studied how autophagy affected viability of NSCLC cells treated with combined drug. Thus, we firstly suppressed autophagy with CQ (20 μM), an autophagy inhibitor, and then determined the viability of tumor cells subjected to the treatments. As shown in Figure 4A, immunoblot results indicated that CQ increased p62 expression level but reduced the level of LC3-II in A549 cells, whereas Rap (100 nM) presented the opposite effect. MTS results revealed that inhibition of autophagy by CQ (EGCG + Gef + CQ) further augmented cell death compared with the EGCG + Gef group (P<0.05) (Figure 4B).
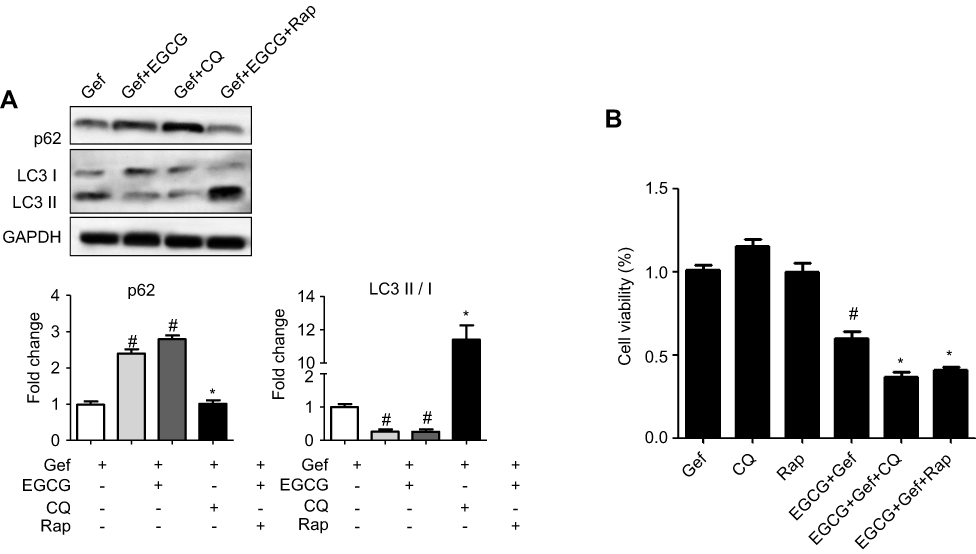 |
Figure 4 Inhibition of autophagy in Gef-resistant A549 cells augmented cell death. (A) Western blot images and statistical results of the expression of p62 and LC3-II/I in A549 cells with different treatments. #P<0.05, vs Gef-alone treatment group, *P<0.05, vs EGCG + Gef treatment group. (B) Cell viability was measured by MTS assay. Data are shown as mean ± SEM of three independent experiments. #P<0.05, vs Gef-alone treatment group, *P<0.05, vs EGCG + Gef-treated group.Abbreviations: EGCG, epigallocatechin gallate; Gef, gefitinib; CQ, chloroquine; Rap, rapamycin. |
Combined EGCG and Gef inhibited ERK phosphorylation
As illustrated in other studies that ERK signaling was essential for Gef resistance and ERK inhibition could alleviated Gef resistance.26 To clarify the mechanism underlying drug combination–induced autophagic inhibition, Raf/MEK/ERK pathway was determined. As illustrated in Figure 5A and B, high concentration of EGCG or Gef treatment (EGCG, 80 µM; Gef, 20 µM) inhibited ERK phosphorylation (p-ERK). Neither EGCG (40 µM) nor Gef (10 µM) alone inhibited p-ERK or p-MEK at lower concentrations, while their combination treatment at lower doses significantly reduced both p-ERK and p-MEK levels, but did not affect total MEK or ERK level.
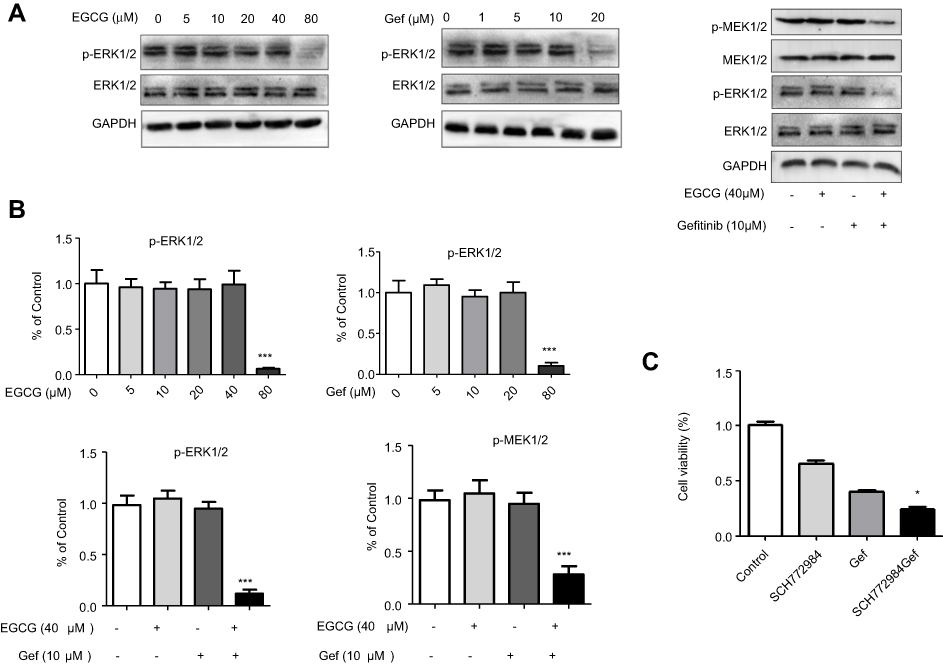 |
Figure 5 Combined EGCG and Gef inhibited ERK phosphorylation. (A) Western blot analysis showed expression of p-ERK1/2 and p-MEK1/2 in A549 cells treated with different concentrations of EGCG (0, 5, 10, 20, 40, 80 µM) or Gef (0, 1, 5, 10, 20 µM) or EGCG (40 µM) plus Gef (10 µM). (B) Bar charts show the quantification of p-ERK1/2 and p-MEK1/2. Values are expressed as the mean ± SEM, ***P<0.001, vs vehicle group (n=4). (C) The viability of cells treated with different drug combinations was measured by MTS assay, control group treated with (0.1% DMSO), *P<0.05, vs Gef-alone group. Abbreviations: EGCG, epigallocatechin gallate; Gef, gefitinib; p-ERK, ERK phosphorylation; p-MEK, MEK phosphorylation. |
We further confirmed the above results using ERK inhibitor, SCH772984. SCH772984 (0.5 µM) or Gef (10 µM) treatment inhibited A549 cell proliferation by 33.08% and 59.15%, respectively, whereas, Gef and SCH772984 co-treatment remarkably inhibited A549 cell proliferation (by 75.59%) compared with cells treated with SCH772984, or Gef alone (Figure 5C). These results indicate that EGCG overcomes Gef resistance through inhibiting Raf/MEK/ERK Pathway.
Discussion
As the basic treatment of advanced NSCLC, platinum-based chemotherapy shows serious side effects such as nausea, diarrhea and neutropenia.27 TKIs, like Gef and erlotinib, were recommended as the second- or third-line therapy after failure of first-line chemotherapy in NSCLC.28 It is well known that TKIs are generally more efficacious and less toxic than the classic cell toxicity-based chemotherapy.29 Unfortunately, the therapeutic effect of TKIs was limited by the emergence of drug resistance caused by the second mutation, for instance, replacing threonine 790 with methionine (T790M) of EGFR accounts for approximately 50% of all resistance to Gef or erlotinib.30 There are currently no effective treatments for EGFR TKI-resistant patients.
A number of natural products have been reported presenting synergetic effect when co-treated with certain types of popular chemotherapy medicine. It has been demonstrated that resveratrol synergizes with Gef could inhibited the proliferation of Gef-resistant NSCLC cells.31 Combination of curcumin and fluorouracil synergistically inhibits cell growth and induces potent apoptosis of the drug-resistant human gastric cancer cells lines in vitro.32 Besides, it is reported that EGCG can mediate human neutrophil elastase-induced migration in A549 cells.22
As a type of polyphenol with antioxidant and anti-inflammatory properties, EGCG has been proven to inhibit the growth of cancer cells but has no toxic effects on normal cells.33 It is reported that EGCG inhibits proliferation and migration of bladder cancer 5637 and T24 cells.34 EGCG inhibits the proliferation and invasiveness and induces apoptosis in nasopharyngeal carcinoma cells.35 EGCG suppresses cell proliferation and promotes apoptosis in Ec9706 and Eca109 esophageal carcinoma cells.36 In the lung cancer xenograft nude mice model, combination of EGCG and curcumin can improve weight loss due to tumor burden and inhibit tumor growth, without causing any serious side effects.32 The combination of EGCG and erlotinib inhibits cell proliferation and colony formation more effectively than either agent alone.37 Overall, EGCG effectively inhibited cell proliferation of several types of tumor cell lines.
As it is known, Gef exerts its anticancer activity at the intracellular domain of EGFR by preventing tyrosine kinase phosphorylation and subsequent activation of the downstream signaling pathway.38 Dragowska et al demonstrated that elevated levels of autophagy were early responses to Gef treatment and that targeting EGFR and autophagy should be a potential therapeutic target in EGFR-expressing breast cancers.39 Further, it was reported that autophagy contributes to the chemoresistance of NSCLC in hypoxic conditions.40 Inhibition of autophagy may be a new means to improve chemotherapy efficiency in breast cancers.41 These indicate that autophagy exhibits a critical role in the survival of tumor cell under extreme stress condition.
As mentioned in the previous paragraph, inhibition of autophagy might contribute to the amelioration of TKIs resistance, and thus targeting autophagy might be a novel therapeutic strategy for cancer treatment.9,10 In our study, we demonstrated that combined EGCG and Gef inhibited Gef-induced autophagy and partially restored Gef sensitivity in A549 cells. Besides, immunoblot assay revealed that the ratio of LC3-II/I dramatically decreased in EGCG treatment cells compared with that in Gef treated cells. Further, inhibiting autophagy with CQ increased p62 expression and reduced LC3 expression in the Gef-resistant A549 cells. Moreover, MTS results demonstrated inhibiting autophagy could augment cell death. What needs to be mentioned is that the longest treatment time of EGCG is 48 hrs in our present study, so experimental findings of the present study are mainly attributed to the regulation by prototype rather than metabolites of EGCG.
In subsequent studies, molecular mechanisms involved in the inhibition of autophagy were analyzed. We know that the Raf/MEK/ERK signaling pathway regulates many biological and pathological processes in cancer cells and normal cells. MEK1 and MEK2 are dual-specificity protein kinases and phosphorylate ERK on specific threonine and tyrosine residues. MEKs are activated by upstream serine kinase, Raf, by phosphorylating catalytic domain on key serine residues.42 In the present study, inhibition of ERK phosphorylation obviously alleviated Gef resistance.
Conclusion
In summary, EGCG overcomes Gef resistance by inhibiting autophagy and augmenting cell death through targeting ERK pathway in NSCLC. This study provides a novel, enhanced targeted therapy for overcoming acquired resistance in NSCLC and may open a window for the treatment of NSCLC.
Abbreviation list
CI, combination index; CQ, chloroquine; EGCG, epigallocatechin gallate; Gef, gefitinib; LC3, microtubule-associated protein 1 light chain 3; NSCLC, non-small cell lung cancer; p-ERK, ERK phosphorylation; PFA, paraformaldehyde; Rap, rapamycin; TKIs, tyrosine kinase inhibitors.
Ethics approval
All animal experiments were performed according to NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Fourth People’s Hospital of Shaanxi (China).
Acknowledgment
This work was supported by the Natural Science Foundation of Shannxi Province (No. 2016SF-285).
Author contributions
Jiao Meng, Cuicui Chang are the co-first authors to this article and contributed equally to this research project. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
3. Yang JJ, Chen HJ, Yan HH, et al. Clinical modes of EGFR tyrosine kinase inhibitor failure and subsequent management in advanced non-small cell lung cancer. Lung Cancer. 2013;79(1):33–39. doi:10.1016/j.lungcan.2012.09.016
4. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi:10.1056/NEJMoa0810699
5. Sholl LM, Aisner DL, Varella-Garcia M, et al. Multi-institutional oncogenic driver mutation analysis in lung adenocarcinoma: the lung cancer mutation consortium experience. J Thorac Oncol. 2015;10(5):768–777. doi:10.1097/JTO.0000000000000516
6. Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–1180. doi:10.1158/1078-0432.CCR-10-2277
7. Okon IS, Coughlan KA, Zhang M, Wang Q, Zou MH. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290(14):9101–9110. doi:10.1074/jbc.M114.631580
8. Aljohani H, Koncar RF, Zarzour A, Park BS, Lee SH. Bahassi el M. ROS1 amplification mediates resistance to gefitinib in glioblastoma cells. Oncotarget. 2015;6(24):20388–20395. doi:10.18632/oncotarget.3981
9. Zhao L, Yang G, Shi Y, Su C, Chang J. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J Nanobiotechnology. 2015;13:57. doi:10.1186/s12951-015-0121-5
10. Lv X, Liu F, Shang Y, Chen SZ. Honokiol exhibits enhanced antitumor effects with chloroquine by inducing cell death and inhibiting autophagy in human non-small cell lung cancer cells. Oncol Rep. 2015;34(3):1289–1300. doi:10.3892/or.2015.4091
11. Jain SK, McVie R, Duett J, Herbst JJ. Erythrocyte membrane lipid peroxidation and glycosylated hemoglobin in diabetes. Diabetes. 1989;38(12):1539–1543. doi:10.2337/diab.38.12.1539
12. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. doi:10.1016/j.bbamcr.2006.10.001
13. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6(11):827–837. doi:10.1038/nrm1743
14. Hatzivassiliou G, Liu B, O’Brien C, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154. doi:10.1158/1535-7163.MCT-11-1010
15. Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2(10):934–947. doi:10.1158/2159-8290.CD-12-0103
16. Abrams SL, Steelman LS, Shelton JG, et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010;9(9):1781–1791. doi:10.4161/cc.9.9.11483
17. Johnson R, Bryant S, Huntley AL. Green tea and green tea catechin extracts: an overview of the clinical evidence. Maturitas. 2012;73(4):280–287. doi:10.1016/j.maturitas.2012.08.008
18. Khurana S, Venkataraman K, Hollingsworth A, Piche M, Tai TC. Polyphenols: benefits to the cardiovascular system in health and in aging. Nutrients. 2013;5(10):3779–3827. doi:10.3390/nu5103779
19. Meydani M, Hasan ST. Dietary polyphenols and obesity. Nutrients. 2010;2(7):737–751. doi:10.3390/nu2070737
20. Shankar S, Ganapathy S, Srivastava RK. Green tea polyphenols: biology and therapeutic implications in cancer. Front Biosci. 2007;12:4881–4899.
21. Furst R, Zundorf I. Plant-derived anti-inflammatory compounds: hopes and disappointments regarding the translation of preclinical knowledge into clinical progress. Mediators Inflamm. 2014;2014:146832. doi:10.1155/2014/146832
22. Xiaokaiti Y, Wu H, Chen Y, et al. EGCG reverses human neutrophil elastase-induced migration in A549 cells by directly binding to HNE and by regulating alpha1-AT. Sci Rep. 2015;5:11494. doi:10.1038/srep11494
23. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi:10.1124/pr.58.3.10
24. Pan Y, Zhong LJ, Zhou H, et al. Roles of vimentin and 14-3-3 zeta/delta in the inhibitory effects of heparin on PC-3M cell proliferation and B16-F10-luc-G5 cells metastasis. Acta Pharmacol Sin. 2012;33(6):798–808. doi:10.1038/aps.2012.42
25. Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi:10.4161/auto.5338
26. Qi M, Tian Y, Li W, et al. ERK inhibition represses gefitinib resistance in non-small cell lung cancer cells. Oncotarget. 2018;9(15):12020–12034. doi:10.18632/oncotarget.24147
27. Garon EB, Ciuleanu TE, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665–673. doi:10.1016/S0140-6736(14)60845-X
28. Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa survival evaluation in lung cancer). Lancet. 2005;366(9496):1527–1537. doi:10.1016/S0140-6736(05)67625-8
29. Burotto M, Manasanch EE, Wilkerson J, Fojo T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: a meta-analysis of toxicity and efficacy of randomized clinical trials. Oncologist. 2015;20(4):400–410. doi:10.1634/theoncologist.2014-0154
30. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
31. Zhu Y, He W, Gao X, et al. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci Rep. 2015;5:17730. doi:10.1038/srep17730
32. Zhou DH, Wang X, Yang M, Shi X, Huang W, Feng Q. Combination of low concentration of (−)-epigallocatechin gallate (EGCG) and curcumin strongly suppresses the growth of non-small cell lung cancer in vitro and in vivo through causing cell cycle arrest. Int J Mol Sci. 2013;14(6):12023–12036. doi:10.3390/ijms140612023
33. Chen ZP, Schell JB, Ho CT, Chen KY. Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett. 1998;129(2):173–179.
34. Luo KW, Lung WY, Chun X, Luo XL, Huang WR. EGCG inhibited bladder cancer T24 and 5637 cell proliferation and migration via PI3K/AKT pathway. Oncotarget. 2018;9(15):12261–12272. doi:10.18632/oncotarget.24301
35. Fang CY, Wu CC, Hsu HY, et al. EGCG inhibits proliferation, invasiveness and tumor growth by up-regulation of adhesion molecules, suppression of gelatinases activity, and induction of apoptosis in nasopharyngeal carcinoma cells. Int J Mol Sci. 2015;16(2):2530–2558. doi:10.3390/ijms16022530
36. Liu L, Zuo J, Wang G. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis in Ec9706 and Eca109 esophageal carcinoma cells. Oncol Lett. 2017;14(4):4391–4395. doi:10.3892/ol.2017.6712
37. Milligan SA, Burke P, Coleman DT, et al. The green tea polyphenol EGCG potentiates the antiproliferative activity of c-Met and epidermal growth factor receptor inhibitors in non-small cell lung cancer cells. Clin Cancer Res. 2009;15(15):4885–4894. doi:10.1158/1078-0432.CCR-09-0109
38. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–26. doi:10.1016/j.ijrobp.2003.11.041
39. Dragowska WH, Weppler SA, Wang JC, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS One. 2013;8(10):e76503. doi:10.1371/journal.pone.0076503
40. Lee JG, Shin JH, Shim HS, et al. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res. 2015;16:138. doi:10.1186/s12931-015-0285-4
41. Lefort S, Joffre C, Kieffer Y, et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy. 2014;10(12):2122–2142. doi:10.4161/15548627.2014.981788
42. Kang NJ, Lee KW, Kwon JY, et al. Delphinidin attenuates neoplastic transformation in JB6 Cl41 mouse epidermal cells by blocking Raf/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling. Cancer Prev Res (Phila). 2008;1(7):522–531. doi:10.1158/1940-6207.CAPR-08-0071
Supplementary material
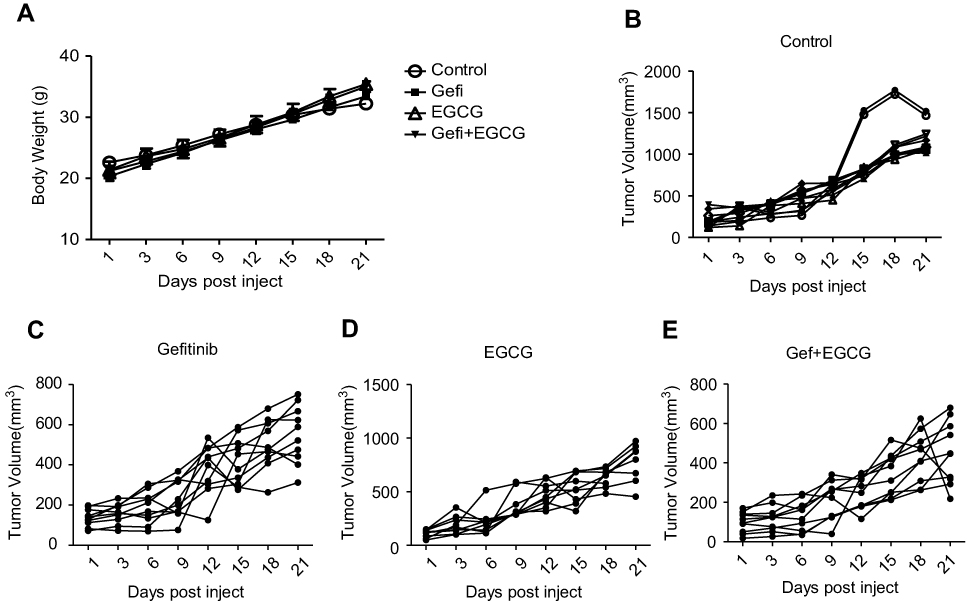 |
Figure S1 Animal mass and individual tumor curve in the xenograft mouse model. (A) Body weight of a xenograft mouse model with different treatments. Inhibitory effects of control (normal saline) (B), Gef (10 mg/kg/day, p.o.) (C), EGCG (200 mg/kg/day, p.o.) (D) and Gef + EGCG (10 mg/kg/day Gef +200 mg/kg/day EGCG, p.o.) (E) on tumor volume in a xenograft mouse model. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.