")
Back to Journals » International Journal of Nanomedicine » Volume 16
Efficient Delivery of Antibodies Intracellularly by Co-Assembly with Hexahistidine-Metal Assemblies (HmA)
Authors Wei S, Zhou S, Huang W, Zan X , Geng W
Received 2 August 2021
Accepted for publication 27 September 2021
Published 6 November 2021 Volume 2021:16 Pages 7449—7461
DOI https://doi.org/10.2147/IJN.S332279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Shaoyin Wei,1 Sijie Zhou,1 Wenjuan Huang,1,2 Xingjie Zan,1,3 Wujun Geng4
1School of Ophthalmology and Optometry, Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou, 325035, Zhejiang Province, People’s Republic of China; 2Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, 317000, Zhejiang Province, People’s Republic of China; 3Oujiang Laboratory, Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, 325001, Zhejiang Province, People’s Republic of China; 4Department of Anesthesiology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, Zhejiang Province, People’s Republic of China
Correspondence: Xingjie Zan; Wujun Geng Email [email protected]; [email protected]
Purpose: There has been a substantial global market for antibodies, which are based on extracellular targets. Binding intracellular targets by antibodies will bring new chances in antibody therapeutics and a huge market increase. We aim to evaluate the efficiency of a novel delivery system of His6-metal assembly (HmA) in delivering intracellular antibodies and biofunctions of delivered antibodies.
Methods: In this study, the physicochemical properties of HmA@Antibodies generated through co-assembling with antibodies and HmA were well characterized by dynamic light scatter. The cytotoxicity of HmA@Antibodies was investigated by Cell Counting Kit-8 (CCK-8). The endocytic kinetics and lysosome escape process of HmA@Antibodies were studied by flow cytometry and fluorescent staining imaging, respectively. Compared to the commercialized positive control, the intracellular delivery efficiency by HmA@Antibodies and biofunctions of delivered antibodies were evaluated by fluorescent imaging and CCK-8.
Results: Various antibodies (IgG, anti-β-tubulin and anti-NPC) could co-assemble with HmA under a gentle condition, producing nano-sized (∼ 150 nm) and positively charged (∼+30 eV) HmA@Antibodies particles with narrow size distribution (PDI ∼ 0.15). HmA displayed very low cytotoxicity to divers cells (DCs, HeLa, HCECs, and HRPE) even after 96 h for the feeding concentration ≤ 100 μg mL− 1, and fast escape from endosomes. In the case of delivery IgG, the delivery efficiency into alive cells of HmA was better than a commercial protein delivery reagent (PULSin). For cases of the anti-β-tubulin and anti-NPC, HmA showed comparable delivery efficiency to their positive controls, but HmA with ability to deliver these antibodies into alive cells was still superior to positive controls delivering antibodies into dead cells through punching holes.
Conclusion: Our results indicate that this strategy is a feasible way to deliver various antibodies intracellularly while preserving their functions, which has great potential in various applications and treating many refractory diseases by intracellular antibody delivery.
Keywords: antibody, intracellular delivery, coordination polymer, nanocarrier, peptide assembly
Introduction
Antibodies are designed to bind various extracellular targets to specifically block certain pathways, effects that cannot be achieved by small molecules.1 Due to their strong specificities and potency, antibodies are broadly used in diagnostic tools, basic research and treating many refractory diseases, such as neovascular diseases, cancer and autoimmune diseases.2,3 In past decades, antibody-based therapy has experienced rapid growth and occupied an important position in the drug market, with forecasted future scale up to $190 billion by 2022.3 More than a hundred monoclonal antibodies have been approved by the FDA for clinical use,2 and over two thousand antibodies are currently under preclinical or clinical development, indicating a very promising future for antibody therapeutics.4
As known, compared to targets at extracellular, there are more intracellular targets, which are involved in the development of various diseases, and these targets can also be activated or blocked by specific antibodies.5,6 Accumulating evidence suggested that intracellular targeting antibodies could bring new chances in antibody therapeutics and huge market increase.7 Recently, increasing attention has been given to intracellular functional antibody fragments targeting and tracing the intracellular components in living cells.8 However, intracellular delivery of antibodies is severely limited by their poor permeability into cell membrane and trap in endosomes and/or lysosomes after internalization, due to their intrinsic properties, such as large molecular weights, hydrophilicity and limited positive charges.9 These features seriously limit their practical applications and inhibit the desired intracellular biological activities of antibodies. To date, antibodies binding intracellular targets have mostly been utilized in scientific laboratories for immunostaining assays on fixed cells.
Motivated by the potential of intracellular targeting antibodies, various methods, including direct injection, intrabodies, fusion to the protein domain, cell penetrating peptides (CPPs), and nanocarriers, have been developed with the aim of delivering antibodies into the cytosol. Although these methods have successfully targeted the antibodies into the cytosol, they are still facing many challenges, such as a delicate design and complicated process for constructing the delivery vehicle, deactivation or loss of the antibody’s functionality during the production of the delivery vehicle, being stuck inside endosomes after endocytosis, etc. For example, the intracellular expression of antibodies was reported, but only a few of the antibody fragments could be properly folded and functionalized in the reducing environment of the cytosol.10 Fusion of a transmembrane with the antibody was helpful for intracellular antibody delivery, but it was limited by uncertainty of the functionality of the fused protein.7 Conjugating CPPs showed high intracellular delivery efficiency, but a low lysosomal escape efficiency and no cell targeting led to a low intracellular targeting efficiency.11,12 Obviously, designing a universal and efficient intracellular delivery carrier with strong ability to escape from endosomes and/or lysosomes is of great significance but very challenging.
With advances in nanotechnologies, nanocarriers such as liposomes, polymersomes, virus-like particles, and inorganic nanoparticles have been extensively explored as antibody carriers and have showed great potential in the intracellular targeting of antibodies.13–16 Previously, we reported a promising drug delivery system of His6-metal assembly (HmA) generated from the coordination of His6 and zinc ions, featuring quick internalization, the ability to bypass lysosomes and rapid intracellular drug release.17 In this work, we found that various antibodies could be co-assembled with HmA (HmA@Abs), due to high encapsulating efficiency of HmA. Importantly, HmA@Abs exhibited higher penetration ability through cell membrane and quicker escaping from the endosomes compared with commercialized intracellular delivery agent, with preservation of the intracellular functionality of the antibodies. Our finding suggested this strategy may advance the cytosolic delivery of antibodies for therapeutic targets and bring hopes to many refractory disease treatments and new applications based on intracellular antibody delivery.
Materials and Methods
Materials
Polyvinylpyrrolidone (PVP, Mw∼58k), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, ≥ 99%) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Aladdin (China). Hexahistidine (His6, ≥99%) was synthesized by KareBay Biochem, Inc. and used without further purification. Mouse anti-nuclear pore complex protein antibody (anti-NPC) (ab50008), goat anti-rabbit IgG H&L (Alexa Fluor®488) (ab150077) (IgG-488), anti-EEA1 antibody-early endosome marker (ab2900) (EER), recombinant anti-M6PR (cation independent) antibody [EPR6599]-lysosome membrane marker (ab124767) (LyR), and goat anti-mouse IgG H&L (Alexa Fluor®594) (ab150116) (IgG-594) were purchased from Abcam. Mouse anti-β-tubulin antibody (T5201) and Triton X-100 were purchased from Sigma-Aldrich. Cy3-labelled goat anti-mouse IgG (IgG-Cy3), FITC-labelled goat anti-mouse IgG (IgG-FITC), Alexa Fluor 647-labelled goat anti-rabbit IgG (IgG-647), and DAPI were purchased from Beyotime (China). Protein delivery reagent (PULSin) was purchased from Polyplus-transfection® (France). Cell Counting Kit-8 (CCK-8) reagent was purchased from Dojindo (Japan). Dendritic cells (DCs), HeLa cells, human corneal epithelial cells (HCECs), and human retinal pigment epithelium cells (HRPEs) were provided by ATCC. Mammalian cell culture media, foetal bovine serum (FBS), and the ProLong® Gold anti-fade kit were purchased from Thermo Scientific.
The Synthesis of Antibody-Encapsulated HmA (HmA@Antibodies) Particles
The synthesis of the HmA@Antibodies (HmA@Abs) was conducted by following the drug encapsulation procedure of HmA.17 Antibodies, including IgG-Cy3, IgG-488, anti-NPC, and anti-β-tubulin, were encapsulated into the HmA particles separately. In brief, 10 μL of Zn(NO3)2·6H2O (30 mg mL−1) was added dropwise to 200 μL solution (antibodies, His6 (0.84 mg) and PVP (1 mg)) buffered with 50 mM HEPES at pH 8.0. The whole encapsulating procedure was conducted at 4°C under sonication. After the HmA@Antibody (HmA@Ab) particles were generated, they were collected by centrifugation (10,000 rpm, 10 min) and washed three times with deionized H2O. The particles were stored at 4°C and sonicated for 1 min before characterization and cellular testing. The assembly procedure was conducted in a dark room. The generated HmA@Ab particles were analyzed by DLS (size and zeta potential; Malvern, UK) and Scanning electron microscope (morphology; SU8010 HITACHI SEM).
Cell Culture and the Evaluation of Cytotoxicity
In general, DCs were cultured in 1640 medium containing 10% foetal bovine serum, 100 U mL−1 penicillin and 100 U mL−1 streptomycin at 37°C in a 5% CO2 atmosphere. In addition, HeLa cells and HRPE cells were both cultured in Dulbecco’s modified Eagle medium (DMEM), and HCECs were cultured in DMEM with 5 μg mL−1 insulin under the same culture conditions. Cytotoxicity tests were performed by using the Cell Counting Kit-8 (CCK-8) assay. The standard procedure was performed as follows: First, cells (DCs, HeLa cells, HRPE and HCECs) were seeded in 96-well plates at an appropriate density for set-up time (8000 cells per well for 24 h, 5000 cells per well for 48 h, and 3000 cells per well for 96 h). After incubation for set-up time, the culture medium in the plates was removed, and the cells were washed once with PBS buffer. Then, the fresh medium with particles suspended at various concentrations was added. Cells without treatment were used as the control. After being further incubated for the indicated time, the plates were washed with PBS buffer three times to remove the treatments. Then, 100 μL of fresh medium supplemented with 10% CCK-8 solution was added to each testing well, and the cells were incubated for another 2 h. Finally, the plates were read at 450 nm by a microplate reader (Varioskan LUX Multimode Thermo, USA). Five repeats were conducted for each experiment.
Evaluation of Intracellular Antibody Delivery Efficiency of HmA
Flow Cytometry Assays
DCs were seeded in 24-well plates at a density of 1 × 105 cells/well and allowed to attach for 24 h. The culture media were removed, and the cells were washed with PBS buffer. Then, the cells were treated with 1640 media containing the HmA@IgG-Cy3 (HmA: 50 μg mL−1, IgG-Cy3: 1.5 μg mL−1) particles. Cells treated with free IgG-Cy3 were used as the control. After incubation, the treatments were removed, and the cells were washed with PBS three times. The cells were then detached from the culture plates with 0.05% trypsin-EDTA solution and redispersed in ice-cold PBS. The cell suspension was washed three times before analysis by flow cytometry (CytoFLEX Beckman Coulter, USA).
Fluorescence Imaging of the Intracellular Delivery Efficiency of HmA@IgG-Cy3
First, different cell lines were used to evaluate the intracellular antibody delivery ability of HmA. DCs, HeLa cells, HCECs, and HRPE were seeded on 24-well glass plates at a density of 4 × 104 cells/well. HmA@IgG-Cy3 (HmA: 50 μg mL−1, IgG-Cy3: 1.5 μg mL−1) or free IgG-Cy3 (1.5 μg mL−1) was incubated with DCs, HeLa cells, HCECs, and HRPE for 6 h. The treated cell lines were imaged by fluorescence microscopy.
Then, DCs were seeded on 24-well glass plates at a density of 4 × 104 cells/well. After incubation for 24 h, the culture medium was removed, and the cells were washed with PBS buffer. Then, the cells were treated with 1640 media containing the HmA@IgG-Cy3 (HmA: 50 μg mL−1) particles, in which the IgG-Cy3 was added at the indicated various concentrations. After incubation for 4 h, all of the treatments were removed, and the cells were washed with PBS three times. Finally, the cells were observed by laser scanning confocal microscopy (LSCM) (A1 Nikon, Japan) and quantitatively measured by the ImageJ program (NIH, USA). Three repeats were conducted for each sample. A commercial protein transfection reagent (PULSin) was used as a positive control and used according to the manufacturer’s protocol. Briefly, IgG-Cy3 was mixed with 2.5 μL of PULSin reagent at different concentrations and incubated for 15 min at room temperature before adding it to the cells.
Fluorescent Imaging of Intracellular Localization of the Delivered Antibodies
The intracellular localization of delivered antibodies was tested according to previous reports,18,19 and the protocol provided by manufacturer was rigorously followed. In brief, DCs were pre-incubated with the HmA@IgG-FITC particles (HmA: 50 μg mL−1, IgG-FITC: 1.5 μg mL−1) for a period of time. The cells were washed three times with cold PBS before immunofluorescence. The samples were pre-treated with 1% Triton X-100 and 5% goat serum in PBS. Anti-EEA1 antibody-early endosome marker (1:100) and lysosome membrane marker (1:100) were added to the cells and incubated overnight at 4°C. Then, the cells were washed three times, and a secondary antibody (IgG-647) was added. After incubating for another 1 h at 25°C, the samples were mounted with a ProLong® Gold anti-fade kit and imaged by LSCM. Three experiments were performed using three samples each.
Fluorescent Imaging of the Intracellular Activities of the Delivered Antibodies
DCs were seeded on 24-well glass plates at a density of 4 × 104 cells/well. After incubation for 24 h, the culture medium was removed, and the cells were washed with PBS buffer. Then, the cells were treated with 1640 medium with suspended HmA@Ab particles. After incubating for the indicated time, the treatments were removed, and the cells were washed with cold PBS three times. The fluorescence staining of the cells was as follows: the cells were fixed with 4% paraformaldehyde and pre-treated with 1% Triton X-100 and 5% goat serum in PBS. Then, the cells were incubated with the secondary antibody (IgG-594) (1:500) for 1 h at room temperature. Finally, the cells were imaged by LSCM. Three repeats were conducted for each sample.
The fluorescence staining of positive control was carried out by untreated DC cells. Similar to the above methods, DCs were seeded on 24-well glass plates at a density of 4 × 104 cells/well. After incubation for 24 h, the culture medium was removed, and the cells were washed with PBS buffer. Then, the cells were fixed with 4% paraformaldehyde about 15 minutes. The fluorescence staining of the cells was as follows: the cells were pre-treated with 1% Triton X-100 and 5% goat serum in PBS. Then, the cells were incubated with the antibody (1:1000) overnight at 4°C. After washing with PBS, the cells were incubated with the secondary antibody (IgG-594) (1:500) for 1 h at room temperature. Finally, the cells were imaged by LSCM. Three repeats were conducted for each sample.
Statistical Analysis
Statistical analysis was performed using SPSS software (IBM SPSS statistics 22.0). Student’s t-test was used to evaluate the significant differences in means between the two groups. “*”, “**”, “***” and “****” represent P < 0.05, P < 0.01, P < 0.001 and P < 0.0001, respectively. P < 0.05 was considered a statistically significant difference. Triplicate experiments were performed using triplicate samples.
Results and Discussion
The Synthesis and Characterization of the HmA@Abs
Three representative antibodies (IgG-Cy3, anti-β-tubulin and anti-NPC) were chosen to investigate the potency of this strategy of intracellular antibody delivery. Depending on the experimental purpose, different antibodies were selected. All HmA@Abs were generated by a facile procedure under mild conditions: zinc ions were dropwise added into the mixture of His6 and antibodies at pH 8 under slight sonication, as schematically illustrated in Figure 1A. Dynamic light scattering (DLS) was employed to test their size distribution and zeta potential. As listed in Figure 1B, the average sizes of HmA@Abs particles were different from each other, but the sizes were in the range of 130–155 nm with a narrow PDI range (0.1–0.2), dependent on the encapsulated antibody. Zeta potentials of HmA@Abs were closed to each other, around 30 mV, suggesting slightly positively charged particle surfaces. The IgG-Cy3 was chosen for investigating the encapsulation of antibodies, cell endocytosis process, and potency of HmA@antibodies for delivering antibodies into cells because it is cheaper and fluorescent compared to other two antibodies. In the UV–vis spectra (Figure 1C), for the free IgG-Cy3, the absorbances at 280 nm and 552 nm were clearly observed, which were attributed to the protein and its labelled fluorescent molecules Cy3. For HmA@IgG-Cy3, the protein signal at 280 nm was not very remarkable, but it could still be observed. The light scattering on nano-sized HmA@IgG-Cy3 particles was the reason for the increased baseline, and the red-shift of absorbance of Cy3 suggested most IgG-Cy3 was encapsulated inside the HmA@IgG-Cy3, which was consistent with previous reports.20 Under SEM (Figure 1D), these HmA@Abs particles showed very similar morphologies, nano-sized and irregular shaped particles. All these data suggested HmA@Abs particles were generated, with antibodies encapsulated inside HmA particles.
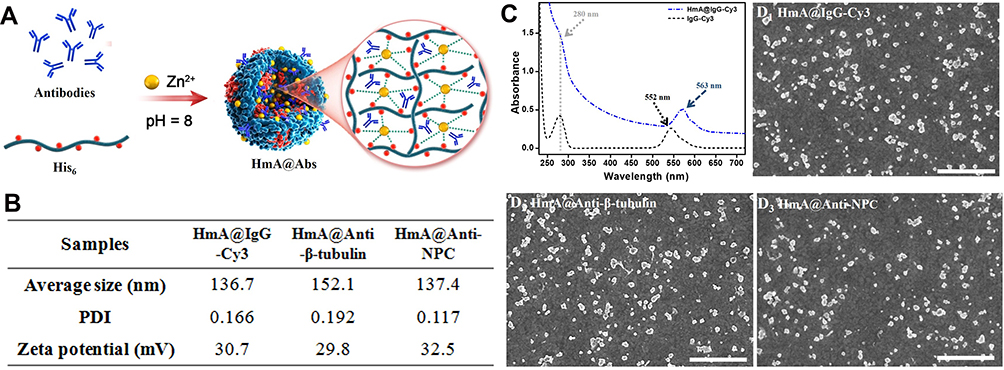 |
Figure 1 (A) Schematic illustration of the formation of HmA@Abs particles; (B) Average size, PDI and zeta potential of HmA@Abs particles (HmA@IgG-Cy3, HmA@Anti-β-tubulin and HmA@Anti-NPC); (C) UV-vis spectra of free IgG-Cy3 and HmA@IgG-Cy3; (D) SEM images of HmA@Abs particles (HmA@IgG-Cy3, HmA@Anti-β-tubulin and HmA@Anti-NPC); The scale bars in (D) were 1 μm. |
The cytotoxicity of the HmA particles was evaluated by different cell lines, including cancer cells (HeLa), immune cells (DCs), and epithelial cells (HCECs and HRPEs), on which HmA particles with different concentrations of the (20–100 μg mL−1) were performed for 24, 48 and 96 h. Using CCK-8 assay, the cell viabilities were available and shown in Figure 2A–D. For all tested cells (DCs, HeLa, HCEC and RPE), their cell viabilities were higher than 90%, even at the highest tested concentration (100 μg mL−1) within extension to 96 h, which indicated no or low cytotoxicity of the HmA particles to the tested cells.
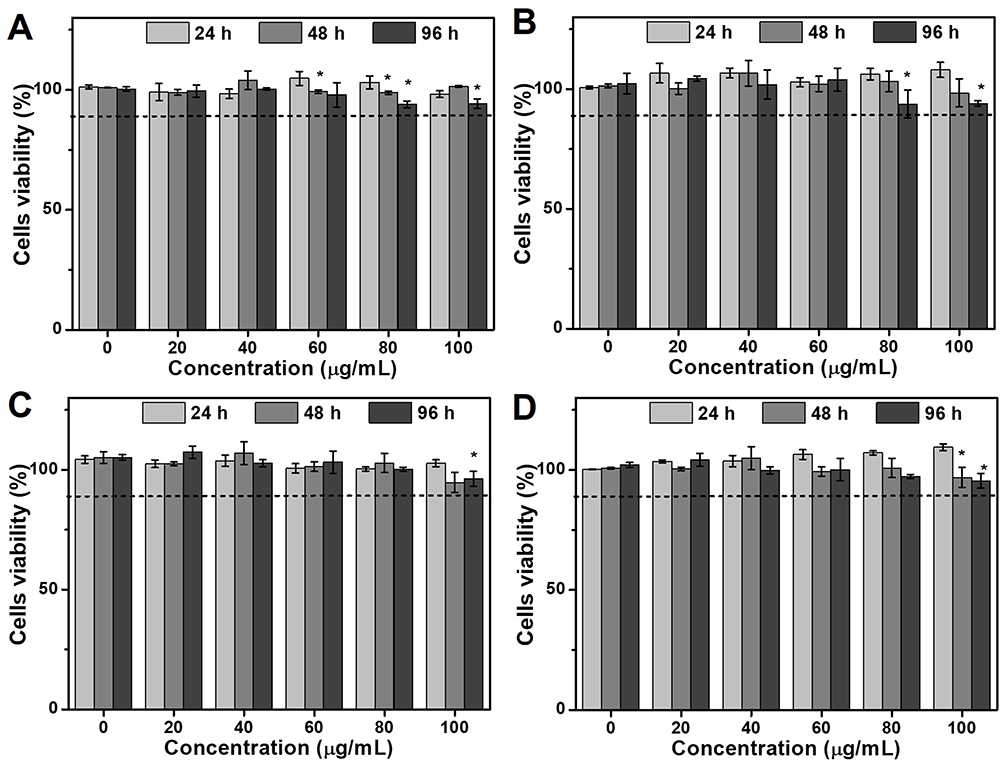 |
Figure 2 Cell viability of (A) immune cells (DCs), (B) cancer cells (HeLa), and epithelial cells ((C) HCECs and (D) HRPEs) after treatment with different concentrations of the HmA particles (0–100 μg mL−1) for 24 h. (P values: *P < 0.05, all the values are expressed as mean ± SD, n = 3). |
Evaluation of Intracellular Antibody Delivery Efficiency of HmA
Different cellular phenotypes, including cancer cells (HeLa), immune cells (DCs), and ocular epithelial cells (HCECs and HRPEs), were chosen to evaluate the ability of HmA@IgG-Cy3 to deliver IgG-Cy3. After incubating with HmA@IgG-Cy3 particles for 6 h, the living cells were imaged, and a red colour from Cy3 was clearly observed for all cell lines (top panel in Figure 3A). A comparison with the cells treated with free IgG-Cy3 (bottom panel in Figure 3A), which showed no red colours for all tested cells, strongly suggested that IgG-Cy3 was delivered inside the cells by the carrier. Roughly judging from the colour intensity, different cells exhibited a different cellular uptake ability of HmA@IgG-Cy3, following the uptake sequence HeLa > HRPEs ~ DCs > HCECs.
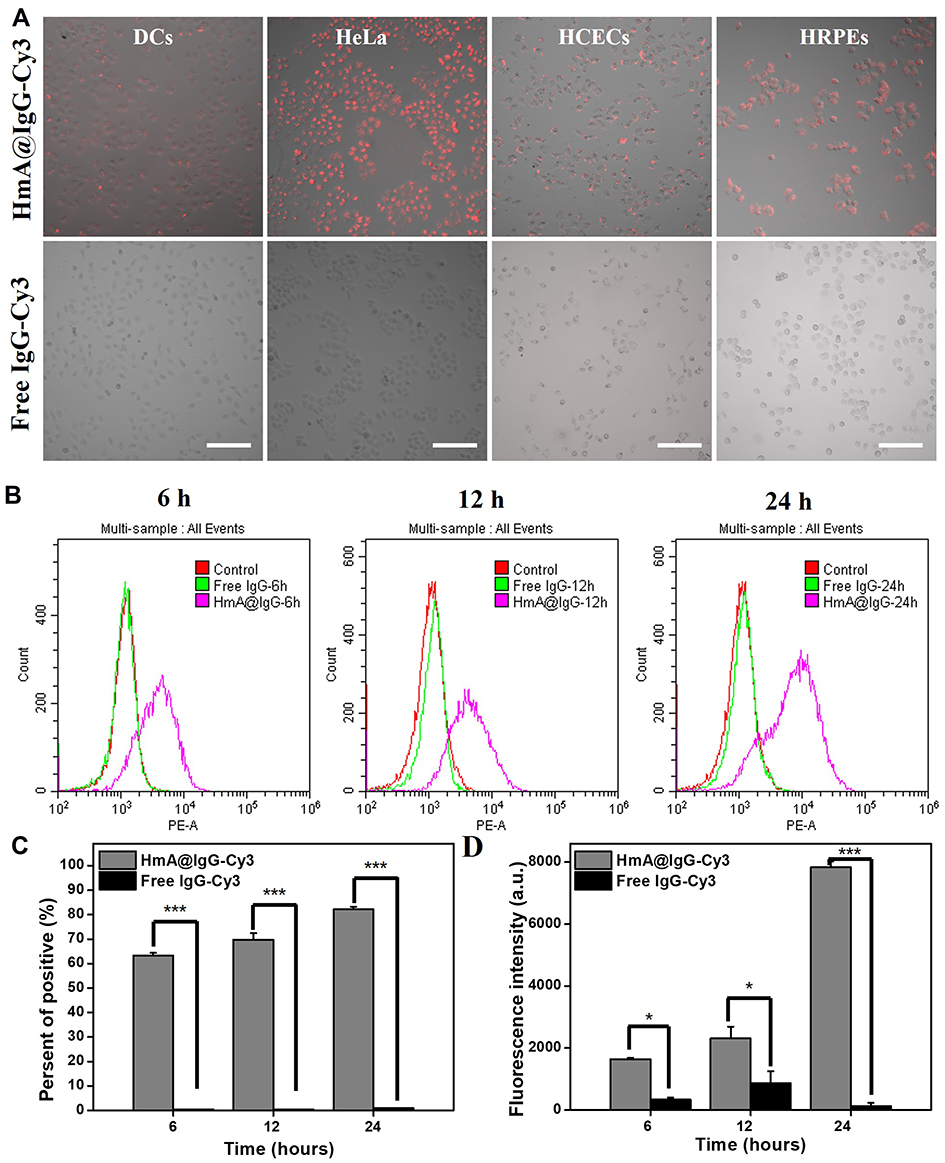 |
Figure 3 (A) Fluorescence images of DCs, HeLa cells, HCECs, and HRPE after treatment with HmA@IgG-Cy3 or free IgG-Cy3 for 6 h; IgG-Cy3 (red); (B) Flow cytometry histogram of the DCs after treatment with HmA@IgG-Cy3 or free IgG-Cy3 for 6 h, 12 h, and 24 h; Endocytosis-positive cell rate graph (C) and average fluorescence intensity graph (D) with HmA@IgG-Cy3 or free IgG-Cy3; *Indicates the differences between the experimental group and the control group: *p < 0.01, and ***p < 0.001. The scale bars were 200 μm. |
DCs are the most powerful antigen-presenting cells in the human body. They play a key role in antigen uptake, processing and presentation.21 Recently, researchers have discovered that the DC system exhibits a wide range of dysfunctions in the tumour microenvironment (TME), which ultimately affects the anti-tumour immune response.22 DC-based therapy demonstrated promising potential in tumour therapy by restoring the function of DCs in the TME.23 In addition, the uptake efficiency of DCs was moderate among these cells. Therefore, we chose DCs as the representative to evaluate the intracellular antibody delivery efficiency in the following experiments.
DCs were incubated with HmA@IgG-Cy3 particles (HmA: 50 μg mL−1) for 6 h, 12 h and 24 h. The intracellular fluorescence was tested by flow cytometry. The histogram results are shown in Figure 3B, and the quantitative analyses are shown in Figure 3C and D. The proportion of fluorescent DCs gradually increased from 63% at 6 h to 82% at 24 h after incubation with HmA@IgG-Cy3 particles, but less than 1% of fluorescent DCs for all tested time points were seen when incubated with free IgG-Cy3. The mean fluorescence intensity of the HmA@IgG-Cy3-treated DCs increased with an increasing incubation time over the whole test period, while there was no obvious fluorescent change for DCs treated with free IgG-Cy3. At 6 h, the intensity of HmA@IgG-Cy3 was 5-fold higher than that of free IgG-Cy3, and it became 60-fold higher at 24 h. All of these results suggested a rapid and potent intracellular uptake of HmA@IgG-Cy3 particles. It is worthy to be noted that these data in Figure 3 were obtained from the living cells, which was definitely advantageous to antibodies delivery by HmA, compared to widely and currently applied strategy to delivery antibodies inside dead cells.
To further objectively investigate the intracellular delivery efficiency of HmA, a commercial protein delivery reagent (PULSin) was used for comparison, as PULSin is highly effective for protein transfection in living cells and is commonly used in scientific research.24 Under fixed concentrations of carriers, IgG-Cy3 at different concentrations (from 0.125 μg mL−1 to 1.5 μg mL−1) was delivered by HmA and PULSin. After incubation for 4 h, LSCM images of the living DCs were taken and are shown in Figure 4A. Obviously, the uptake amount of IgG-Cy3 was concentration dependent and increased uptake was seen with an increasing IgG-Cy3 concentration. However, the increasing tendency of the intracellular fluorescence of HmA@IgG-Cy3 was much faster than that of PULSin@IgG-Cy3. In addition, at the same time point, the uptake amount of IgG-Cy3 delivered by HmA was much higher than that delivered by PULSin. The delivery difference between HmA and PULSin increased with increasing IgG-Cy3 concentration. The images were quantified by Image J, and the data is shown in Figure 4B. At lower IgG-Cy3 concentrations (0.125 μg mL−1 and 0.5 μg mL−1), there was no significant difference in the mean fluorescence intensity between the HmA and PULSin groups. When the concentration increased to 1 μg mL−1, the mean fluorescence intensity of the HmA group was approximately 3 times that of the PULSin group. At 1.5 μg mL−1, the difference increased to 4 times. These results favourably supported the potential of HmA as an efficient intracellular antibody delivery system, even compared to the commercial protein delivery agent, PULSin. One had to be mentioned, DCs intrinsically uptake various antigens, and therefore such uptake could be unspecific. In addition, it was not clear whether HmA@IgG-Cy3 were taken up inside the cell as a part of the endo-lysosomal complex, or they indeed escaped from the endosomal entrapment and localized in the cytosol. Hence, the endo-lysosomal escape process of HmA@IgG-Cy3 had to be further investigated. Considering the biofunctions of endocytic antibodies (such as β-tubulin and Anti-NPC) only exhibited after successful endo-lysosomal escape, intracellular biofunctions of β-tubulin and Anti-NPC delivered by HmA were further evaluated to confirm whether such endocytic antibodies were endosomal entrapment or endosomal escape.
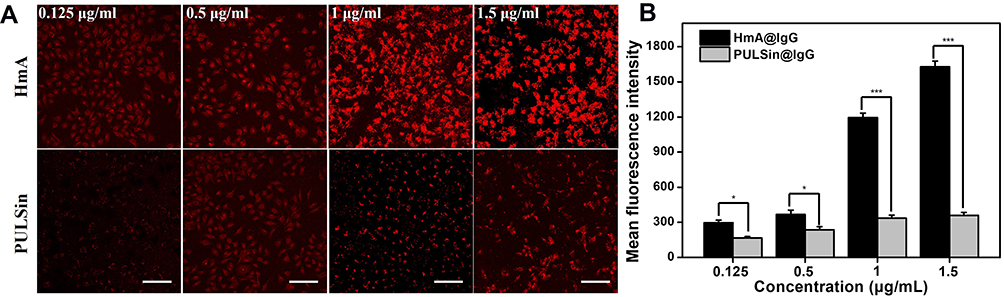 |
Figure 4 (A) Fluorescent photos of DCs after treatment with different concentrations of IgG-Cy3-loaded HmA or PULSin for 4 h. IgG-Cy3 (red), the scale bars were 100 μm; (B) Quantified fluorescence intensity graph using Image J. *Indicates the differences between the experimental group and the positive control group: *p < 0.01, and ***p < 0.001. |
Evaluation of the Intracellular Localization and Endolysosomal Escape of HmA@IgG-FITC
After being delivered into the cells, the fate of antibodies is crucial for their intracellular function preservation. To clarify the intracellular localization of the delivered antibodies, we used an antibody-early endosome marker (EER) and lysosome membrane marker (LyR) to perform fluorescent staining of subcellular organelles. The former was used for the localization of early endosomes, and the latter was used for lysosomal localization. Figure 5 shows the localization of IgG-FITC (green) and the staining of early endosomes (red) and lysosomes (red) in DCs after treatment with HmA@IgG-FITC particles for 4 h. The images demonstrated a moderate overlap between the red fluorescence of the early endosomes and the green fluorescence of IgG-FITC. In contrast, the fluorescence of lysosomes and IgG-FITC only slightly overlapped. The colocalization coefficients (Pearson correlation coefficients) of the early endosomes and lysosomes with antibodies were 0.12 and −0.05, respectively. The relatively large colocalization coefficient of the early endosomes and delivered antibodies indicated a portion of the delivered antibodies in the early endosomes after endocytosis. In contrast, a smaller colocalization coefficient of lysosomes and delivered antibodies verified a smaller portion of delivered antibodies were in lysosomes. This result demonstrated the endocytic pathway of HmA@IgG-FITC and indicated an effective escape of delivered antibodies from the endosome into the cytosol.
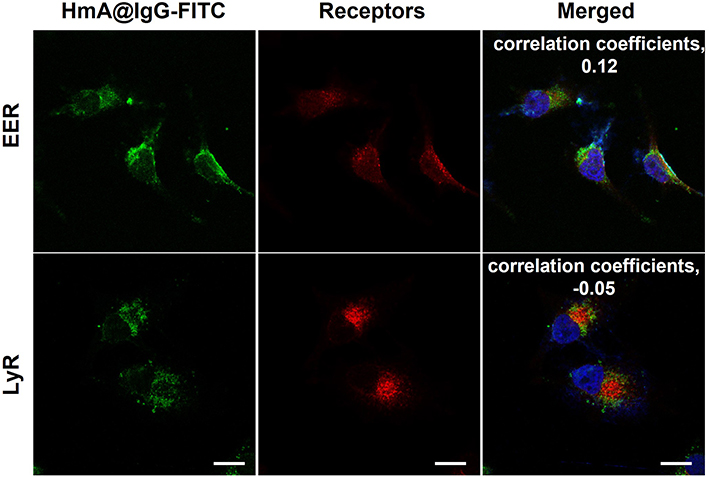 |
Figure 5 LSCM images of early endosome (EER)- and lysosome (LyR)-localized receptors in DC cells after treatment with different concentrations of HmA@IgG-FITC particles for 4 h; HmA@IgG-FITC particles (green); EER and LyR (red); DAPI (blue); The scale bars were 20 μm. |
Intracellular Target Delivery of Functional Antibodies
The intracellular functional preservation of antibodies after being delivered into cells is of great importance for intracellular targeting. Anti-β-tubulin and anti-NPC antibodies were chosen as the model antibodies for the functional protection tests of the antibodies. Anti-β-tubulin antibody targets intracellular β-tubulin, which is a component of the cellular skeleton. It is commonly used as the primary antibody for the immunofluorescence staining of microtubules in fixed and permeabilized cells. In this work, anti-β-tubulin antibodies were encapsulated by HmA under mild conditions and transported into live DCs. The LSCM images in Figure 6A show the anti-β-tubulin immunofluorescence staining images of DCs after incubating with HmA@anti-β-tubulin (HmA: 50 μg mL−1; anti-β-tubulin: 1 μg mL−1) or free anti-β-tubulin (anti-β-tubulin: 1 μg mL−1) for different times. Free anti-β-tubulin was used as the control, and conventional immunofluorescence staining of microtubules in the fixed and permeabilized DCs were used as the positive control. At 6 h, the HmA@Anti-β-tubulin group showed a high density of microtubular staining, which was similar to the positive control group. The free anti-β-tubulin group showed no fluorescent staining in the cytosol even after incubation with DCs for 24 h. In detail observation, the phenomenon indicated that HmA could not only transport the anti-β-tubulin antibodies into live DCs efficiently but also they were able to escape from the endosome and their specific binding function was preserved.
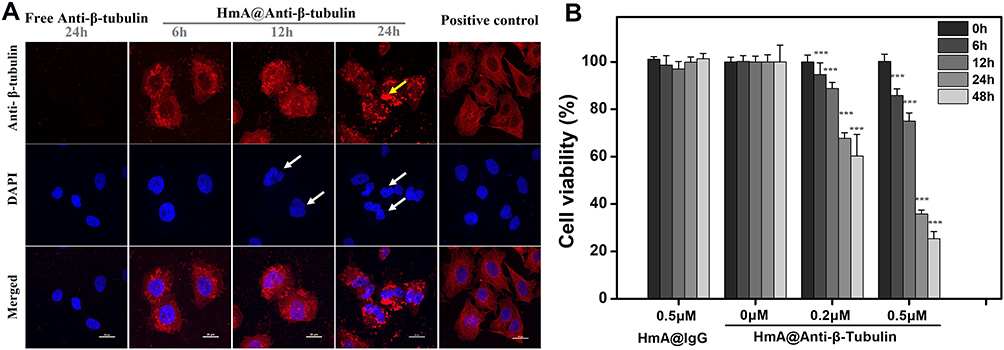 |
Figure 6 (A) LSCM images of anti-β-tubulin immunostained DCs after treating with the same concentration of HmA@Anti-β-tubulin for 6 h, 12 h and 24 h or free anti-β-tubulin (1 μg mL−1) for 24 h; Positive control: LSCM images of anti-β-tubulin immunostained untreated DCs; Anti-β-tubulin (red), DAPI (blue); The white arrow points to the deformed nucleus, the yellow arrow points to the aggregated tubulin; The scale bars were 20 μm; (B) The cell viability of DCs after treating with HmA@Anti-β-tubulin (0 μM, 0.2 μM, 0.5 μM) and HmA@IgG (0.5 μM) at different times. (P values: ***P <0.001, all the values are expressed as mean ± SD, n = 3). |
The images of the HmA@Anti-β-tubulin group demonstrated strong fluorescence aggregation (yellow arrow) and nuclear deformation (white arrow) when the incubation time was extended, which confirmed the dysfunction of microtubules in DCs and their gradual cell death. In contrast to the positive control, in which the anti-β-tubulin was penetrated inside dead cells through the punched holes, there was no nuclear deformation. Inside living cells, the microtubules, an essential element of the eukaryotic cytoskeleton, are formed by polymerizing α- and β-tubulin proteins into long filaments or chains.25 These microtubules play a crucial role in cell support and cell movement, making tubulin imperative to normal cell function. Once the β-tubulin protein was targeted by Anti-β-tubulin, the polymerization was stopped, which resulted in the disassembly of microtubules and further failure in supporting cell and nuclear deformation. In addition, it has been reported that dysfunctional microtubules can prevent the formation of endosomes and lysosomes, thereby affecting the metabolism of cells, leading to cell death.26,27 To further verify this result, CCK-8 assays were conducted to evaluate the cell viabilities of the DCs after treatment with different concentrations of HmA@anti-β-tubulin for different times (Figure 6B). The histogram demonstrated a dominant reduction in DC viability after treatment with HmA@anti-β-tubulin for 48 h (40% decrease for 0.2 μM; 75% decrease for 0.5 μM). However, the HmA@IgG-Cy3 group did not show any cytotoxicity. This result indicated that cell death was induced by the dysfunction of microtubules in DCs after the intracellular delivery of anti-β-tubulin by HmA, not the cytotoxicity of the HmA themselves.
After successful encapsulation and intracellular delivery of nonspecific IgG-Cy3 and specific anti-β-tubulin antibodies, the generality of functional antibody delivery to the cytosol by HmA was investigated. Anti-NPC antibody could specifically target the nuclear membrane pore protein complex, which showed conventional immunofluorescence staining of the nuclear membrane in fixed and permeabilized DCs (positive control). As shown in Figure 7, bright red fluorescence circles around the nuclei were demonstrated in the positive control group (white arrow), which indicated the specific targeting of the anti-NPC antibody to the nuclear membrane pore protein complex. The free anti-NPC group showed no red fluorescence in the cytosol, which indicated that no anti-NPC antibodies were delivered into living DCs. However, it is worth noting that the HmA@Anti-NPC group showed bright red fluorescence circles around the nuclei in living DCs (white arrow). This phenomenon was similar to that of the positive control group, but the anti-NPC antibodies were delivered to live DCs instead of dead DCs. These results validated the potential of HmA as generally applicable for efficient intracellular delivery of antibodies and a good functional preservation ability.
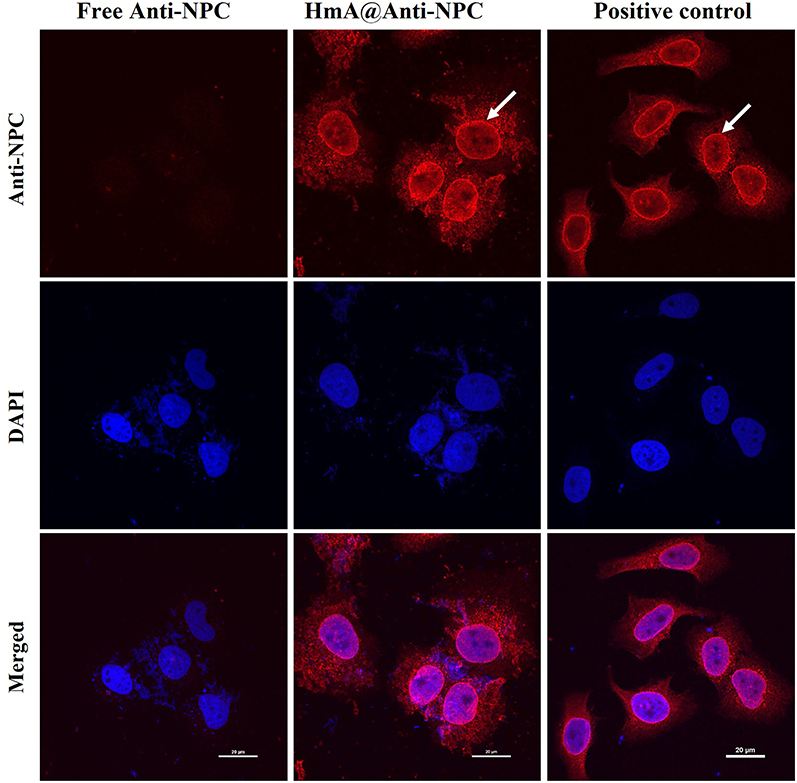 |
Figure 7 LSCM images of anti-NPC immunostained DCs after treatment with the same concentration of HmA@ Anti-NPC and Free Anti-NPC for 6 h; positive control: the anti-NPC immunostained untreated DCs; anti-NPC antibodies (red), the white arrows indicate the nuclear membrane, and the scale bars were 20 μm. |
Gathering the above data, HmA was a potent vehicle to deliver various antibodies (IgG, anti-β-tubulin and anti-NPC), exhibiting not only the high efficiency but also the tremendous advantages in delivering antibodies into living cells. Besides, the profile of endosomal escape displayed by HmA make it a promising candidate for delivering intracellular antibodies. The strong evidence on endosomal escape was insufficient due to the improper method or the lake of effective means. For example. Triton X-100, a permeabilization agent that allows penetration of antibodies to the cytosol, had to be used to allow the secondary antibody penetrating through for staining the EER and LyR in Figure 5 or the intracellular antibodies (anti-β-tubulin in Figure 6A and anti-NPC in Figure 7). Nevertheless, some phenomena still spied into the endosomal escape process of HmA@Antibodies, eg, in Figure 4A, much fewer punctate staining (an indicator to endosomal entrapment) of IgG-Cy3 delivered into living cells by HmA than that by PULSin; evenly distributed signal of anti-β-tubulin obtained from HmA (Figure 6A). From another aspect, the endosomal escape ability of HmA@Antibodies was able to be traced in many reports.28–30 In HmA@Antibodies, the imidazole groups of hexa-histidine and/or other groups (carboxyl, amino, and imidazole) of antibodies coordinated with zinc ions to form co-assemble particles.17 Such coordinative interaction was highly pH sensitive due to the protonation of imidazole (pKa ~ 6.8), reducing rapidly with the decreased pH and totally lost at pH around 4.5.31,32 After endocytosis, the HmA@Antibodies were wrapped into endosomes, in which the degrading enzymes were recruited and pH value gradually decreased with the endosomes maturing to pH ~ 4.5 ultimately.28,33 Obviously, the decreased pH in maturing endosomes finally leads to the disassembly of HmA@Antibodies into free Hisd to the disassembly of HmA@Antibodies into free His6 and antibodies. Meanwhile, the His6 was positively charged (the pH in matured endosomes was lower than its pKa value), which activated the endosomal escape and finally led to the destruction of the inner plastid membrane.25,26 Such pH sensitive profile of HmA propelled the trapped antibodies released into cytosol in a relatively short period to avoid the denature and/or enzymolysis of endocytic antibodies, ensuring their biofunctions played properly.
As reported, HmA displayed a very high efficiency to encapsulate model drugs with diverse physiochemical properties, no matter the hydrophilicity and molecular weight of drugs.17 Importantly, the mild conditions to generate HmA were advantageous to load various biomolecules (peptide, protein, antibodies, DNA, etc.), especially to biomolecules sensitive to harsh environments.19,34,35 And the fast endocytosis and endosomal escape of HmA are beneficial for the proper biofunctions of intracellular delivered biomolecules. All these profiles make HmA a promising vehicle for biomolecules delivery. Recently, we have developed a facile strategy to generate subunit vaccines based on HmA, in which the antigen (ovalbumin peptides) and adjuvant (cytosine-phosphate-guanine oligodeoxynucleotides, CpG ODNs) were co-delivered into antigen presenting cells (RAW264.7 and DC2.4) and elicited strong humoral and cellular immunities. Here, we demonstrated HmA effectively delivered different antibodies into living cells, and antibodies can exhibit bioactivities properly. This is of great significance for imaging of intracellular structure and/or cellular organelles in the living cell. Besides, because of their strong specificities and potency, antibodies have been widely used for treating various refractory diseases, such as neovascular diseases, cancer and autoimmune diseases.36 All antibodies-based therapeutics on current market have been explored based on targeting extracellular receptors (less than 30%). However, there were much longer list of intracellular receptors (more than 70%) which could be targeted by the corresponding antibodies. Unfortunately, intracellular receptors remained unexplored, not due to the technical difficulties in engineering corresponding antibodies, but the limited plasmid penetration of antibodies into cells. From this point, HmA can bring new chances in intracellular antibody therapeutics and huge market increase. Moreover, there are many intracellular receptors involved in various biological process (such as signal transduction, gene expression, proliferation and apoptosis of cells, etc.), which can be tuned by antibodies as well. In this case, HmA can shed the light on revealing intracellular signal transduction in life science.
Last but not least, the biotoxicity of HmA should get enough attention before it runs in the clinical trial, which may be approximately derived from its components, zinc ions and His6. His6-tag has been widely used for purification of fusion protein, then normally cut off after finishing the purification since it may cause unnecessary immune response to its tagged proteins.37 Although the His6 used here was a small molecule (0.84 kDa), unlike the tag in the fusion protein, it is still unclear whether such small His6 itself would cause adverse immunological responses or not. Zinc ions, as the second richest metal element in the body after iron, play critical roles in various life activities, such as differentiation, proliferation and apoptosis of cells, signal transduction, gene expression and nucleic acid metabolism.38 The deficiency of zinc ions normally leads to metabolic and organic dysfunctions.39 As recommended by Dietary Nutrient Reference, 12.5 mg/day zinc should be intake for adults aged 18–49, and the highest tolerable intake is about 40 mg/day. The highest amount of zinc ions used in this work for cellular cytotoxicity test was 50 μg/mL, which is far below recommended uptake, and very low cytotoxicity was found (Figure 2). In recent works, the injection of HmA into mice with different disease model did not bring any damage to their organs (kidney, liver, spleen, heart and lung) in the experimental time (about 28 days).20,40 However, many metal ions have long-term cytotoxicity and lead to irreversible disease, especially dangerous to neurocyte, which may bring HmA into very controversies when it applies in clinical treatment. Although zinc ions have a faster metabolic rate and a higher safe concentration range compared to other metal ions,39 the long-term cytotoxicity of HmA must be evaluated before any further clinical trial.
Conclusion
In conclusion, we successfully constructed a carrier based on co-assembling antibodies with His6-metal assembly (HmA), by which various antibodies (IgG, anti-β-tubulin and anti-NPC) could be delivered inside living cells. Antibodies could be encapsulated in situ into nanocarriers with a narrow size distribution and a positive charge under very gentle conditions and delivered into different cells (DCs, HeLa, HCECs, and HRPE). The intracellular delivery efficiency was even better than that of a commercial protein delivery reagent (PULSin). Most importantly, the antibodies could escape from the lysosomes after endocytosis and properly carry out their functions inside living cells. Our results indicate that this strategy is a feasible way to deliver various antibodies intracellularly while preserving their functions, which has great potential in treating many refractory diseases by intracellular antibody delivery.
Acknowledgments
This work was supported by the Natural Science Foundation of China (81774109 and 81973620), start-up funding from Wenzhou Institute of UCAS [WIUCASQD2019009 and WIUCASQD2021017].
Disclosure
There are no conflicts of interest to declare.
References
1. Chiu ML, Gilliland GL. Engineering antibody therapeutics. Curr Opin Struct Biol. 2016;38:163–173. doi:10.1016/j.sbi.2016.07.012
2. Mueller KL. Antibodies. The future is now. Introduction. Science. 2013;341(6151):1191. doi:10.1126/science.341.6151.1191
3. Awwad S, Angkawinitwong U. Overview of antibody drug delivery. Pharmaceutics. 2018;10(3):83–107. doi:10.3390/pharmaceutics10030083
4. Aggarwal SR. A survey of breakthrough therapy designations. Nat Biotechnol. 2014;32(4):323–330. doi:10.1038/nbt.2864
5. Marschall ALJ, Frenzel A, Schirrmann T, et al. Targeting antibodies to the cytoplasm. Mabs. 2011;3(1):3–16. doi:10.4161/mabs.3.1.14110
6. Trenevska I, Li DM, Banham AH. Therapeutic antibodies against intracellular tumor antigens. Front Immunol. 2017;8:12. doi:10.3389/fimmu.2017.01001
7. Slastnikova TA, Ulasov AV, Rosenkranz AA, Sobolev AS. Targeted intracellular delivery of antibodies: the state of the art. Front Pharmacol. 2018;9:1208. doi:10.3389/fphar.2018.01208
8. Kaiser PD, Maier J, Traenkle B, Emele F, Rothbauer U. Recent progress in generating intracellular functional antibody fragments to target and trace cellular components in living cells. Biochim Biophys Acta. 2014;1844(11):1933–1942. doi:10.1016/j.bbapap.2014.04.019
9. Postupalenko V, Desplancq D, Orlov I, et al. Protein delivery system containing a nickel-immobilized polymer for multimerization of affinity-purified his-tagged proteins enhances cytosolic transfer. Angew Chem Int Ed Engl. 2015;54(36):10583–10586. doi:10.1002/anie.201505437
10. Auf der Maur A, Tissot K, Barberis A. Antigen-independent selection of intracellular stable antibody frameworks. Methods. 2004;34(2):215–224. doi:10.1016/j.ymeth.2004.04.004
11. Fischer PM. Cellular uptake mechanisms and potential therapeutic utility of peptidic cell delivery vectors: progress 2001–2006. Med Res Rev. 2007;27(6):755–795. doi:10.1002/med.20093
12. Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157(2):195–206. doi:10.1111/j.1476-5381.2009.00057.x
13. Lin C, Zhang X, Chen H, et al. Dual-ligand modified liposomes provide effective local targeted delivery of lung-cancer drug by antibody and tumor lineage-homing cell-penetrating peptide. Drug Deliv. 2018;25(1):256–266. doi:10.1080/10717544.2018.1425777
14. Lim SI, Lukianov CI, Champion JA. Self-assembled protein nanocarrier for intracellular delivery of antibody. J Control Release. 2017;249:1–10. doi:10.1016/j.jconrel.2017.01.007
15. Abraham A, Natraj U, Karande AA, et al. Intracellular delivery of antibodies by chimeric Sesbania mosaic virus (SeMV) virus like particles. Sci Rep. 2016;6:21803. doi:10.1038/srep21803
16. Chiu HY, Deng W, Engelke H, et al. Intracellular chromobody delivery by mesoporous silica nanoparticles for antigen targeting and visualization in real time. Sci Rep. 2016;6:25019. doi:10.1038/srep25019
17. Huang W, Hao P, Qin J, et al. Hexahistidine-metal assemblies: a promising drug delivery system. Acta Biomater. 2019;90:441–452. doi:10.1016/j.actbio.2019.03.058
18. Zhang J, He M, Nie C, et al. Biomineralized metal-organic framework nanoparticles enable a primer exchange reaction-based DNA machine to work in living cells for imaging and gene therapy. Chem Sci. 2020;11(27):7092–7101. doi:10.1039/D0SC00339E
19. Huang W, Zhou S, Tang B, et al. Efficient delivery of cytosolic proteins by protein-hexahistidine-metal co-assemblies. Acta Biomater. 2021;129:199–208. doi:10.1016/j.actbio.2021.05.001
20. Zhang T, Zhang L, Wu X, et al. Hexahistidine-metal assemblies: a facile and effective codelivery system of subunit vaccines for potent humoral and cellular immune responses. Mol Pharm. 2020;17(7):2487–2498. doi:10.1021/acs.molpharmaceut.0c00212
21. Luckashenak N, Eisenlohr LC. Dendritic cells. Cancer Immunother. 2013;2013:55–70.
22. Wang JB, Huang X, Li FR. Impaired dendritic cell functions in lung cancer: a review of recent advances and future perspectives. Cancer Commun (Lond). 2019;39(1):43. doi:10.1186/s40880-019-0387-3
23. Zhu S, Yang N, Wu J, et al. Tumor microenvironment-related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharmacol Res. 2020;159:104980. doi:10.1016/j.phrs.2020.104980
24. Wang J, Ptacek JB, Kirkegaard K, Bullitt E. Double-membraned liposomes sculpted by poliovirus 3AB protein. J Biol Chem. 2013;288(38):27287–27298. doi:10.1074/jbc.M113.498899
25. Binarova P, Tuszynski J. Tubulin: structure, functions and roles in disease. Cells. 2019;8(10):1294–1301. doi:10.3390/cells8101294
26. Cole NB, Lippincott-Schwartz J. Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol. 1995;7(1):55–64. doi:10.1016/0955-0674(95)80045-X
27. Schafer DA. Coupling actin dynamics and membrane dynamics during endocytosis. Curr Opin Cell Biol. 2002;14(1):76–81. doi:10.1016/S0955-0674(01)00297-6
28. Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. doi:10.1016/j.jconrel.2010.11.004
29. Moreira C, Oliveira H, Pires LR, et al. Improving chitosan-mediated gene transfer by the introduction of intracellular buffering moieties into the chitosan backbone. Acta Biomater. 2009;5(8):2995–3006. doi:10.1016/j.actbio.2009.04.021
30. Benjaminsen RV, Mattebjerg MA, Henriksen JR, et al. The possible “proton sponge “ effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21(1):149–157. doi:10.1038/mt.2012.185
31. Lopez-Laguna H, Unzueta U, Conchillo-Sole O, et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomaterialia. 2019;83:257–264. doi:10.1016/j.actbio.2018.10.030
32. Pogostin BH, Malmendal A, Londergan CH, Akerfeldt KS. pKa determination of a histidine residue in a short peptide using raman spectroscopy. Molecules. 2019;24(3):405. doi:10.3390/molecules24030405
33. Logan R, Kong AC, Krise JP. Time-dependent effects of hydrophobic amine-containing drugs on lysosome structure and biogenesis in cultured human fibroblasts. J Pharm Sci. 2014;103(10):3287–3296. doi:10.1002/jps.24087
34. Liu Y, Huang Q, Wang J, et al. Microfluidic generation of egg-derived protein microcarriers for 3D cell culture and drug delivery. Sci Bull. 2017;62(18):1283–1290. doi:10.1016/j.scib.2017.09.006
35. Maus A, Strait L, Zhu D. Nanoparticles as delivery vehicles for antiviral therapeutic drugs. Eng Regener. 2021;2:31–46. doi:10.1016/j.engreg.2021.03.001
36. Malachowski T, Hassel A. Engineering nanoparticles to overcome immunological barriers for enhanced drug delivery. Eng Regener. 2020;1:35–50.
37. Cheung RCF, Wong JH, Ng TB. Immobilized metal ion affinity chromatography: a review on its applications. Appl Microbiol Biotechnol. 2012;96(6):1411–1420. doi:10.1007/s00253-012-4507-0
38. Sengupta J, Ghosh S, Datta P, et al. Physiologically important metal nanoparticles and their toxicity. J Nanosci Nanotechnol. 2014;14(1):990–1006. doi:10.1166/jnn.2014.9078
39. Baltaci AK, Yuce K, Mogulkoc R. Zinc metabolism and metallothioneins. Biol Trace Elem Res. 2018;183(1):22–31. doi:10.1007/s12011-017-1119-7
40. Shi P, Tang B, Zhou S, et al. Sheltering proteins from protease-mediated degradation and a de novo strategy for preventing acute liver injury. Biomater Sci. 2021;9(12):4423–4427. doi:10.1039/D1BM00063B
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.