")
Back to Journals » International Journal of Nanomedicine » Volume 12
Efficient co-delivery of immiscible hydrophilic/hydrophobic chemotherapeutics by lipid emulsions for improved treatment of cancer
Authors Zhang B, Song YM, Wang TQ, Yang SM, Zhang J, Liu YJ, Zhang N, Garg S
Received 30 November 2016
Accepted for publication 24 February 2017
Published 7 April 2017 Volume 2017:12 Pages 2871—2886
DOI https://doi.org/10.2147/IJN.S129091
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Bo Zhang,1 Yunmei Song,2 Tianqi Wang,1 Shaomei Yang,1 Jing Zhang,1 Yongjun Liu,1 Na Zhang,1 Sanjay Garg2
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Shandong University, Jinan, Shandong Province, People’s Republic of China; 2Centre for Pharmaceutical Innovation and Development (CPID), School of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA, Australia
Abstract: Combinational nanomedicine is becoming a topic of much interest in cancer therapy, although its translation into the clinic remains extremely challenging. One of the main obstacles lies in the difficulty to efficiently co-deliver immiscible hydrophilic/hydrophobic drugs into tumor sites. The aim of this study was to develop co-loaded lipid emulsions (LEs) to co-deliver immiscible hydrophilic/hydrophobic drugs to improve cancer therapy and to explore the co-delivery abilities between co-loaded LEs and mixture formulation. Multiple oxaliplatin/irinotecan drug–phospholipid complexes (DPCs) were formulated. Co-loaded LEs were prepared using DPC technique to efficiently encapsulate both drugs. Co-loaded LEs exhibited uniform particle size distribution, desired stability and synchronous release profiles in both drugs. Co-loaded LEs demonstrated superior anti-tumor activity compared with the simple solution mixture and the mixture of single-loaded LEs. Furthermore, co-loaded nanocarriers could co-deliver both drugs into the same cells more efficiently and exhibited the optimized synergistic effect. These results indicate that co-loaded LEs could be a desired formulation for enhanced cancer therapy with potential application prospects. The comparison between co-loaded LEs and mixture formulation is significant for pharmaceutical designs aimed at co-delivery of multiple drugs.
Keywords: cancer, combination therapy, co-delivery, lipid emulsions, drug–phospholipid complex
Introduction
Cancer is a major public health problem worldwide with increasing incidence and mortality.1 Combination chemotherapy is widely employed in clinics,2,3 with the objective of improving therapeutic effect and decreasing drug resistance, by targeting a single oncogenic pathway through different modes of action or across multiple interrelated pathways.4–6 Taking into account that the traditional administration in clinics is just a simple cocktail, the uncoordinated pharmacokinetics and uncontrolled release properties of different drugs restrict their applications, leading to the uncertainty of treatment.7,8 Nanomedicine approach of synergistic drug combinations can normalize pharmacokinetics and pharmacodynamics of the active agents, control the synchronous release in target site and provide superior therapeutic effects.5,9
Several different types of nanocarriers have been developed to co-deliver multiple drugs such as liposomes, micelles and mesoporous silica.10,11 Liposomes could encapsulate both hydrophilic and hydrophobic drugs due to the special core/shell structure.12,13 Micelles could co-load different cargoes by both encapsulation and conjugation.14,15 Mesoporous silica could trap different drugs in the pores due to their tunable pore sizes and large pore volumes.16,17 Nevertheless, the translation of nanocarrier-based combination therapy into the clinic remains extremely challenging, and none of the products have been approved until now.5,9 Alongside the complexity of physicochemical variability, safety concerns and regulatory and manufacturing issues,18 major obstacles also lie in the difficulty in achieving efficient co-delivery of multiple chemotherapeutics, accurate drug-loading ratio and controlled synchronous release, especially for immiscible hydrophilic/hydrophobic drugs.19–21
Consequently, development of nanocarriers that can provide efficient co-delivery of immiscible hydrophilic/hydrophobic drugs with mature technology for industrial production is crucial. Keeping this in mind, lipid emulsions (LEs) were selected as the desired carriers to achieve the co-delivery ability of multiple drugs and the improvement of cancer therapeutic effect. LEs, also named as fat emulsions or lipid microspheres, generally include emulsifiers, co-emulsifiers, stabilizers and iso-osmotic adjusting agents to form a homogeneous oil-in-water formulation.22–24 LEs could entrap the drug in the inner oil phase and hence avoid the drug leaking, precisely controlling the loading ratio of multiple drugs, and co-deliver the drugs into the tumor sites. Compared with the other emerging nanocarriers, LEs possess several advantages, including mature manufacturing and scalable technology, desired stability and reliable safety of excipients.25,26 Furthermore, LEs have been widely used for several decades with many products available in clinics, such as diazepam, vitamin, propofol, prostaglandin, etomidate and flurbiprofen.27–29
To evaluate the co-delivery profiles of LEs, oxaliplatin (OXA) and irinotecan (IRI) were selected as model drugs in this study. Both chemotherapeutics are widely used for the treatment of colorectal cancer in clinics, and their combination regimen has been recommended as a first-line treatment as per the National Comprehensive Cancer Network (NCCN) guidelines.30 These drugs are immiscible with notably different water solubility, and co-delivery of both hydrophilic and hydrophobic chemotherapeutics was expected to be more challenging.
Another objective of this study was to evaluate the differences between a mixture of single LEs (separated-loading pattern) and co-loaded LEs (synchronous-loading pattern). It is expected that the single-loaded formulation would be more controllable in manufacturing and more convenient in controlling the drug ratios via simple mixing. Due to the similar release and biodistribution profiles attributed by the same nanocarriers, the delivery abilities for both drugs are expected to be closely matching. On the contrary, many previous studies described co-loaded nanoparticles exhibiting superior anti-tumor effect compared to the simple mixture.12,31–35 Therefore, the differences in the therapeutic effect and the co-delivery ability were also compared between synchronous-loading and separated-loading formulations, which will add to the knowledge on co-delivery of multiple drugs.
The aim of this study was to develop LEs as the nanocarriers for co-delivery of immiscible hydrophilic/hydrophobic chemotherapeutics. Oxaliplatin and irinotecan co-loaded LEs (OXA/IRI-LEs) were prepared using the drug–phospholipid complex (DPC) technique, characterized and evaluated by in vitro and in vivo tests. Physicochemical properties of the emulsions were characterized with respect to particle size, zeta potential, morphology and stability. In vitro release study was performed to investigate the synchronized release profiles of both drugs. To assess the therapeutic benefits, in vitro cytotoxicity was tested using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay and in vivo anti-tumor activity was performed in CT-26-bearing BALB/c mice. Near infrared fluorophore (NIRF) imaging was applied to mimic the biodistribution of the drugs. To assess the co-delivery ability into the same cells between the co-loaded emulsions and the physical mixture formulation, the in vitro cellular uptake and in vivo confocal laser scanning microscopy (CLSM) images of cryo-sections were analyzed.
Methods
Materials
OXA and IRI were purchased from Boyuan Pharmaceutical Co., Ltd. (Jinan, China) and Knowshine Pharmachemicals Inc. (Shanghai, China), respectively. Egg phosphatidylcholine (EPC) and Pluronic F68 were from AVT Pharmaceutical Technology Co., Ltd. (Shanghai, China) and Sigma-Aldrich (St Louis, MO, USA), respectively. MTT was purchased from Solarbio® Life Science (Beijing, China). Medium chain triglyceride (MCT) was provided by Luoxin Pharmaceutical Co., Ltd. (Linyi, China). All the other reagents were of analytical purity grade or higher, obtained commercially.
Cell cultures
Murine colon carcinoma cells (CT-26) and human colon cancer cells (HCT-116) were purchased from Chinese Academy of Sciences (Shanghai, China). Both cell lines were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS), 1% penicillin and 1% streptomycin at 37°C in an environment containing 5% CO2.
Animals
Female BALB/c mice (weight: 18±2 g) were supplied by the Medical Animal Test Center of Shandong University (Jinan, China). The animals were fed with a standard diet and allowed water ad libitum. All experiments were carried out in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China (Document No 55, 2001). This study and the experiments were formally approved by the Animal Ethics Committee of Shandong University (201002050).
Formulation of DPC
Oxaliplatin–phospholipid complex (OPPC): 20 mg OXA and 300 mg EPC were dissolved in 20 mL methanol/dichloromethane (DCM) (v/v 9:1) and reacted at 40°C for 3 h to form the complex. The organic solvent was removed using a rotary evaporator to get OPPC.
Irinotecan–phospholipid complex (IPPC): 50 mg IRI and 200 mg EPC were dissolved in 10 mL DCM and reacted at 40°C for 3 h to form the complex. The organic solvent was removed using a rotary evaporator to get IPPC.
Preparation of OXA/IRI-LEs
The preparation method for co-loaded emulsion is illustrated in Figure 1. OPPC and IPPC were re-dissolved in DCM and 2 g MCT and 50 mg oleic acid (OA) were added; DCM was removed using a rotary evaporator, and the oil phase was obtained. A total of 200 mg Pluronic F68 and 800 mg glycerol were dissolved in 40 mL water to get the aqueous phase. The oil phase was added dropwise into the aqueous phase under shearing at 60°C. The formed coarse emulsion was further circulated through a high-pressure homogenizer (Panda 1K NS1001L; Niro Soavi SpA, Parma, Italy). For single-drug-loaded LEs, similar preparation procedure was performed while only OPPC or IPPC was added.
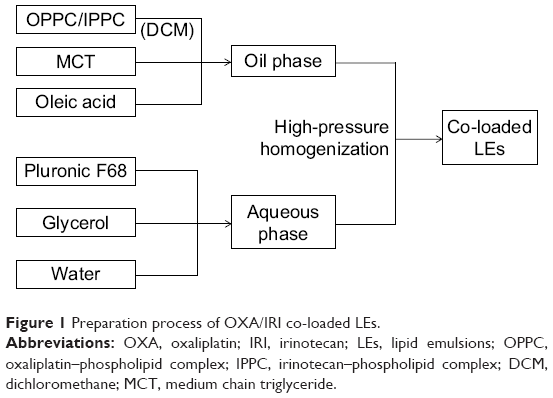 | Figure 1 Preparation process of OXA/IRI co-loaded LEs. |
Characterization
DPC was characterized by differential scanning calorimetry (DSC) method. Samples were sealed in aluminum crucible and heated from 20°C to 350°C at a flow of 50 mL/min under nitrogen atmosphere (TGA/SDTA851e; Mettler Toledo, Greifensee, Switzerland). DSC signals were recorded at a scanning rate of 10°C per minute heating process.
Size distribution, polydispersity index (PDI) and zeta potential of the emulsions were measured by the dynamic light scattering method using Zetasizer Nano ZS90 (Malvern, Worcestershire, UK). All measurements were performed in triplicate (n=3) and values represented as mean ± standard deviation (SD).
The morphologies of emulsions were visualized by transmission electronic microscopy (TEM) (JEM-1200EX, Japan). The samples were added to the surface of copper grids and stained with phosphotungstic acid (1%, w/v). The accelerating voltage was 120 kV.
Physical stability of emulsions was evaluated according to the stability constant (Ke) values.36 Samples (1 mL) were centrifuged at 3,000 rpm for 15 min in a centrifuge (Eppendorf AG 22331, Hamburg, Germany). A sample (50 μL) was extracted from the bottom and diluted with deionized water. Absorbance of the sample (A) was measured using a UV-visible spectrometer (Persee TU-1810, Beijing, China) at a wavenumber of 500 nm. Moreover, the absorbance of LEs without centrifugation (A0) was also measured at the same procedure. Ke was calculated using Equation (1):
|
|
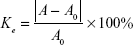
To evaluate the storage stability of the formulation, co-loaded LEs were passed through 220 nm filters for sterilization, and 0.5 mL sample was extracted into different tubes and kept in a refrigerator (4°C) or room temperature (20°C±2°C). Each sample was taken out at predetermined days, and the size distribution and PDI were measured using Zetasizer Nano ZS90 (Malvern).
In vitro release
In vitro release of OXA and IRI from LEs was determined by the dialysis method. Tween-80 (0.5% w/v) was added in phosphate-buffered saline (PBS) (pH 7.4) as the release medium due to the poor solubility of IRI and sink condition requirement of the release test.37 Briefly, 1 mL sample was added to the dialysis bag and incubated with 20 mL release medium in a plastic tube at 37°C. At the predetermined time intervals, 2 mL of release medium from outside of the dialysis bag was withdrawn and replaced with fresh medium. The cumulative amounts of OXA and IRI in release medium were determined by high-performance liquid chromatography (HPLC) and UV spectrophotometer, respectively. All measurements were carried out in triplicate.
In vitro cytotoxicity
In vitro cytotoxicity of different formulations was tested in CT-26 and HCT-116 cells using MTT assay. Cells were seeded in 96-well plates at a density of 5,000 per well in 150 μL of RPMI-1640 medium. After overnight incubation, the cells were treated with each formulation and incubated for 48 h. MTT (20 μL, 5 mg/mL) was added to each well and incubated for another 4 h. The cell plates were centrifuged at 3,000 rpm for 10 min, and the culture medium was discarded; 150 μL of dimethyl sulfoxide (DMSO) was added to dissolve formazan crystals. The absorbance of the obtained DMSO solution was measured at a wavelength of 570 nm with a microplate reader (Model 680; Bio-Rad, Hercules, CA, USA). The relative cell viability (%) was calculated using Equation (2):
|
|
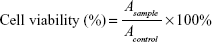
where Acontrol and Asample represent the absorbance of negative control and sample, respectively.
Eight groups were set as 1) blank lipid emulsions (Blank-LEs); 2) oxaliplatin solution (OXA-Sol); 3) irinotecan solution (IRI-Sol); 4) oxaliplatin lipid emulsions (OXA-LEs); 5) irinotecan lipid emulsions (IRI-LEs); 6) oxaliplatin plus irinotecan solution (OXA/IRI-Sol); 7) mixture of oxaliplatin lipid emulsions and irinotecan lipid emulsions (OXA-LEs/IRI-LEs) and 8) OXA/IRI-LEs.
In vivo anti-tumor activity
CT-26-bearing female BALB/c mice xenograft model was used to investigate the in vivo anti-tumor efficacy. The mice were subcutaneously injected at the right axillary space with 0.1 mL of cell suspension containing 1×106 CT-26 cells. After cell inoculation, a solid tumor was allowed to grow ≥100 mm3. After 1 week, mice were divided into nine groups (six mice per group): 1) 5% glucose solution (Control); 2) Blank-LEs; 3) OXA-Sol; 4) IRI-Sol; 5) OXA-LEs; 6) IRI-LEs; 7) OXA/IRI-Sol; 8) OXA-LEs/IRI-LEs and 9) OXA/IRI-LEs.
The dosage of OXA and IRI was selected as 5 and 12.5 mg/kg according to our previous study and the publications.38–41 The mice in each group were injected with the abovementioned formulations in the tail vein, once per week for 3 weeks. After the first administration, the tumor diameters were measured with a caliper and the body weights of the mice were determined using an electronic balance every 2 days. After 3 weeks, the mice were sacrificed, and the tumors were excised and weighed. The tumor volume was calculated using the following Equation (3):
|
|
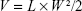
where L and W represent the length and width of the tumor, respectively.
In vivo and ex vivo NIRF imaging
Real-time NIRF imaging was applied to observe the biodistribution of the drugs. DiR was selected as the NIRF dye due to the excellent penetration ability from skin. DiR-loaded LEs were prepared via the same method as earlier, and the final concentration for DiR was 50 μg/mL (Supplementary materials).
CT-26-bearing female BALB/c mice were used for NIRF imaging. When the tumor volume reached >300 mm3, mice were administered 0.2 mL of free DiR and DiR-loaded LEs via the tail vein injection. After 1, 4, 8 and 24 h, mice were anesthetized with 10% chloral hydrate (intraperitoneally) and imaged. At the end of imaging, the mice were sacrificed, and heart, liver, spleen, lung, kidney and tumor were harvested for further ex vivo imaging. The real-time NIRF images were acquired using Xenogen IVIS Lumina system (Caliper Life Sciences, Hopkinton, MA, USA) with the ICG filter (excitation: 745 nm, emission: 835 nm, exposure time: 3 s). Results were analyzed using Living Image 3.1 software (Caliper Life Sciences).
In vitro cellular uptake
DiI (red fluorescence) and DiO (green fluorescence) were selected as the fluorescent dyes for labeling the LEs. DiI-and/or DiO-loaded LEs were prepared using the same procedure as described earlier (Supplementary materials).
CT-26 cells and HCT-116 cells were cultured overnight. Labeled LEs were added to the cells at the final concentration of 5 μg/mL for both DiI and DiO. Two groups were studied as 1) mixture group: the mixture of DiI-LEs and DiO-LEs; and 2) co-loaded group: DiI and DiO co-loaded LEs. After incubation for 0.5, 2 and 4 h, the cells were washed three times with cold PBS, fixed with 4% paraformaldehyde for 20 min, and the nuclei were stained with Hoechst 33342 for 15 min. Cellular uptake of the co-loaded LEs was visualized using a confocal laser scanning microscopy (LSM-780; Carl Zeiss, Germany).
For quantifying the cellular uptake efficiency, the cells were digested and re-suspended in 0.1 mL PBS after incubating with DiI- and DiO-labeled LEs, and cellular uptake was quantified using flow cytometry (FACS Calibur; BD Biosciences, San Jose, CA, USA).
In vivo co-delivery study
In order to evaluate the in vivo co-delivery ability of LEs into the tumor, cryo-section observations were carried out in CT-26-bearing female BALB/c mice. The mice were subcutaneously injected at the right axillary space with 0.1 mL of cell suspension containing 1×106 CT-26 cells. When the tumor volume reached 200–300 mm3, mice were injected with 1) DiI and DiO mixture solution, 2) mixture of DiI-LEs and DiO-LEs and 3) DiI/DiO co-loaded LEs. The administration concentration for both DiI and DiO was 125 μg/mL. After the mice were sacrificed at 12 h post intravenous (i.v.) injection, the tumors were collected and cryo-sectioned with the thickness of 10 μm. The nuclei were stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and then imaged using a confocal laser scanning microscope (LSM 780; Carl Zeiss).
Statistical analysis
All experimental data are presented as the mean ± SD. Statistical differences were evaluated using Student’s t-test (Excel 2007, Microsoft), and P<0.05 was considered to be statistically significant.
Results
DSC thermograms of DPCs
Both OPPC and IPPC were formulated for further drug loading, and DSC thermograms were employed to confirm the formation of the complexes. DSC is a reliable way to screen drug and excipient compatibility, and it provides maximum information about possible interactions.42,43 As shown in Figure 2, both DSC curves of OPPC and IPPC showed that original peaks of the drugs and phospholipid disappeared. The significant differences between the pure drug and the complex indicated weak interactions such as the hydrogen bonds or van der Waals force between the drugs and phospholipid molecules leading to the formation of the DPCs.44
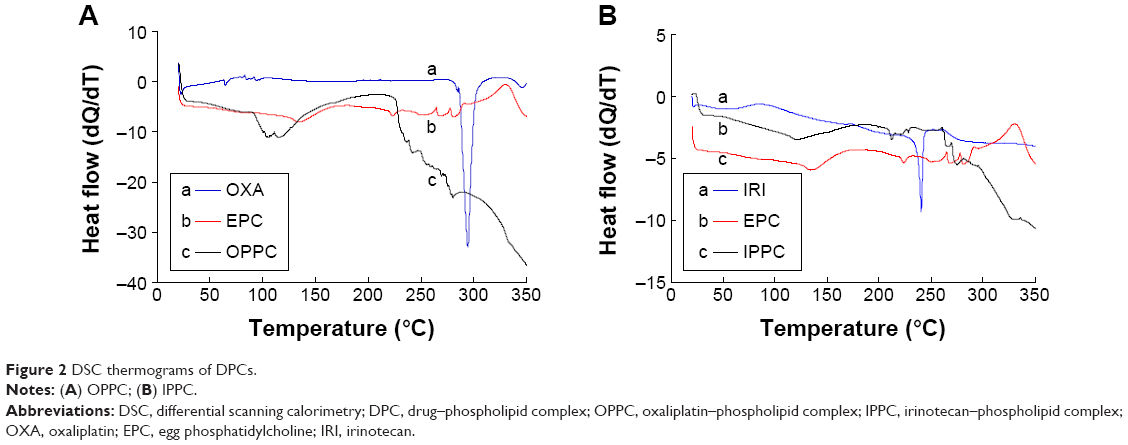 | Figure 2 DSC thermograms of DPCs. |
Preparation and characterization of co-loaded LEs
The optimized formulation of co-loaded LEs is listed in Table 1. MCT was selected as the oil due to its better miscibility of the drugs than long-chain oil, and Pluronic F68 was added as the emulsifier in aqueous phase for better emulsification. Both lipid and F68 as emulsifiers are approved by the US Food and Drug Administration for intravenous (IV) injection, indicating good safety of the excipients. OA and glycerol were used as stabilizer and iso-osmotic adjusting agent, respectively. The mass ratio of OXA/IRI was optimized at 1:2.5 based on our previous attempts, equal to molar ratio 1:1.5.38
 | Table 1 Optimized formulation of OXA/IRI co-loaded LEs |
The particle sizes of all emulsion droplets were ~100 nm with PDI <0.2 (Figure 3; Table 2), indicating that the prepared LEs were uniform with narrow particle size distribution and suitable for IV injection. It is known that the particle size ~100–200 nm could promote the extravasation from blood vessels and interstitial transport into the tumor issue via the enhanced permeation and retention (EPR) effect.45,46 The particle size of co-loaded LEs was slightly increased in comparison to the single-loaded LEs, which might be caused by stereo-specific occupation. The morphologies of co-loaded LEs in Figure 3C showed that the formulation was well dispersed with spherical shape. In addition, the average zeta potential of the prepared LEs was around −20 mV; this negative charge would avoid the recognition by plasma and decrease the impact on the blood clearance.47
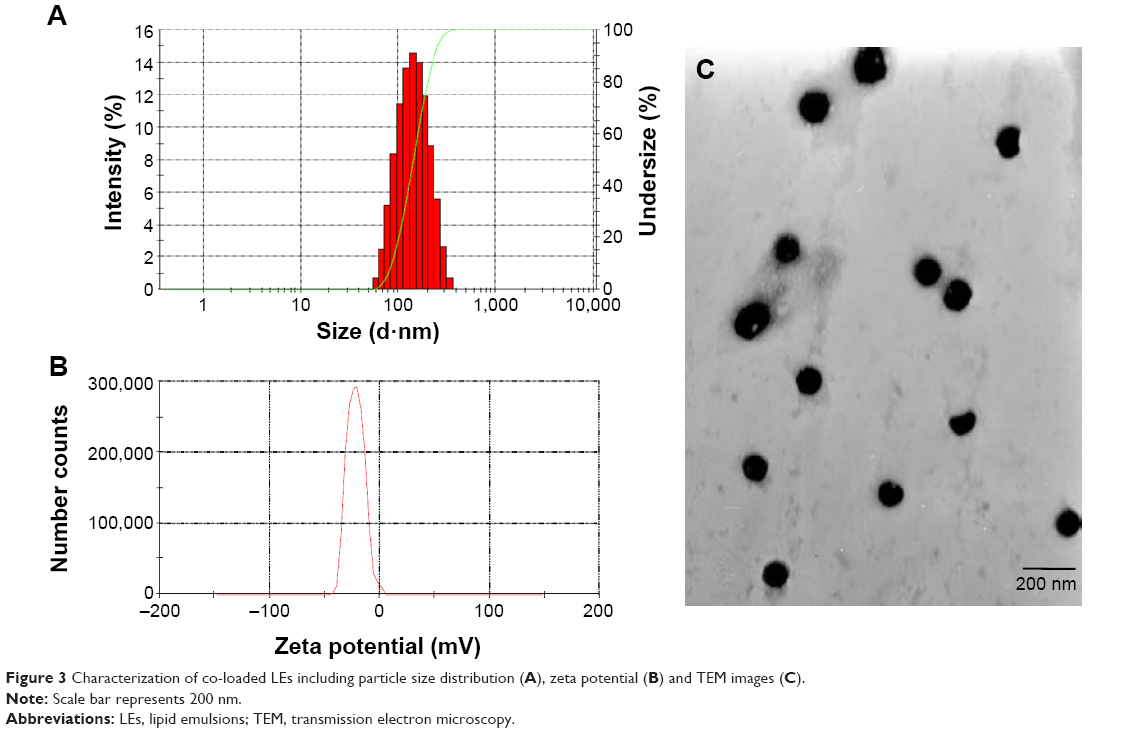 | Figure 3 Characterization of co-loaded LEs including particle size distribution (A), zeta potential (B) and TEM images (C). |
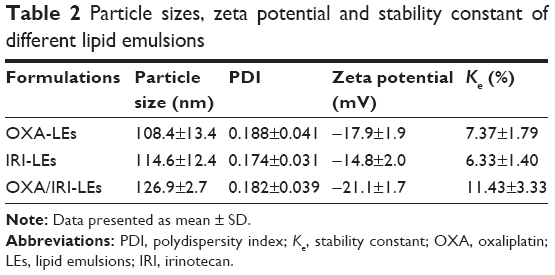 | Table 2 Particle sizes, zeta potential and stability constant of different lipid emulsions |
The stability constant (Ke), evaluated by the change in the degree of absorbance, is a quantitative method to determine the physical stability of emulsions. The smaller Ke value indicates a more stable emulsion. Although the Ke value of co-loaded LEs was slightly increased, it was still only ~10% indicating good physical stability of LEs.
The storage stability of co-loaded LEs was evaluated by measuring the particle size and PDI changes for 91 days. The sample stored at 4°C was stable without obvious size and PDI changes. Meanwhile, the sample at ambient temperature (20°C±2°C) exhibited significant size increase to nearly 300 nm after 91 days indicating the changes to LEs’ structure (Figure 4). It was concluded that the LE formulation should be stored at 4°C.
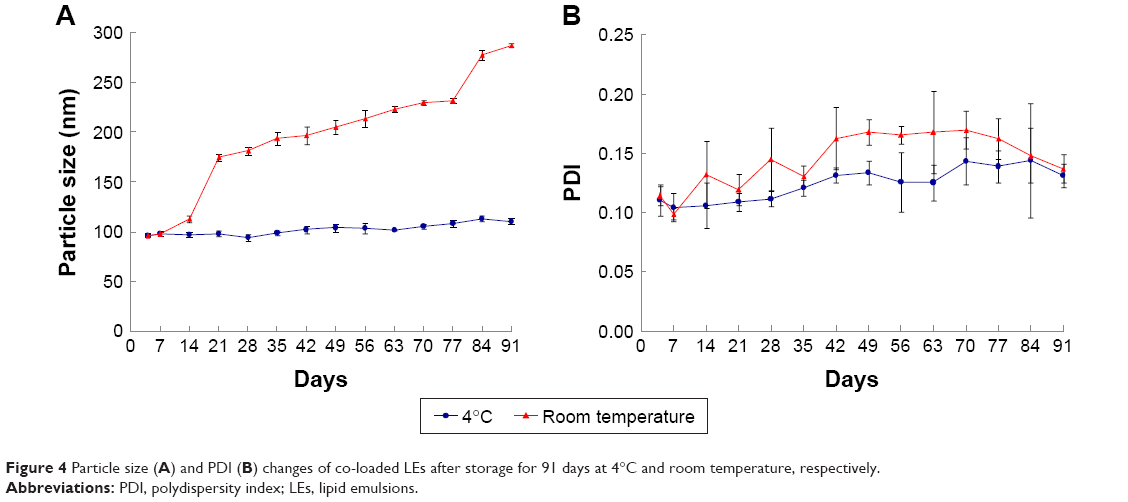 | Figure 4 Particle size (A) and PDI (B) changes of co-loaded LEs after storage for 91 days at 4°C and room temperature, respectively. |
In vitro release
In vitro release study was carried out using dynamic membrane dialysis method. As illustrated in Figure 5, OXA from OXA-LEs and OXA/IRI-LEs had similar release behaviors: nearly 40% burst release within 30 min, followed by a sustained release and ~80% release after 48 h. While IRI exhibited sustained release behaviors at all time intervals without obvious initial burst release, only 20% released within 2 h due to the hydrophobicity. Both drugs showed similar release profiles after 4 h indicating the synchronous release of both drugs.
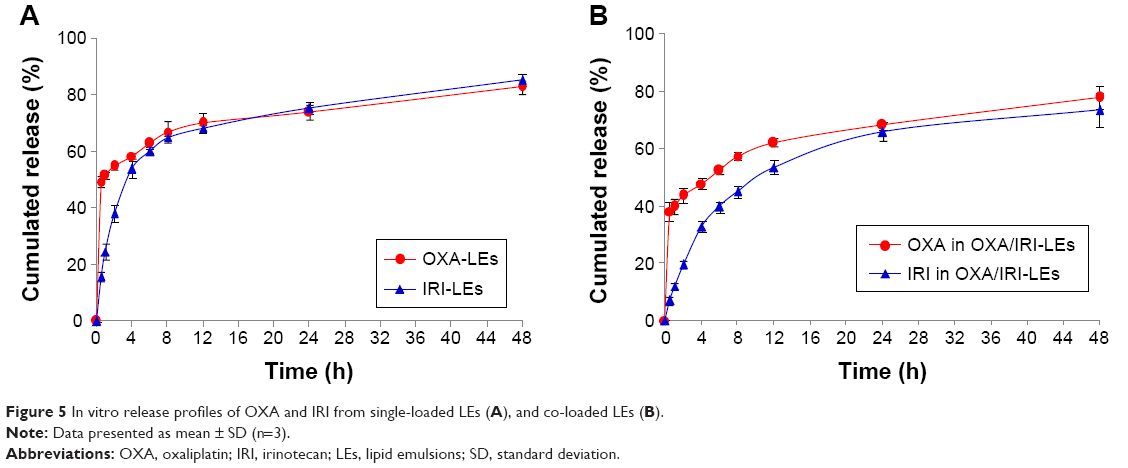 | Figure 5 In vitro release profiles of OXA and IRI from single-loaded LEs (A), and co-loaded LEs (B). |
We previously verified that the maximum synergism existed at the IRI/OXA molar ratios of 1:1–1.5:1.38 The released IRI/OXA molar ratios at different time intervals were further calculated to investigate the synergistic effect in this study (Table 3). Mixture and co-loaded LEs could reach the optimal synergism after 2 and 4 h, respectively. The release rate of co-loaded LEs lagged behind that of single LEs, which might be caused by the competition of both drugs diffusing from the inner oil phase. On the other hand, the delayed release of co-loaded LEs might be beneficial for decreasing the leakage during the circulation process. Overall, these results indicate that the co-loaded LEs have equivalent release profiles with little influence compared with single formulations.
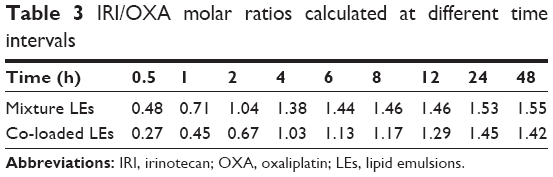 | Table 3 IRI/OXA molar ratios calculated at different time intervals |
In vitro cytotoxicity
In vitro cytotoxicity of different formulations was evaluated by MTT assay (Figure 6). Blank LEs exhibited no significant toxicity in CT-26 cells, while slight toxicity was seen at higher concentrations in HCT-116 cells. The difference might be caused by the varying sensitivity of the cells owing to the different gene expressions, especially for different murine and human-derived cells. OXA-LEs and IRI-LEs showed comparative cytotoxicity compared with their solutions in both cells. All the combination groups exhibited stronger cell-killing ability than single groups. Furthermore, co-loaded LEs showed similar cytotoxicity with both mixture solutions and mixture LEs (P>0.05).
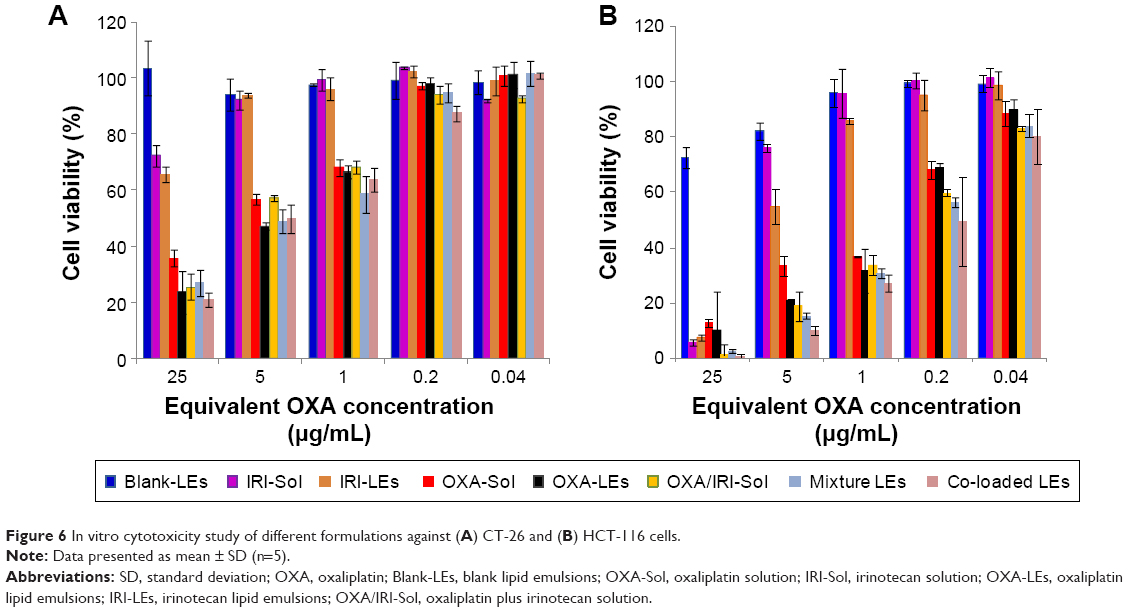 | Figure 6 In vitro cytotoxicity study of different formulations against (A) CT-26 and (B) HCT-116 cells. |
The calculated half maximal inhibitory concentration (IC50) values of combination groups, based on the cell viability results (Table 4), was lower than single groups, indicating the benefits of combinational strategy. Although the co-loaded LEs had lower IC50 values than mixture solution and mixture LEs, no statistical difference was observed (P>0.05).
 | Table 4 Calculated IC50 values of different formulations in both CT-26 and HCT-116 cells |
In vivo anti-tumor activity
In vivo anti-tumor efficacy of different formulations was evaluated in CT-26-bearing female BALB/c mice. As shown in Figure 7, Blank-LEs did not exhibit obvious anti-tumor activity, which had equivalent tumor volumes compared with the control group. Although slight in vitro cytotoxicity was observed in HCT-116 cells, no tumor inhibition or weight loss was seen in mice, which means that the nanocarriers were well tolerated.
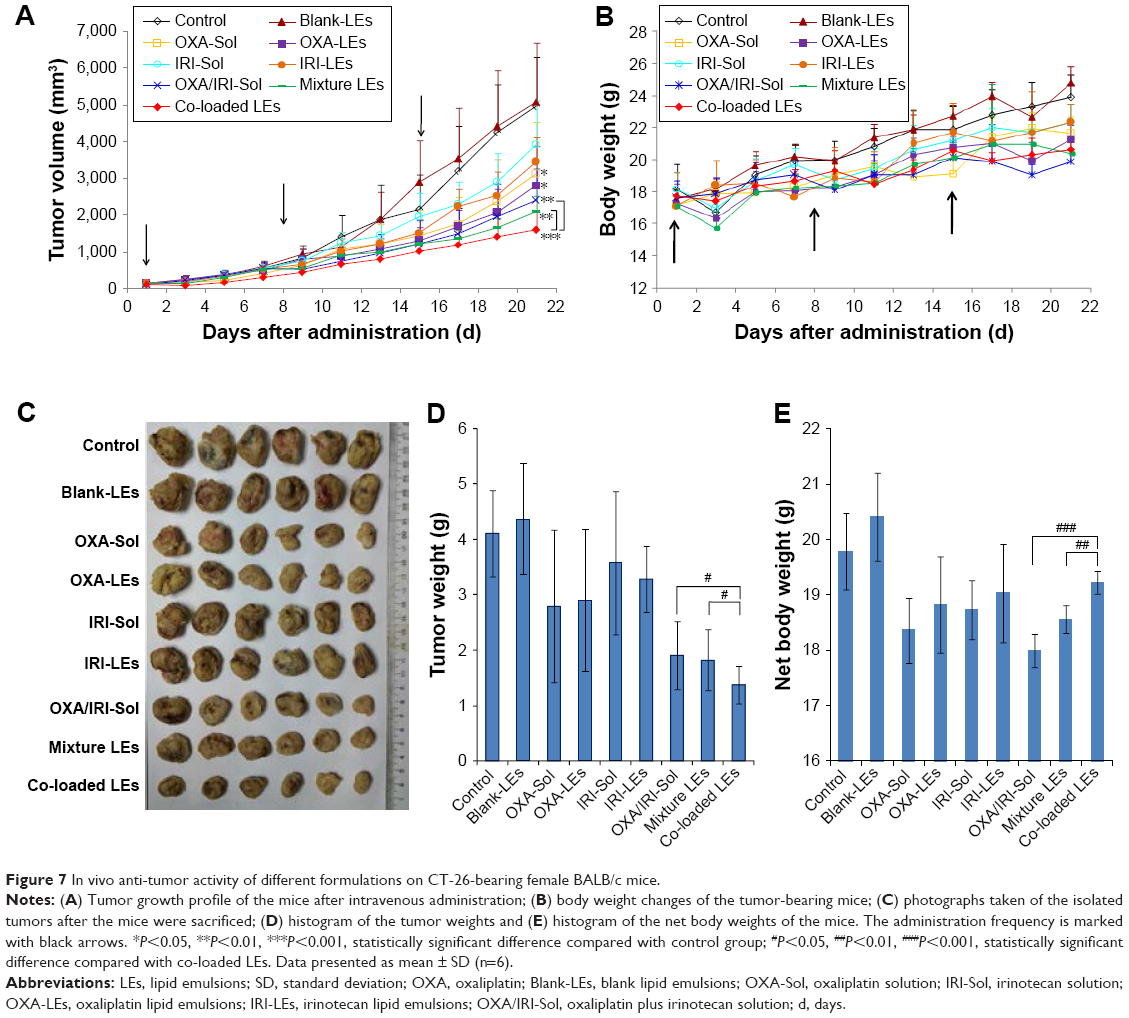 | Figure 7 In vivo anti-tumor activity of different formulations on CT-26-bearing female BALB/c mice. |
For single therapy, both OXA-LEs and IRI-LEs had slightly better anti-tumor activity than the corresponding drug solution. OXA showed significant difference compared with the control group, while the P-values for both IRI formulations were >0.05. IRI did not exhibit obvious therapeutic effect, because the selected dosage of IRI in mice was equivalent to the dosage in combination scheme, which is insufficient for the treatment of cancer for single IRI.38
In case of combination therapy, the tumor growth of the combinational groups was inhibited compared to that of the single formulations. The co-loaded LEs (OXA/IRI-LEs) showed superior anti-tumor activity than their solutions (OXA/IRI-Sol) and the mixture LEs (OXA-LEs plus IRI-LEs) with statistical significance (P<0.05). The results were also confirmed by the tumor weights that were much lower with co-loaded LEs than mixture LEs and mixture solutions (P<0.05). On the other hand, compared with control group, co-loaded LEs had very significant differences with P<0.001, while the P-values for mixture solutions and mixture LEs were <0.01.
The body weights of mice were monitored to assess the safety of the formulations. As shown in Figure 7B, the body weights of the mice in all the groups showed overall increasing trend, and no severe body weight loss was observed during the treatment process. Additionally, in order to eliminate the influence of tumor weight, the net body weight of each group was calculated after correcting for the tumor weight. Co-loaded LEs had higher body weight than the mixture solutions and mixture LEs, indicating the safety and minimum side effects of LEs (Figure 7E).
In vivo NIRF imaging
The in vivo biodistribution is crucial to explain the mechanism of enhanced anti-tumor effects. NIRF imaging is now widely accepted as one of the convenient methods to mimic the drug biodistribution in vivo.48,49 NIRF imaging was applied to trace the biodistribution of LEs. As shown in Figure 8, most of the free DiR were distributed in the liver and spleen, while scarcely in the tumor at all time intervals. For DiR-LEs, the liver is also the main accumulation organ, while accumulation in the tumor was obviously elevated compared with free DiR. NIRF signals of DiR-LEs sustained for 24 h in tumor. The ex vivo data directly illustrated the increased accumulation of dye in tumor for LE formulation.
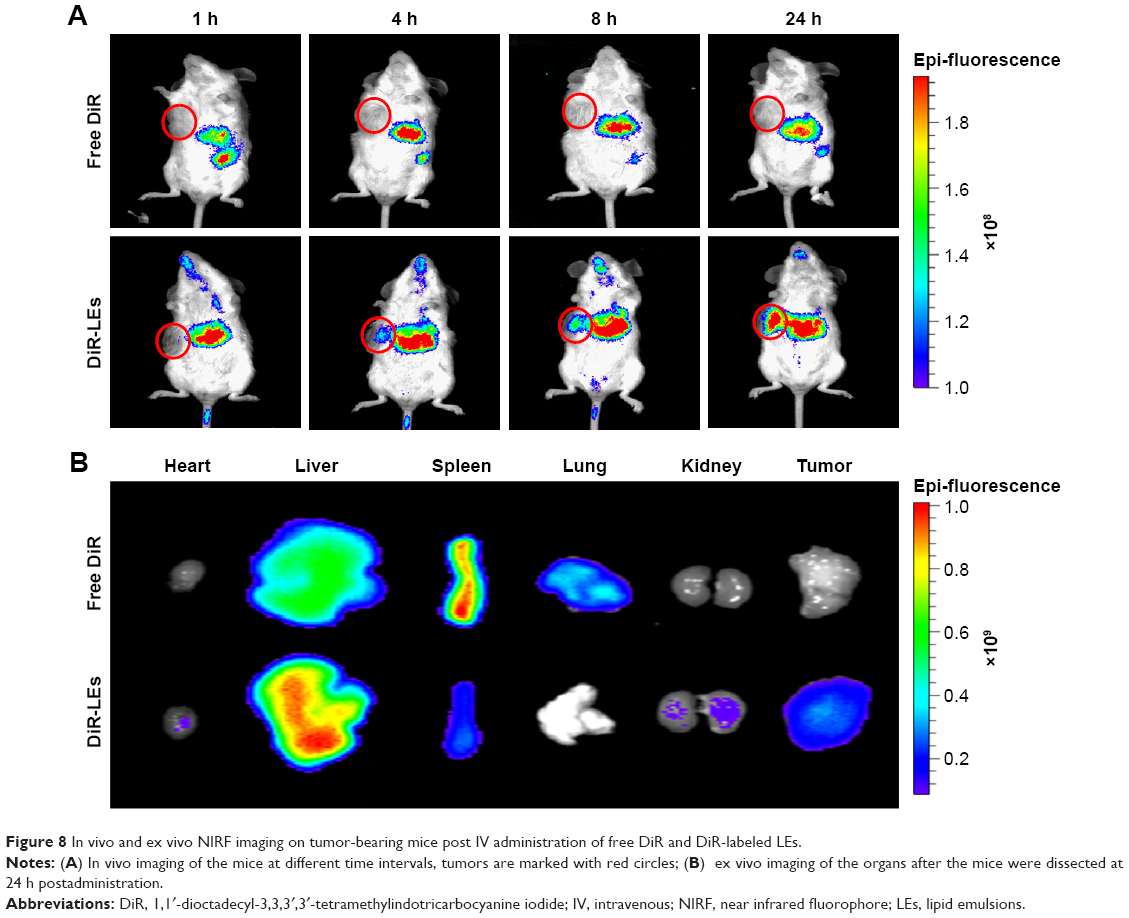 | Figure 8 In vivo and ex vivo NIRF imaging on tumor-bearing mice post IV administration of free DiR and DiR-labeled LEs. |
In vitro cellular uptake
Dye as the substitute for tracking the carriers to mimic the in vitro and in vivo behaviors is widely applied in pharmaceutical field.50–52 Assuming that the dye leakage from carriers would be unavoidable, in vitro release studies of both dyes from single or co-loaded LEs were first investigated. As shown in Figure S1, both dyes showed similar sustained release profiles from prepared LEs and <20% leaked within 24 h. The results indicated that most of the dye was retained in the inner oil phase of LEs, reflecting the distribution of the carriers.
Co-delivery efficiency of the LEs was evaluated by the cellular uptake behaviors in both CT-26 cells and HCT-116 cells. Cells incubated with LEs were positive with green (FL1-H, DiO) and red fluorescence (FL2-H, DiI), and the co-delivery efficiency was indicated by yellow fluorescence due to merging of green and red fluorescence. As shown in Figure 9A and B, both DiI and DiO cargoes were distributed in cytoplasm after 4 h of incubation. The intensity of the yellow fluorescence was boosted up with the increase of incubation time in all groups. Obvious strong yellow fluorescence could be seen in co-loaded LEs, while more red and green fluorescence could be seen in the mixture group compared with co-loaded group, indicating higher co-delivery efficiency for co-loaded LEs. Similar results were obtained in both CT-26 and HCT-116 cells.
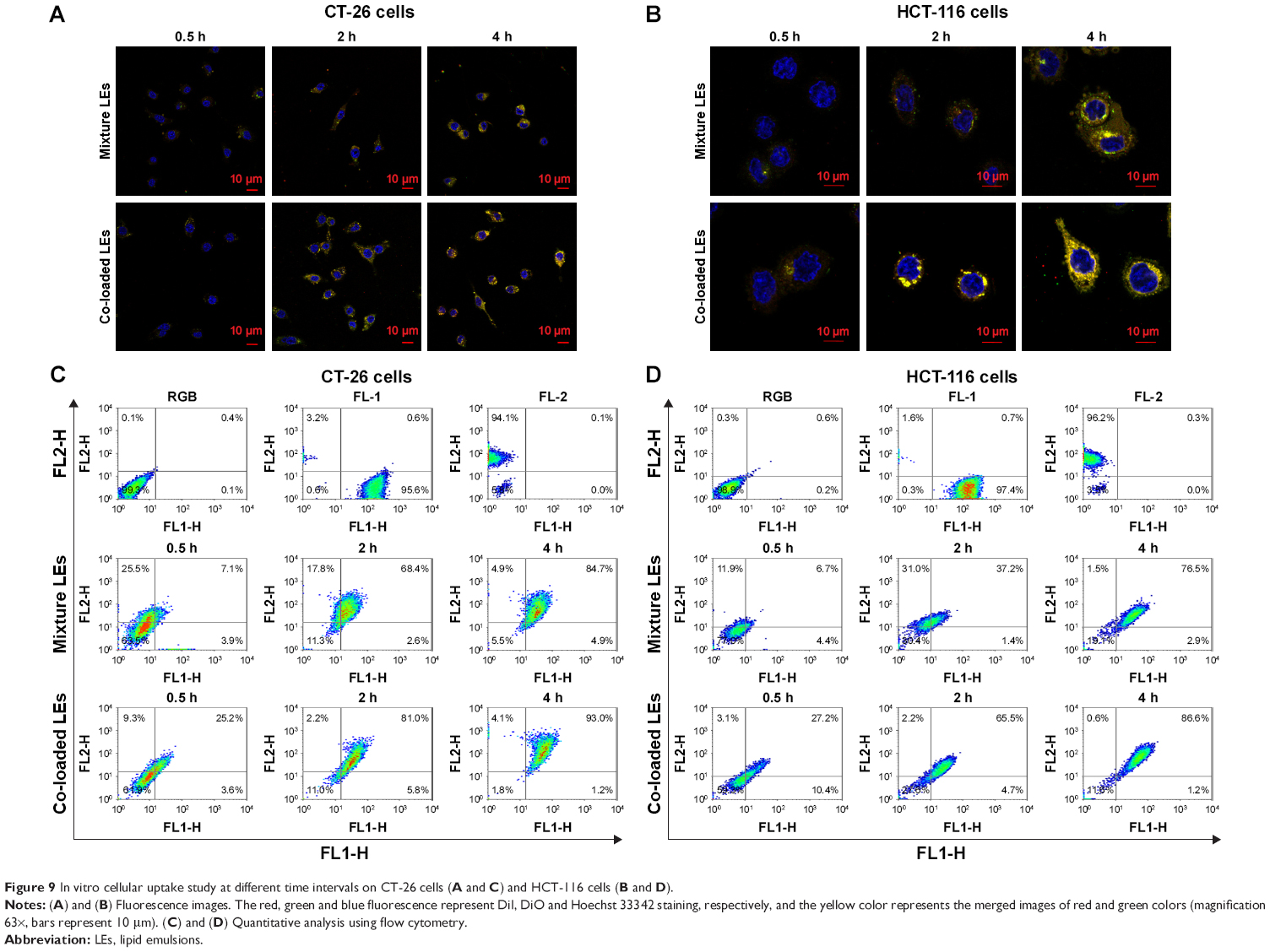 | Figure 9 In vitro cellular uptake study at different time intervals on CT-26 cells (A and C) and HCT-116 cells (B and D). |
Cellular uptake was further quantified using flow cytometry to evaluate the co-delivery ability of the LEs. In CT-26 cells (Figure 9C), the co-delivery efficiency for co-loaded group was 25.2% after 0.5 h incubation, which is ~3.55 times higher than that of mixture group (7.1%); the results were 68.4% for mixture group and 81.0% for co-loaded group at 2 h; while at 4 h, they were 84.7% and 93.0%, respectively, with no significant differences. Similar results existed in HCT-116 cells, as shown in Figure 9D, the co-delivery efficiency for co-loaded group (27.2%) was ~4.05 times higher than mixture group (6.7%) at 0.5 h and also 1.76 times higher even at 2 h (37.2% for mixture group and 65.5% for co-loaded group), while at 4 h, they were 76.5% and 86.6% with no significant differences.
In vivo co-delivery study
In order to explore the in vivo co-delivery ability of the formulations into the tumor, CLSM images of the tumor cryo-sections were observed after the mice were administered DiI- and DiO-labeled formulations. As shown in Figure 10, the fluorescent intensity of red and green colors in the solution group was much lower than both LE groups. Obvious red and green colors were seen in the solution group, indicating the accumulation of free dyes into the tumor to be less than the LE formulations, and the in vivo distribution of the free drug was nonspecific. Both LE groups exhibited more merged yellow color than the solution, and co-loaded LEs had top-level yellow color in the merged picture, indicating that both dyes accumulated in the same sites much better. These results are in accordance with the in vitro cellular uptake study and revealed that co-loaded LEs had superior ability to co-deliver both the cargoes into the same tumor cells.
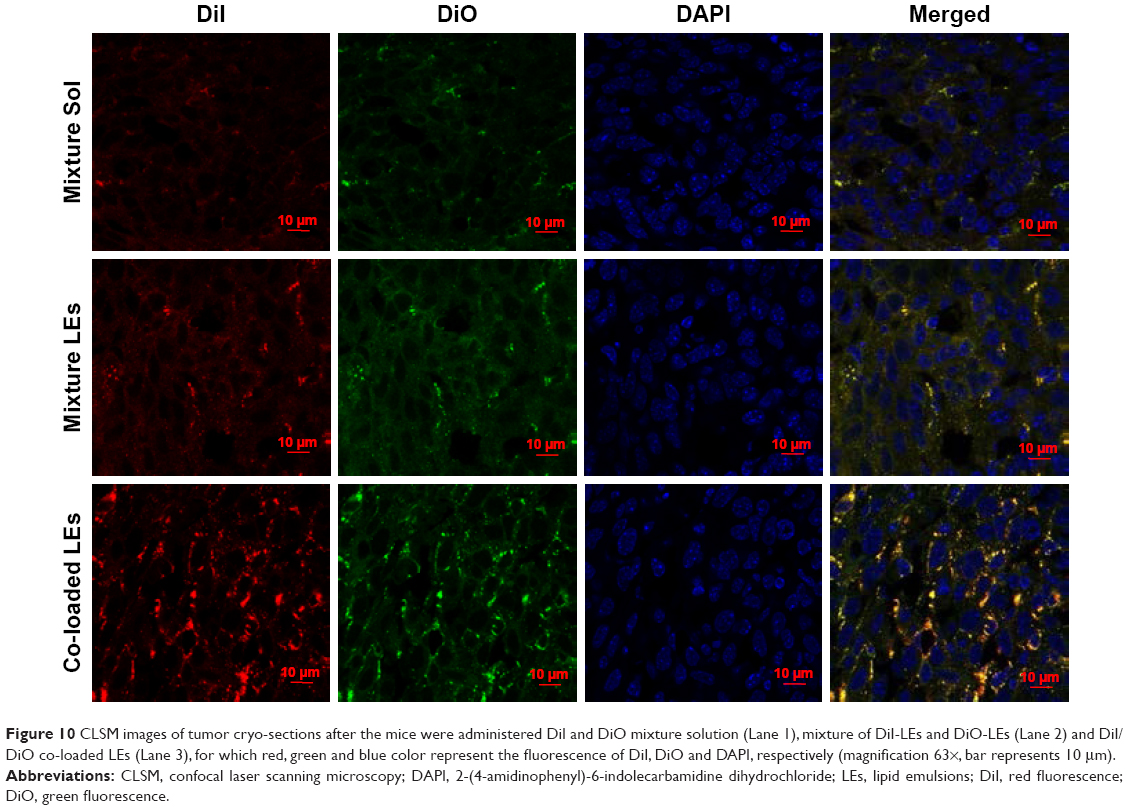 | Figure 10 CLSM images of tumor cryo-sections after the mice were administered DiI and DiO mixture solution (Lane 1), mixture of DiI-LEs and DiO-LEs (Lane 2) and DiI/DiO co-loaded LEs (Lane 3), for which red, green and blue color represent the fluorescence of DiI, DiO and DAPI, respectively (magnification 63×, bar represents 10 μm). |
Discussion
OXA/IRI co-loaded LEs were prepared using DPC technique. DPC has been demonstrated to be useful for enhancing the lipophilicity and facilitating the encapsulation of hydrophilic molecules into hydrophobic core. It can also sustain the drug release due to the molecular interactions between the drug and phospholipid.53 In this study, OXA as a hydrophilic drug is difficult to be encapsulated into the LEs; thus, it was transformed into hydrophobic pattern by producing OPPC, which proved to be very useful. In our previous attempt, it was found that IPPC in the formulation would be more stable than free IRI in the oil phase. Furthermore, multiple DPCs can coordinate the release profiles of different drugs and achieve synchronous release by reducing the hydrophobic discrepancy between different drugs, thus generating the optimized synergistic effect. Therefore, both DPCs were introduced into the formulation for further study. Although many researchers have reported utilizing DPC technique to control the sustained release of the drug, there are no reports concerning multiple DPCs to coordinate the synchronous release of multiple drugs.
Co-loaded LEs had desired size distribution and zeta potential, which would be suitable for IV injection. In vitro release study showed that both drugs could be synchronously released from co-loaded LEs with potential for the optimized synergism. Moreover, co-loaded LEs demonstrated superior in vivo anti-tumor activity compared with mixed solution and the mixture LEs. All these results indicate that LEs would be the excellent nanocarriers for co-delivery of multiple drugs. With the mature technology, safe excipients and ease of scale up, LEs are a good choice to bridge the gap between the unmet clinical demands and uncertain translational prospects of co-delivery nanomedicine.
In vivo and ex vivo NIRF imaging study was carried out to illustrate the superior anti-tumor activity by mimicking the biodistribution of the drugs (Figure 8). The results showed that the drug accumulation in tumor for LE formulation was improved when compared to the free drug. Although it is just a preliminary evaluation and cannot entirely substitute for real drug biodistribution due to the differences between fluorescent dye and drug, it indicates the tumor accumulation capacity to a certain extent. These results demonstrated that LEs increased drug accumulation in tumor, which would be beneficial for the enhanced anti-tumor activity.
The cellular uptake process was further investigated to explore the co-delivery efficiency for co-loaded LEs (Figure 9). The significant differences in the early stage might be caused by the mutual competition of endocytosis process between different nanoparticles.54 In the beginning, different nanoparticles would randomly distribute around the cells and then be swallowed into the cytoplasm; thus, the molar ratios on both drugs in the same cell could be in any proportion, leading to the generation of individual therapeutic effect. At the late stage when the endocytosis process nearly saturated, both kinds of LEs tended to reach the balance and distribute homogeneously; thus, the co-location in the cell (yellow color) could be observed. The differences of the cellular uptake behaviors between mixture group and co-loaded group diminished in the end.
The in vivo CLSM imaging of tumor cryo-sections also demonstrated better co-delivery ability of co-loaded LEs (Figure 10). Although the opportunity of accumulation in tumor for both mixture and co-loaded groups would be equivalent, their endocytosis process was very different. When the mixture of single-loaded LEs entered the cells, the molar ratios of both drugs could be in any proportion. Meanwhile, the co-loaded LEs would deliver both drugs into the cells simultaneously with the fixed ratio, and thus, the optimized synergistic effect could be achieved. In brief, we believe that the enhanced therapeutic effect of co-loaded LEs was attributed to the higher efficiency on co-delivering both drugs into the same cells and generated the maximum synergistic effect. All the earlier results reflect that co-loaded formulations would be the desired carriers for delivering different cargoes, due to the excellent co-delivery ability.
Conclusion
OXA/IRI-LEs were successfully developed to efficiently encapsulate and coordinate synchronous release of both drugs. In vitro release profiles illustrated that both drugs could achieve the sustained release from co-loaded LEs, and their molar ratios could be well controlled. Co-loaded LEs demonstrated superior in vivo anti-tumor activity compared with the solution and the mixture LEs. Furthermore, co-loaded LEs could co-deliver both drugs into the same cells more efficiently, which might be crucial for improving the therapeutic effect. These results confirm that the OXA/IRI-LEs can be an efficient formulation for enhanced colorectal cancer therapy, with great application prospects. The comparison between co-loaded LEs and mixture group would be valuable for pharmaceutical designs in co-delivery of multiple drugs.
Acknowledgment
This work was supported by the China-Australia Centre for Health Sciences Research (CACHSR, 2014GJ10) and the Science and Technology Development Project of Shandong Province (2014GGE27121).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19–34. | ||
Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33(36):4451–4463. | ||
Kummar S, Chen HX, Wright J, et al. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9(11):843–856. | ||
Kemp JA, Shim MS, Heo CY, Kwon YJ. “Combo” nanomedicine: co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Deliv Rev. 2016;98:3–18. | ||
Yuan Y, Wang Z, Cai P, et al. Conjugated polymer and drug co-encapsulated nanoparticles for chemo- and photo-thermal combination therapy with two-photon regulated fast drug release. Nanoscale. 2015;7(7):3067–3076. | ||
Nowak-Sliwinska P, Weiss A, Ding X, et al. Optimization of drug combinations using feedback system control. Nat Protoc. 2016;11(2):302–315. | ||
Xiao B, Han MK, Viennois E, et al. Hyaluronic acid-functionalized polymeric nanoparticles for colon cancer-targeted combination chemotherapy. Nanoscale. 2015;7(42):17745–17755. | ||
Zhang RX, Wong HL, Xue HY, Eoh JY, Wu XY. Nanomedicine of synergistic drug combinations for cancer therapy – strategies and perspectives. J Control Release. 2016;240:489–503. | ||
Mignani S, Bryszewska M, Klajnert-Maculewicz B, Zablocka M, Majoral JP. Advances in combination therapies based on nanoparticles for efficacious cancer treatment: an analytical report. Biomacromolecules. 2015;16(1):1–27. | ||
Jang B, Kwon H, Katila P, Lee SJ, Lee H. Dual delivery of biological therapeutics for multimodal and synergistic cancer therapies. Adv Drug Deliv Rev. 2016;98:113–133. | ||
Assanhou AG, Li W, Zhang L, et al. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. | ||
Gao M, Xu Y, Qiu L. Enhanced combination therapy effect on paclitaxel-resistant carcinoma by chloroquine co-delivery via liposomes. Int J Nanomedicine. 2015;10:6615–6632. | ||
Pathak RK, Dhar SA. Nanoparticle cocktail: temporal release of predefined drug combinations. J Am Chem Soc. 2015;137(26):8324–8327. | ||
Zhang W, Li C, Shen C, et al. Prodrug-based nano-drug delivery system for co-encapsulate paclitaxel and carboplatin for lung cancer treatment. Drug Deliv. 2016;23(7):2575–2580. | ||
Castillo RR, Colilla M, Vallet-Regi M. Advances in mesoporous silica-based nanocarriers for co-delivery and combination therapy against cancer. Expert Opin Drug Del. 2017;14(2):229–243. | ||
Croissant JG, Zhang D, Alsaiari S, et al. Protein-gold clusters-capped mesoporous silica nanoparticles for high drug loading, autonomous gemcitabine/doxorubicin co-delivery, and in-vivo tumor imaging. J Control Release. 2016;229:183–191. | ||
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. | ||
Teo PY, Cheng W, Hedrick JL, Yang YY. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv Drug Deliv Rev. 2016;98:41–63. | ||
Su CW, Chiang CS, Li WM, Hu SH, Chen SY. Multifunctional nanocarriers for simultaneous encapsulation of hydrophobic and hydrophilic drugs in cancer treatment. Nanomedicine (Lond). 2014;9(10):1499–1515. | ||
Hu SH, Chen SY, Gao X. Multifunctional nanocapsules for simultaneous encapsulation of hydrophilic and hydrophobic compounds and on-demand release. ACS Nano. 2012;6(3):2558–2565. | ||
Hormann K, Zimmer A. Drug delivery and drug targeting with parenteral lipid nanoemulsions – a review. J Control Release. 2016;223:85–98. | ||
Negi P, Singh B, Sharma G, Beg S, Raza K, Katare OP. Phospholipid microemulsion-based hydrogel for enhanced topical delivery of lidocaine and prilocaine: QbD-based development and evaluation. Drug Deliv. 2016;23(3):951–967. | ||
Jiang SP, He SN, Li YL, et al. Preparation and characteristics of lipid nanoemulsion formulations loaded with doxorubicin. Int J Nanomedicine. 2013;8:3141–3150. | ||
Ma J, Teng H, Wang J, et al. A highly stable norcantharidin loaded lipid microspheres: preparation, biodistribution and targeting evaluation. Int J Pharm. 2014;473(1–2):475–484. | ||
Nakano M. Places of emulsions in drug delivery. Adv Drug Deliv Rev. 2000;45(1):1–4. | ||
Mizushima Y. Lipid microspheres (lipid emulsions) as a drug carrier – an overview. Adv Drug Deliv Rev. 1996;20:113–115. | ||
Yamaguchi T. Lipid microspheres as drug carriers, a pharmaceutical point of view. Adv Drug Deliv Rev. 1996;20:117–130. | ||
Collins-Gold LC, Lyons RT, Bartholow LC. Parenteral emulsions for drug delivery. Adv Drug Deliv Rev. 1990;5:189–208. | ||
Benson AB, Venook AP, Cederquist L, et al. Colon Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Net. 2017;15(3): 370–398. | ||
Lammers T, Subr V, Ulbrich K, et al. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30(20):3466–3475. | ||
Dai W, Jin W, Zhang J, et al. Spatiotemporally controlled co-delivery of anti-vasculature agent and cytotoxic drug by octreotide-modified stealth liposomes. Pharm Res. 2012;29(10):2902–2911. | ||
Yang T, Wang Y, Li Z, et al. Targeted delivery of a combination therapy consisting of combretastatin A4 and low-dose doxorubicin against tumor neovasculature. Nanomedicine. 2012;8(1):81–92. | ||
Luo S, Gu Y, Zhang Y, et al. Precise ratiometric control of dual drugs through a single macromolecule for combination therapy. Mol Pharm. 2015;12(7):2318–2327. | ||
Sun R, Liu Y, Li SY, et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015;37:405–414. | ||
Xiong F, Xiong C, Yao J, Chen X, Gu N. Preparation, characterization and evaluation of breviscapine lipid emulsions coated with monooleate-PEG-COOH. Int J Pharm. 2011;421(2):275–282. | ||
Gao W, Xiang B, Meng TT, Liu F, Qi XR. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34(16):4137–4149. | ||
Zhang B, Wang T, Yang S, et al. Development and evaluation of oxaliplatin and irinotecan co-loaded liposomes for enhanced colorectal cancer therapy. J Control Release. 2016;238:10–21. | ||
Abu Lila AS, Doi Y, Nakamura K, Ishida T, Kiwada H. Sequential administration with oxaliplatin-containing PEG-coated cationic liposomes promotes a significant delivery of subsequent dose into murine solid tumor. J Control Release. 2010;142(2):167–173. | ||
Nakamura H, Doi Y, Abu Lila AS, Nagao A, Ishida T, Kiwada H. Sequential treatment of oxaliplatin-containing PEGylated liposome together with S-1 improves intratumor distribution of subsequent doses of oxaliplatin-containing PEGylated liposome. Eur J Pharm Biopharm. 2014;87(1):142–151. | ||
Abu Lila AS, Kizuki S, Doi Y, Suzuki T, Ishida T, Kiwada H. Oxaliplatin encapsulated in PEG-coated cationic liposomes induces significant tumor growth suppression via a dual-targeting approach in a murine solid tumor model. J Control Release. 2009;137(1):8–14. | ||
Guo B, Liu H, Li Y, et al. Application of phospholipid complex technique to improve the dissolution and pharmacokinetic of probucol by solvent-evaporation and co-grinding methods. Int J Pharm. 2014;474(1–2):50–56. | ||
Zhao YQ, Wang LP, Ma C, Zhao K, Liu Y, Feng NP. Preparation and characterization of tetrandrine-phospholipid complex loaded lipid nanocapsules as potential oral carriers. Int J Nanomedicine. 2013;8:4169–4181. | ||
Li Y, Jin W, Yan H, Liu H, Wang C. Development of intravenous lipid emulsion of vinorelbine based on drug-phospholipid complex technique. Int J Pharm. 2013;454(1):472–477. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Nehoff H, Parayath NN, Domanovitch L, Taurin S, Greish K. Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int J Nanomedicine. 2014;9:2539–2555. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. | ||
Kobayashi H, Longmire MR, Choyke PL. Polychromatic in vivo imaging of multiple targets using visible and near infrared light. Adv Drug Deliv Rev. 2013;65(8):1112–1119. | ||
Bouchaala R, Mercier L, Andreiuk B, et al. Integrity of lipid nanocarriers in bloodstream and tumor quantified by near-infrared ratiometric FRET imaging in living mice. J Control Release. 2016;236:57–67. | ||
Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano. 2013;7(4):2935–2947. | ||
Chen Z, Li Z, Lin Y, Yin M, Ren J, Qu X. Biomineralization inspired surface engineering of nanocarriers for pH-responsive, targeted drug delivery. Biomaterials. 2013;34(4):1364–1371. | ||
Lee JS, Zhou W, Meng F, Zhang D, Otto C, Feijen J. Thermosensitive hydrogel-containing polymersomes for controlled drug delivery. J Control Release. 2010;146(3):400–408. | ||
Peng Q, Zhang ZR, Gong T, Chen GQ, Sun X. A rapid-acting, long-acting insulin formulation based on a phospholipid complex loaded PHBHHx nanoparticles. Biomaterials. 2012;33(5):1583–1588. | ||
Hocherl A, Dass M, Landfester K, Mailander V, Musyanovych A. Competitive cellular uptake of nanoparticles made from polystyrene, poly(methyl methacrylate), and polylactide. Macromol Biosci. 2012;12(4):454–464. |
Supplementary materials
Preparation of the dyes-loaded lipid emulsions (LEs)
DiO/DiI-loaded LEs
A total of 50 mg DiO (or DiI) was dissolved in 10 mL dichloromethane (DCM) with 250 mg egg phosphatidylcholine (EPC) and then reacted at 40°C for 3 h to form the complex. The organic solvent was removed using a rotary evaporator to get DiO-DPC or DiI-DPC.
For co-loaded LEs, DiO-DPC and DiI-DPC were re-dissolved in DCM and 2 g medium chain triglyceride (MCT) and 50 mg oleic acid (OA) were added; DCM was removed using a rotary evaporator, and the oil phase was obtained. A total of 200 mg Pluronic F68 and 800 mg glycerol were dissolved in 40 mL water to get the aqueous phase. After that, oil phase was added dropwise into the aqueous phase under shearing at 60°C. The formed coarse emulsion was further circulated through a high-pressure homogenizer (Panda 1K NS1001L; Niro Soavi SpA, Parma, Italy).
For single LEs, DiO-DPC (or DiI-DPC) was re-dissolved in DCM, and extra 250 mg EPC, 2 g MCT and 50 mg OA were added. The same procedure was performed in the following operations.
1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide (DiR)-loaded LEs
A total of 50 mg DiR was dissolved in 10 mL DCM with 250 mg EPC and then reacted at 40°C for 3 h to form the complex. The organic solvent was removed using a rotary evaporator to get DiR-DPC.
DiR-DPC was re-dissolved in DCM, and extra 250 mg EPC, 2 g MCT and 50 mg OA were added. DCM was removed using a rotary evaporator, and the oil phase was obtained. A total of 200 mg Pluronic F68 and 800 mg glycerol were dissolved in 40 mL water to get the aqueous phase. After that, oil phase was added dropwise into the aqueous phase under shearing at 60°C. The formed coarse emulsion was further circulated through a high-pressure homogenizer (Panda 1K NS1001L; Niro Soavi SpA).
In vitro release of DiI/DiO from LEs
In vitro release of DiI and DiO from LEs was determined by the dialysis method. Tween-80 0.5% (w/v) was added in phosphate-buffered saline (PBS) (pH 7.4) as the release medium for the sink condition requirement of the release test. Briefly, 1 mL sample was added to the dialysis bag and incubated with 20 mL release medium in a plastic tube at 37°C. At the predetermined time intervals, 2 mL of release medium was withdrawn and replaced with fresh medium. The cumulative amounts of DiI and DiO in release medium were determined using a fluorescence spectrofluorometer (F-7000, HITACHI, Japan). All measurements were carried out twice.
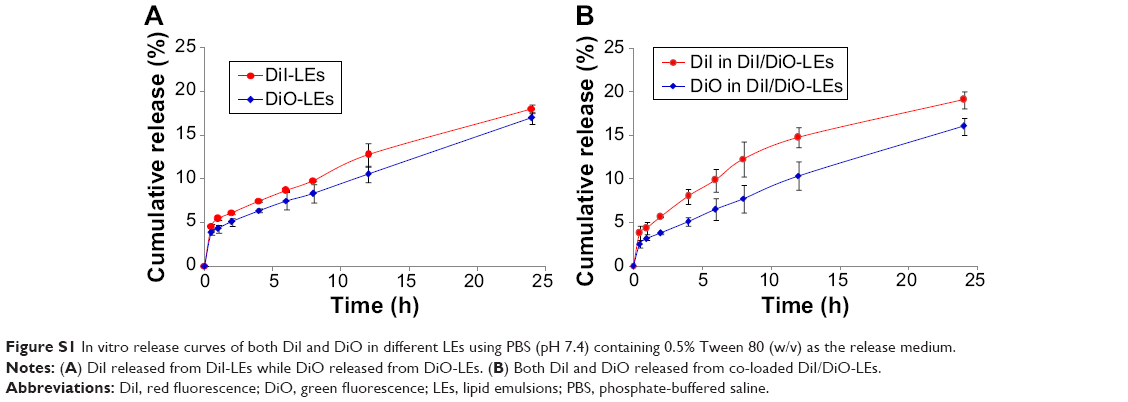 | Figure S1 In vitro release curves of both DiI and DiO in different LEs using PBS (pH 7.4) containing 0.5% Tween 80 (w/v) as the release medium. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.