")
Back to Journals » Infection and Drug Resistance » Volume 11
Efficacy of φkm18p phage therapy in a murine model of extensively drug-resistant Acinetobacter baumannii infection
Authors Wang JL , Kuo CF, Yeh CM, Chen JR, Cheng MF , Hung CH
Received 10 July 2018
Accepted for publication 4 October 2018
Published 15 November 2018 Volume 2018:11 Pages 2301—2310
DOI https://doi.org/10.2147/IDR.S179701
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Jiun-Ling Wang,1,* Chih-Feng Kuo,2,* Che-Ming Yeh,3 Jung-Ren Chen,4 Ming-Fang Cheng,5–7 Chih-Hsin Hung3
1Department of Internal Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan, ROC; 2Department of Nursing, I-Shou University, Kaohsiung, Taiwan, ROC; 3Department of Chemical Engineering and Institute of Biotechnology and Chemical Engineering, I-Shou University, Kaohsiung, Taiwan, ROC; 4Department of Biological Science and Technology, I-Shou University, Kaohsiung, Taiwan, ROC; 5Department of Pediatrics, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan, ROC; 6School of Medicine, Faculty of Medicine, National Yang-Ming University, Taipei, Taiwan, ROC; 7Department of Nursing, Fooyin University, Kaohsiung, Taiwan, ROC
*These authors contributed equally to this work
Purpose: Few effective antibiotics are available for treating extensively drug-resistant Acinetobacter baumannii (XDRAB) sepsis. Phage therapy may show potential in treating XDRAB infections.
Materials and methods: We studied ϕkm18p phage therapy in BALB/c and C57BL/6 mice models of XDRAB bacteremia.
Results: We observed survival rates of nearly 100% in groups given phage therapy concurrent with XDRAB at different multiplicities of infection. In mice that received phage therapy after a 1-hour delay, the survival rate decreased to about 50%. The bacterial load in the blood decreased from 108 to 102 and 103 colony-forming units (CFU)/mL in the concurrent treatment group. In the phage therapy group, the levels of the cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), were low at 3 hours after infection. Although some phage-resistant mutants were isolated after phage therapy, a cytotoxicity study showed that they had reduced fitness.
Conclusion: Phage therapy in XDRAB bacteremia increased the animal survival rates, decreased the bacteremia loads, and decreased the levels of inflammatory markers TNF-α and IL-6. However, the reduced therapeutic effect with delayed administrations may be a concern in developing a successful phage therapy for treating acute infections of multidrug-resistant pathogens.
Keywords: Acinetobacter baumannii, survival rates, bacteria, multiple antimicrobial drug resistance, phage therapy
Introduction
Global outbreaks of infections caused by Acinetobacter baumannii strains with multiple antimicrobial drug resistance represent a growing public-health problem.1 It is difficult to treat multidrug-resistant A. baumannii (MDRAB) and extensively drug-resistant A. baumannii (XDRAB),2 and a lack of response to therapy for A. baumannii bacteremia is a strong predictor of death. XDRAB has shown a relatively low rate of developing drug resistance in vitro to two currently available antibiotics, such as colistin and tigecycline. Only a few small trials without randomization have been conducted to evaluate the efficacy of colistin therapy.3–6 Colistin had two main drawbacks: it provides suboptimal results and shows potential for renal toxicity with prolonged use, particularly in patients with chronic renal disease. Animal studies have suggested that a combination therapy with colistin and either carbapenem or rifampin may be able to increase the treatment success, but the hypothesis lacks support from solid clinical data.6,7
Tigecycline shows good activity against MDRAB in vitro but has not been approved for use against A. baumannii infections. Several retrospective studies have demonstrated the efficacy of tigecycline, but the results remain controversial. Tigecycline therapy is limited by the possibility of a superinfection and the emergence of resistance in XDRAB during therapy.8,9 New alternative approaches have been recommended by some experts to control A. baumannii infections, such as antimicrobial peptides and immunization with an inactivated whole-cell vaccine.10,11
Alternatively, bacteriophage therapy has been used in Russia and Poland for decades. This therapy provides new hope in addressing the difficult treatment conditions faced in multidrug-resistant bacterial infections.12–14 Several animal studies have demonstrated that phages can kill clinical multidrug-resistant bacterial isolates, including extended spectrum beta-lactamase (ESBL) Escherichia coli,15,16 Pseudomonas aeruginosa,17,18 and Klebsiella pneumoniae.19,20 More recently, several studies have isolated phages showing good lytic activity against MDRAB in vitro.21–23
Nevertheless, before introducing phage therapy as an alternative therapy for XDRAB infections in humans, it is necessary to evaluate its efficacy in an animal model. Animal studies are important for evaluating the efficacy of XDRAB treatments due to the difficulties in performing randomized controlled trials with human subjects. Several animal models have been developed to compare combinations of antibiotic therapies with monotherapies in MDRAB infections.24,25 No animal model is currently available for testing phage therapies in XDRAB bacteremia infections. Thus, the aim of this study is to evaluate the efficacy of bacteriophage therapy as an antimicrobial treatment for XDRAB infections in a mouse model.
Materials and methods
Bacteria
KM18 is a clinical bacterial isolate of XDRAB that has previously been tested to determine the minimum inhibitory concentrations of various drugs based on guidelines from the Clinical and Laboratory Standards Institute (National Committee for Clinical Laboratory Standards, USA). The isolate KM18 was found to be resistant to all antibiotics used to treat A. baumannii infection except for tigecycline and polymyxins.23
Phage
Phages were isolated from hospital sewage, which showed a narrow host spectrum. The phage ϕkm18p could effectively lyse most XDRAB strains. Bacterial populations decreased from 108 to 103 colony-forming units (CFU)/mL within 30 minutes of exposure to ϕkm18p. The safety of phage therapy was investigated by determining the toxicity of phages to A549 human lung epithelial cells. The details of these methods are described in a previous study.23
Mice
BALB/c and C57BL/6 (B6) mice were purchased from the National Laboratory Animal Center and National Cheng Kung University, Taiwan, ROC. We used the following two inbred strains of laboratory mice with different inflammatory responses to bacteria: BALB/c and C57BL/6 mice. In a previous lung infection model, BALB/c mice had a more intense neutrophil influx and more severe lung damage than C57BL/6 mice.26
The animals were raised and cared for according to the guidelines established by the National Science Council of the Republic of China. All procedures, care, and animal handling protocols were reviewed and approved by the Institutional Animal Care and Use Committees of both I-Shou University and National Cheng Kung University. All experiments used 8–10-week-old male mice.
Protection of murine macrophage and macrophage-like RAW 264.7 cell lines
Mouse RAW 264.7 cell lines were purchased from the Bioresource Collection and Research Center (ATCC TIB-71). The cells were seeded at 1.25×105 cells/well and incubated for 12 hours. Different concentrations of phage and bacteria were infected to evaluate the phage cytotoxicity and bacterial clearance.
Animal survival and bacteremia test
Mice were inoculated intraperitoneally with XDRAB isolate (2–3×108 CFU KM18in BALB/c and 6×108 CFU KM18 in C57BL/6). The ϕkm18p phage was administered intraperitoneally with different multiplicities of infection (MOIs) 10, 1, and 0.1 at 10 minutes (immediate phage therapy) and 1 hour (delayed phage therapy) at different inoculation sites postinfection. After 3 hours of treatment, blood (100–200 µL) was aseptically collected from the orbital areas of the BALB/c mice and mixed with heparin. The blood from each group was then serially diluted, poured on Lysogeny broth (LB) agar plates, and incubated at 37°C overnight. The colony count was measured on the LB agar and recorded in CFU per milliliter.
Cytokine assay in BALB/c mice
At 3 hours postinfection, mouse sera were collected and examined for cytokines. Tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) were detected using a DuoSet ELISA Development kit for mouse TNF-α/TNFSF1A (cat no DY410 R&D Systems, IL-6 cat no DY410 R&D Systems).
Phage-resistant bacterial strains’ isolation
During the phage and bacterium co-incubation period, the concentration of bacterium decreased first and then increased after 3 hours. To isolate the phage-resistant strains, we extended the co-incubation time to ensure the generation of mutants. First, the km18p phage was inoculated in the bacterial culture (OD600 nm =1) and co-cultured for 12 hours. The co-culture was incubated for 12 hours, and then, 100 µL of co-culture medium was serially diluted. Next, 100 µL of the dilution was poured on an agar plate containing the antibiotic ampicillin and incubated for 16 hours. We then randomly selected 100 single colonies and spread them on new agar plates. Then, 10 µL of phage (109 phage-forming units [PFU]/mL) was dropped on the bacteria on the agar and kept at 37°C overnight. The bacteria without lysis were selected and rechecked by a spot test to confirm the phage resistance. These mutant strains were analyzed by pulsed field gel electrophoresis to confirm the identity of the DNA patterns with the wild-type strain.
Protein analysis of the phage-resistant mutants
Phage therapy was administered by injecting 1010 PFU of ϕkm18p into the retroperitoneal area of the BALB/c mice. Blood was then collected 96 hours later from the orbital area to detect anti-ϕkm18p phage antibodies. Sera samples were collected again at 2 weeks after each injection. Anti-ϕkm18p phage serum titers were determined with ELISA.
Proteins were extracted from native KM18 bacteria, the phage-resistant mutant bacterial lines (R1–R5; 100 µL, OD600 nm =1, mixed with 100 µL of 2× protein sample buffer and heated at 100°C for 10 minutes), and the ϕkm18p phage preparations (1011 PFU/mL, 100 µL, mixed with 20 µL of 6× protein sample buffer and heated at 100°C for 10 minutes). The extracts were analyzed on 12% sodium dodecyl sulfate-polyacrylamide gels that were subjected to electrophoresis. The separated proteins in the gels were transferred onto polyvinylidene difluoride membranes. The blots were probed with a ϕkm18p-specific antibody.
Bactericidal test
To test the virulence of the phage-resistant mutants compared to KM18, we tested their susceptibility to white cells in whole human blood. We performed a whole-blood bactericidal test with KM18 and mutant R1 bacteria. Briefly, we mixed 4×104 CFU of bacteria with 1 mL at whole human blood, which contained 5–8×104 white blood cells per milliliter at an MOI of about 0.01 (polymorphonuclear leukocytes:bacteria).
Bacterial suspensions of KM18 and R1 (each containing 4×104, 4×105, or 4×106 organisms) cultured in DMEM were mixed with 10% FBS and antibiotics. After incubations for 6, 12, and 24 hours, we measured the concentration of bacteria at OD600 nm and calculated the bacterial colony counts (CFU per milliliter) after serial dilution.
The effect of bovine serum and triad antibiotics
The effect of the bovine serum after removing the complement on the growth of KM18 and R1 was also evaluated. A bacteria concentration of 2×104 CFU was added to 24-well plates, and 10% FBS was added to the culture liquid. One group was heated to 56°C for 10 minutes, while another group was heated at the same temperature for 20 minutes, and the control group was not subjected to heating. We checked the OD600 nm absorbance value of the 10× diluted culture liquid after 24 hours.
Statistical analysis
The data are presented as mean±SD. Differences in quantitative measurements were assessed using Student’s t-test or a one-way or two-way ANOVA. Differences were considered statistically significant when the P-values were <0.05.
Results
Survival test for the murine RAW 264.7 macrophage cell line
KM18 infections significantly reduced the macrophage cell survival. However, KM18 did not affect the survival when cells received phage therapy (Figure 1A). Increases in ϕm18p phage MOIs were associated with increases in bacterial clearance (Figure 1B).
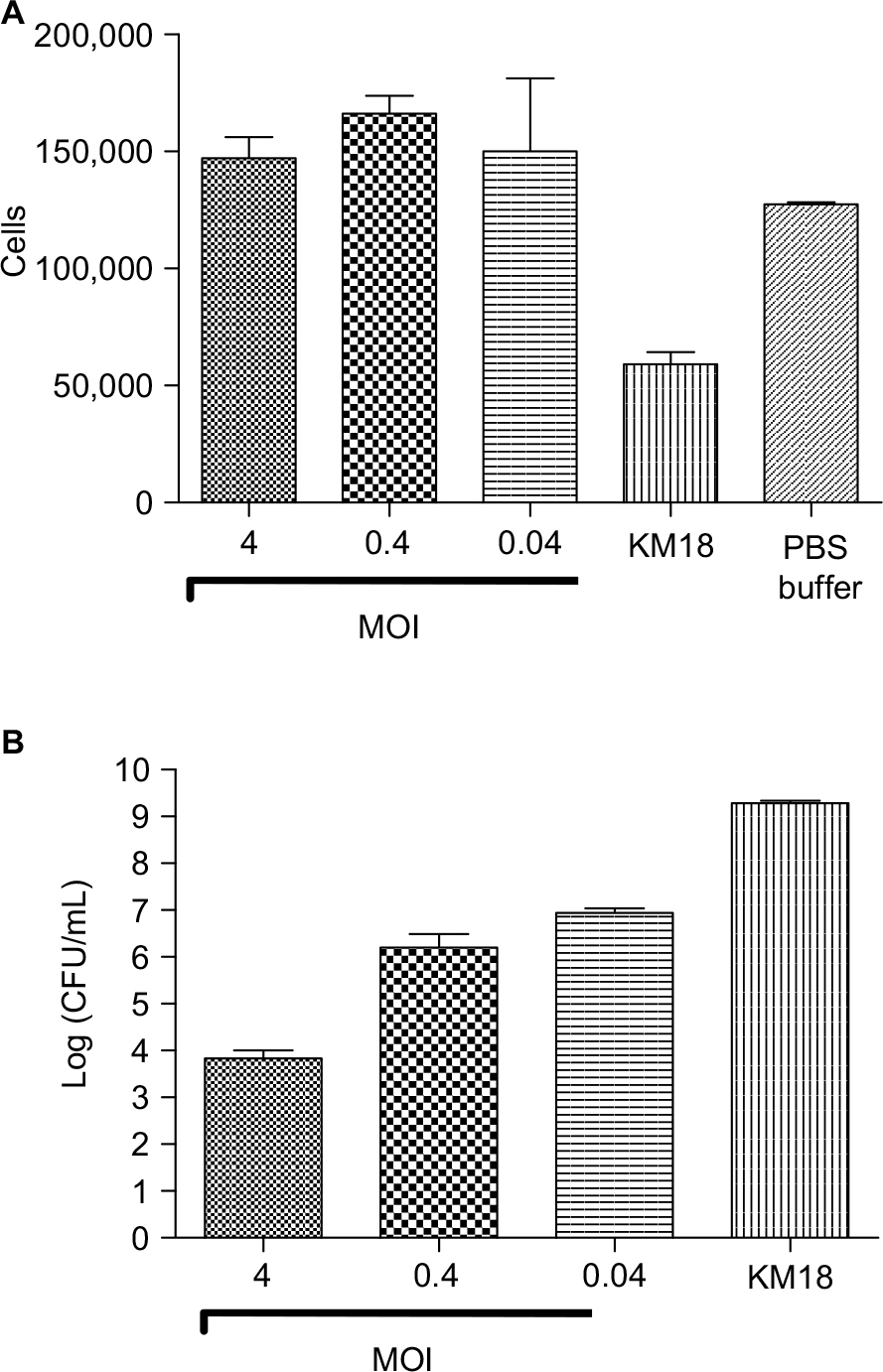 | Figure 1 Murine RAW 264.7 macrophage cell line survival test. Notes: (A) Numbers of surviving RAW 264.7 cells after infections with KM18 and different MOIs of ϕkm18p. (B) Bacterial clearance (CFU/mL) in cells infected with different MOIs of ϕkm18p. Abbreviations: CFU, colony-forming units; MOIs, multiplicities of infection. |
Survival in the mouse model
We infected BALB/c mice with intraperitoneal injections of different concentrations of A. baumannii, and the mouse survival was measured for 14 days. All mice died after the injections of 2–2.5×108 CFU of KM18. After the injections of 2–3×107 CFU, the survival rate was 50%. After the injections of 2–3×106 CFU, the survival rate was 100%. Therefore, the lethal dose in BALB/c mice was LD100=2–2.5×108 CFU (P=0.0047; data not shown).
We concurrently administered different MOIs (MOI =10, 1, or 0.1) of ϕkm18p phage to treat BALB/c mice with an intraperitoneal injection of 2–3×108 CFU of KM18. All three MOIs of phage protected the mice, and the survival rate was 100%, in contrast to the control group without phage therapy (P≤0.0001). However, the survival rate decreased to 56% when the mice received ϕkm18p phage therapy with a 1-hour delay after the KM18 injection (Figure 2A).
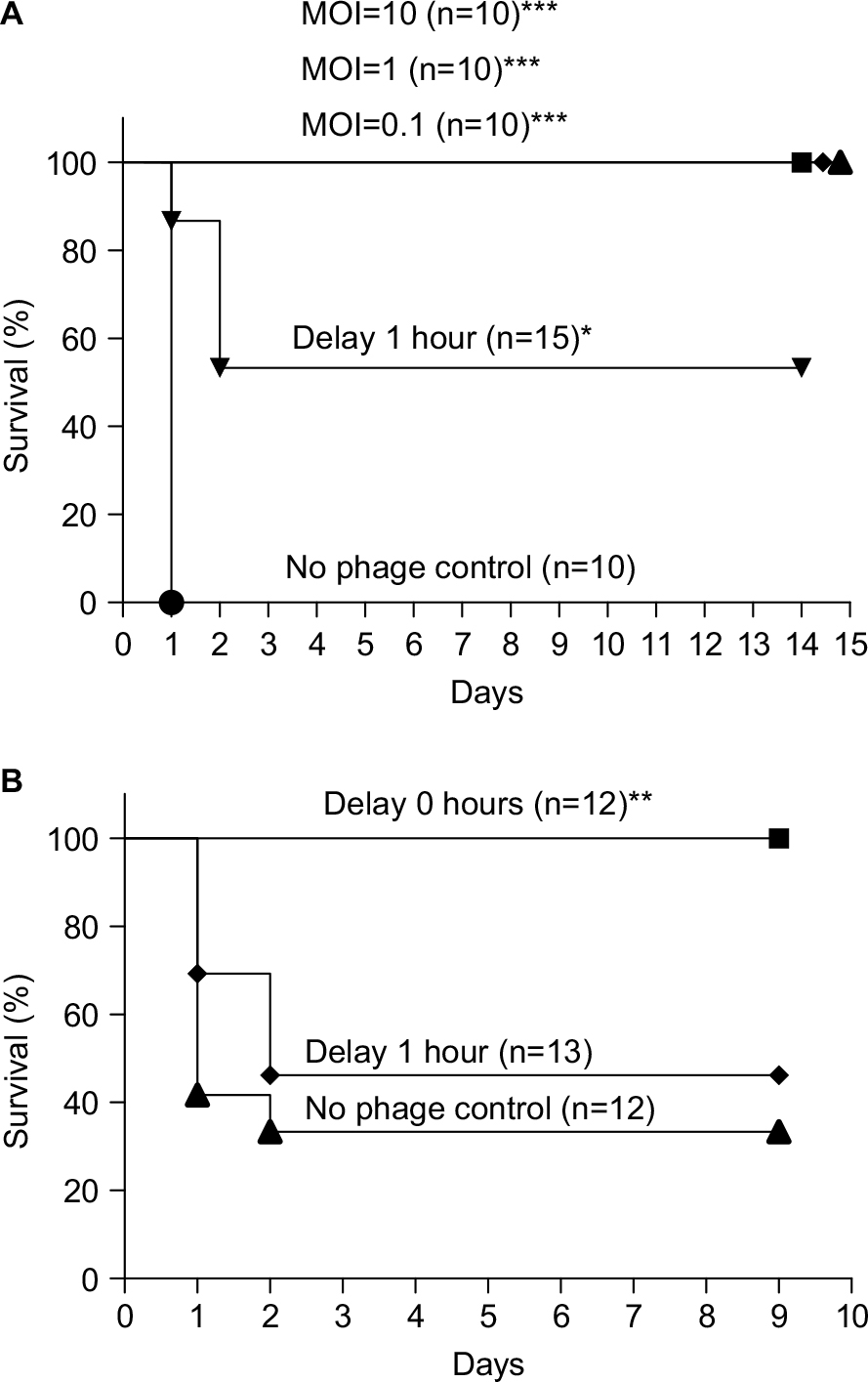 | Figure 2 (A) Survival of BALB/c mice with XDRAB infection after treatment with different MOIs (10, 1, and 0.1) of phage ϕkm18p. Kaplan–Meier survival curves were obtained from mice infected with a lethal dose of ip KM18 (3×108 CFU) in BALB/c mice. ***P-value <0.001 compared to the no phage control. Mice that received ϕkm18p therapy at a 1-hour delay after KM18 injection (MOI=1) showed a significantly different Kaplan–Meier survival curve compared to mice that received immediate phage therapy; *P=0.014. (B) Survival of C57BL/6 mice with ip KM18 (6×108 CFU) (MOI =1); **P=0.0106. The survival rate of mice was observed after 10 days. The control group was injected with PBS (no phage control). Kaplan–Meier survival rate analysis shows that the immediate injection of bacteriophages has a significantly different survival than the 1-hour delayed group and the no phage control. Abbreviations: CFU, colony-forming units; ip, intraperitoneal; MOIs, multiplicities of infection; XDRAB, extensively drug-resistant Acinetobacter baumannii. |
The lethal dose of A. baumannii in C57BL/6 mice was 6×108 CFU (data not shown). We administered an intraperitoneal injection of 6×108 CFU of KM18 to the C57BL/6 mice. The survival was 88%–100% in the mice that received different MOIs of ϕkm18p (10, 1, or 0.1), which was significantly higher than the survival of mice without phage therapy. In mice that received delayed phage therapy, the survival rate decreased to 46% (MOI=1; Figure 2B; P=0.0106).
Bacterial load in mice
In the control group with no phage therapy, the bacterial load was 108 CFU/mL. In the groups that received phage therapy immediately, the bacterial load decreased significantly to below 103 CFU/mL, even at the low MOI (P=0.0078). In the delayed phage therapy group, the bacterial load was also significantly lower (about 103 CFU/mL) than in the control group (P=0.0012; Figure 3A). We determined the bacterial load in the C57BL/6 mice at 3 hours after KM18 infections (Figure 3B). In mice without phage therapy, the mean bacterial load was 108 CFU/mL. The loads were lower in mice received delayed phage therapy (mean bacterial load=106 CFU/mL) and much lower in mice received concurrent phage therapy (102 CFU/mL).
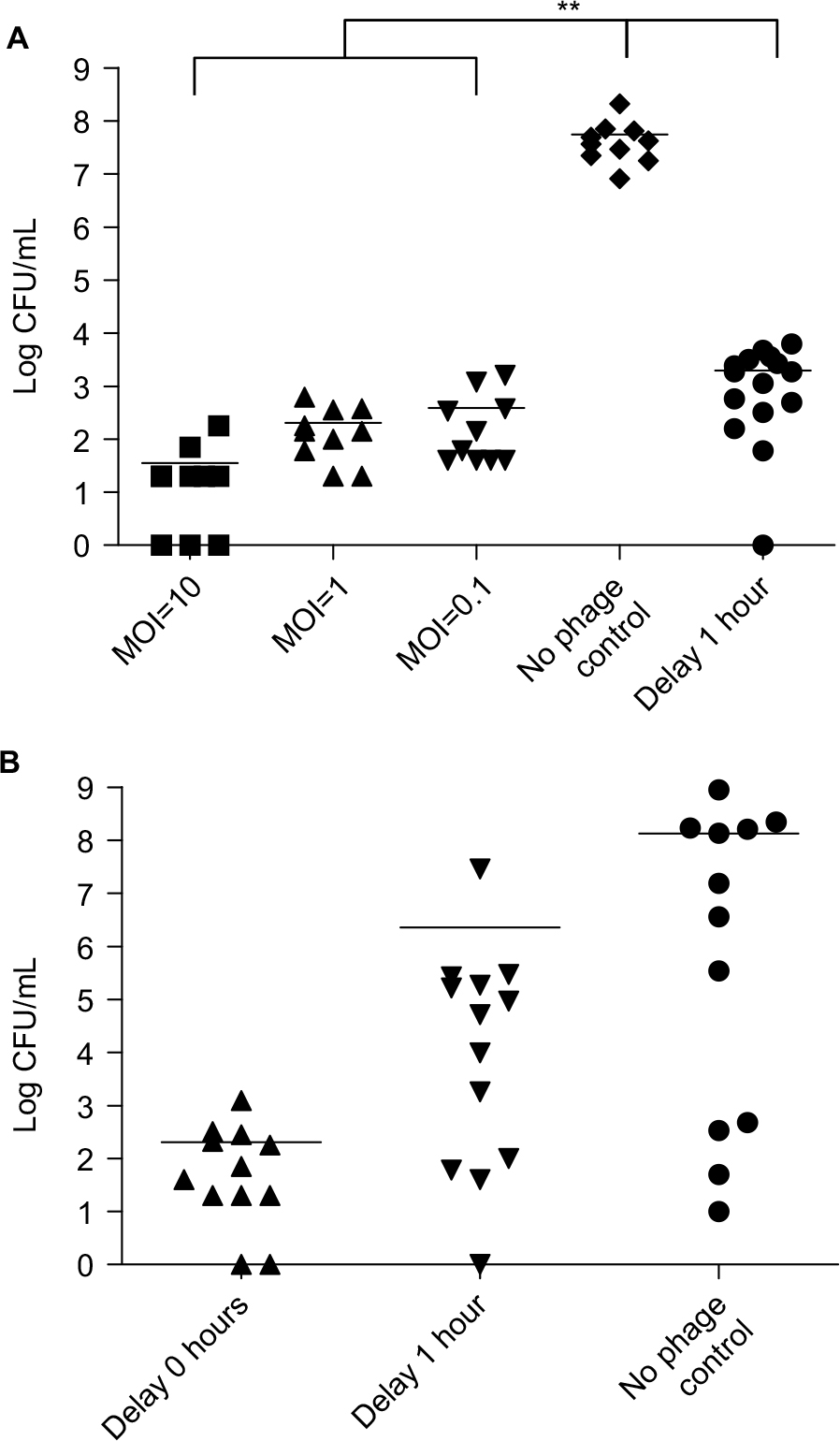 | Figure 3 (A) Bacterial load (log CFU/mL) in the blood of BALB/c mice after intraperitoneal injection of KM18 (3×108 CFU). Five groups were compared: three groups treated with immediate ϕkm18p phage therapy (MOIs: 10, 1, and 0.1), the control group (PBS injection), and one group treated with 1-hour delayed phage therapy (MOI: 1); **P<0.01. (B) Bacterial load in C57BL/6 mice after intraperitoneal injections of 6×108 CFU KM18. The groups received phage therapy (MOI: 1) either immediately after KM18 injection (0-hour delay) or after a 1-hour delay. The bacterial load (CFU/mL) was measured at 3 hours after KM18 injections. Statistical analysis: ANOVA. Abbreviations: CFU, colony-forming units; MOIs, multiplicities of infection. |
Cytokine assay in BALB/c mice
The cytokine assay (Figure 4A) of BALB/c mice revealed high levels of TNF-α and IL-6 (TNF-α: 3,000 pg/mL, IL-6: 35,000 pg/mL) in mice without phage therapy and low levels of TNF-α and IL-6 (TNF-α: 250 pg/mL, IL-6: 1,000 pg/mL) in the phage therapy groups, even at an MOI of 0.1. This effect was also observed in the group with the 1-hour delayed treatment (TNF-α: 400 pg/mL, IL-6: 12,000 pg/mL). The cytokine assay (Figure 4B) of the C57BL/6 mouse model showed high mean levels of TNF-α and IL-6 (TNF-α: 2,000 pg/mL, IL-6: 12,000 pg/mL) in C57BL/6 mice without phage therapy. Both TNF-α and IL-6 levels decreased with phage therapy, even at an MOI of 0.1 (TNF-α: 150 pg/mL, IL-6: 5,500 pg/mL). In the delayed treatment group, the mean TNF-α decreased to 250 pg/mL, but the mean IL-6 level remained high (20,000 pg/mL).
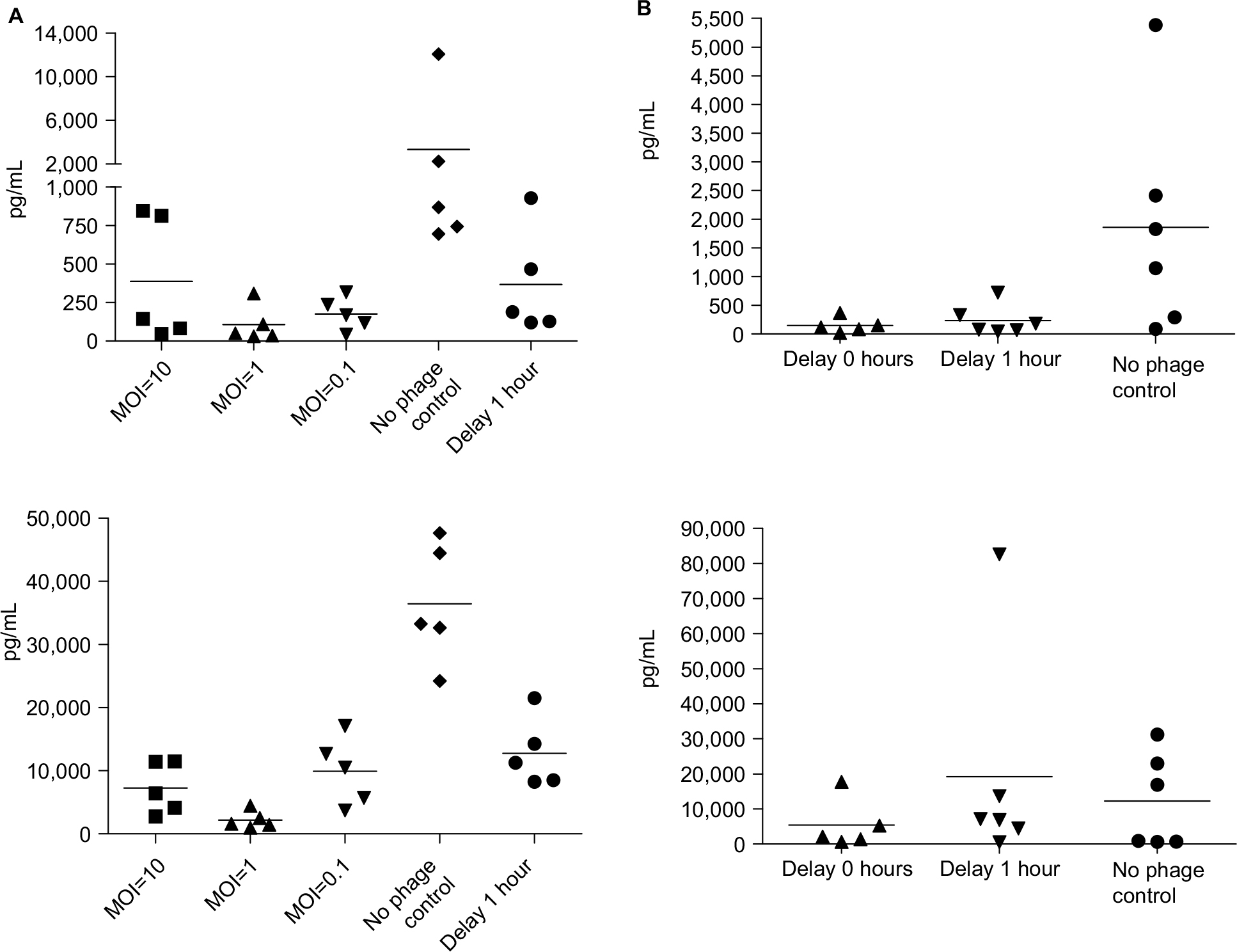 | Figure 4 Cytokine levels were reduced with phage therapy in BALB/c mice. Notes: (A) Top: TNF-α. Bottom: IL-6 levels (pg/mL) measured in mice infected with KM18. The five groups shown received immediate phage therapy (MOIs: 10, 1, and 0.1), control injections (PBS), or 1-hour delayed phage therapy (MOI: 1). (B) In a C57BL/6 mouse model, phage km18p (7.2×108 PFU) was injected immediately and after a delay for of 1 hour to treat mice infected with KM18 (6×108 CFU) (MOI=1). We checked the cytokine concentration 3 hours after phage injection. The contents of TNF (top) and IL-6 (bottom) were measured in blood from the orbital area. Each dot represents the cytokine concentrations in an isolated mouse. Abbreviations: CFU, colony-forming units; IL-6, interleukin-6; MOIs, multiplicities of infection; PFU, phage forming units; TNF-α, tumor necrosis factor alpha. |
Phage-resistant XDRAB mutants
Phage-resistant XDRAB mutants (R1–R5) were isolated from mouse sera after phage therapy. The phage-resistant strains were confirmed in vitro in an overnight culture of ϕkm18p and A. baumannii. The proportion of resistant or sensitive bacteria was about 26%±4%. Southern blotting showed that the R1–R5 mutants carried no phage gene in either the A. baumannii chromosome or plasmid form. These mutant strains were analyzed by pulsed field gel electrophoresis to confirm the identity of DNA patterns with the wild-type strain (data not shown).
Protein lysates extracted from the native KM18, the R1–R5 mutants, and the ϕkm18p phage displayed the same two proteins nearly 170 and 57 kDa (Figure 5). However, the KM18 and R1–R5 mutant isolates did not produce three specific proteins found in the ϕkm18p phage nearly 180, 120, and 50 kDa. Additionally, the R1–R5 phage-resistant mutant isolates displayed a specific protein nearly 36 kDa, which was not observed in the native KM18.
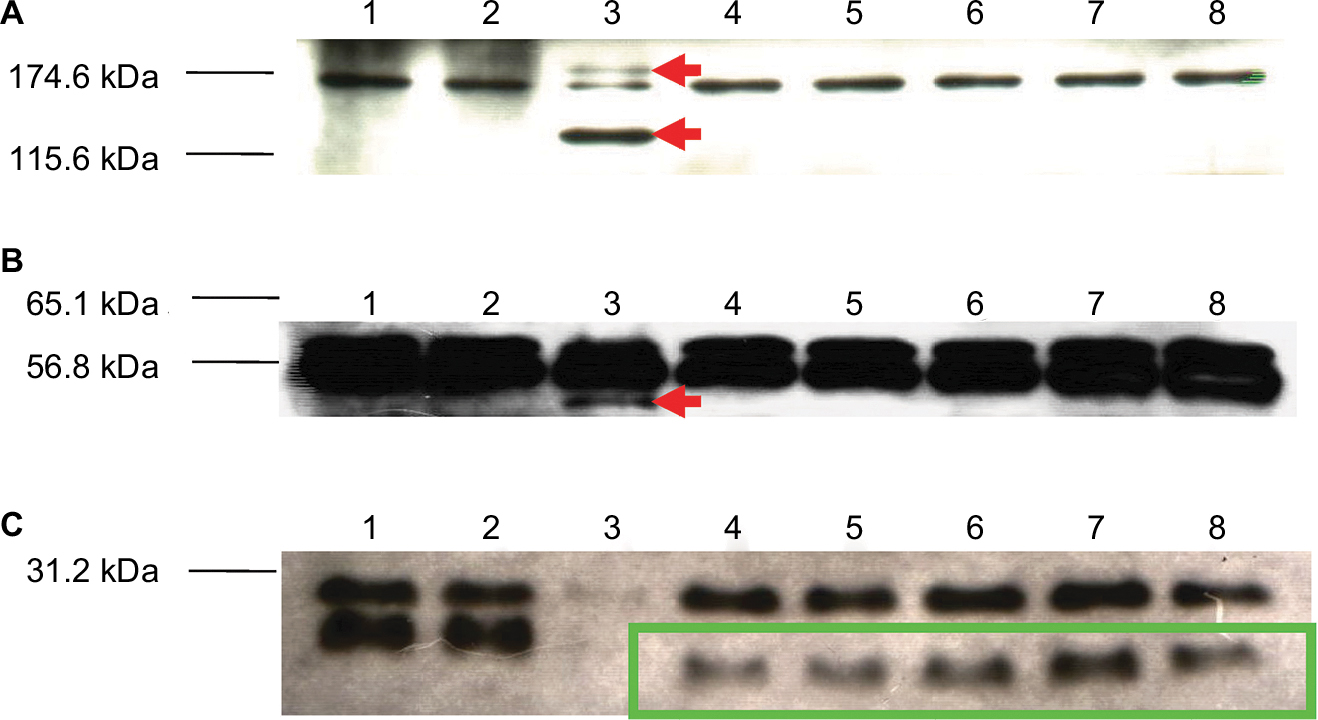 | Figure 5 Western blot analysis showing proteins of different bacterial strains detected with a ϕkm18p-specific antibody. Notes: Lane 1: nonmultidrug-resistant Acinetobacter baumannii isolates (A35). Lane 2: KM18 strain. Lane 3: ϕkm18p phage. Lanes 4–8: phage-resistant mutant A. baumannii isolates R1–R5. Red arrows indicate phage-specific proteins. Green box indicates specific proteins found in phage-resistant mutants. |
Bactericidal assay of a phage-resistant mutant
We performed a serum bactericidal test in whole blood from mice injected with native KM18 or the phage-resistant mutant R1. We used the whole blood from a human infection to explore whether the immune system in whole human blood can effectively remove the mutant strain R1. The wild and variant strains were infected with MOI=0.01 in whole human blood and compared in terms of the difference in the sterilization ability of whole blood. As shown in Figure 6, the wild strain KM18 had a strong antiphagocytosis ability and affected more than 90% of the remaining bacteria. The mutant strain R1 affected only less than 1% of bacteria.
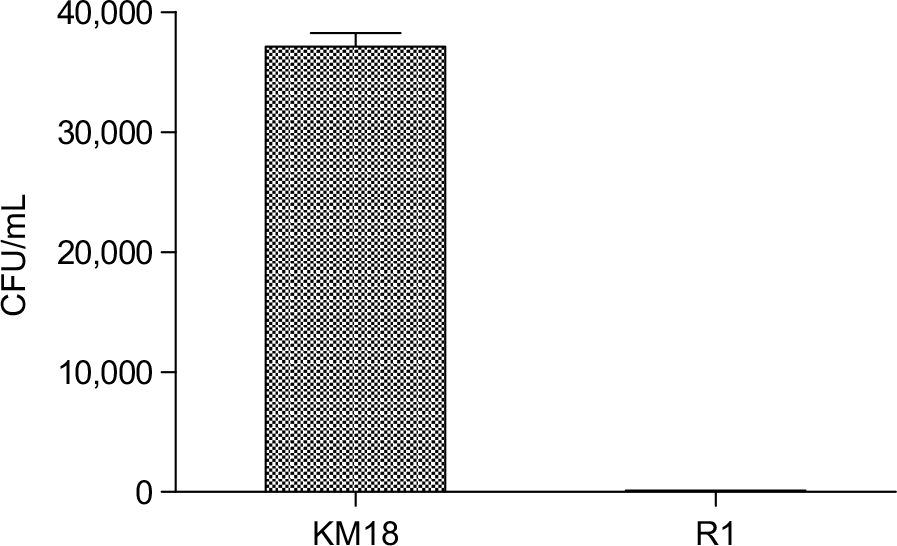 | Figure 6 The number of residual bacteria in whole human blood: R1 mutant strains vs wild strain KM18. Notes: A concentration of 4×104 CFU was mixed with 1 mL of whole human blood (white blood cell: 5–8×104 cells/mL; MOI: ~0.01). After mixing and even distribution, the number of bacteria was determined using a dilution plate. Abbreviations: CFU, colony-forming units; MOI, multiplicity of infection. |
To identify the determinant of the serum bactericidal test, we tested bacterial suspensions of KM18 and R1 cultured in DMEM. We measured the concentration of bacteria at OD600 nm after incubation for 6, 12, and 24 hours and calculated the colony counts (CFU per milliliter) after serial dilution. We found no bacterial growth (Figure 7) when R1 isolates were diluted to a concentration of 4×104 CFU/mL. Moreover, the colony count was higher in cultures of KM18 than R1 that were diluted to the bacterial concentrations of 4×105 or 4×106 CFU/mL.
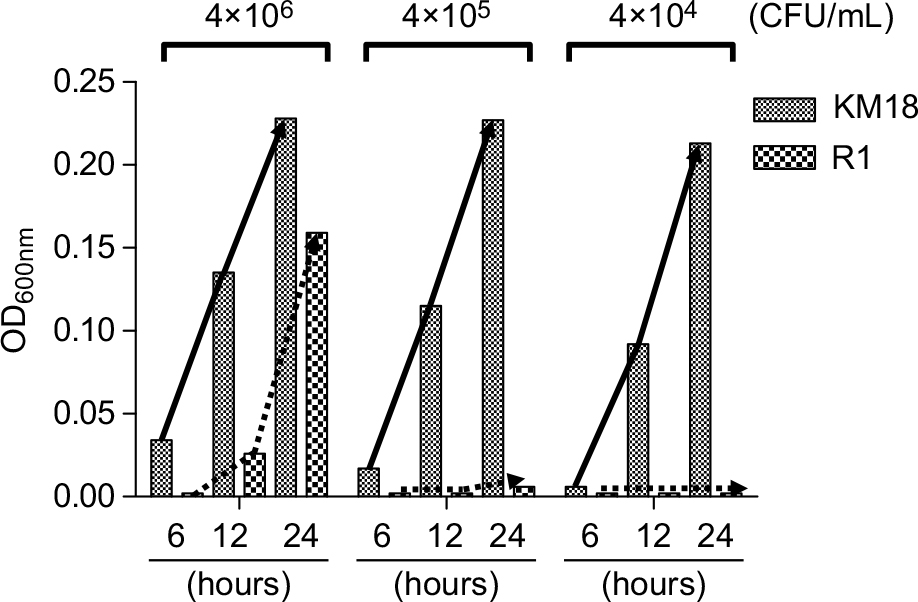 | Figure 7 The growth of KM18 and phage-resistant strains of wild strains R1 in DMEM culture, broth containing 10% FBS, and triad antibiotics. Notes: We added different concentrations of the bacteria and took measurements at 6, 12, and 24 hours to determine the absorbance value at OD600 nm. The solid arrow indicates the growth trend line of KM18, and the dashed arrows indicate the growth trend line of the mutant R1 (phage-resistant strain). |
An analysis of the separate effects of antibiotics and FBS showed that the presence of FBS determined the bactericidal effects on R1 isolates (Figure 8A). The efficacy of phage-resistant cells of pure antibiotics is shown in Figure 8A (third column, FBS [−], antibiotic [+]). The R1 isolates showed a trend of decreased bacterial growth compared to KM18 after pure antibiotic treatment. Moreover, when we used heat-inactivated serum (56°C for 10 or 20 minutes), the R1 isolates showed the same level of bacterial growth as KM18 (Figure 8B).
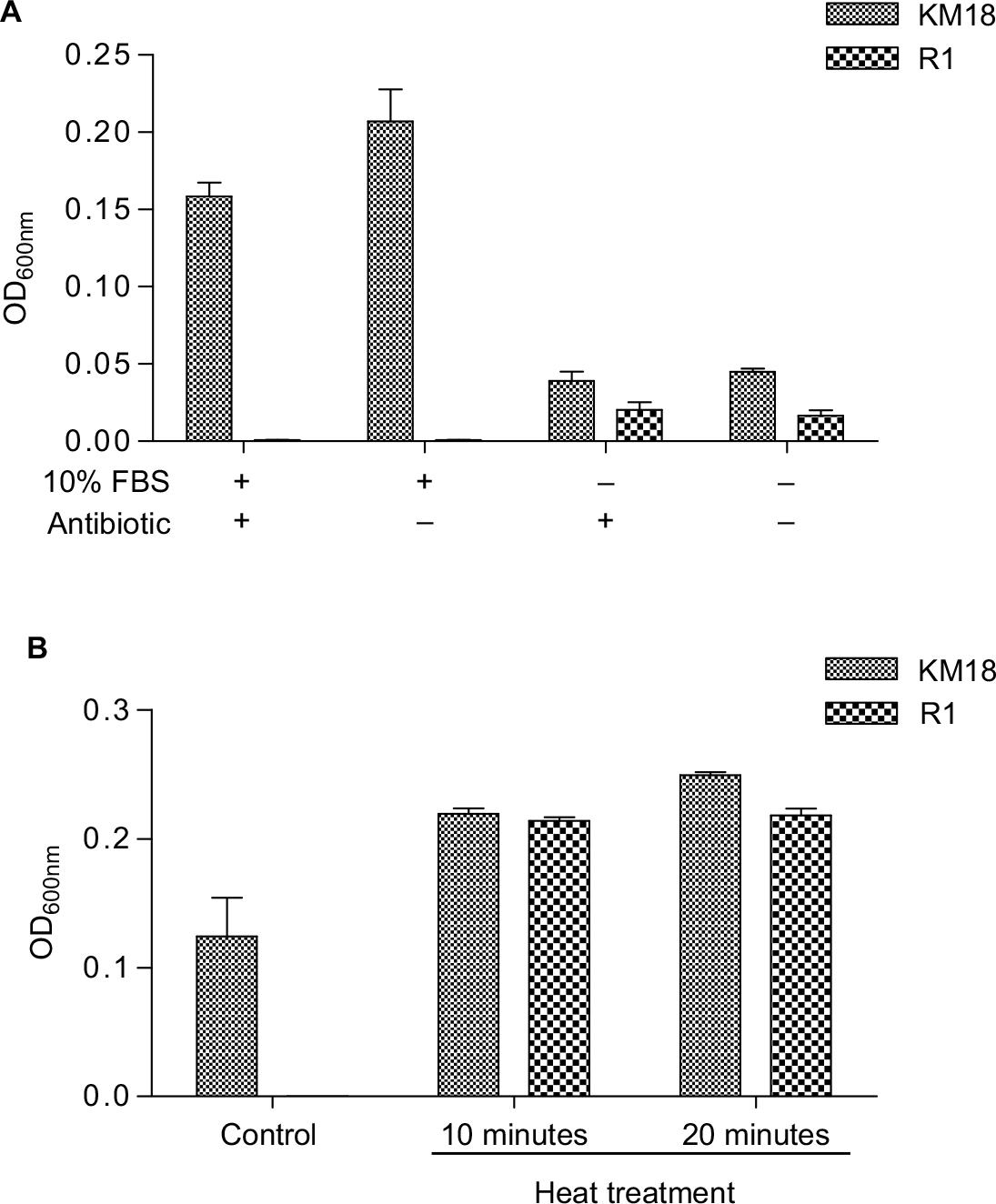 | Figure 8 Separate effects of antibiotic and FBS on bactericidal efficacy in the presence (+) or absence (−) of antibiotic and FBS. Notes: (A) The effect of bovine serum and triad antibiotics on the growth of R1 and KM18 wild strains. The absorbance value was measured after 24 hours. (B) The effect of bovine serum after removing the complement on the growth of KM18 and R1. We checked the OD600 nm absorbance value of the 10× diluted culture liquid after 24 hours. When we used heat-inactivated serum (56°C for 10 or 20 minutes), R1 isolates displayed the same growth as KM18. |
Discussion
Studies on phage therapy against A. baumannii in vitro and in animal models have not been as abundant as studies on other pathogens, such as S. aureus, P. aeruginosa, and E. coli.27–29 In our two mouse models of sepsis (BALB/c and C57BL/6), we showed that immediate ϕkm18p phage therapy could effectively reduce bacterial counts in the blood and increase the survival rate compared to untreated animals. This increase in survival rate correlated with a reduced inflammatory response elicited by the KM18 infection. Although the bacteria developed resistance after phage therapy, the mutant-resistant strains displayed decreased fitness (ie, lower resistance to sera).
This study is the first to test complete phage therapy in animals with acute XDRAB infections. Soothill30 performed a small study on antibiotic susceptible A. baumannii infections. The study showed that an Acinetobacter phage that was active in vitro was protective in vivo at low dose ratios (1 PFU to 106 bacterial CFUs). The present results were consistent with the results from previous studies showing that a single intraperitoneal injection of the appropriate phage could rescue mice with Gram-negative bacilli bacteremia due to E. coli, P. aeruginosa, or K. pneumoniae infections.16,18,31 Phage therapy can decrease the infection, epithelization period, and wound contraction in animal models with a wound infected with MDRAB32,33 and can help MDRAB clearance in the lung.34
Mendes et al35 showed that topical bacteriophage treatment effectively decreased the bacterial load and improved wound healing in two animal models of diabetes mellitus infected with S. aureus and P. aeruginosa but not those infected with A. baumannii. A more recent study showed that treatment with phage lysin could kill A. baumannii in a mouse model.36 Our animal model study showed that phage therapy may represent a therapeutic alternative for treating XDRAB sepsis. The survival rates were 100% for animals treated with ϕkm18p at MOIs 0.1 and 1 (Figure 1). The survival rate was not dependent on the MOI concentrations of phage therapy in our model. However, the inflammatory response was slightly higher with high MOIs.
This animal study showed that the timing of phage therapy was important. A delayed injection of phage led to reduced survival. This result was similar to the results from a previous study that used a diabetic mouse model of Pseudomonas bacteremia. The results showed that protection was reduced when the phage treatment was delayed for 4 and 6 hours after a lethal bacterial challenge.37 Nevertheless, the phage could rescue nondiabetic bacteremic mice even when the treatment was delayed to 20 hours after a lethal bacterial challenge.
In mice infected with methicillin-resistant Staphylococcus aureus, an intraperitoneal phage injection at 6 hours postinfection reduced the severity of the infection and rescued the mice.38 In a rat pup model of ST131 E. coli sepsis,15 early phage therapy administered within 7 hours of infection ensured 100% survival, but survival decreased to 50% when the phage was administered at 24 hours postinfection. The emergence of phage-resistant bacteria may represent a limitation to phage therapy. Bacteria can become resistant through several mechanisms. For example, receptors may become altered and no longer bind phages, or adaptive immunity may develop by interfering with DNA sequences known as clustered regularly interspaced short palindromic repeats.39 Some experts have suggested that resistance may be a temporary trait of bacteria, and thus, the evolution of phage-resistant superbugs is not likely to occur.40
Many studies have shown that bacteriophage resistance in Salmonella, E. coli, and Pseudomonas infections was associated with a reduction in bacterial virulence.41 Similarly, the present study showed that resistant bacteria had reduced virulence in the whole-blood killing assay and in the serum-resistant assay. However, one study on phage therapy on pseudomonas infections showed that phage-resistant variants had a greater tendency to cause membrane damage and produce increased levels of secreted virulence factors.42
The present study showed that phage-resistant mutant bacteria expressed some specific proteins, but the identity and functions of these proteins are unknown. This phenomenon of phage resistance developing in XDRAB treated with phage therapy should be explored in more detail in the future. Alternatively, this problem may be overcome by using phage cocktails to avoid bacterial resistance.38
Based on previous experiences with phage therapies, there are some concerns about using phage therapy in clinical practice. In 2017, a personalized bacteriophage-based therapeutic treatment was reported for a 68-year-old diabetic patient in USA with necrotizing pancreatitis complicated by MDRAB infection.14,43 However, several practical issues have remained unaddressed, including regulation, the limited host range, the development of bacterial resistance to phages, manufacturing difficulties, side effects of bacterial lysis, pharmacokinetics and pharmacodynamics, and the MOI, valency, and delivery in humans.44 These gaps must be resolved before bacteriophage therapy can be successfully introduced in clinical settings. High-throughput methods must be developed for isolating, characterizing, engineering, manufacturing, and delivering phages in a clinical setting.44 Because the narrow host range is a concern, a potential solution maybe the use of a combination of phages as a cocktail.23
Conclusion
In this animal study, we demonstrated that phage therapy decreased serum cytokines and we observed reduced cytotoxicity in phage-resistant mutants. However, the reduced therapeutic effect with delayed administrations may be a concern in developing a successful phage therapy for treating acute infections of multi-drug resistant pathogens. Phage therapy showed good effects in the mouse models of XDRAB bacteremia. The therapy increased the animal survival rates, decreased bacteremia loads, and decreased the levels of the inflammatory markers TNF-α and IL-6. Although phage-resistant mutants developed after phage therapy, they tended to show reduced resistance to human sera in cytotoxicity assays.
Acknowledgments
We are grateful to the members of the medical laboratory for their excellent work. This work was supported by research grants from the Taiwan Ministry of Science and Technology (MOST 102-2313-B-214-001-MY3, 107-2313-B-214 -001 -MY3, and 103-2314-B-214-001). This work was supported by research grants from the Taiwan National Science Council (NSC96-2313-B214-002). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Karageorgopoulos DE, Falagas ME. Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect Dis. 2008;8(12):751–762. | ||
Landman D, Georgescu C, Martin DA, Quale J. Polymyxins revisited. Clin Microbiol Rev. 2008;21(3):449–465. | ||
Sirijatuphat R, Thamlikitkul V. Preliminary study of colistin versus colistin plus fosfomycin for treatment of carbapenem-resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother. 2014;58(9):5598–5601. | ||
Durante-Mangoni E, Signoriello G, Andini R, et al. Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: a multicenter, randomized clinical trial. Clin Infect Dis. 2013;57(3):349–358. | ||
Betrosian AP, Frantzeskaki F, Xanthaki A, Douzinas EE. Efficacy and safety of high-dose ampicillin/sulbactam vs. colistin as monotherapy for the treatment of multidrug resistant Acinetobacter baumannii ventilator-associated pneumonia. J Infect. 2008;56(6):432–436. | ||
Bassetti M, Repetto E, Righi E, et al. Colistin and rifampicin in the treatment of multidrug-resistant Acinetobacter baumannii infections. J Antimicrob Chemother. 2008;61(2):417–420. | ||
Pachón-Ibáñez ME, Docobo-Pérez F, López-Rojas R, et al. Efficacy of rifampin and its combinations with imipenem, sulbactam, and colistin in experimental models of infection caused by imipenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. 2010;54(3):1165–1172. | ||
Ni W, Han Y, Zhao J, et al. Tigecycline treatment experience against multidrug-resistant Acinetobacter baumannii infections: a systematic review and meta-analysis. Int J Antimicrob Agents. 2016;47(2):107–116. | ||
Karageorgopoulos DE, Kelesidis T, Kelesidis I, Falagas ME. Tigecycline for the treatment of multidrug-resistant (including carbapenem-resistant) Acinetobacter infections: a review of the scientific evidence. J Antimicrob Chemother. 2008;62(1):45–55. | ||
Neonakis IK, Spandidos DA, Petinaki E. Confronting multidrug-resistant Acinetobacter baumannii: a review. Int J Antimicrob Agents. 2011;37(2):102–109. | ||
Vila J, Pachón J. Therapeutic options for Acinetobacter baumannii infections: an update. Expert Opin Pharmacother. 2012;13(16):2319–2336. | ||
Hanlon GW. Bacteriophages: an appraisal of their role in the treatment of bacterial infections. Int J Antimicrob Agents. 2007;30(2):118–128. | ||
Burrowes B, Harper DR, Anderson J, Mcconville M, Enright MC. Bacteriophage therapy: potential uses in the control of antibiotic-resistant pathogens. Expert Rev Anti Infect Ther. 2011;9(9):775–785. | ||
Lyon J. Phage therapy’s role in combating antibiotic-resistant pathogens. JAMA. 2017;318(18):1746–1748. | ||
Pouillot F, Chomton M, Blois H, et al. Efficacy of bacteriophage therapy in experimental sepsis and meningitis caused by a clone O25b:H4-ST131 Escherichia coli strain producing CTX-M-15. Antimicrob Agents Chemother. 2012;56(7):3568–3575. | ||
Wang J, Hu B, Xu M, et al. Therapeutic effectiveness of bacteriophages in the rescue of mice with extended spectrum beta-lactamase-producing Escherichia coli bacteremia. Int J Mol Med. 2006;17(2):347–355. | ||
Alemayehu D, Casey PG, Mcauliffe O, et al. Bacteriophages ΦMR299-2 and ΦNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. MBio. 2012;3(2):e00029–00012. | ||
Vinodkumar CS, Kalsurmath S, Neelagund YF. Utility of lytic bacteriophage in the treatment of multidrug-resistant Pseudomonas aeruginosa septicemia in mice. Indian J Pathol Microbiol. 2008;51(3):360–366. | ||
Cao F, Wang X, Wang L, et al. Evaluation of the efficacy of a bacteriophage in the treatment of pneumonia induced by multidrug resistance Klebsiella pneumoniae in mice. Biomed Res Int. 2015;2015:752930–9. | ||
Vinodkumar CS, Neelagund YF, Kalsurmath S. Bacteriophage in the treatment of experimental septicemic mice from a clinical isolate of multidrug resistant Klebsiella pneumoniae. J Commun Dis. 2005;37(1):18–29. | ||
Popova AV, Zhilenkov EL, Myakinina VP, Krasilnikova VM, Volozhantsev NV. Isolation and characterization of wide host range lytic bacteriophage AP22 infecting Acinetobacter baumannii. FEMS Microbiol Lett. 2012;332(1):40–46. | ||
Yang H, Liang L, Lin S, Jia S. Isolation and characterization of a virulent bacteriophage AB1 of Acinetobacter baumannii. BMC Microbiol. 2010;10:131. | ||
Shen GH, Wang JL, Wen FS, et al. Isolation and characterization of Φkm18p, a novel lytic phage with therapeutic potential against extensively drug resistant Acinetobacter baumannii. PLoS One. 2012;7(10):e46537. | ||
Montero A, Ariza J, Corbella X, et al. Efficacy of colistin versus beta-lactams, aminoglycosides, and rifampin as monotherapy in a mouse model of pneumonia caused by multiresistant Acinetobacter baumannii. Antimicrob Agents Chemother. 2002;46(6):1946–1952. | ||
Montero A, Ariza J, Corbella X, et al. Antibiotic combinations for serious infections caused by carbapenem-resistant Acinetobacter baumannii in a mouse pneumonia model. J Antimicrob Chemother. 2004;54(6):1085–1091. | ||
Preston JA, Beagley KW, Gibson PG, Hansbro PM. Genetic background affects susceptibility in nonfatal pneumococcal bronchopneumonia. Eur Respir J. 2004;23(2):224–231. | ||
Iwano H, Inoue Y, Takasago T, et al. Bacteriophage ΦSA012 has a broad host range against Staphylococcus aureus and effective lytic capacity in a mouse mastitis model. Biology. 2018;7(1):8. | ||
Furusawa T, Iwano H, Hiyashimizu Y, et al. Phage therapy is effective in a mouse model of bacterial equine keratitis. Appl Environ Microbiol. 2016;82(17):5332–5339. | ||
Holguín AV, Rangel G, Clavijo V, et al. Phage ΦPan70, a putative temperate phage, controls pseudomonas aeruginosa in planktonic, biofilm and burn mouse model assays. Viruses. 2015;7(8):4602–4623. | ||
Soothill JS. Treatment of experimental infections of mice with bacteriophages. J Med Microbiol. 1992;37(4):258–261. | ||
Hung CH, Kuo CF, Wang CH, Wu CM, Tsao N. Experimental phage therapy in treating Klebsiella pneumoniae-mediated liver abscesses and bacteremia in mice. Antimicrob Agents Chemother. 2011;55(4):1358–1365. | ||
Shivaswamy VC, Kalasuramath SB, Sadanand CK, et al. Ability of bacteriophage in resolving wound infection caused by multidrug-resistant Acinetobacter baumannii in uncontrolled diabetic rats. Microb Drug Resist. 2015;21(2):171–177. | ||
Regeimbal JM, Jacobs AC, Corey BW, et al. Personalized therapeutic cocktail of wild environmental phages rescues mice from Acinetobacter baumannii wound infections. Antimicrob Agents Chemother. 2016;60(10):5806–5816. | ||
Jeon J, Ryu CM, Lee JY, Park JH, Yong D, Lee K. In vivo application of bacteriophage as a potential therapeutic agent to control OXA-66-like carbapenemase-producing Acinetobacter baumannii strains belonging to sequence type 357. Appl Environ Microbiol. 2016;82(14):4200–4208. | ||
Mendes JJ, Leandro C, Corte-Real S, et al. Wound healing potential of topical bacteriophage therapy on diabetic cutaneous wounds. Wound Repair Regen. 2013;21(4):595–603. | ||
Lood R, Winer BY, Pelzek AJ, et al. Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother. 2015;59(4):1983–1991. | ||
Shivshetty N, Hosamani R, Ahmed L, et al. Experimental protection of diabetic mice against Lethal P. aeruginosa infection by bacteriophage. Biomed Res Int. 2014;2014:793242–11. | ||
Takemura-Uchiyama I, Uchiyama J, Osanai M, et al. Experimental phage therapy against lethal lung-derived septicemia caused by Staphylococcus aureus in mice. Microbes Infect. 2014;16(6):512–517. | ||
Hyman P, Abedon ST. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol. 2010;70:217–248. | ||
Ormala AM, Jalasvuori M. Phage therapy: Should bacterial resistance to phages be a concern, even in the long run? Bacteriophage. 2013;3(1):e24219. | ||
León M, Bastías R. Virulence reduction in bacteriophage resistant bacteria. Front Microbiol. 2015;6:343. | ||
Hosseinidoust Z, van de Ven TG, Tufenkji N. Evolution of Pseudomonas aeruginosa virulence as a result of phage predation. Appl Environ Microbiol. 2013;79(19):6110–6116. | ||
Schooley RT, Biswas B, Gill JJ, et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant acinetobacter baumannii Infection. Antimicrob Agents Chemother. 2017;61(10). | ||
Lu TK, Koeris MS. The next generation of bacteriophage therapy. Curr Opin Microbiol. 2011;14(5):524–531. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.