")
Back to Journals » International Journal of Nanomedicine » Volume 12
Efficacy of neutral and negatively charged liposome-loaded gentamicin on planktonic bacteria and biofilm communities
Authors Alhariri M, Majrashi MA, Bahkali AH, Almajed FS , Azghani AO , Khiyami MA, Alyamani EJ, Aljohani SM , Halwani MA
Received 13 May 2017
Accepted for publication 29 August 2017
Published 18 September 2017 Volume 2017:12 Pages 6949—6961
DOI https://doi.org/10.2147/IJN.S141709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Moayad Alhariri,1 Majed A Majrashi,2 Ali H Bahkali,3 Faisal S Almajed,4 Ali O Azghani,5 Mohammad A Khiyami,2 Essam J Alyamani,2 Sameera M Aljohani,6 Majed A Halwani1
1Nanomedicine Department, King Abdullah International Medical Research Center (KAIMRC), King Saud bin Abdulaziz University for Health Sciences, 2National Centre for Biotechnology, Life Sciences and Environment Research Institute, King Abdulaziz City for Science and Technology (KACST), 3Botany and Microbiology Department, College of Science, King Saud University, 4Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud bin Abdulaziz University for Health Sciences, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia; 5Department of Biology, The University of Texas at Tyler, Tyler, TX, USA; 6College of Science and Health Professions, King Saud bin Abdulaziz University for Health Sciences, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia
Abstract: We investigated the efficacy of liposomal gentamicin formulations of different surface charges against Pseudomonas aeruginosa and Klebsiella oxytoca. The liposomal gentamicin formulations were prepared by the dehydration–rehydration method, and their sizes and zeta potential were measured. Gentamicin encapsulation efficiency inside the liposomal formulations was determined by microbiologic assay, and stability of the formulations in biologic fluid was evaluated for a period of 48 h. The minimum inhibitory concentration and the minimum bactericidal concentration were determined, and the in vitro time kill studies of the free form of gentamicin and liposomal gentamicin formulations were performed. The activities of liposomal gentamicin in preventing and reducing biofilm-forming P. aeruginosa and K. oxytoca were compared to those of free antibiotic. The sizes of the liposomal formulations ranged from 625 to 806.6 nm in diameter, with the zeta potential ranging from –0.22 to –31.7 mV. Gentamicin encapsulation efficiency inside the liposomal formulation ranged from 1.8% to 43.6%. The liposomes retained >60% of their gentamicin content during the 48 h time period. The minimum inhibitory concentration of neutral formulation was lower than that of free gentamicin (0.25 versus 1 mg/L for P. aeruginosa and 0.5 versus 1 mg/L for K. oxytoca). The negatively charged formulation exhibited the same bacteriostatic concentration as that of free gentamicin. The minimum bactericidal concentration of neutral liposomes on planktonic bacterial culture was twofold lower than that of free gentamicin, whereas the negatively charged formulations were comparable to free gentamicin. The killing time curve values for the neutral negatively charged formulation against planktonic P. aeruginosa and K. oxytoca were better than those of free gentamicin. Furthermore, liposomal formulations prevent the biofilm-formation ability of these strains better than free gentamicin. In summary, liposomal formulations could be an effective lipid nanoparticle to combat acute infections where planktonic bacteria are predominant.
Keywords: drug delivery, stability, antibacterial activity, biofilm
Introduction
Antibiotic resistance of bacterial pathogens has become a serious public health problem since the introduction of the first antibiotics, such as sulfa drugs and penicillin, into clinical use.1,2 Multidrug-resistant pathogens present a serious challenge in the treatment of infectious diseases; they are classified as the second leading cause of mortality globally and the third leading cause in developed countries.2–4 To avoid and prevent the emergence and spread of resistant bacteria, it is important to modify conventional antibiotics and develop new antibacterial agents.2,5,6
It has been reported that many strains of Staphylococcus aureus are already resistant to all antibiotics, except vancomycin. However, some strains did exhibit resistance to vancomycin, defining the problem of multi-antibiotic resistance.7 A Gram-negative bacterium Pseudomonas aeruginosa has already evaded most existing antibiotics.8 The multidrug-resistant strains of P. aeruginosa are often isolated from nosocomial infections of patients who are admitted to intensive care units.9,10 Moreover, Acinetobacter species showed increased antimicrobial resistance levels in the past decade, and Acinetobacter baumannii is recognized as a multidrug-resistant species that is difficult to control and treat.11,12 The clinical isolates of Klebsiella oxytoca is an active K1 β-lactamase enzymes which can cause resistance to cephalosporin and aztreonam antibiotics.13,14 In addition, Proteus vulgaris has the capacity to resist antibiotic treatment due to the inducible expression of chromosomal β-lactamases,15 while Listeria monocytogenes showed resistance to antibiotic treatment that could be acquired through self-transferable plasmids, mobilizable plasmids, and conjugative transposons.16
Biofilm formation is an important factor in bacterial virulence. It follows the adherence of the bacteria to abiotic or biotic surfaces, and increases the ability of bacteria to survive environmental stress and enhances protection against bactericidal effects.17 In addition, biofilm formation is crucial for bacteria to resist antibiotic chemotherapy.18 P. aeruginosa and K. oxytoca biofilms are negatively charged; therefore, binding of the positively charged antibiotics to the negatively charged biofilm matrix polymers prevents the penetration of antibiotics such as gentamicin into the bacteria.19–22
Gentamicin, a member of aminoglycoside antibiotics, is commonly used for treating severe nosocomial infection caused by Gram-negative bacteria.23 Aminoglycosides exert their effect on bacteria by increasing the cell membrane permeability through irreversible binding of aminoglycosides to divalent cations and inhibiting normal protein synthesis through binding to the highly conserved A-site of bacterial 16 S subunit of 30 S rRNA.24 This binding leads to disruption of proofreading in the protein synthesis process, as a result, bacterial virulence factors production including biofilm can be disrupted.25,26 The disruption results in the instability of the membranes, greater penetration of bactericidal agents, and finally cell death.27 Aminoglycosides can be used synergistically with different types of antibiotics such as β-lactams to provide a broad coverage for bacterial infections.28 The use of aminoglycosides in clinical settings has been challenged by their high toxicity and emergence of drug-resistant strains.29 The use of novel liposomal formulations appears to be a viable option in combating this universal problem.
Liposomes are spherical lipid nanoparticles composed of one or more phospholipid bilayers.30 A diverse group of biologic molecules such as proteins, DNA, RNA, drugs, antioxidants, as well as vitamins can be encapsulated within the liposome bilayers.31 Similar to a cellular membrane, liposomal formulations can incorporate hydrophilic and hydrophobic molecules and are biocompatible and biodegradable.32 The protective nature of liposomes is another attractive feature of this nanoparticle. Encapsulated drugs and molecules within liposome compartments are protected from the biologic milieu and destructive host defense molecules, such as interleukins and the proteases of the complement system.33 Liposomal particles are characterized by slow and continued release of encapsulated drugs from their compartment, ultimately improving their bioavailability and distribution in targeted tissues.32 Liposome-encapsulated bactericidal agents can be delivered to the lung for direct effect on pulmonary infection.34 Liposomes are an attractive option for delivery of bactericidal agents to the lung, since they can be prepared from ingredients compatible to the lung.35
The loading capacity of the therapeutic agent inside the liposomal nanoparticles is critical for achieving an effective and sustainable drug release at the target site. Therefore, the objective of the study was to make and test the effectiveness of neutral and negatively charged liposomal formulations. We found that the neutral formulation has a potent activity against planktonic bacteria, whereas the negatively charged liposomes exhibit a potent antibiofilm activity.
Materials and methods
Microorganisms
P. aeruginosa ATCC 27853, Escherichia coli ATCC 25922, A. baumannii BAA747, K. oxytoca 700324, P. aeruginosa BAA1744, P. aeruginosa 9721, Pr. vulgaris 6380, S. aureus BAA976, S. aureus BAA977, L. monocytogenes BAA751, and S. aureus ATCC 29213 were kindly provided by Dr Essam Alyamani at the National Center for Biotechnology, King Abdulaziz City for Science and Technology. These organisms were stored at −80°C in a Mueller–Hinton broth supplemented with 50% (v/v) glycerol. For experimentation, these strains were inoculated onto Mueller–Hinton agar plates and incubated for 18 h at 37°C.
Chemicals
The negatively charged phospholipid 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DMPG), cholesterol (Chol), and the neutral phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Triton X-100, chloroform, and methanol were purchased from Sigma-Aldrich (St Louis, MO, USA). Gentamicin sulfate was purchased from Amresco (Solon, OH, USA). The Difco Mueller–Hinton agar and broth and Luria-Bertani agar were purchased from BD (Sparks, MD, USA).
Preparation of liposomal formulations
Different formulations of negatively charged and neutral lipids and Chol in different molar ratios with a total of 20 mg/mL of lipid content, that is, 7.5 mg of DPPC:10.5 mg of DMPG:2 mg of Chol in a molar ratio 2:3:1 (NELG-1), 6.3 mg of DPPC:12 mg of DMPG:1.7 mg of Chol in a molar ratio 2:4:1 (NELG-2), and 15.8 DPPC:4.2 mg of Chol in a molar ratio 2:1 (NLG), were prepared through the dehydration–rehydration vesicle method.36 Lipids were weighed and placed in round-bottomed flasks. An appropriate amount of chloroform:methanol (2:1) was added, until the lipids were completely dissolved. Then, the organic solvents were evaporated in a rotary evaporator (BÜCHI Rotavapor R-114) at 45°C, until a thin layer of lipid was formed. High-grade nitrogen gas was used to flush out any trace of chloroform, and the flasks were left at room temperature overnight to ensure that there was no trace of chloroform. The lipid film was then rehydrated with 1 mg/mL gentamicin dissolved in PBS at pH 7.4 or rehydrated with PBS to form empty liposomes without antibiotics. The samples were sonicated (Model 3000 ultrasonic homogenizer; Biologics, Inc., Manassas, VA, USA) for 5 min (40 s run, 20 s pause) in order to homogenize liposomal particle size before lyophilizing the preparation in a freeze dry system (FreeZone Triad Cascade Benchtop; Labconco Corporation, Kansas City, MO, USA). The lyophilized liposomes were kept at −80°C until use.
For rehydration, 10% of PBS was added at 10% of the original volume and vortexed. This step was repeated three times before adding more PBS to bring the solution to its original volume. Excess unencapsulated gentamicin was removed by three rounds of washing with cold PBS using an Eppendorf centrifuge at 16,000× g for 20 min at 4°C (5804R; Eppendorf, San Diego, CA, USA). Size and z-potential were measured in triplicate using a Zetasizer Nano ZSP (Malvern, Worcestershire, UK).
Encapsulation efficiency (EE) of gentamicin
The quantity of encapsulated gentamicin was measured through an agar diffusion assay using S. aureus (ATCC 29213) as a reference microorganism according to the Clinical and Laboratory Standards Institute standards.37 Briefly, the overnight bacterial culture was adjusted to 1.5×108 colony forming units (CFU)/mL (equivalent to 0.5 McFarland standard). The bacteria were then suspended in 300 mL warm (45°C) Mueller–Hinton agar, poured into a sterilized disposable square culture plate (45×45 cm), and left to solidify at room temperature. Using a vacuum puncher, wells of 5 mm in diameter were made on the agar. Standard curves of diluted gentamicin as well as samples of liposomes were prepared. Liposome samples were lysed with 0.2% Triton X-100. This level of Triton X-100 has no effect on the assay performance as tested. Triplicate samples (50 μL) were transferred into the agar plate holes. The plate was covered and incubated for 18 h at 37°C. The inhibition zones were measured, and the average of the triplicate measures was used in the data analysis. The standard curve was utilized to calculate the concentration of encapsulated gentamicin that was released from the liposomes by 0.2% Triton X-100. The EE was calculated as a percentage of encapsulated gentamicin in the liposome relative to the initial concentration of gentamicin. Measuring the EE was performed in triplicate.
|

Biologic fluids collection
Sputum
Sputum samples were obtained from the patients following informed consent, after the regular daily routine laboratory tests were performed by the Microbiology Laboratory at King Abdulaziz Medical City National Guard Hospital Riyadh, Saudi Arabia (protocol approved by King Abdullah International Medical Research Center [KAIMRC] Institutional Review Board: SP16/312/R). Personal information of patients who signed the consents, including age, name, treatments, and exacerbation records, was kept confidential. Samples were diluted 1:10 (w/v) in PBS, autoclaved, aliquoted in 50 mL tubes, and frozen at −80°C until use, as previously described.38
Bronchoalveolar lavage
Male rats (250–300 g; rats were obtained from Animals’ Experiment Care Unit, College of Pharmacy, King Saud University, Riyadh, Kingdom of Saudi Arabia) were maintained according to the animal welfare guidelines of KAIMRC and used for bronchoalveolar lavage (BAL) collection according to the protocol approved by KAIMRC’s Institutional Review Board: SP16/312/R. Briefly, the rats were euthanized by using CO2. The lungs and trachea were collected and then lavaged with 3 mL cold PBS using a scalp vein (butterfly syringe). The lungs were gently filled with PBS and withdrawn by 10 mL syringe to a total volume of 30 mL, and stored in aliquots at −80°C until use, as previously described.39
Stability of liposomal formulation in different environments
We tested the novel liposomal gentamicin formulations for their retention of gentamicin in different physiologic fluids for the time intervals 0, 2, 4, 6, 8, 12, 24, and 48 h. This test would give more information about the release of the content of nanoparticles in biologic milieus and with different routes of administration. Samples were washed as described previously36 and suspended in 1 mL of PBS, BAL, plasma, or sputum. The mixtures were incubated with a mild agitation at 150 rpm at either 4°C for PBS or 37°C for the PBS, BAL, plasma, or sputum. Samples were harvested after the designated time intervals and centrifuged at 16,000× g at 4°C for 15 min (centrifuge 5804R, Eppendorf). The supernatants were tested for gentamicin release using an agar diffusion assay as described previously.37 Results were collected for the three separate experiments in duplicate. Retention of gentamicin in percentage was calculated as follows:
|

Antibacterial activity assay
The minimum inhibitory concentrations (MICs) of the free gentamicin and the liposomal gentamicin formulation were determined using a microbroth dilution method for P. aeruginosa ATCC 27853, E. coli ATCC 25922, A. baumannii BAA747, K. oxytoca 700324, P. aeruginosa BAA1744, P. aeruginosa 9721, Pr. vulgaris 6380, S. aureus BAA976, S. aureus BAA977, and L. monocytogenes BAA751, as reported previously.40 Serial dilutions of gentamicin in free or liposomal formulations at 0.125, 0.25, 0.5, 1, 2, 4, 8, 16, 32, and 64 mg/L were prepared in the Mueller–Hinton broth. Empty liposomes were also suspended in Mueller–Hinton broth to be tested on bacteria. The overnight bacterial suspension was adjusted to follow the 0.5 McFarland standard. The bacteria were then diluted to achieve a final inoculum of 5×105 CFU/mL. The lowest concentration of the liposomal gentamicin formulations and the free gentamicin that inhibited the visible bacterial growth (inhibition ≥80%) after 24 h at 37°C was defined as the MIC.41 Media with and without the same amount of inoculum were used as positive and negative controls, respectively. The minimum bactericidal concentration (MBC) was defined as the lowest concentration of either free or liposomal formulations that resulted in eliminating bacterial growth. Three separate experiments were performed in duplicate to confirm the results.
Time kill assays
We selected two of the bacterial strains, P. aeruginosa BAA1744 and K. oxytoca 700324, as model microbes to compare the optimal bactericidal activity time of the liposomal formulations and the free gentamicin in vitro. The killing time assay was performed for the liposomal gentamicin formulations and the free antibiotic using P. aeruginosa and K. oxytoca as described previously.42 Briefly, bacterial isolates at a final inoculum of 5×105 CFU/mL were suspended in Mueller–Hinton broth and incubated at 37°C for 2, 4, 6, 8, and 24 h in the presence of either free or liposomal formulation–encapsulated gentamicin at one, two, or four times the MICs. Samples were taken at each designated time interval and plated on Mueller–Hinton agar in triplicate for the bacterial count. Three separate experiments were performed and plates with CFUs between 30 and 300 colonies were included in this assay.
Biofilm assays
We evaluated the efficacy of our liposomal formulations to prevent and reduce bacterial biofilm formed by P. aeruginosa BAA1744 and K. oxytoca 700324. Biofilm assay was performed to test the ability of liposomal formulations at sub-MIC to prevent biofilm communities, as described previously.43 Briefly, a standard static biofilm assay was performed by using an overnight bacterial culture adjusted to 0.5 McFarland standard and diluted to 1.5×107 CFU/mL. Samples were seeded in a 96-well polystyrene plate in the presence of 1/2× MIC of NLG, NELG-1, NELG-2, or free gentamicin. The plate was then incubated for 18 h at 37°C. After the incubation period, unattached bacteria were removed and plates were washed three times with sterile PBS. The biofilm formed was stained with 0.1% crystal violet for 10 min and washed once to remove excess stain, and left to dry. Ethanol (95%) was added to the plates to solubilize the crystal violet for 15 min. Each plate was mixed, and solubilized biofilm was transferred to a new 96-well flat-bottomed plate. The OD was measured at 590 nm using a microplate reader PowerWav XS2 (BioTek Instruments Inc., Winooski, VT, USA). The biofilm reduction assay was performed to determine whether the liposomal formulations at a concentration of 16× MIC could inhibit the formation of biofilm, as described previously.44 For biofilm reduction, bacteria were allowed to attach to the surface of a 96-well polystyrene plate and allowed to form a biofilm for 18 h. The next day, unadhered bacteria were washed and the biofilm was incubated again for 18 h with treatments at 16× MIC. Biofilm formation was measured as above. All tests were performed three times in duplicate.
Statistical analysis
The data are presented as the mean and standard error of the mean of the three independent experiments. Comparisons of groups were made through one-way analysis of variance using InStat3 from GraphPad Software (version 5.0) followed by a Tukey-Kramer multiple comparisons test. P-values of *P<0.05, **P<0.01, and ***P<0.001 were considered as statistically significant.
Results
Particle size and polydispersity index of liposomal formulations
We compared the physicochemical properties of our formulations in terms of their size, charge, polydispersity, and EE. The NLG yielded 625±5.5 nm in size with relatively low EE, that is, 1.8%±0.15% (20±1.5 mg/L). The formula net charge was neutral (−0.22±0.17 mV). The sizes of negatively charged NELG-1 and NELG-2 were 691.3±3.3 and 806.6±3.4 nm with zeta potentials of −25.3±2.5 and −31.7±1.7 mV, respectively. The gentamicin loads in these formulations were 37.2%±0.46% (370±4.6 mg/L) for NELG-1 and 43.6%±0.65% (440±6.5 mg/L) for NELG-2, as shown in Table 1.
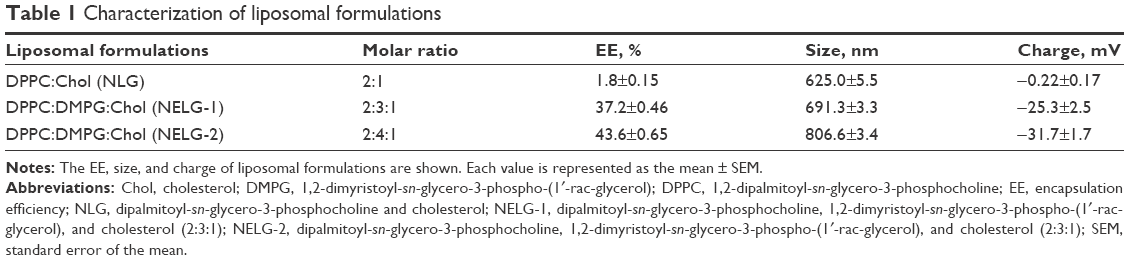 | Table 1 Characterization of liposomal formulations |
Stability of liposomal formulations
To test the stability of the liposomal formulations to hold gentamicin within the phospholipid vehicle, we exposed the NLG, NELG-1, and NELG-2 formulations to different conditions and measured gentamicin release as an indicator of stability. The liposomal formulation NLG in PBS at 4°C retained 92.5%±1.2%, NELG-1 retained 94.3%±0.4%, and NELG-2 retained 95.7%±0.7% of gentamicin after 48 h. Liposomal formulations in PBS at 37°C retained gentamicin as follows: 75.0%±2.8% for NLG, 74.3%±0.7% for NELG-1, and 75.2%±1.5% for NELG-2 after 48 h. In the presence of BAL at 37°C, NLG retained 67.5%±1.2%, NELG-1 retained 57.1%±4.0%, and NELG-2 retained 45.2%±2.6% of gentamicin after 48 h. In the presence of plasma at 37°C, the NLG formulation retained 55.0%±0.9% of encapsulated gentamicin, whereas NELG-1 and NELG-2 formulations retained only 31.4%±0.8% and 22.7%±3.2% of gentamicin, respectively, after 48 h. In the presence of sputum at 37°C, NLG retained 61.7%±2.4% of the encapsulated gentamicin, whereas NELG-1 and NELG-2 retained 48.3%±4.0% and 40.2%±1.1%, respectively, after 48 h. After 4 h of incubation from the initial period, the liposomal formulations’ retention was decreased dramatically and then stabilized in all physiologic conditions (Figures 1–3).
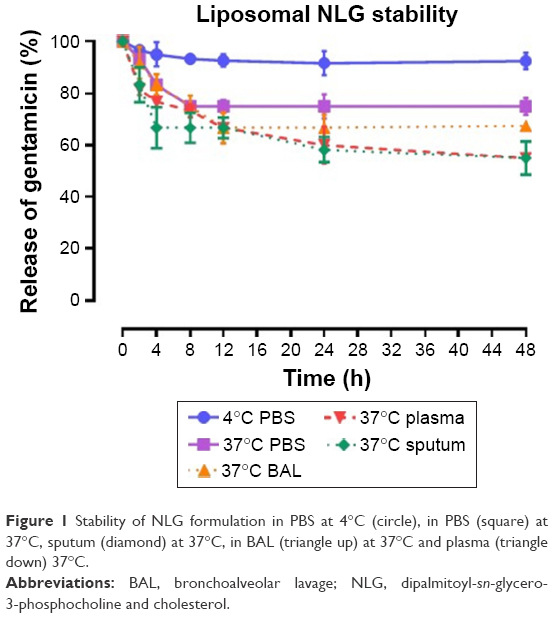 | Figure 1 Stability of NLG formulation in PBS at 4°C (circle), in PBS (square) at 37°C, sputum (diamond) at 37°C, in BAL (triangle up) at 37°C and plasma (triangle down) 37°C. |
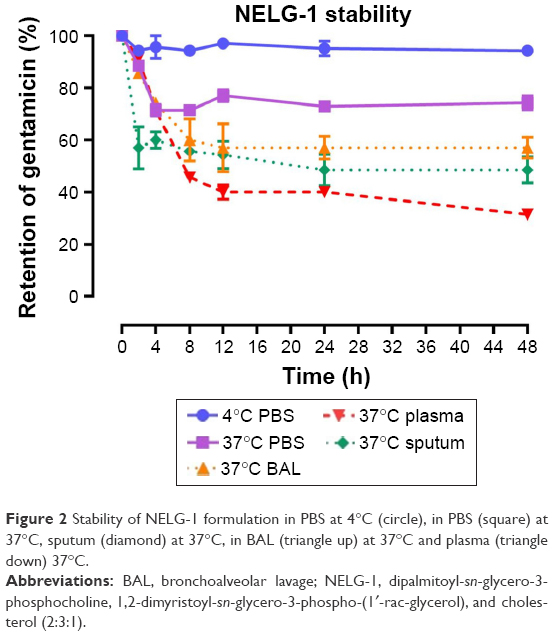 | Figure 2 Stability of NELG-1 formulation in PBS at 4°C (circle), in PBS (square) at 37°C, sputum (diamond) at 37°C, in BAL (triangle up) at 37°C and plasma (triangle down) 37°C. |
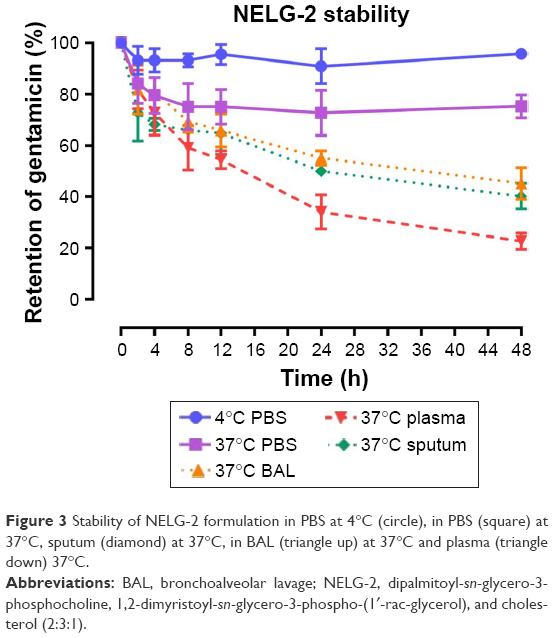 | Figure 3 Stability of NELG-2 formulation in PBS at 4°C (circle), in PBS (square) at 37°C, sputum (diamond) at 37°C, in BAL (triangle up) at 37°C and plasma (triangle down) 37°C. |
Antimicrobial activity of liposomal gentamicin
We examined the MIC and MBC of encapsulated gentamicin in both neutral and negatively charged formulations and compared them to those of the free drug against several Gram-negative and Gram-positive bacterial strains. Empty liposomes consisting of DPPC:DMPG:Chol (2:3:1) and (2:4:1) and DPPC:Chol (2:1) had no effect on bacteria, as compared to the positive control. As shown in Table 2, the MICs and MBCs of the encapsulated gentamycin were significantly lower than those of free gentamicin in inhibiting the growth of bacterial strains tested. In particular, the MBCs of liposomal formulations were considerably lower than free gentamicin which exhibited a relatively high MBC of 8 mg/L against K. oxytoca 700324.
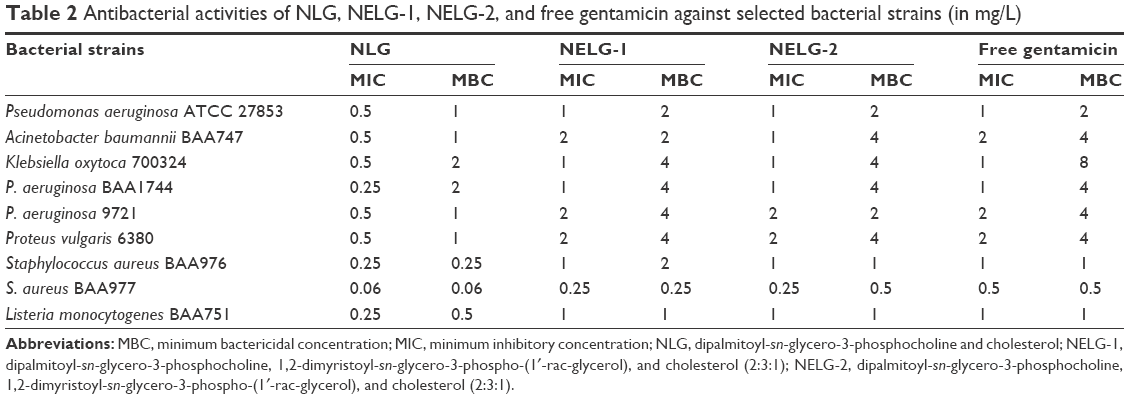 | Table 2 Antibacterial activities of NLG, NELG-1, NELG-2, and free gentamicin against selected bacterial strains (in mg/L) |
Time kill assays
Time kill of P. aeruginosa
The neutral liposomal gentamicin formulation at 1–4× MIC (0.25–1 mg/L) was more effective in killing planktonic P. aeruginosa in terms of time and dose. NLG (0.25 mg/L) and free gentamicin (1 mg/L) at 1× MIC had a comparable bacteriostatic effect at different time intervals between 0 and 24 h. However, the kill time at 2× MIC for NLG (0.5 mg/L) was slightly shorter than the free drug (2 mg/L), that is, 6 versus 8 h.
Considering the differences in MICs for NLG and free gentamicin (0.5 versus 2 mg/L), however, it was evident that the liposomal formulation exhibited a similar kill time for P. aeruginosa at a concentration lower than that of free gentamicin (Figure 4).
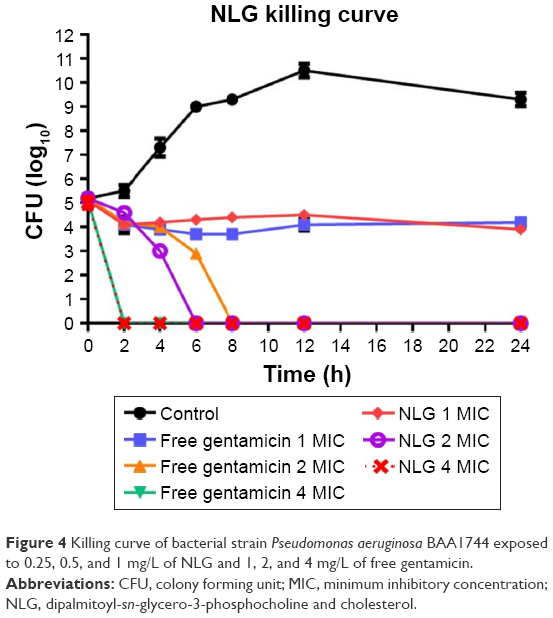 | Figure 4 Killing curve of bacterial strain Pseudomonas aeruginosa BAA1744 exposed to 0.25, 0.5, and 1 mg/L of NLG and 1, 2, and 4 mg/L of free gentamicin. |
NELG-1 formulation and free gentamicin had comparable bacteriostatic and bactericidal effects on P. aeruginosa. NELG-1 and free gentamicin eliminated the bacteria at 2× MIC (2 versus 2 mg/L) in 8 h and inhibited their growth at 1× MIC (1 versus 1 mg/L) during a 24 h study period. Both forms of the drug had comparable MIC as well (Figure 5).
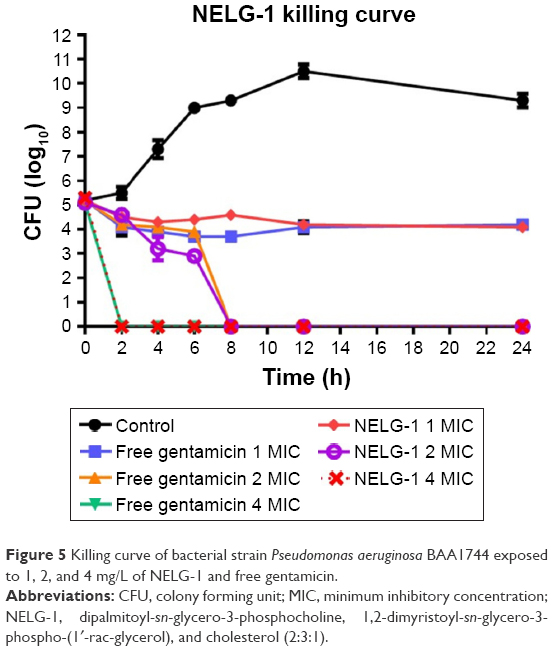 | Figure 5 Killing curve of bacterial strain Pseudomonas aeruginosa BAA1744 exposed to 1, 2, and 4 mg/L of NELG-1 and free gentamicin. |
Finally, free gentamicin at concentrations 1, 2, 4 mg/L was equivalent to NELG-2 (1, 2, 4 mg/L) formulation in terms of preventing bacterial growth with the amount of antibiotic, but performed better in killing the organism in 8 h compared to 12 h time kill of liposomal gentamicin (Figure 6).
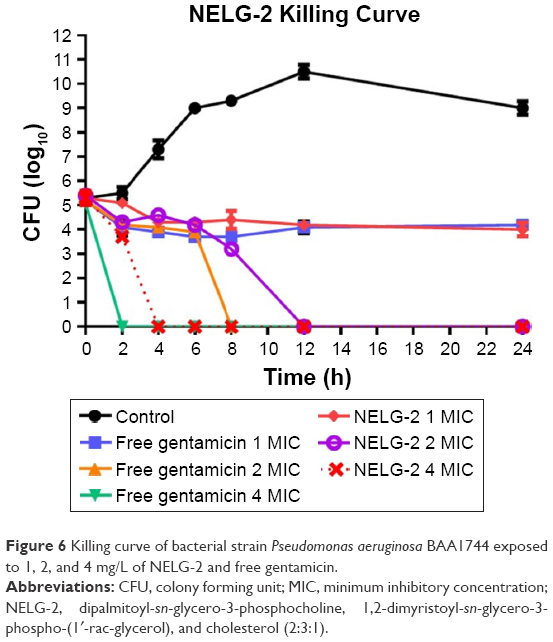 | Figure 6 Killing curve of bacterial strain Pseudomonas aeruginosa BAA1744 exposed to 1, 2, and 4 mg/L of NELG-2 and free gentamicin. |
Time kill of K. oxytoca
This assay was also used to measure the killing time interval for NLG formulation against planktonic K. oxytoca 700324 strain in comparison to free gentamicin. The NLG formulation at 0.5 mg/L (1× MIC) exhibited suppression of bacterial growth at log10 4.3 CFU/mL in 24 h. Free gentamicin at 1 mg/L (1× MIC) did not reach the suppression potency of NLG, since it was only able to suppress bacterial growth up to log10 6.6 CFU/mL in 24 h. Likewise, the NLG formulation at 1 mg/L (2× MIC) showed a decrease in bacterial colonies to log10 2.3 after 24 h, whereas free gentamicin at 2 mg/L (2× MIC) failed to reach this suppression potency of the NLG formulation over 24 h. Killing activity for NLG appeared at 2 mg/L (4× MIC), eradicating the bacteria at 2 h, whereas free gentamicin at 4 mg/L (4× MIC) exhibited killing activity against K. oxytoca 700324 after 4 h (Figure 7).
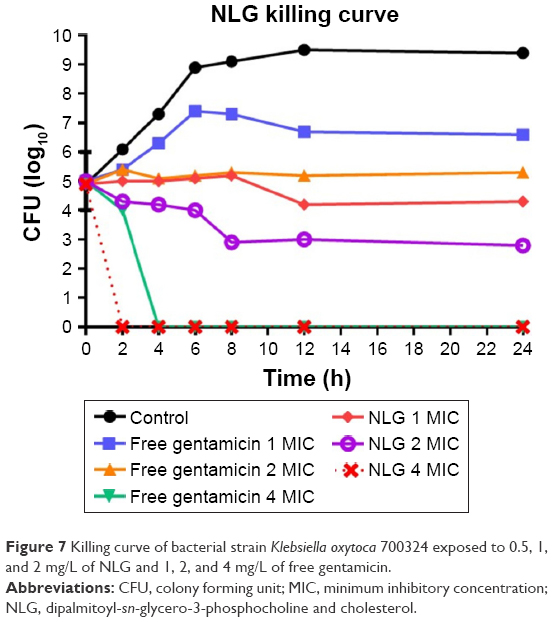 | Figure 7 Killing curve of bacterial strain Klebsiella oxytoca 700324 exposed to 0.5, 1, and 2 mg/L of NLG and 1, 2, and 4 mg/L of free gentamicin. |
Bacterial growth was reduced to log10 4.3 CFU/mL by the NELG-1 formulation at 1 mg/L (1× MIC) in 24 h. However, free gentamicin at 1 mg/L (1× MIC) only decreased bacterial growth to log10 6.6 CFU/mL in 24 h. Similarly, the NELG-1 formulation at 2 mg/L (2× MIC) reduced bacterial growth even more to log10 3.6 CFU/mL at 24 h. Free gentamicin at 2 mg/L (2× MIC) failed to reduce bacterial growth to the same level as the NELG-1 formulation; with the killing activity for both NELG-1 formulation and free gentamicin starting at 4 mg/L (4× MIC), the NELG-1 formulation was faster by one-time interval than the free gentamicin (2 versus 4 h), as shown in Figure 8.
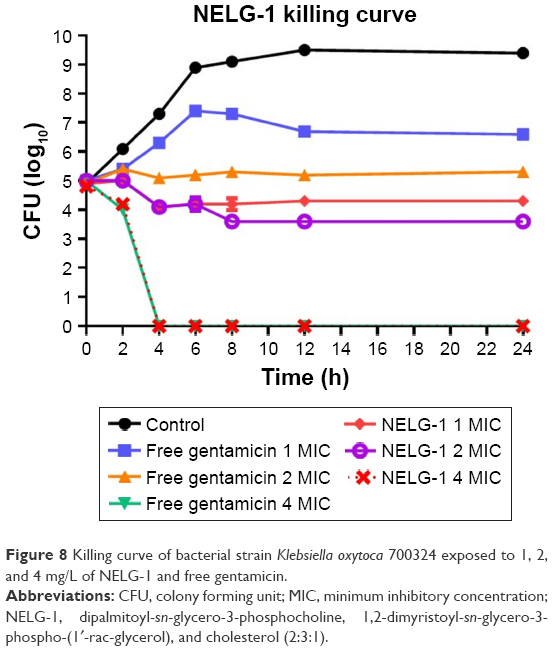 | Figure 8 Killing curve of bacterial strain Klebsiella oxytoca 700324 exposed to 1, 2, and 4 mg/L of NELG-1 and free gentamicin. |
The NELG-2 formulation at 1 mg/L (1× MIC) exhibited a bacteriostatic activity lower than that of free gentamicin after 24 h. Similarly, the NELG-2 formulation at 2 mg/L (2× MIC) suppressed bacterial growth better than free gentamicin at the same concentration after 24 h. In contrast, the NELG-2 formulation and free gentamicin killed the bacterial strain at 4 mg/L (4× MIC). However, the killing time of free gentamicin was faster than that of NELG-2 (Figure 9).
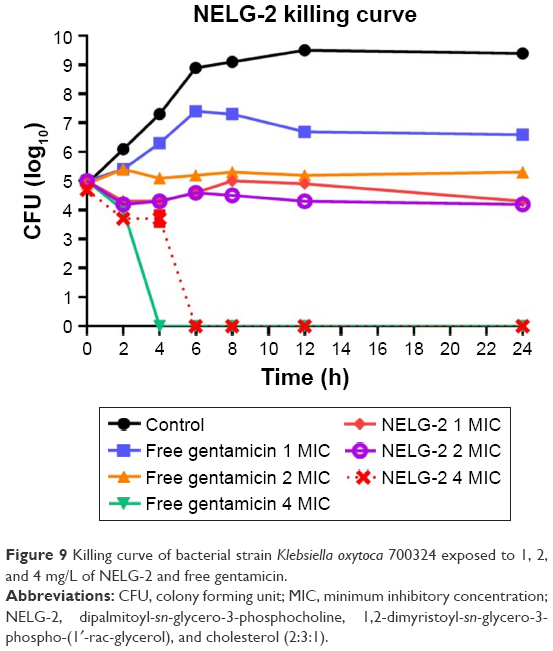 | Figure 9 Killing curve of bacterial strain Klebsiella oxytoca 700324 exposed to 1, 2, and 4 mg/L of NELG-2 and free gentamicin. |
Biofilm assays
In the P. aeruginosa BAA1744 prevention assay, free gentamicin at 0.5 mg/L prevented biofilm formation less when compared to the control (P<0.05). However, NLG at 1/2× MICs (0.125 mg/L) was highly significant in preventing the formation of biofilm compared to the control (P<0.001), and NELG-1 at 0.5 mg/L also exhibited significantly higher biofilm prevention compared to the control (P<0.01). NELG-2 was able to prevent biofilm formation significantly as free gentamicin when compared to the control (P<0.05), as shown in Figure 10. However, when NLG (0.125 mg/L) and free gentamicin (0.5 mg/L) were compared, NLG (0.125 mg/L) was significantly better in preventing biofilm formation (P<0.01). In the K. oxytoca 700324 prevention assay, free gentamicin (0.5 mg/L) exhibited slight prevention of biofilm formation, but not significantly compared to the control. However, NLG (0.25 mg/L), NELG-1 (0.5 mg/L), and NELG-2 (0.5 mg/L) were able to prevent biofilm formation significantly better compared to the control (P<0.001). When comparing NLG to NELG-1 and NELG-2, NLG more significantly prevented biofilm formation by K. oxytoca (P<0.01), as shown in Figure 11.
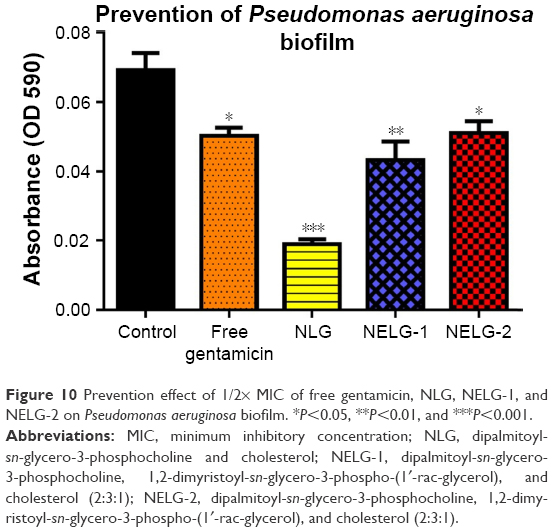 | Figure 10 Prevention effect of 1/2× MIC of free gentamicin, NLG, NELG-1, and NELG-2 on Pseudomonas aeruginosa biofilm. *P<0.05, **P<0.01, and ***P<0.001. |
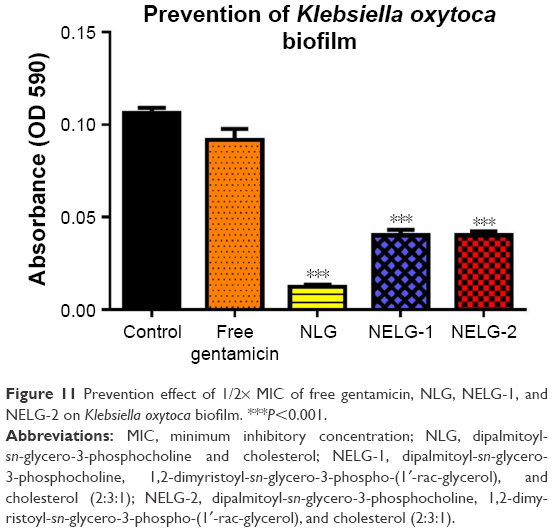 | Figure 11 Prevention effect of 1/2× MIC of free gentamicin, NLG, NELG-1, and NELG-2 on Klebsiella oxytoca biofilm. ***P<0.001. |
The reduction effect of liposomal formulations and free gentamicin on structured biofilm formed by P. aeruginosa is shown in Figure 12. Free gentamicin at 16× MIC (16 mg/L) reduced biofilm, but the reduction was not significant when compared to the control, whereas NLG (4 mg/L) showed a significant reduction of biofilm compared to the control (P<0.05). NELG-1 (16 mg/L) also exhibited a significant reduction of biofilm, which was comparable to NLG (P<0.05). Furthermore, the increased reduction effect of NELG-2 (16 mg/L) on the biofilm was significant when compared to the control (P<0.01). In the K. oxytoca 700324 reduction assay, free gentamicin (16 mg/L) reduced the biofilm, but not significantly compared to the control. However, all liposomal formulations reduced structured biofilm formation compared to the control. For instance, NLG (8 mg/L) reduced biofilm significantly compared to the control (P<0.05). Notably, the negatively charged liposome formulations effectively reduced the biofilm. NELG-1 (16 mg/L) reduced the biofilm formation better than NLG and the difference was significant (P<0.01); NELG-2 (16 mg/L) reduced the biofilm formation better than NLG and NELG-1 when compared to the control (P<0.001), as shown in Figure 13.
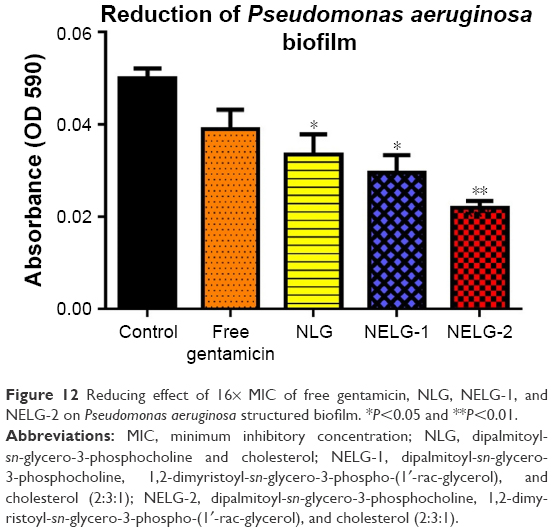 | Figure 12 Reducing effect of 16× MIC of free gentamicin, NLG, NELG-1, and NELG-2 on Pseudomonas aeruginosa structured biofilm. *P<0.05 and **P<0.01. |
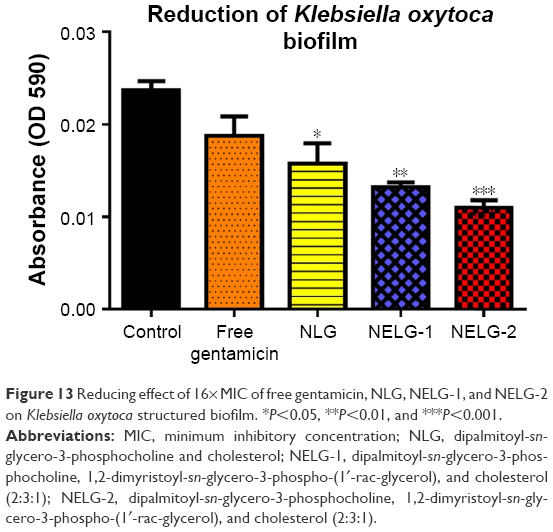 | Figure 13 Reducing effect of 16× MIC of free gentamicin, NLG, NELG-1, and NELG-2 on Klebsiella oxytoca structured biofilm. *P<0.05, **P<0.01, and ***P<0.001. |
Discussion
It is challenging to achieve a high percentage of encapsulation of aminoglycosides within liposomal nanoparticles. Electrostatic interaction between negatively and positively charged molecules is a particularly interesting approach to enhance the loading of drugs in nanoparticles.45 In this study, we encapsulated gentamicin in different liposomal formulations including neutral and negatively charged liposomes. Increasing the negatively charged phospholipids ratio in different liposomal formulations (NELG-1 and NELG-2) played a role in increasing the EE of gentamicin compared to the neutral liposomes, due to an interaction between the positively charged gentamicin and the negatively charged liposomes. These results can be explained by the initiation of an electrostatic interaction force between the positively charged molecules such as gentamicin and the negatively charged phospholipids.46 Our results are in agreement with a study that reported the enhancement of EE of the positively charged clarithromycin by negatively charged liposomes.30
The size of the liposomes is an important characteristic parameter. The particle diameter of the negatively charged liposome gentamicin was larger than the diameter of the neutral liposomes. This might be due to the inclusion of the charge, which originates in an enlargement of space between the adjacent bilayers.47 This aspect can be explained by the attraction of the positively charged gentamicin to the negatively charged liposomes and moving the phospholipid head group apart.48
The stability of the liposome plays an important role in the development of a nanocarrier containing sufficient drug. Therefore, we evaluated our formulations in different biologic environments to mimic physiologic conditions. The stability of liposomal formulations was found to be largely dependent on temperature. The neutral and negatively charged formulations retained >91% of encapsulated gentamicin in PBS at 4°C after 48 h. However, in PBS at 37°C, the stabilization of the loaded cargo was slightly reduced throughout the study period, compared to PBS at 4°C. The stability of the liposomal formulations can be explained by the extension in the acyl chain length of lipid compositions, and the presence of DPPC in our formulations contributed to a higher transition temperature. A previous study reported that saturated phospholipids with a higher transition temperature, such as DPPC, are more stable due to an increase of acyl chain.49 Also, the presence of Chol in our formulation reduced the bilayer permeability of the liposomal membrane, resulting in high drug retention at a higher temperature.50 In the presence of BAL, sputum, and human plasma, the release varied. NLG maintained a reasonable amount of gentamicin in these environments. NELG-1 and NELG-2 lost their gentamicin content whenever the negative lipid ratio was increased. This is explained by the avidity of negative formulations to interact with lipoproteins, albumin, immunoglobulins, and phospholipases present in BAL, sputum, and plasma, which have the ability to destabilize the liposomes depending on the lipid composition or the electrostatic charge.46,51,52 A study reported that liposomes containing DPPC bound less protein and were more stable than a negatively charged liposome.53 Maximum release of gentamicin was noted after 4 h of incubation at 4°C and 37°C, and gentamicin was constant over the 48 h study period. This is due to the osmotic equilibrium of gentamicin concentration between the inside and outside of the liposomes.49
In this in vitro study, we extensively examined the antimicrobial effect of liposomal gentamicin formulations on different bacterial strains. The results of MIC and MBC demonstrated that NLG enhanced the antimicrobial activity against tested bacteria (Table 2). Negatively charged liposomes exhibited MIC and MBC close to free gentamicin. These results are in agreement with a previous study which found that negatively charged liposome gentamicin had less activity on Brucella abortus.54
To confirm our present MIC and MBC findings, time kill studies were performed (Figures 4–6). The NLG formulation at 2× MIC and 4× MIC improved the killing activity of gentamicin, eradicating the bacterial strains (P. aeruginosa BAA1744 and K. oxytoca 700324) at concentrations fourfold and twofold lower than free gentamicin, respectively. Our results are in agreement with a previous study that confirmed the enhanced activity of gentamicin by encapsulating it into a neutral liposomal formulation against P. aeruginosa.42 Although NELG-1 at 2× MIC exhibited the same killing time compared to free gentamicin at 2× MIC, NELG-1 inhibited P. aeruginosa by log10 1 CFU at 6 h compared to free gentamicin (log10 3 CFU versus log10 4 CFU) as well as reduced K. oxytoca growth by log10 1 CFU throughout the study period. However, NELG-1 at 4× MIC eliminated K. oxytoca faster than free gentamicin (2 versus 4 h). A study demonstrated that negatively charged liposomes consisting of DPPC/DMPG and loaded with aminoglycoside exhibited an enhanced aminoglycoside antibacterial activity.55 In our experiments, the negative formulations containing gentamicin showed a bactericidal efficacy equal to that of the free drug. Drulis-Kawa et al noted similar results and concluded that neutral and cationic liposomes interacted more with the Gram-negative negatively charged bacterial cell wall or the Gram-positive peptidoglycan than the negatively charged liposomes.56 The interaction affinity between the negatively charged liposomes and the bacterial cell membrane could be delayed as a result of the repulsive force between the nanoparticles and bacteria.
We investigated the ability of our formulations to prevent bacterial biofilm formation. Free gentamicin at 1/2× MIC exhibited a preventive effect on P. aeruginosa biofilm formation; however, NLG and NELG-1 significantly prevented biofilm formation. Similarly, free gentamicin did not prevent K. oxytoca 700324 biofilm formation significantly compared to the control, whereas the liposomal formulations exhibited a potent prevention of biofilm formation. A previous study reported that the enhanced delivery of antibiotics to bacterial cells using liposomes is through a fusion mechanism,40 thereby allowing the liposome-entrapped antibiotic to be injected directly into the bacterial cytoplasm. Our data showed an enhanced activity of liposomal formulations probably due to the increased delivery of gentamicin to the bacterial cytoplasm. We noticed a weak activity of NELG-2 with a strong negative charge, possibly because of limitation of liposomal fusion with a negatively charged bacterial cell wall, due to repulsive forces at close proximity.56 Interestingly, the negatively charged liposomes exhibited a potent antibiofilm activity compared to NLG and free gentamicin against P. aeruginosa and K. oxytoca. Previous research reported an increase in activity of negatively charged liposome–encapsulated clarithromycin against P. aeruginosa biofilm.30 Another study found that the encapsulated tobramycin in negatively charged liposomes becomes immobilized in close proximity to a biofilm cluster which attracts the liposomes electrostatically leading to penetration and killing of bacteria.57
Conclusion
The data presented here indicate that neutral liposomes enhance the antimicrobial activity of gentamicin against planktonic bacteria, while negatively charged liposomes increase encapsulation of gentamicin and significantly improve the activity of gentamicin against the biofilm community.
Acknowledgments
This work has been funded by King Abdulaziz City for Science and Technology (KACST) research grant number: GSP-34-279 which was awarded to Majed A Majrashi. Nevertheless, KACST had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. In addition, we would like to acknowledge Mr Ahmad Saleh Abu Jaffal for his cooperation and allowing our team to use his laboratory space at the College of Applied Medical Sciences, King Saud bin Abdulaziz University for Health Sciences to conduct some of the experiments. We would also like to acknowledge Dr Susanna Wright at the Publication Office, King Abdullah International Medical Research Center for her assistance in editing and revising the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Cohen ML. Changing patterns of infectious disease. Nature. 2000;406(6797):762–767. | ||
Yoneyama H, Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem. 2006;70(5):1060–1075. | ||
Fauci AS. Infectious diseases: considerations for the 21st century. Clin Infect Dis. 2001;32(5):675–685. | ||
Nathan C. Antibiotics at the crossroads. Nature. 2004;431(7011):899–902. | ||
Projan SJ. New (and not so new) antibacterial targets – from where and when will the novel drugs come? Curr Opin Pharmacol. 2002;2(5):513–522. | ||
Leeb M. Antibiotics: a shot in the arm. Nature. 2004;431(7011):892–893. | ||
Chambers HF. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin Microbiol Rev. 1997;10(4):781–791. | ||
Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem. 2014;6:25–64. | ||
Tassios PT, Gennimata V, Spaliara-Kalogeropoulou L, et al. Multiresistant Pseudomonas aeruginosa serogroup O:11 outbreak in an intensive care unit. Clin Microbiol Infect. 1997;3(6):621–628. | ||
Al-Marzoqi A, Al-Taee Z. Pseudomonas aeruginosa: Antibiotic resistance pattern to different isolates in Al-Hillah city, Iraq. J Natl Sci Res. 2013;3(3):69–74. | ||
Lockhart SR, Abramson MA, Beekmann SE, et al. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J Clin Microbiol. 2007;45(10):3352–3359. | ||
Maragakis LL, Perl TM. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin Infect Dis. 2008;46(8):1254–1263. | ||
Fournier B, Arlet G, Lagrange PH, Philippon A. Klebsiella oxytoca: resistance to aztreonam by overproduction of the chromosomally encoded beta-lactamase. FEMS Microbiol Lett. 1994;116(1):31–36. | ||
Babini GS, Livermore DM. Antimicrobial resistance amongst Klebsiella spp. collected from intensive care units in Southern and Western Europe in 1997–1998. J Antimicrob Chemother. 2000;45(2):183–189. | ||
Yang YJ, Livermore DM. Chromosomal beta-lactamase expression and resistance to beta-lactam antibiotics in Proteus vulgaris and Morganella morganii. Antimicrob Agents Chemother. 1988;32(9):1385–1391. | ||
Lungu B, O’Bryan CA, Muthaiyan A, et al. Listeria monocytogenes: antibiotic resistance in food production. Foodborne Pathog Dis. 2011;8(5):569–578. | ||
Kim H, Ryu JH, Beuchat LR. Attachment of and biofilm formation by Enterobacter sakazakii on stainless steel and enteral feeding tubes. Appl Environ Microbiol. 2006;72(9):5846–5856. | ||
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35(4):322–332. | ||
Tseng BS, Zhang W, Harrison JJ, et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ Microbiol. 2013;15(10):2865–2878. | ||
Cao B, Christophersen L, Thomsen K, et al. Antibiotic penetration and bacterial killing in a Pseudomonas aeruginosa biofilm model. J Antimicrob Chemother. 2015;70(7):2057–2063. | ||
Martino PD, Fursy R, Bret L, Sundararaju B, Phillips RS. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can J Microbiol. 2003;49(7):443–449. | ||
Tang X, Flint SH, Bennett RJ, Brooks JD, Morton RH. Biofilm growth of individual and dual strains of Klebsiella oxytoca from the dairy industry on ultrafiltration membranes. J Ind Microbiol Biotechnol. 2009;36(12):1491–1497. | ||
Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother. 1999;43(4):727–737. | ||
Dozzo P, Moser HE. New aminoglycoside antibiotics. Expert Opin Ther Pat. 2010;20(10):1321–1341. | ||
Schroeder M, Brooks BD, Brooks AE. The Complex Relationship between Virulence and Antibiotic Resistance. Genes (Basel). 2017;8(1):E39. | ||
Morita Y, Tomida J, Kawamura Y. Responses of Pseudomonas aeruginosa to antimicrobials. Front Microbiol. 2014;4:422–430. | ||
Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010;8(6):423–435. | ||
Brilene T, Soeorg H, Kiis M, et al. In vitro synergy of oxacillin and gentamicin against coagulase-negative staphylococci from blood cultures of neonates with late-onset sepsis. APMIS. 2013;121(9):859–864. | ||
Aldahmash BA, El-Nagar DM, Ibrahim KE. [Reno-protective effects of propolis on gentamicin-induced acute renal toxicity in swiss albino mice.] Nefrologia. 2016;36(6):643–652. Spanish. | ||
Alhajlan M, Alhariri M, Omri A. Efficacy and safety of liposomal clarithromycin and its effect on Pseudomonas aeruginosa virulence factors. Antimicrob Agents Chemother. 2013;57(6):2694–2704. | ||
Mozafari M, Flanagan J, Matia-Merino L, et al. Recent trends in the lipid-based nanoencapsulation of antioxidants and their role in foods. J Sci Food Agr. 2006;86(13):2038–2045. | ||
Alhariri M, Omri A. Efficacy of liposomal bismuth-ethanedithiol-loaded tobramycin after intratracheal administration in rats with pulmonary Pseudomonas aeruginosa infection. Antimicrob Agents Chemother. 2013;57(1):569–578. | ||
Alhariri M, Azghani A, Omri A. Liposomal antibiotics for the treatment of infectious diseases. Expert Opin Drug Deliv. 2013;10(11):1515–1532. | ||
Marier JF, Brazier JL, Lavigne J, Ducharme MP. Liposomal tobramycin against pulmonary infections of Pseudomonas aeruginosa: a pharmacokinetic and efficacy study following single and multiple intratracheal administrations in rats. J Antimicrob Chemother. 2003;52(2):247–252. | ||
Justo OR, Moraes AM. Incorporation of antibiotics in liposomes designed for tuberculosis therapy by inhalation. Drug Deliv. 2003;10(3):201–207. | ||
Mugabe C, Azghani AO, Omri A. Preparation and characterization of dehydration-rehydration vesicles loaded with aminoglycoside and macrolide antibiotics. Int J Pharm. 2006;307(2):244–250. | ||
Halwani M, Yebio B, Suntres ZE, Alipour M, Azghani AO, Omri A. Co-encapsulation of gallium with gentamicin in liposomes enhances antimicrobial activity of gentamicin against Pseudomonas aeruginosa. J Antimicrob Chemother. 2008;62(6):1291–1297. | ||
Halwani M, Mugabe C, Azghani AO, Lafrenie RM, Kumar A, Omri A. Bactericidal efficacy of liposomal aminoglycosides against Burkholderia cenocepacia. J Antimicrob Chemother. 2007;60(4):760–769. | ||
Halwani M, Hebert S, Suntres ZE, Lafrenie RM, Azghani AO, Omri A. Bismuth-thiol incorporation enhances biological activities of liposomal tobramycin against bacterial biofilm and quorum sensing molecules production by Pseudomonas aeruginosa. Int J Pharm. 2009;373(1–2):141–146. | ||
Mugabe C, Halwani M, Azghani AO, Lafrenie RM, Omri A. Mechanism of enhanced activity of liposome-entrapped aminoglycosides against resistant strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50(6):2016–2022. | ||
European Committee for Antimicrobial Susceptibility Testing of the European Society of Clinical M, Infectious D. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin Microbiol Infect. 2003;9(8):ix–xv. | ||
Rukholm G, Mugabe C, Azghani AO, Omri A. Antibacterial activity of liposomal gentamicin against Pseudomonas aeruginosa: a time-kill study. Int J Antimicrob Agents. 2006;27(3):247–252. | ||
Naparstek L, Carmeli Y, Navon-Venezia S, Banin E. Biofilm formation and susceptibility to gentamicin and colistin of extremely drug-resistant KPC-producing Klebsiella pneumoniae. J Antimicrob Chemother. 2014;69(4):1027–1034. | ||
Hasanin A, Omri A. Liposomal N-acetylcysteine modulates the pathogenesis of P. aeruginosa isolated from the lungs of cystic fibrosis patient. J Nanomed Nanotechnol. 2014;5(4):219–230. | ||
Villasmil-Sánchez S, Rabasco AM, González-Rodriguez ML. Thermal and 31P-NMR studies to elucidate sumatriptan succinate entrapment behavior in phosphatidylcholine/cholesterol liposomes. Comparative 31P-NMR analysis on negatively and positively-charged liposomes. Colloids Surf B Biointerfaces. 2013;105:14–23. | ||
Li Y, Huang L, Tang C, Zhang E, Ding L, Yang L. Preparation and characterisation of the colistin-entrapped liposome driven by electrostatic interaction for intravenous administration. J Microencapsul. 2016;33(5):427–437. | ||
Chougule M, Padhi B, Misra A. Nano-liposomal dry powder inhaler of tacrolimus: preparation, characterization, and pulmonary pharmacokinetics. Int J Nanomedicine. 2007;2(4):675–688. | ||
Hosny KM. Ciprofloxacin as ocular liposomal hydrogel. AAPS PharmSciTech. 2010;11(1):241–246. | ||
Solleti VS, Alhariri M, Halwani M, Omri A. Antimicrobial properties of liposomal azithromycin for Pseudomonas infections in cystic fibrosis patients. J Antimicrob Chemother. 2015;70(3):784–796. | ||
Anderson M, Omri A. The effect of different lipid components on the in vitro stability and release kinetics of liposome formulations. Drug Deliv. 2004;11(1):33–39. | ||
Bonté F, Juliano RL. Interactions of liposomes with serum proteins. Chem Phys Lipids. 1986;40(2–4):359–372. | ||
Simões S, Moreira JN, Fonseca C, Düzgüneş N, de Lima MC. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56(7):947–965. | ||
Hernández-Caselles T, Villalain J, Gómez-Fernández JC. Influence of liposome charge and composition on their interaction with human blood serum proteins. Mol Cell Biochem. 1993;120(2):119–126. | ||
Vitas AI, Diaz R, Gamazo C. Protective effect of liposomal gentamicin against systemic acute murine brucellosis. Chemotherapy. 1997;43(3):204–210. | ||
Beaulac C, Sachetelli S, Lagace J. In-vitro bactericidal efficacy of sub-MIC concentrations of liposome-encapsulated antibiotic against gram-negative and gram-positive bacteria. J Antimicrob Chemother. 1998;41(1):35–41. | ||
Drulis-Kawa Z, Gubernator J, Dorotkiewicz-Jach A, Doroszkiewicz W, Kozubek A. A comparison of the in vitro antimicrobial activity of liposomes containing meropenem and gentamicin. Cell Mol Biol Lett. 2006;11(3):360–375. | ||
Messiaen AS, Forier K, Nelis H, Braeckmans K, Coenye T. Transport of nanoparticles and tobramycin-loaded liposomes in Burkholderia cepacia complex biofilms. PLoS One. 2013;8(11):e79220. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.