")
Back to Archived Journals » Journal of Neurorestoratology » Volume 2
Efficacy of fetal stem cells in Duchenne muscular dystrophy therapy
Authors Sych N, Klunnik M , Ivankova O, Matyaschuk I, Demchuk M , Novytska A, Arkhipenko I, Shalita I, Siniscalco D
Received 9 November 2013
Accepted for publication 10 December 2013
Published 26 February 2014 Volume 2014:2 Pages 37—46
DOI https://doi.org/10.2147/JN.S57276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Nataliia Sych,1 Mariya Klunnik,1 Olena Ivankova,1 Irina Matyaschuk,1 Mariya Demchuk,1 Alla Novytska,1 Inna Arkhipenko,2 Iuliia Shalita,2 Dario Siniscalco3
1Clinical Department, 2Biotechnology Department, Cell Therapy Center EmCell, Kiev, Ukraine; 3Biomedical Centre for Autism Research and Therapy, Bari, Italy
Abstract: The absence of effective treatment methods for Duchenne muscular dystrophy (DMD) calls for new therapeutic approaches. One of the promising treatment methods for DMD is stem cell therapy. This study demonstrates the impact of fetal stem cells (FSCs) on functional capacity and life quality of DMD patients and the ability of FSCs to prevent DMD-related complications in order to inhibit the disease progression. FSC therapy substantially improves functional capacity, life quality, left ventricular ejection fraction, and forced vital capacity of the lungs of DMD patients; this was confirmed by comparison of neurological, laboratory (aspartate aminotransferase, alanine aminotransferase, creatine phosphokinase, and lactate dehydrogenase), and morphofunctional findings (left ventricular ejection fraction and forced vital capacity) in DMD patients before the treatment, and 6 and 12 months after the FSC treatment.
Keywords: life quality, heart failure, respiratory failure, cell therapy
Introduction
Duchenne muscular dystrophy (DMD) is a genetic disorder affecting 3.3 in 100,000 people, 14 in 100,000 newborns and one in 3,600–6,000 live male births.1 Since DMD is an X-linked disorder, it mainly affects boys and very rarely girls.1–3
DMD occurs as a result of mutations in the dystrophin gene (locus Xp21.2) resulting in dystrophin deficiency or structural changes leading to progressive muscular degeneration.4,5 Clinical symptoms of DMD usually appear during the first 3 years of life, and by the age of 6–9 years most patients have contractures of the iliotibial tract, hip flexors, and Achilles tendons. By the age of 10 years, most DMD patients lose independent ambulation or even the ability to stand and require the use of a wheelchair. The upper extremities are affected by 14–15 years of age. Intellectual impairment is present in 10% of cases. Average life expectancy for patients afflicted with DMD is ≤20 years; the main causes of death are lung infections, respiratory failure, gastrointestinal diseases, or cardiomyopathy.2,6,7
Cardiovascular disease is one of the serious complications in DMD, and often these patients develop cardiomyopathy – the main cause of death in this disease. Its symptoms are dilatation of the left ventricle, arrhythmia, and heart failure.8,9 The myocardium is affected long before the symptoms appear, and the pathologic process starts with pseudohypertrophy followed by dilatation as result of atrophy, necrosis, and substitution of cardiomyocytes with fibrous tissue.1,10,11
Weakness of respiratory muscles is another debilitating complication, limiting chest mobility (muscle weakness, skeletal deformities) and thus causing chronic respiratory failure, which markedly reduces life expectancy of myopathy patients.5,12,13
Thus, the condition of the myocardium and respiratory muscles of DMD patients determines the course of the disease and its outcome. The severity of cardiac and respiratory muscle affection is proportional to the severity of neurological symptoms.13,14
Currently, there is no cure for DMD; available treatments are limited to corticosteroids.15 Large-scale clinical trials aiming to tackle this disease are in progress throughout the world. They mainly aim to use gene replacement therapy by production of synthetic genes capable of dystrophin production, or they use gene repair therapy by correction of transcriptional processes of the dystrophin gene (ie, targeted exon skipping through RNA to exclude specific sites of RNA splicing, or using antisense oligonucleotides to induce the restoration of the open reading frame and the production of a functional dystrophin protein). However, while several years ago gene therapy represented the hope for DMD care, these gene-based approaches are still far from effectiveness.16 Indeed, several issues need to be further determined, evaluated, and implemented, such as the optimal mode of gene delivery, the host immune rejection, increased exon skipping efficiency, and an optimized oligonucleotide sequence.
Cell-based therapy could represent the optimal treatment for this disease. The rationale for cell therapy is to promote muscle regeneration.17 Indeed, the patients could be offered myoblast transplantation: donor cells are injected into the affected muscles with the hope that they will fuse with those muscles and produce several healthy muscle fibers capable of dystrophin production. Transplanted cells lead to a correct and functional dystrophin gene and allogeneic transplantation (in which cells are extracted from healthy donors) offers the possibility to overcome ex vivo gene repair.18 However, the efficacy of this approach is still questionable.19–21 Besides, treatment aimed solely at muscle restoration will most likely result in temporary relief, if any. It can yet make the situation worse by quicker depletion of muscle stem cells.22 It is believed that the earlier DMD treatment starts (ideally, during the first years of life), the better the results will be, as the treatment will show effect until the terminal stage of tissue damage is developed.23
In principle, several type of cells could be used in DMD. It has been demonstrated that injected myoblast cells produce new muscle fibers, even if poor cell survival and limited migratory ability have been reported.24
Stem cells represent the great promise for regenerative medicine,25,26 and offer a new treatment modality for DMD.27 These cells are preferable for DMD applications due to their capacity of self-renewal and differentiative potential.28,29 Several studies have focused on applying stem cells for myopathy treatment.30,31 Adult stem cells have shown interesting results in treatment of muscular dystrophies,24 whereas some studies attempting to treat different types of hereditary myopathy with embryonic stem cells turned out to be futile. When transplanted into rats, immature stem cells are able to differentiate into muscle fibers, transform into different tissue cells, and form potentially malignant tumors.
In order to prevent tumor formation, researchers started using more specialized cells, differentiating only into definite cell types. In stem cells, pre-activation of the Pax3 gene is responsible for muscle cell formation, which results in the formation of myoblasts, precursors of striated muscles.32 Myoblasts, isolated from cell mass by using fluorescent substances selectively binding with receptors of muscle cell membranes, were engrafted in the hind limbs of rodents. One month later, the cells spread throughout the muscle tissue by multiplying and forming new muscle fibers. The mobility of the hind limbs markedly improved, which was proved by immunofluorescence testing. Apart from this, coordination also improved. Three months after the injections, the rodents were tumor-free.30,33
In an experimental murine model of muscle dystrophy, it has been shown that dystrophin loss caused continuous muscle damage alternating with restoration until stem cell capacity was depleted, and this was when the symptoms appeared.22 Transplantation of healthy mice stem cells to the diseased mice alleviated the symptoms in the latter.22
Among the stem cell population, fetal stem cells (FSCs) have also been used in neurological diseases.34 These cells are more specialized compared to embryonic stem cells, but at the same time have high proliferative potential and are effectively being used in a number of diseases, including neurological ones, without the need for human leukocyte antigen (HLA) matching.33,35,36 FSCs are safe, easily obtainable, and have higher proliferative and expansion potential than adult bone marrow cells.35,37,38 They have not shown in vivo teratoma formation or uncontrollable cell proliferation as embryonic stem cells.39 In addition, FSCs possess strong immunomodulatory characteristics, stable phenotypes, and less senescence; they are able to engraft and target damaged tissue.34 Taken together, these powerful capacities indicate that FSCs are useful candidates for clinical cell-based DMD therapy.
The absence of effective methods for DMD treatment on the one hand and the positive experience of FSC application in neurological diseases on the other hand both indicate that FSC application in DMD may possibly be effective.
The aim of this study was to examine FSC treatment safety and efficacy in DMD children. We studied clinical, neurological, laboratory, morphofunctional, and lung capacity changes in DMD patients after FSC therapy (FSCT).
Materials and methods
Patients
Subjects were recruited from Great Britain (3.70%); Ukraine (18.52%); Iran (7.41%); USA (7.41%); Georgia (3.70%); Russia (7.41%); United Arab Emirates (3.70%); Romania (3.70%); Slovakia (3.70%); Livya (3.70%); Poland (18.52%); Italy (7.41%); Serbia (7.41%); and Turkey (3.70%). Before the treatment, all the patients, or their legal representatives, signed the informed consent. This study was performed pursuant to the principles of the Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable law of Ukraine on clinical trials, as it was approved by the Health Ministry of Ukraine (approval number 599817/A⌈). This study was registered at the public registry of the Ministry of Education and Science of Ukraine, Ukrainian Institute of Scientific, Technical and Economic Information. The registration number is 0113U000957.
The study group consisted of 27 male patients aged 4–27 years (mean age 12.86±1.78 years) with confirmed DMD and the following distribution by stage: II – 12, III – three, IVa – ten, and IVb – two.
The control group included 15 patients aged 5–25 years (mean age 11.93±1.49 years) with confirmed DMD and the following distribution by stage: II – six, III – four, IVa – three, and IVb – two.
DMD was confirmed by imaging analysis, past history, positive findings of electroneuromyography, laboratory findings (alanine aminotransferase [ALT], aspartate aminotransferase [AST], creatine phosphokinase [CPK], lactate dehydrogenase [LDH]), genetic testing, and biopsy. In 95.24% cases it was confirmed with genetic testing; in 90.47% by muscle biopsy; and in 44.44% by electroneuromyography, genetic testing, and biopsy.
The functional status of the DMD patients was assessed with the Brooke’s scale. Evaluation is based on upper and lower extremities assessment tests performed in accordance with the standard protocol (walking, climbing stairs, sitting down on a chair, getting up from the floor or a chair, and wrist power) and graded from 0 to 4. The total score on this scale can vary from 0 (normal) to 22 (bedridden, absence of voluntary motions in the arms, unable to speak and swallow). Quality of life (LQ) was evaluated with the SF-36 Health Status Survey.
Cardiovascular status was evaluated by means of routine clinical methods, electrocardiography (ECG), and echocardiogram (echoCG). A UCARD 100 electrocardiograph (UTAS Co, Kiev, Ukraine) was used for the 12-lead ECG, while a Toshiba SSA-380-A Powervision (Tokyo, Japan) with a 2.5–3.5 mHz variable frequency transducer was used for the echoCG performed on the left side in accordance with the approved protocols.
Heart failure was diagnosed on the basis of typical symptoms confirmed by specific echoCG evidence (left ventricular ejection fraction [LVEF] <55%).
Respiratory capacity was measured on a BTL-08 Spiro (BTL Industries, Stevenage, UK) in accordance with the approved protocols.40
Serum ALT, AST, CPK, and LDH were measured by enzyme-linked immunoassay.41
Along with routine therapy, all patients of the study group underwent transplantation of mesenchymal and ectodermal FSC suspension containing both hematopoietic stem cells and other stem cell types.
Stem cell procedure
We used suspensions containing stem cells harvested from 5–9-week-old human fetuses. One suspension was made of hematopoietic stem cells from fetal livers, while the other was made from fetal brain stem cells.
Fetal material was harvested in the surgery room pursuant to all aseptic and antiseptic requirements. Tissue was collected, upon written informed consent of the woman donor, from liver and brain nervous tissue of 5–9-week-old human fetuses aborted for family planning purposes and found to have no developmental abnormalities or infections. Fetal tissue was then placed into sterile transport medium made of Hanks solution and an antibiotic. Tissues were aseptically separated and homogenized in Hanks solution. The stem cell suspension was then filtered. Dimethyl sulfoxide was used as a cryoprotectant. Cryopreservation of cell suspensions was performed in the controlled-rate freezer chamber pursuant to the selected program.
In order to ensure safety, both the women donors and the ready-made hematopoietic stem cells from the fetal liver suspension were tested for bacterial and fungal safety, and viral infections and parasites such as human immunodeficiency virus 1 and 2, hepatitis B virus, hepatitis C virus, syphilis (Treponema pallidum), toxoplasmosis (Toxoplasma gondii), rubella, cytomegalovirus, herpes simplex virus 1 and 2, Epstein–Barr virus, Mycoplasma genitalium, Ureaplasma urealyticum, Ureaplasma parvum, and Chlamydia trachomatis.
Suspensions containing hematopoietic stem cells from fetal liver and brain (nervous) stem cells were stored in liquid nitrogen at −196°C in a properly arranged cryobank.
The stem cell procedure consisted of transplantation of the suspension containing cryopreserved fetal stem cells preceded by premedication – infusion of diphenylhydramine 10 mg and prednisone 15 mg on day 1 and a specially prepared solution on day 2.
On day 1, we used hematopoietic stem cells from fetal liver harvested from the tissues of 5–9-week-old human fetuses. A suspension containing cryopreserved hematopoietic stem cells was administered via intravenous drip-feed in the amount of 1.75±0.51 mL with nucleated cell count >58.74 × 106/mL per transplantation.
On day 2, we administered ectodermal homogenate containing nervous cell precursors subcutaneously, in the amount of 2.12±0.49 mL with nucleated cell count >7.9 × 106/mL per transplantation.
CD34+ were counted by using flow cytofluorometry (Becton Dickinson, Franklin Lakes, NJ, USA) with fluorescent tagged antibodies (Santa Cruz Biotechnology, Dallas, TX, USA).
Colony-forming activity
Culturing was performed in 24-hole trays in an atmosphere with 5% CO2 in the air and 100% humidity at 37°C in the following semisolid medium: agar 33% (Difco; Becton Dickinson), glutamine 4.0 mM/L (Sigma-Aldrich, St Louis, MO, USA), penicillin-streptomycin (100 U + 100 mcg/mL) (Sigma-Aldrich), granulocyte-macrophage colony-stimulating factor 100 U/mL (Sigma-Aldrich), interleukin 3 100 U/mL (Behringwerke AG, Marburg, Germany), stem cell factor 50 ng/mL (R&D Systems, Minneapolis, MN, USA), and erythropoietin 10 U/mL (epoetin beta; Boehringer Manheim Pharmaceuticals, Ingelheim, Germany). The media was prepared on Dulbecco’s Modified Eagle’s Medium (PAA Laboratories GmbH, Pasching, Austria). The colony (CFU-E [colony-forming unit erythrocyte], CFU-GM [colony-forming unit granulocyte monocyte], and CFU-GEMM [colony forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte]) number and type calculation was performed on the 14th day of culturing.
Progenitor cells and stem cells of fetal neural tissue were characterized by culturing them in vitro. Thus, neurospheres were formed, and therefore their number pointed to the functional potential of fetal neural stem cells and strongly suggested their subsequent use (Figure 1).
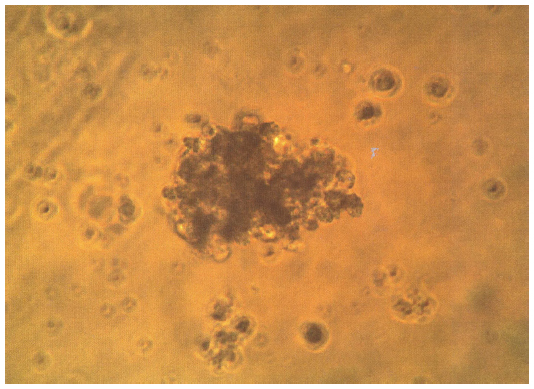 | Figure 1 Formation of colonies of fetal liver stem cells on day 7. |
Control group patients underwent only routine therapy including muscle supporting medications (L-carnitine, vitamins, lipoic acid, amino acids, vasoactive medications, and biostimulators) and medications reducing muscle cell membrane damage (antioxidants).
Neurological evaluation, laboratory tests (ALT, AST, CPK, and LDH) and instrumental examinations were performed before FSCT and 6 and 12 months after it.
Before the treatment, all of the patients, or their legal representatives, signed the informed consent. This study was registered as a clinical trial at the public registry of the Ministry of Education and Science of Ukraine, Ukrainian Institute of Scientific, Technical and Economic Information. The trial registration number is 0113U000957. Approval was obtained by the local ethics committee.
Statistics
Statistical analysis of the data was performed with Statistika v.6.0 (StatSoft, Inc.) with calculation of averages and standard deviation. Student’s t-test criterion was used for reliability evaluation. P<0.05 was considered statistically significant.
Results
Patients presented such clinical symptoms as weakness of the proximal regions of the arms (44.44%); wrist weakness (37.04%); weakness of lower extremities (48.15%); shoulder blade wasting (33.33%); wasting of the proximal regions of the arms (33.33%); back muscle wasting (29.63%); wasting of pelvic muscles (23.63%); wasting of proximal regions of lower extremities (25.93%); chest deformity (14.81%); scoliosis (14.81%); hyperlordosis (70.37%); calve pseudohypertrophy (77.78%); Achilles tendon shortening (25.93%); feet deformity (40.74%); ankle, knee, and elbow contractures (44.44%); macroglossia (18.52%); and cognitive disturbances (14.81%). None of the patients underwent hormonal treatment and tendon surgeries.
No adverse effects were observed after FSCT: there was not an increase in body temperature, or diarrhea, rash, or pruritus after stem cell transplantation. In the study group, FSC transplantation resulted in neurologic improvements on Brooke’s scale: average total score before the treatment was 8.57±0.66 compared with 7.75±0.54 half a year later (P<0.05); 85.19% of patients saw improvement by 0.5 point, 11.11% by 1.0 point and 3.7% by 1.5 points. One year after FSCT, the average total score on this scale was 6.85±0.57, which demonstrates a reliable decrease in comparison with the baseline (P<0.05). In 70.37% of patients, functional capacity improved by 0.5 points on Brooke’s scale, in 18.52% by 1.0 point, and in 11.11% by 1.5 points. In the control group, a significant reduction of the total score on the Brooke’s scale was reported 1 year after the treatment – 7.15±0.43 versus baseline of 8.82±0.59 (P<0.05); 25% of the control group patients reported improvement by 0.5 points, 58.33% by 1.0 point, and 16.67% by 1.5 points.
The LQ of DMD patients was assessed before and after FSCT (Figure 2). Before the treatment, patients of both groups had lower scores of physical functioning, mental health, emotional role functioning, vitality, and social role functioning, which negatively affected their LQ.
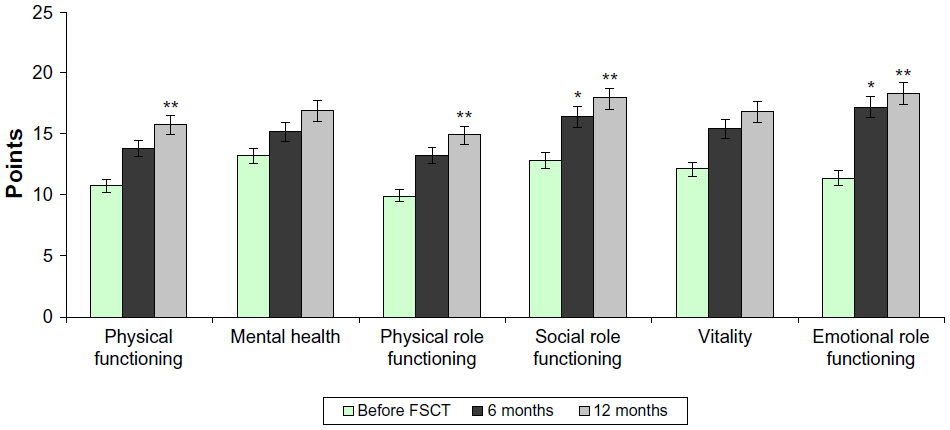 | Figure 2 Quality of life results on the SF-36 Health Status Survey: study group. |
Six and 12 months after FSCT, LQ evaluation by the study group patients revealed improvement in all areas 6 and 12 months after the treatment (Figure 2). Six months after FSCT, significant improvement was reported in the emotional role functioning (15.2±0.39) and social role functioning (16.4±0.59), while 1 year after the treatment, significant improvement was also reported in physical functioning (13.2±0.38) and physical role functioning (13.8±0.5) (P<0.05). Patients also had higher self-confidence and improved spiritual realization. FSCT resulted in marked improvement of the general LQ perception by DMD patients.
In the control group, significant improvement was reported 12 months after the treatment in the social role functioning (16.9±0.59) versus baseline (12.0±0.49) and mental health (18.1±0.48) versus baseline (11.0±0.38) (P<0.05; Figure 3).
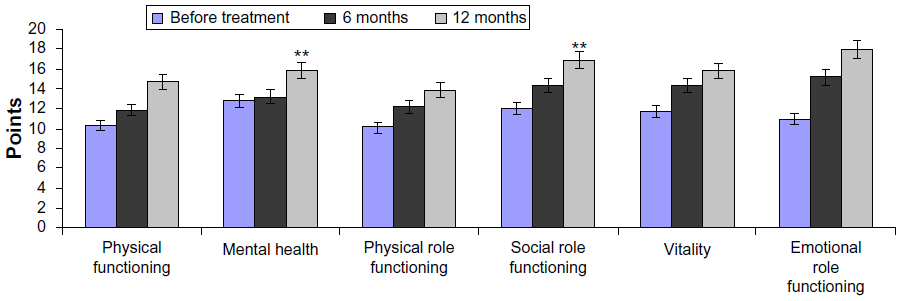 | Figure 3 Quality of life results on the SF-36 Health Status Survey: control group. |
It is known that DMD causes enzymatic abnormalities. Even though most enzymes are intercellular, plasma and serum tests show abnormalities in the cells and tissues.
Despite the absence of organ specificity, serum ALT and AST is of high diagnostic value, and elevation of these enzymes in DMD is suggestive, first of all, of muscle tissue necrosis.
The FSCT resulted in reliable ALT decrease in the study group in 6 months (0.94±0.10 U/L versus 1.58±0.19 U/L; P<0.05). AST also reduced 6 months after the treatment (0.61±0.06 U/L versus 0.84±0.07 U/L); however, this decrease was not reliable (P>0.05). One year after the FSCT, serum ALT and AST were 0.81±0.20 U/L and 0.50±0.07 U/L, respectively, which confirms a reliable (P<0.05) decrease in comparison with the baseline (Figure 4).
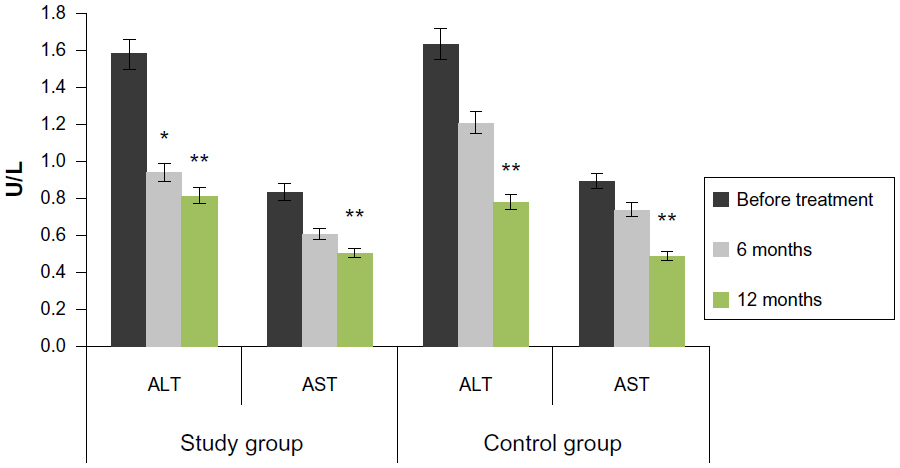 | Figure 4 Effect on serum ALT and AST levels. |
The situation in the control group was different, and significant ALT and AST reduction was reported 12 months after the treatment (0.78±0.15 U/L and 0.49±0.04 U/L, respectively) versus baseline (1.63±0.21 U/L and 0.89±0.03 U/L, respectively; P<0.05).
Dystrophin deficiency in muscle fibers leads to massive CPK penetration from the damaged muscle into the bloodstream. High CPK is suggestive of myolysis, but its clinical specificity is low because there is no 100% guarantee of exact diagnosis (muscle disease type).
We studied effect of FSC on CPK in DMD patients (Figure 5).
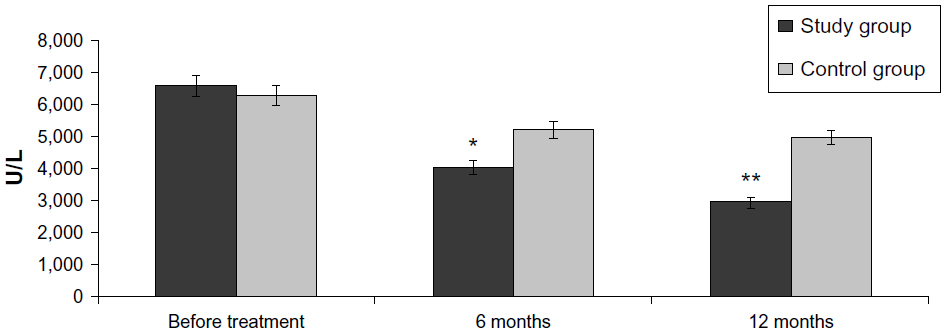 | Figure 5 Effect on CPK levels. |
A reliable CPK reduction in comparison with the baseline was reported in the study group 6 months after the treatment (6,583.4±58.21 U/L before FSCT versus 4,012.3±41.14 U/L at 6 months versus 2,918.0±29.37 U/L at 12 months; P<0.05). In the control group, CPK also reduced to 5,212.3±34.15 U/L 6 months after the treatment and 4,988.0±23.31 U/L in 12 months versus the baseline of 6,273.4±48.26 (P>0.05).
The LDH test helps identify tissue damage, and in DMD its elevation is found several years before onset. The average LDH before FSCT was 526.4±18.24 U/L in the study group and 533.2±17.15 U/L in the control group (Figure 6).
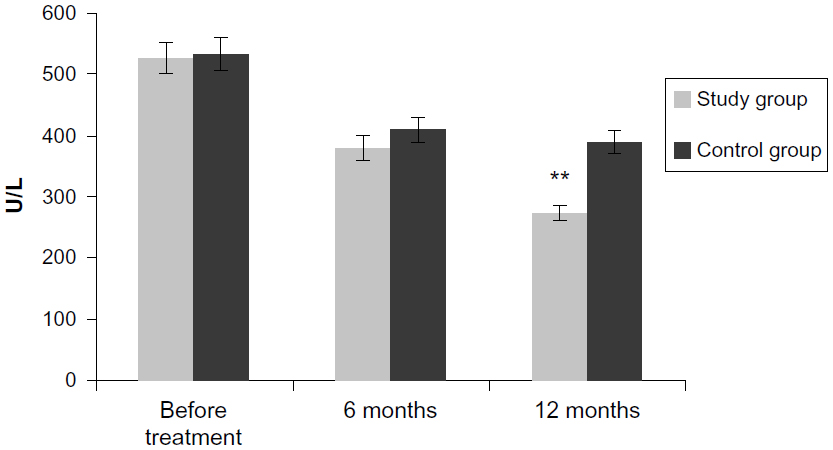 | Figure 6 Effect on serum LDH levels. |
LDH was reduced 6 months after the treatment to 379±21.14 U/L in the main group and to 409±19.12 U/L in the control group. In the study group, a significant decrease was obvious 12 months after FSCT (284±19.2 U/L; P<0.05). In the control group, LDH was 389±17.21 U/L 12 months after FSCT (P>0.05).
ECG revealed the following cardiovascular problems in 74.10% of DMD patients: rhythm irregularity (40.74%), sinus tachycardia (96.30%), auricular extrasystole (32.00%), isolated early ventricular extrasystole (5.00%), bundle block (79.00%), PQ interval shortening (60.10%), QT interval prolongation (57.80%), negative T-wave (25.10%), ST segment depression (7.40%), and left ventricular hypertrophy (25.90%). None of the patients had a past history of treatment with angiotensin-converting-enzyme inhibitors and beta-adrenoblockers. The patients’ morphofunctional data are presented in Table 1.
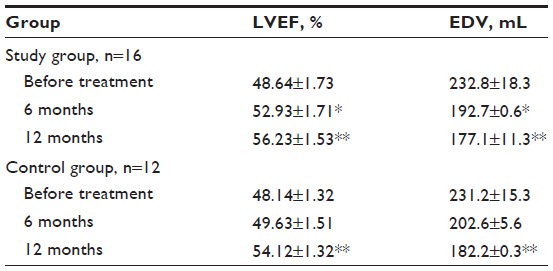 | Table 1 Morphofunctional changes in patients of both groups |
We studied morphofunctional heart parameters in 16 patients of the study group, for all of whom FSCT resulted in a reliable LVEF increase by 11.10% and 18.03% 6 and 12 months after the treatment, respectively (P<0.05). Treatment also resulted in end-diastolic volume (EDV) reduction by 15.6% and 25.8% 6 and 12 months later, respectively (P<0.05). In the control group, a significant LVEF increase by 12.42% and EDV reduction by 21.19% was reported 12 months after the treatment (P<0.05). The results are presented in Table 1.
In order to diagnose respiratory failure, 16 patients of the study group and 12 patients of the control group with different DMD stages underwent spirography, which showed that nine patients of the study group and six patients of the control group with stage II DMD had no respiratory problems, three patients of the study group and two patients of the control group with stage III DMD had marked respiratory failure, two patients of the study group and two patients of the control group with stage IVa DMD had very advanced respiratory failure, and two patients of the study group and two patients of the control group with IVb DMD stage used a ventilator at night. Thus, it was quite obvious that respiratory failure advanced with the stage.
After FSCT, the study group patients reported improved respiratory status as early as day 2 after the treatment. Six months after FSCT, forced vital capacity (FVC) increased by a mean of 4.2% and forced expiratory volume (FEV1) by 4.5% (P>0.05). One year after FSCT, we saw a reliable increase of FVC and FEV1 by 13.2% and 15.3%, respectively.
In the control group, a significant FEV1 increase, by 10.5% (P<0.05%) was reported 12 months after the treatment.
Discussion
In this study, we used FSCT to verify the potential beneficial effects of stem cells in DMD patients.
Currently, DMD is an untreatable lethal recessive disease. Beyond gene replacement therapy, cell-based therapy is offering new horizons for DMD management.42
FSCs are more specialized than embryonic stem cells; moreover, they show higher proliferative and expansion potential and lower immunogenicity than other stem cell types.43 FSCs are more rapidly, easily, and efficiently reprogrammed to pluripotency than neonatal and adult cells.44,45 In addition, they exert strong immunomodulatory effects,46 possess a stable phenotype, and demonstrate less senescence. These properties render them as useful candidates for cell transplantation and particularly for DMD, as verified in several preclinical studies.44 In addition, FSCs, unlike embryonic stem cells, have no potential to form in vivo tumors, still retaining their primitive properties and expansion capacity.44 At 4–5 days of embryonic development, at the blastocyst stage of its inner cell mass, embryonic stem cells can be isolated. Subsequently, after this division, FSCs can be isolated. Indeed, by day 7–10, gastrulation occurs and the embryonic cells have committed to a specific lineage: meso, ecto, endo. By 4 weeks, the neural tube has formed and by 5–7 weeks, the major organs are all formed. We used FSCs from 5–9 weeks.
The FSC transplantation we performed was safe and well-tolerated. No immediate or long-term side effects, as well as immunological reactions, were observed during the 1-year follow-up period.
How FSCs work in DMD is still to be clarified and is beyond the scope of this study. However, FSCs could exert their beneficial role through two mechanisms. These cells show an ability to 1) differentiate to form myogenic cells; and 2) offer a trophic effect. It has been demonstrated that in an experimental model reproducing human DMD obtained using a dystrophic mdx mouse, transplanted FSCs are able to differentiate at low levels into skeletal and myocardial muscle.47 This differentiation level was not considered to be curative, indicating that cell replacement theory probably is not the major mechanism involved by which FSC provide functional benefits.
FSCs show strong paracrine trophic effects on host tissues.34 Indeed, FSCs are able to secrete diffusible neurotrophic factors and several growth factors.
It is noteworthy to consider that the FSC-mediated positive mechanisms could be also related to the capacity of the transplanted cells to restore dystrophin gene expression.48,49 This mechanism of action could be confirmed by FSC-mediated CPK reduction as seen in our study.
Indeed, transplanted stem cells are able to systemically migrate within the host, where they could home to multiple muscle compartments, engraft, and differentiate into functional expressing dystrophin muscle cells, or they could fuse with myofibrils and restore dystrophin synthesis.7 In addition, engraftment and homing is increased in damaged tissue and FSCs show higher engraftment potential than adult stem cells.50
Migration and engraftment capacity could also be the cause of the improvement in cardiovascular and respiratory parameters seen in our study. It is known that respiratory failure in DMD patients is caused by restrictive changes in the lungs. Stem cells are able to migrate to the affected areas of the lungs, regenerate alveoli, and improve blood circulation in the lung tissue.31
Treatment of cardiomyopathy in DMD patients is based on the stem cells’ capacity to restore the cardiomyoblast population. It has been shown that mesenchymal stem cells are able to replace connective tissue cells with functional cardiomyocytes. Ejection fraction increase and higher contractile activity of myocardium was reported.8,10,11 In addition, we cannot exclude the possibility that FSCs’ ameliorative mechanisms are due to their capacity to restore, rebuild, and enhance the neuromuscular system. In an in vitro model, as well as in a murine model, it has been demonstrated that FSCs are able to recruit into the myogenic lineage and to form mature myotubes, contributing to muscle formation.51 Interestingly, it has been reported that human stem cells are able to integrate into normal and infarcted rat cardiac tissue and to differentiate into cardiomyocyte-like cells, improving ventricular function and capillary density.52
Thus, our research has shown that, irrespective of DMD stage, FSCT inhibits progression of the disease and markedly improves patient conditions.
In DMD, FSCT results in higher LQ, subsidence of neurologic symptoms, and prevention of cardiovascular and respiratory complications (heart and respiratory failure). However, optimal results can be achieved by combined treatment – FSCT, physical therapy, massage, and breathing exercises.
Conclusion
In DMD, FSCT results in inhibition of the disease progression and improvements in different areas, such as:
- Reliable improvement of functional capacity and LQ after 6 and 12 months of treatment compared with the control group.
- Reliable reduction of serum ALT, AST, CPK, and LDH 6 months after FSCT in the study group. In the control group, significant reduction of ALT and AST was reported 12 months after the treatment, while CPK and LDH reduction was insignificant both 6 and 12 months after the treatment.
- Significant morphofunctional improvements of cardiac status (LVEF increase and EDV reduction) 6 months after FSCT in the study group and 12 months after the treatment in the control group.
- Reliable improvement of forced lung capacity (FVC, FEV1) 1 year after the FSCT in the study group, while in the control group only FEV1 improvement was significant.
For inhibition of rapid progression of DMD, LQ improvement, and prevention of disease-related cardiovascular and respiratory complications, it is recommended to combine FSCT with physical therapy, massage, and breathing exercises.
Future larger randomized and placebo-controlled trials will be needed to further characterize long-term FSC improvements in DMD; as well, more research will be needed in order to fully understand the FSC-mediated mechanisms underlying the cardiovascular, respiratory, LQ, and neurological improvements obtained using FSCT in DMD. However, personalized stem cell therapy could be the most effective treatment for a specific DMD patient, opening a new era of DMD management in the future.
Disclosure
The authors, excluding Dario Siniscalco, are EmCell employees. No grants were obtained. The authors report no other conflicts of interest in this work.
References
Bushby K, Bourke J, Bullock R, Eagle M, Gibson M, Quinby J. The multidisciplinary management of Duchenne muscular dystrophy. Current Paediatrics. 2005;15(4):292–300. | |
Gorbunova VN, Savelyeva-Vasilyeva EA, Krasilnikov VV. Molecular neurology. Neuromuscular Diseases. 2000:8. | |
Schneider NA, et al. Myotonia: Doctor’s Manual M.:NMF “MBN”; 2005. | |
Hu XY, Ray PN, Worton RG. Mechanisms of tandem duplication in the Duchenne muscular dystrophy gene include both homologous and nonhomologous intrachromosomal recombination. EMBO J. 1991;10(9):2471–2477. | |
Nikolaeva IA, et al. Genetic neurological diseases. Yakut Medical Journal. 2006;2:52–54. | |
Baranov VS, Baranov AN. Modern age and perspectives of genetic therapy for Duchenne muscular dystrophy in the world and Russia. Genetics. 2009;8:1046–1053. | |
Illarioshkin SN, Ivanova-Smolenskaya IA, Markova ED. DNA-Diagnostics and Genetic Consulting in Neurology (Illarioshkin S.N.), 2002; 78–95. | |
Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy:10 years’ follow-up. Am Heart J. 2007;154(3):596–602. | |
Zuev AA, et al. Mexidol in Hereditary Neuromuscular Diseases. J Exper Biol Med. 2006;1:22–27. | |
Makino S, Fukuda K, Miyoshi S, et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest. 1999;103(5):697–705. | |
Rees W, Schüler S, Hummel M, Hetzer R. Heart transplantation in patients with muscular dystrophy associated with end-stage cardiomyopathy. J Heart Lung Transplant. 1993;12(5):804–807. | |
Finder JD, Birnkrant D, Carl J, et al; American Thoracic Society. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170(4):456–465. | |
Yevtushenko SK, et al. Sohan stimol and singlet oxygen in combined treatment for progressive muscular dystrophy and amyotrophy with concomitant cardiomyo- and pneumopathy in children and adolescents. Int J Neurol. 2008;4(20):132–141. | |
Schneider NA, et al. Role of selene in the diet of patients with myotonic dystrophy. NSU Journal. 2008;1:91–96. | |
Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(1):CD003725. | |
Goyenvalle A, Seto JT, Davies KE, Chamberlain J. Therapeutic approaches to muscular dystrophy. Hum Mol Genet. 2011;20(R1):R69–R78. | |
Meregalli M, Farini A, Colleoni F, Cassinelli L, Torrente Y. The role of stem cells in muscular dystrophies. Curr Gene Ther. 2012;12(3):192–205. | |
Konieczny P, Swiderski K, Chamberlain JS. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve. 2013;47(5):649–663. | |
Kozlova SI, Demikova NS. Genetic Syndromes and Genetic Consulting (Kozlova S.I.), 2007. | |
Lobzin VS, Saykova LA. Neuromuscular Diseases. Hippocrates. 1998:75–208. | |
Mikhailov VM, Zhestianikov VD, Savel’eva GE. [Myocardium of mdx mice contains factor(s) that damage DNA structure and retard DNA reparation after gamma-irradiation (experiences in modeling system)]. Tsitologiia. 2003;45(4):418–421. Russian. | |
Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 1983;80(15):4856–4860. | |
Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456(7221):502–506. | |
Meregalli M, Farini A, Parolini D, Maciotta S, Torrente Y. Stem cell therapies to treat muscular dystrophy: progress to date. BioDrugs. 2010;24(4):237–247. | |
Siniscalco D, Giordano A, Galderisi U. Novel insights in basic and applied stem cell therapy. J Cell Physiol. 2012;227(5):2283–2286. | |
Siniscalco D, Pandolfi A, Galderisi U. State-of-the-art on basic and applied stem cell therapy; Stem Cell Research Italy-International Society for Cellular Therapy Europe, Joint Meeting, Montesilvano (PE)-Italy, June 10–12, 2011. Stem Cells Dev. 2012;21(5):668–669. | |
Wilschut KJ, Ling VB, Bernstein HS. Concise review: stem cell therapy for muscular dystrophies. Stem Cells Transl Med. 2012;1(11):833–842. | |
Nishiyama T, Takeda S. [Induced pluripotent stem (iPS) cell-based cell therapy for muscular dystrophy: current progress and future prospects]. Brain Nerve. 2012;64(1):39–46. Japanese. | |
Meregalli M, Farini A, Belicchi M, et al. Perspectives of stem cell therapy in Duchenne muscular dystrophy. FEBS J. 2013;280(17):4251–4262. | |
Bushby K, Finkel R, Birnkrant DJ, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. | |
Kazakov VM. Clinical molecular genetic classification of muscular dystrophies (scientific summary with comments). J Neurol. 2001;3:7–12. | |
Diao Y, Guo X, Li Y, et al. Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell. 2012;11(2):231–241. | |
Montarras D, Morgan J, Collins C, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309(5743):2064–2067. | |
Siniscalco D, Bradstreet JJ, Antonucci N. Cell therapies for autism spectrum disorders. In: Autism Spectrum Disorders: New Research. Hauppauge, NY: Nova Science Publishers; 2012:129–142. | |
Musina RA, Egorov EE, Beliavskii AV. [Stem cells: properties and perspectives of therapeutic use]. Mol Biol (Mosk). 2004;38(4):563–577. Russian. | |
Petrenko AY, Grischenko VI. Stem Cell Transplantation – Therapy of XXI Century. Stem Cell Properties and Description. Microbiology Issues. 2001;2:3–12. | |
Darabi R, Gehlbach K, Bachoo RM, et al. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med. 2008;14(2):134–143. | |
Siniscalco D, Bradstreet JJ, Sych N, Antonucci N. Perspectives on the Use of Stem Cells for Autism Treatment. Stem Cells Int. 2013;2013:262438. | |
Siniscalco D. Suspended Life – Stem Cells: Are Treatments Possible? J Regen Med. 2013;2:1. | |
Spirometry Standardization. ATS/ERS Target Group: Function Testing Standardization. Eur Respire. 2005;26:319–338. | |
Bazarnova MP. A guide to practical training in clinical laboratory. 1991. | |
Negroni E, Vallese D, Vilquin JT, Butler-Browne G, Mouly V, Trollet C. Current advances in cell therapy strategies for muscular dystrophies. Expert Opin Biol Ther. 2011;11(2):157–176. | |
O’Donoghue K, Fisk NM. Fetal stem cells. Best Pract Res Clin Obstet Gynaecol. 2004;18(6):853–875. | |
Abdulrazzak H, Moschidou D, Jones G, Guillot PV. Biological characteristics of stem cells from foetal, cord blood and extraembryonic tissues. J R Soc Interface. 2010;7 Suppl 6:S689–S706. | |
Galende E, Karakikes I, Edelmann L, et al. Amniotic fluid cells are more efficiently reprogrammed to pluripotency than adult cells. Cell Reprogram. 2010;12(2):117–125. | |
Chen PM, Yen ML, Liu KJ, Sytwu HK, Yen BL. Immunomodulatory properties of human adult and fetal multipotent mesenchymal stem cells. J Biomed Sci. 2011;18:49. | |
Chan J, Waddington SN, O’Donoghue K, et al. Widespread distribution and muscle differentiation of human fetal mesenchymal stem cells after intrauterine transplantation in dystrophic mdx mouse. Stem Cells. 2007;25(4):875–884. | |
Gussoni E, Soneoka Y, Strickland CD, et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401(6751):390–394. | |
Torrente Y, Tremblay JP, Pisati F, et al. Intraarterial injection of muscle-derived CD34(+)Sca-1(+) stem cells restores dystrophin in mdx mice. J Cell Biol. 2001;152(2):335–348. | |
Taylor PA, McElmurry RT, Lees CJ, Harrison DE, Blazar BR. Allogenic fetal liver cells have a distinct competitive engraftment advantage over adult bone marrow cells when infused into fetal as compared with adult severe combined immunodeficient recipients. Blood. 2002;99(5):1870–1872. | |
Chan J, O’Donoghue K, Gavina M, et al. Galectin-1 induces skeletal muscle differentiation in human fetal mesenchymal stem cells and increases muscle regeneration. Stem Cells. 2006;24(8):1879–1891. | |
Zhao P, Ise H, Hongo M, Ota M, Konishi I, Nikaido T. Human amniotic mesenchymal cells have some characteristics of cardiomyocytes. Transplantation. 2005;79(5):528–535. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.